Back to Journals » Orthopedic Research and Reviews » Volume 11
Hereditary Multiple Exostoses: Current Insights
Authors D'Arienzo A, Andreani L, Sacchetti F, Colangeli S ![]() , Capanna R
, Capanna R
Received 1 July 2019
Accepted for publication 11 October 2019
Published 13 December 2019 Volume 2019:11 Pages 199—211
DOI https://doi.org/10.2147/ORR.S183979
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Clark Hung
Antonio D’Arienzo, Lorenzo Andreani, Federico Sacchetti, Simone Colangeli, Rodolfo Capanna
Department of Translational Research on New Surgical and Medical Technologies, University of Pisa, Pisa, Italy
Correspondence: Federico Sacchetti
Department of Orthopedics and Trauma Surgery, University of Pisa, Via Paradisa 2, Pisa 56124, Italy
Email [email protected]
Abstract: Hereditary multiple exostoses (HME), also called hereditary multiple osteochondromas, is a rare genetic disorder characterized by multiple osteochondromas that grow near the growth plates of bones such as the ribs, pelvis, vertebrae and especially long bones. The disease presents with various clinical manifestations including chronic pain syndromes, restricted range of motion, limb deformity, short stature, scoliosis and neurovascular alteration. Malignant transformation of exostosis is rarely seen. The disease has no medical treatment and surgery is only recommended in symptomatic exostoses or in cases where a malignant transformation is suspected. HME is mainly caused by mutations and functional loss of the EXT1 and EXT2 genes which encode glycosyltransferases, an enzyme family involved in heparan sulfate (HS) synthesis. However, the peculiar molecular mechanism that leads to the structural changes of the cartilage and to osteochondroma formation is still being studied. Basic science studies have recently shown new insights about altering the molecular and cellular mechanism caused by HS deficiency. Pediatricians, geneticists and orthopedic surgeons play an important role in the study and treatment of this severe pathology. Despite the recent significant advances, we still need novel insights to better specify the role of HS in signal transduction. The purpose of this review was to analyze the most relevant aspects of HME from the literature review, give readers an important tool to understand its clinical features and metabolic-pathogenetic mechanism, and to identify an effective treatment method. We focused on the aspects of the disease related to clinical management and surgical treatment in order to give up-to-date information that could be useful for following best clinical practice.
Keywords: EXT1, EXT2, heparan sulfate synthesis, hereditary multiple exostoses, medical and surgical treatment, osteochondromas
Introduction
Hereditary multiple exostoses (HME) is a rare genetic disorder where several benign cartilaginous tumors arise from the perichondrium and flank the cartilage growth. It has a prevalence of 1:50,000 in Western countries.1–3 This autosomal dominant disease has also been called hereditary multiple osteochondromas, hereditary deforming dyschondroplasia, diaphyseal aclasis and multiple cartilaginous exostoses. It exhibits almost complete penetrance particularly in males.1,2,4–8 In more than 50% of patients, the disease can be passed down directly from an affected parent, usually the father. Normally, an unaffected male does not transmit the disease, however, an unaffected female may have a latent or suppressed form of the disease and may transmit it.9 The disease occurs in only about 5% to 10% of the cases when compared to solitary exostosis, is more frequently seen in males (1.5:1) and is usually diagnosed before the patients reach the age of 10.2,4 Osteochondromas may arise from every segment excluding facial bones. The disease may involve any bone which grows from endochondral ossification. Although exostosis origins more frequently in the scapulae, ribs, pelvis, and vertebrae, the most affected regions are the metadiaphyseal segments of long bones. No involvement of the skull, carpal and tarsal bones have been reported.4 Given the high variety of location, size and number, exostoses can cause several clinical problems.1,10 HMEs usually impact the quality of life and physical activity levels since childhood, in addition to mental health, especially in female patients.11–13 Resection, limb shortening and surgical deformity correction are the only available treatments and are advised when the cases are symptomatic or a malignant transformation is suspected.14
Epidemiology
The prevalence of HME in Western population has been assessed between 0.4 to 1 out of 50,000.2,15 However, in the Chamorro people, a small population in Guam, the prevalence of HME reaches as high as 50 out of 50,000. The incidence of this pathology in the Western countries was reported as 1.5% per year.3 However, this information could be erroneous since these data were taken from hospital reports and health databases of the affected patients and relatives’ families, with a focus on people who reported the symptoms and were classified as affected. Since genetic screenings have been employed to our day, there may be several asymptomatic or paucisymptomatic patients who could be affected by HME, but not detected by hospitals or health databases. Moreover, 10% of the HME patients have not reported any family history of the pathology.1 Schmale2 found a penetrance of 96%, but a more accurate report by Wicklund1 reported the HME penetrance as 100%. Although some reports16 have detected a male predominance in HME, these findings were proven wrong by other authors.1,15
Pathogenesis
The main symptom of HME is the formation of exostosis. Progenitor cells of the chondrocytes in the growth plate that cause osteochondromas have recently been detected in the inner layer of the perichondrium.17 Genetic, molecular biology and clinical investigations are fundamental to understand how osteochondromas arise in patients with HME. Gene sequencing in patients affected by HME led to the discovery of the EXT1 gene, located on Chromosome 8 and at a later stage to the discovery of the EXT2 gene, located on Chromosome 11.18 EXT1 and EXT2 mutations have been detected respectively in 28 to 65% and in 21 to 61% of the affected patients.19–21 EXT1 genes appear to be more susceptible to mutations, while EXT2 can support a higher mutation burden before leading to the development of the disease.22 Patients who have mutations in the EXT1 gene are also more prone to having higher number of exostoses, more cases of limb malalignment, a shorter stature and more pelvic involvement.20 Surprisingly, in 5 to 34% of the HME patients, mutations in the EXT1 or EXT2 genes were not detected.19–21,23 An explanation might be found in the mosaicism of the EXT genes. As a matter of fact, the mutation can lead to insufficient activity of the EXT1 and EXT2 genes in some patients.24 Genetic transmission of HME follows a dominant autosomal pattern, loss of heterozygosis (LOH), haploinsufficiency and other models including mutations in the EXT regulatory genes, and thus post-transcriptional regulation pathways25 have been proposed. In a mouse model, Jones26 demonstrated a LOH in vivo with exostosis development, with the findings of several cells with homozygosis in EXT-allele in the exostosis tissue.27 Reijinders28 proved the hypothesis of LOH development in human osteochondromas, rejecting the haploinsufficiency model. In their study, a second hit mutation was found in 63% of the analyzed osteochondromas. Other studies that analyzed exostosis specimens did not find a second hit mutation in the studied cells. Furthermore, some population studies demonstrated that EXT1 added to EXT2 heterozygous mutations could result in exostosis formation in the affected patients.25,29 In an effort to explain these findings, some authors hypothesized that a possible presence of not yet discovered epigenetic factors like post-translational EXT-products/inhibitors, heparan sulfate (HS) polymerase inactivation agents or miRNAs that could inhibit EXT-translation.30 However, as Matsumuto suggested, the most likely hypothesis is the heterogeneity in the osteochondroma cells, which are usually a mixture of mutated and wild type cells that remain single. It would be reasonable to think that osteochondromas develop by the mutation of some cells only.28,31 The molecular pathway that leads to HME and exostosis formation is strictly linked to the HS molecule. Several studies have asserted over the years that EXT-mutations lead to an insufficient elongation of the HS chains.23,30,32,33 Products of the EXT1 and EXT2 genes form a Golgi reticulum-based complex that is essential for the production and activity of glycosyltransferase enzyme which is finally responsible for chain polymerization of HS. This explains why isolated mutations of EXT1 or EXT2 are sufficient to cause the pathology; in fact, the Golgi complex does not work appropriately without its components from EXT1 or EXT2.32–34 The HS chains have several functions in the extracellular matrix. It is believed that the absence of HS chains or the presence of shorter ones could produce serious variation in chondrocyte differentiation or proliferation pathways. Extracellular HS chains could be involved in the Indian hedgehog (IHH) diffusion in the growth plate.23 IHH is an important factor which regulates the growth and differentiation of chondrocytes.35 HS chains also influence the distribution, range of action, stability and action on target of several other factors such as fibroblastic growth factors (FGFs), bone morphogenetic proteins (BMPs), Wnt signaling pathway (Wnts) and parathyroid hormone-related proteins (PTHrPs).33,36–38 A severe interference to these signaling pathways leads to the development of osteochondromas and also to other manifestations of HME like a shorter stature.39 Osteochondromas derive from an imbalance in the extracellular matrix of factors and signals that enhances chondrocyte proliferation or differentiation. Impairment of the HS chain has also been observed in autistic mice with a normal brain anatomy, and a function in glutamatergic synapses stability has been suggested for HS chains (Figure 1).40
Pathology
Macroscopically, exostosis is covered by a cartilage cap that, until the end of the growth process, entirely covers the lesion. In the first years of life, instead, almost all of the lesions are constituted of a cartilage fused at the base with initial ossification tissue in continuity with the normal bone of the metaphysis. In a child, the cartilage cap is covered with a hyaline cartilage with blue light reflexes, similar to the normal child cartilage, with a variable thickness up to 2–3 cm. Along with the ageing of patients, the thickness of the cartilage cap tends to decrease and disappear in many sites, and the exostosis cap usually appears to be ossified except for some lenticular cartilage residuals with a thickness of few millimeters. Irregular networks of cancellous bone and yellow and red marrows are present under the cap. Insulae of cartilage tissue that ossify with age may be seen in the exostosis base. All osteochondromas are covered by the periosteum. Microscopically, during the growth phase of the exostosis, the cartilage cap has the same structure as the growth plate. Particularly, we can discern the proliferative, columned, hypertrophic and calcified cartilage layers from the cartilage cap surface followed by bone trabeculae generated by the endochondral ossification process. The difference with the normal growth plate is that the trabeculae in that case are irregular in shape and directions and could contain insulae of cartilage tissue. In large sized exostosis, we can also observe islands of necrotic bone trabeculae with precipitations of calcite that derive from local defects in the blood supply.3,5,41–43
Clinical Presentation
Hereditary multiple osteochondromas patients are generally diagnosed before the age of 12. Usually, these patients come to the physician’s attention following the appearance of a palpable mass near the joints. Knees, shoulders, ankles and wrists are the most involved joints during diagnosis. An HME patient is generally burdened with an average of six exostoses.41 In recent years, a clinical classification system for HME has been designed and published to assess the burden and life limitations that follow the disease.44 This system focuses on the deformities and functional limitations associated with HME and classifies the patients under three groups according to the number of involved segments. The effects of HME depend largely on the number and shape of the exostoses, which demonstrate a great variability. Usually, when an osteochondroma has an early onset, the lesion tends to be bigger and causes more problems. This happens because the lesion tends to grow in line with the growth plate and adjusts itself to the evolution of the diaphysis. Sometimes, exostoses could produce disturbances in the growing diaphysis giving rise to limb length discrepancies and characteristic deformations that usually affect the distal femur, radius, ankle and ulna. The ribs and the proximal tibia where exostoses are easily visible and palpable, are the segments which are more prone to esthetic issues.45 The hip joint of the HME patients could be a “coxa valga” due to osteochondromas of the minor trochanter that alter the proximal femur shape by determining a verticalization of the neck-shaft angle, and more rarely the acetabulum exostoses could lead to acetabular dysplasia, femoroacetabular impingement and finally juvenile arthritis that may require arthroplasty.46,47 The knee joint is generally involved, with genu valgum as the most frequently seen deformity that affects approximately one third of HME patients. Genu valgum usually derives from the angulation of the proximal tibia. In some cases, distal femur could also be angulated to form an oblique joint line.48 These findings are associated with early knee arthritis and patellar dislocations or subluxations. The ankle joint is also burdened with valgus deformity. Generally, a relative shortening of the distal fibula compared to the distal tibia leads to an oblique growth plate and joint line with medial dislocation of the talus.49 In the past, spinal exostoses were overlooked, however, it has been assessed that up to 68% of the patients could have spinal osteochondromas. However, data that associate scoliosis to HME are scarce.50 Spinal exostoses could lead to severe and acute neurological syndromes as we have reported in the Prognosis and Diagnosis sections. HME patients, in particular those who have been diagnosed with EXT1 mutations, have a shorter stature compared to the general population. This finding does not seem to be related to limb length discrepancies and bone deformity, but might be a general characteristic of HME. It is well known that HS deficiency could interfere with physiological growth in several ways.51 In case of growth of osteochondromas in adult patients, malignant transformation should be suspected.25,52 Many patients are also diagnosed with chronic inflammation syndromes, limited range of motion or paresthesia and nerve entrapment syndromes. That comes from the final shape of exostoses since it can interfere with nearby structures as nerves, vessels and tendons. Many reports showed that HME patients suffer from poor quality of life. Both children and adult patients reported difficulties with socializing at work and in school. Chronic pain syndromes are common and most of the patients have to abandon sports and leisure activities.13,53,54
Diagnosis
Most of the patients are asymptomatic at birth, thus the diagnosis could be pursued only with genetic screenings of the newborns of affected families. The median age of diagnosis is 3 years.2 Fifty percent of the patients have a visible tumor and get diagnosed by the age of 5 and 80% by the age of 10.3 In general, all patients are diagnosed by the age of 10–12. Conventional roentgenograms have a good accuracy in detecting the shape and presence of osteochondromas in most of the regions. X-rays are also useful to detect and study the HME-associated deformities. Kok55 reported that in radiological studies, 33% of the patients with HME were also diagnosed with ulnar shortening and deformity focusing on bayonet hand or pseudo-Madelung deformities; and in the lower limbs it is generally easier to diagnose a typical distal femur deformity as the “Erlenmeyer flask” deformity. Axial deviations, shortenings and limb length discrepancies are also assessed by X-rays. Axial imaging with computed tomography (CT) scan or magnetic resonance imaging (MRI) is generally mandatory to study anatomical challenging regions such as the pelvis and the thorax. The spine should also be examined in HME patients (see the Prognosis section) with an MRI of the entire spine and the screening should be performed every two years during the follow-up period. An MRI can detect bursitis derived from the mechanical stress around the lesions. Entrapment of the nerves and vessels, their positions, and their anatomical relationships with exostoses are easily shown on the MRI. The nerves that suffer most from entrapment syndromes are the radial and peroneal nerves.55 Performing an MRI is mandatory for planning the surgical removal of exostosis. Magnetic resonance imaging can highlight the shape of the exostosis and its continuity from the cortical bone; but before all, it allows a direct and precise study of the cartilage cap and the soft tissues surrounding the lesions. Cartilage cap can also be analyzed using CT images but an MRI offers a better resolution. Measurement of the cartilage cup is a reliable method to predict malignant transformation of osteochondromas.56 The measurement should be performed from the osseous interface of the exostosis stalk to the edge of the cartilage cap at its thickest portion. During childhood and adolescence, the cap thickness can be more than 3 cm. However, in adults, HME should be suspected when the thickness is more than 1–2 cm. Various authors have set the limit at 1 or 2 cm. Finally, Kok55 stated that a threshold value of 2 cm is enough to diagnose malignancy with 95% specificity and sensitivity. Other radiological signs of malignant transformation are an irregular surface of the cap, a lytic foci inside the exostosis or near the bone, and the presence of soft tissue mass surrounding the exostosis, especially if they contain scattered or irregular calcifications.56 F-18 fluorodeoxyglucose (18F-FDG) positron emission tomography (PET) may have a role in detecting malignancies, especially to evaluate the standardized uptake value (SUV) of glucose in a semi-quantitative manner. However, during childhood and adolescence, an abnormal uptake of 18F-FDG can be detected in absence of malignant lesions as the exostosis is growing. A bone scan is generally considered of no value in the study of HME.55,56 Recently, Sonne-Holm57 has suggested a full body MRI every two years throughout the follow-up period. Heparan sulfate and chondroitin sulfate (CS) could be the molecular target of the diagnostic tests in the future. Faruqi58 used the HS/CS ratio; this could be used as a molecular predictor of HME in children since these patients have a half HS/CS ratio compared to the general population. Differential diagnosis is generally easy. Enchondromas have very different characteristics when compared with exostoses. In very rare syndromes, multiple exostoses could represent an epiphenomenon. To the best of our knowledge, we can cite Langer-Giedion syndrome59 and metachondromatosis. In metachondromatosis, the patients are usually affected by enchondromas and exostoses that regress spontaneously with age. The exostosis histology in metachondromatosis syndrome could be similar to the same lesions in HME, but in certain cases the cartilage cap and in general the lesion differ from the typical osteochondromas in HME patients.60
Prognosis
The burden of HME-related lesions varies according to the patients’ age, gender and specificity. A mean of six exostoses per patient has been reported in previous studies,3,41 with an average of 2–3.5 exostoses removed per patient.1,2 The shape, dimensions and the position of these lesions affect the prognosis in HME patients; limb deformities, involvement of the vessels and nerves, and surgery-related complications are the main factors which determine prognosis and quality of life. Although it is relatively rare, spinal involvement has to be screened since it determines serious damages also in acute settings.61 Malignant transformations and associations to other tumors are relatively rare.52,62 Wiklund1 showed that HME patients have a shorter stature compared to the general population. Thirty-seven percent of males and 44% of females fall under the fifth percentile. These characteristics seem to be intrinsic to the pathology and are not determined by limb length discrepancies and deformities shown by statistical tests. EXT1 and EXT2 mutations also affect patients’ growth. Patients with EXT1 mutations have been more frequently associated with exostoses, exostosis-related complications and a shorter stature.63 Ten percent of the patients reported dimensional changes in exostosis during pregnancy and 63% of the pregnant women who had exostosis-related complications during delivery required a cesarean section.1
Deformity
In the upper limbs, the distal ulna is usually involved with ulnar shortening that can lead to proximal dislocation of the radial head, as reported in 22 to 25% of the patients.3,16 Radial or ulnar deviations were considered as “moderate” or “severe” in 60% of the cases, and as “normal” in the remaining 40%. As for the lower limbs, involvement of the proximal femur led to coxa valga deformity in 25% of the cases.3,16 Cumulative deformities of the lower limbs caused limb length discrepancies in 50% of the patients, 23% of which required correction surgeries. Valgus knee was reported in 33% of the cases.3 Tibia “valga” with obliquity of the distal tibial epiphysis was described in 50 to 54%3,16 of the cases, with 56% of fibular shortening linked to growth defects of the distal fibula.16 Schmale2 reported that 9% of the patients could have severe deformities that impaired their walking ability. Schmale2 also reported lower rates of deformity with 39% of forearm deformities, 8% of varus or valgus knee and 2% of ankle deformity. The hand is rarely involved, but in these cases, the ulnar side is more often affected and particularly the metacarpophalangeal joints. A mean of 12 tumors per hand was reported by Woodside64 with an average of two ray shortenings and five ray angulations.
Neurovascular Complications
Involvement of the peripheral nerves and vessels are major indications for surgery when there is not any malignant transformation. Peripheral nerve compressions and neuropathy are common and observed in 22 to 23% of the cases.1,3 Wiklund1 also reported 11.5% of vessel-related complications. Nerves compression has a subtle clinical presentation with impairing symptoms in the long term. Involvement of the vessels can be more acute with micro-traumatisms that can lead to acute vessels damages.
Pain-Related Complications
Goud53 reported chronic pain in 60% of the children and 80% of the adults (283 patients). Thirty percent of the children and 50% of the adults refrained from playing their preferred sport. Approximately 22% of the adults had to change workplace or get a tailored workplace due to pathology-related complications. Fifty percent of the children had problems with socializing. Darilek65 reported about chronic pain that disturbed 80% of the patients. Thus, the social burden of HME is very high and the quality of life could be strongly impaired by the disease.
Spinal Involvement
Over the past years, several papers highlighted a very rare involvement of the spine in HME patients. In 1995, Wiklund1 showed a very low rate of patients who had spinal cord compressions (0.6%). More recently, however, Roach66 reported that of the patients who were performed axial screening of the spine, 68% had a spinal exostosis involvement. Twenty-seven percent of those patients had lesions that impinged their spinal cord; a much higher percentage than expected. Previous analyses were based on X-ray screening and since this screening method hardly detects spinal exostoses, data about the involvement of the spine were missing. Other authors reported myelopathy in 15% of the patients who had spinal exostosis.61 Ashraf67 showed similar results to those of Roach and added that 5% of the HME patients in his study had compressive intracanal osteochondromas. Since, several patients with minor trauma showed acute neurological syndromes linked to exostosis, the authors proposed a systematic spinal canal screening using axial images (MRI or CT). The authors also reported extracanal lesions that determine pain, dysphagia, sleep-apnea and intracanal lesions that determine paresthesia, myelopathy, general weakness and gait disturbances.
Malignant Transformation
Malignant degeneration has been reported as a rare entity.25,52 Usually a differentiated chondrosarcoma onset on the cartilage cap is diagnosed; however, very rarely, dedifferentiated chondrosarcomas or osteosarcomas from the bone base could arise in HME patients. Clinical suspicion of malignant degeneration comes out when the cartilage cap is more than 1.5–2 cm or when a dimensional growth of the exostosis is reported in adults.25,52 Malignant degeneration is reported to involve 2 to 4% of the patients affected by HME, with a risk of 0.1% per year in the 30–50 year-old population.14 Other reports showed that 0.9% of the patients who were diagnosed with chondrosarcoma and with a lifetime risk of 1–6% or 1–5% develop a malignant degeneration.2,3 Sciubba61 reported a much higher risk of 12% in a patient followed for spinal exostosis and Kivioja found a chondrosarcoma degeneration risk of 9% in a family affected by HME.68 Other authors in the past reported a 25% risk of malignant degeneration, but these data are considered unreliable.
Treatment
To the best of our knowledge, nowadays a medical treatment for HME does not exist. However, there are several papers suggesting that some particular molecules involved in various signaling pathways could represent viable therapeutic options for HME patients in the future. In particular, Huegel69 showed in a mice model that a higher level of heparanase inhibitor (SST0001) in chondrocytes could lead to an inhibition of unregulated chondrogenesis. The presence of heparanase inhibitors could interfere with HS, BMPs and chondrogenesis pathways. This finding suggests that there are more target areas for therapeutic agents other than EXT1, EXT2 and EXT-like genes and products.70,71 Inubushi72 showed that palovarotene (PVO), a selective retinoic acid receptor gamma agonist, investigated as a potential target treatment in fibrodysplasia ossificans progressiva (FOP),73 could be useful in HME. This drug reduced the exostosis formation in mice with EXT1-EXT2 deletion, concluding that palovarotene could be a potential therapeutic agent for HME. This result also showed and remarked the overlaps in pathogenesis between HME and FOP. At the moment, in this scenario it is clear that treatment in HME patients is symptomatic and surgical. Exostosis removal is advised in two cases; when there is a strong suspicion of malignant transformation or when there are symptomatic exostoses. A third case is represented by the intracanal exostoses of the spine which have to be removed due to possible acute neurological syndromes that can derive from microtraumas and represent a severe danger for HME patients. Excision of an osteochondroma is usually an easy process, but in some particular locations (like the proximal femur), it could represent a very challenging operation, especially when there is a complex geometry of the lesion that encompasses nerves or vessel (Figures 2–4, Case #1). Surgical excision was carried out by removing the exostosis at the bone base, with consequential removal of the cartilage cap. Usually, when the cartilage cap is completely removed, there are no recurrences. However, complete removal in some regions could lead to the necessity of reconstruction techniques like bone grafting and internal fixations. Recurrences may be seen when wide excision is not possible. Surgery is also needed in cases with moderate to severe limb length discrepancies, severe limb malalignment or segment shortenings.74 In 2007, Jager75 reported no significant problems after exostosis removal in a cohort of 52 HME patients. However, Wirganowicz76 reported a complication rate of 12.5% after exostosis removal in a cohort of 285 HME patients, with neurapraxia as the most common complication. Given the fact that sporadic and HME-related osteochondromas could spontaneously regress,5,77,78 an aggressive surgical approach is not recommended. A spontaneous regression of the exostosis has been reported in one third of the HME patients. The best management in HME patients should be an accurate follow-up and a wait-and-see approach. Without considering spinal osteochondromas, osteochondromas from other anatomical regions should be treated only in symptomatic patients (Figures 5–10, Case #2). In case of a malignant transformation, an immediate excision is advised. In those cases, usually a wide excision is curative given the low metastatic potential of differentiated chondrosarcoma onset on osteochondroma (more common tumor arising from exostosis). Adjuvant and neoadjuvant therapies are not commonly used in these cases. In case of a dedifferentiated chondrosarcoma, excision is recommended, but radiotherapy and chemotherapy have a poor effect on this kind of malignancy.
Current Trends
In recent years, several techniques in the field of Pediatric Orthopedics have been applied to children affected by HME in order to improve their functional activity levels during adulthood. In particular, we can report knee and radius stapling correction techniques that have been proposed to correct the genu valgus and the wrist deformities associated to HME during the growth process. The first reports have promising results.79,80 Fibular lengthening has also been evaluated in the prevention and initial correction of ankle valgus deformities and talus instability with optimal results.81 Valgus deformities of the proximal femur could be corrected by several approaches with variable results.82 Apart from the removal of symptomatic osteochondromas and correction of established deformities, preventive and less invasive surgeries for the growing children could be proposed in the future more often, given the good results of the first reports. In brief, we prefer not to proceed with exostosis removal, except in suspected cases of malignant transformation. When a “symptomatic exostosis” is diagnosed in cases with neurovascular structure entrapments or damages, we recommend a surgical removal of the lesion. When functional or esthetical problems are raised, we debate with patients and parents to choose the best option. When the prevention or correction of the HME-related deformities are to be addressed in the growing child, we prefer to refer the patient to an orthopedic pediatric center.
Conclusions
Hereditary multiple osteochondromas is a genetic disorder where the orthopedist, the geneticist and the pediatrician should all study and treat the disease. The complexity and unresolved issues related to HME are a challenge for researchers and clinicians even though a significant progress has been made over the last years.83,84 The knowledge about mutational spectrum affecting the EXT genes, especially EXT1, is progressing, showing that these genes are vulnerable and each variant has to be evaluated to arrive at a real evidence of pathogenic relevance.22 The complex genetic mechanism of the interactions between cells and molecules in the growth plate is still being investigated. The role of HS deficiency is well recognized in HME genesis, but we believe that, in the next few years, new discoveries on HS pathways may lead to a medical treatment of this pathology. Furthermore, results from recent mice studies are promising.35,85 An accurate follow-up by experts in sarcoma centers and appropriate surgical indications are nowadays mandatory to guarantee the best possible prognosis for HME patients who have generally a low quality of life and several disabilities due to their pathology.
 |
Figure 1 Brief description of the HME pathogenesis. |
 |
Figure 2 T1-weighted axial MRI showing the osteochondroma at the proximal femur with sciatic nerve entrapment (Case #1). |
 |
Figure 3 Intraoperative image of the osteochondroma (Case #1). |
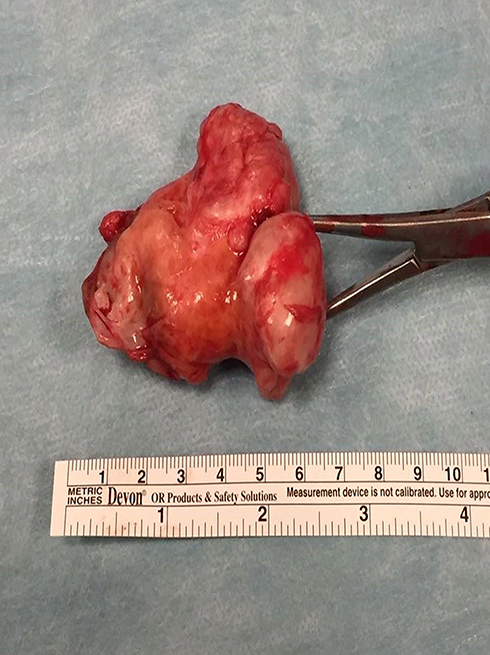 |
Figure 4 Resected specimen (Case #1). |
 |
Figure 5 T1-weighted axial MRI showing the exostosis in the proximal humerus, complicated with a massive brachial artery pseudoaneurysm following microtrauma (Case #2). |
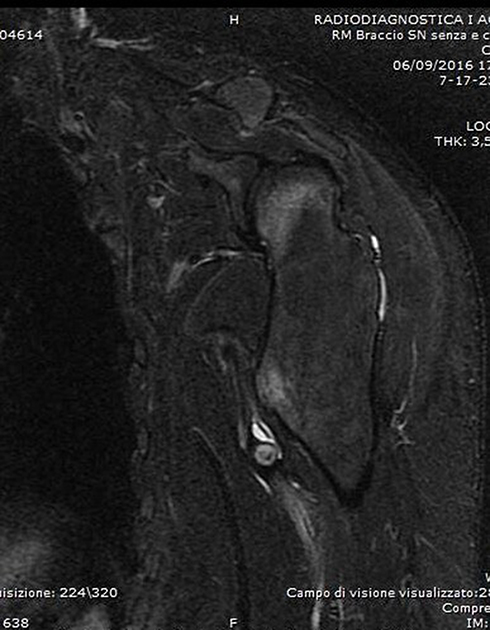 |
Figure 6 MRI with a STIR sequence showing the pseudoaneurysm after drainage of the hematoma (Case #2). |
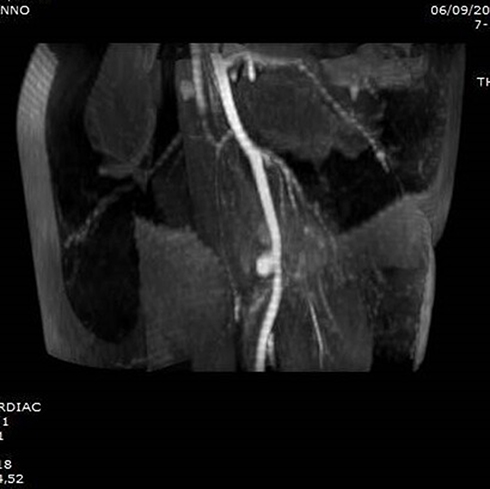 |
Figure 7 A TOF MRI sequence showing the brachial artery pseudoaneurysm (Case #2). |
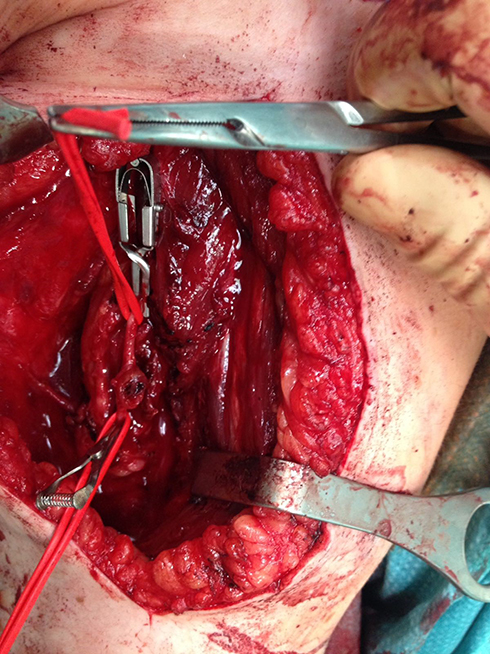 |
Figure 8 Intraoperative photograph showing the brachial artery pseudoaneurysm (Case #2). |
 |
Figure 9 Clinical case 2. Pre-operative radiograph. In the yellow circle, the exostosis that had to be removed. |
 |
Figure 10 Clinical case 2. Post-operative radiograph. |
Disclosure
The authors report no conflicts of interest in this work.
References
1. Wicklund CL, Pauli RM, Johnson DR, Hecht JT. Natural history of hereditary multiple exostoses. Am J Med Genet.1995;55:43–46. doi:10.1002/ajmg.1320550113
2. Schmale GA, Conrad EU, Raskind WH. The natural history of hereditary multiple exostoses. J Bone Joint Surg Am.1994;76:986–992. doi:10.2106/00004623-199407000-00005
3. Ryckx A, Somers JF, Allaert L. Hereditary multiple exostosis. Acta Orthop Belg. 2013;79(6):597–607.
4. Campanacci M. Bone and Soft Tissue Tumors. Bologna: Springer-Verlag Wien GmbH; 1990.
5. Solomon L. Hereditary multiple exostosis. J Bone Joint Surg. 1963;45-B:
6. Matsuno T
7. Krooth RS, Maklin MT, Hilsbish TS. Diaphyseal aclasis (multiple exostoses) on Guam. Am J Hum Genet. 1961;13:340–347.
8. Stocks P, Barrington A. Hereditary Disorders of Bone Development. Vol. 3. Treasury of Human Inheritance. London: Cambridge University Press; 1925.
9. Terry Canale S. Campbell’s Operative Orthopaedics. St.louis: Mosby; 2005.
10. Uchida K, Kurihara Y, Sekiguchi S, et al. Spomtaneous haemotorax caused by costal exostosis. Eur Respir J. 1997;10:735–736.
11. D’Ambrosi RD, Ragone V, Caldarini C, et al. The impact of hereditary multiple exostoses on quality of life, satisfaction, global health status, and pain. Arch Orthop Trauma Surg. 2017;137(2):209–215. doi:10.1007/s00402-016-2608-4
12. D’Ambrosi RD, Caldarini C, Ragone V, Facchini RM. Eff ect of multiple hereditary exostoses on sports activity in children. J Orthop. 2018;15(4):927–930. doi:10.1016/j.jor.2018.08.029
13. Chhina H
14. Porter DE, Lonie L, Fraser M, et al. Severity of disease and risk of malignant change in hereditary multiple exostoses. A genotype phenotype study. J Bone Joint Surg Br. 2004;86:1041e6.
15. Kumar A, Jain VK, Bharadwaj M, et al. Ollier disease: pathogenesis, diagnosis, and management. Orthopedics. 2015;38(6):e497–e506. doi:10.3928/01477447-20150603-58
16. Shapiro F, Simon S, Glimcher HJ. Hereditary multiple exostoses. Anthropometric, roentgenographic, and clinical aspects. J Bone Joint Surg Am. 1979;61(6):815e24.
17. Colnot C, Lu C, Hu D, Helms JA. Distinguishing the contributions of the perichondrium, cartilage, and vascular endothelium to skeletal development. Dev Biol.2004;269:55–69. doi:10.1016/j.ydbio.2004.01.011
18. Stickens D, Clines G, Burbee D, et al. The EXT2 multiple hereditary exostoses gene defines a family of putative tumor suppressor genes. Nat Genet. 1996;14:25–32. doi:10.1038/ng0996-25
19. Jamsheer A, Socha M, Sowińska-Seidler A, et al. Mutational screening of EXT1 and EXT2 genes in polish patients with hereditary multiple exostoses. J Appl Genet. 2014;55(2):183–188. doi:10.1007/s13353-014-0195-z
20. Alvarez C, Tredwell S, De Vera M, et al. The genotype-phenotype correlation of hereditary multiple exostoses. Clin Genet. 2006;70:122–130. doi:10.1111/j.1399-0004.2006.00653.x
21. Ishimaru D, Gotch M, Takayama S, et al. Large scale mutational analysis in the the EXT1 and EXT2 genes for Japanese patients with multiple osteochondromas. BMC Genet. 2016;17:52. doi:10.1186/s12863-016-0359-4
22. Cousminer DL, Arkader A, Voight BF, et al. Assesing the general population frequency of rare coding variants in the EXT1 and –ext2 genes previously implicated in herditary multiple exostoses. Bone. 2016;92:196–200. doi:10.1016/j.bone.2016.09.005
23. Jennes I, Pedrini E, Zuntini M, et al. Multiple osteochondromas: mutation up-date and description of the multiple osteochondromas mutation database (MOdb). Hum Mutat. 2009;30(12):1620–1627. doi:10.1002/humu.v30:12
24. Szuhai K, Jennes I, de Jong D, et al. Tiling resolution array-CGH shows that somatic mosaic deletion of the EXT gene is causative in EXT gene mutation negative multiple osteochondromas patients. Hum Mutat. 2011;32(2):E2036–E2049. doi:10.1002/humu.v32.2
25. Zuntini M, Pedrini E, Parra A, et al. Genetic models of osteochondroma onset and neoplastic progression : evidence for mechanisms alternative to EXT genes inactivation. Oncogene. 2010;29(26):3827–3834. doi:10.1038/onc.2010.135
26. Jones KB, Piombo V, Searby C, et al. A mouse model of osteochondromagenesis from clonal inactivation of Ext1 in chondrocytes. Proc Natl Acad Sci U S A. 2010;107:2054–2059. doi:10.1073/pnas.0910875107
27. Malini K, Gudi NS, Kutty AVM, et al. Mutational analysis of exostosin 1 and 2 genes in multiple osteochondroma. Indian J Pediatr. 2015;82(7):649–650. doi:10.1007/s12098-014-1675-1
28. Reijnders CM, Waaijer CJ, Hamilton A, et al. No haploinsufficiency but loss of heterozygosity for EXT in multiple osteochondromas. Am J Path. 2010;177:1946–1957. doi:10.2353/ajpath.2010.100296
29. Sarrion P, Sangorrin A, Urreizti R, et al. Mutations in the EXT1 and EXT2 genes in Spanish patients with multiple osteochondromas. Sci Rep. 2013;3(1346):1–7. doi:10.1038/srep01346
30. Jones KB. Glycobiology and the growth plate: current concepts in multiple hereditary exostoses. J Pediatr Orthop.2011;31:577–586. doi:10.1097/BPO.0b013e31821c7738
31. Matsumoto K, Irie F, Mackem S, et al. A mouse model of chondrocyte-specific somatic mutation reveals a role for Ext1 loss of hereozygosity in multiple hereditary exostoses. Proc Natl Acad Sci U S A. 2010;107:10932–10937. doi:10.1073/pnas.0914642107
32. Busse- Wicher M, Wicher KB, Kusche-Gullberg M. The exostosin: family proteins with many functions. Matrix Biol. 2014;35:25–33. doi:10.1016/j.matbio.2013.10.001
33. Jones KB, Pacifici M, Hilton MJ. Multiple hereditary exostoses (MHE): elucitading the pathogenesis of a rare skeletal disorder through interdisciplinary research. Connect Tissue Res. 2014;55(2):80–88. doi:10.3109/03008207.2013.867957
34. McCormick C, Duncan G, Goustos KT, Tufaro F. The putative tumor suppressors EXT1 and EXT2 form a stable complex that accumulates in the golgi complex and catalyzes the synthesis of heparan sulfate. Proc Natl Acad Sci U S A.2000;97:668–673. doi:10.1073/pnas.97.2.668
35. Mundy C, Bello A, Sgariglia F, Koyama E, Pacifici M. HhAntag, a hedgehog signaling antagonist, suppresses chondrogenesis and modulates canonical and non-canonical BMP signaling. J Cell Physiol. 2016;231:1033–1044. doi:10.1002/jcp.v231.5
36. Stickens D, Zak BM, Rougier N, et al. Mice deficient in Ext2 lack heparan sulfate and develpoe exostoses. Development. 2005;132:5055–5068. doi:10.1242/dev.02088
37. Yoon BS, Pogue R, Ovchinnikov DA, et al. BMPs regulate multiple aspects of growth-plate chondrogenesis through opposing actions on FGF pathways. Development. 2006;133:4667–4678. doi:10.1242/dev.02680
38. Billings PC, Pacifici M. Interactions of signaling proteins, growth factors and other proteins with heparan sulfate: mechanism and mysteries. Connect Tissue Res.2015;56:272–280. doi:10.3109/03008207.2015.1045066
39. Jochmann K, Bachvarova V, Vortkamp A. Reprint of: heparane sulfate as a regulator of endochondral ossification and osteochondroma development. Matrix Biol. 2014;35:239–247. doi:10.1016/j.matbio.2014.04.001
40. Irie F, Badie-Mahdavi H, Yamaguchi Y. Autism-like socio-communicative deficits and stereotypies in mice lacking heparan sulfate. Proc Natl Acad Sci U S A. 2012;109:5052–5056. doi:10.1073/pnas.1117881109
41. Hennekam RC. Hereditary multiple exostoses. J Med Genet. 1991;28:262e268. doi:10.1136/jmg.28.4.262
42. Jaffe ML. Heriditary multiple exostosis. Arch Pathol. 1943;36:335–357.
43. Sugiura Y, Sugiura I, Iwata H. Heriditary multiple exostoses. Jpn J Hum Genet. 1976;21:149–167.
44. Mordenti M, Ferrari E, Pedrini E, et al. Validation of a new multiple osteochondromas classification through switching neural networks. Am J Med Genet. 2013;161 A:556–560. doi:10.1002/ajmg.a.35819
45. Ali S, Kaplan S, Kaufman T, et al. Madelung deformity and madelung-type deformities: a review of the clinical and radiological characteristics. Pediatr Radiol. 2015;45:1856–1863. doi:10.1007/s00247-015-3390-0
46. Wang YZ, Park KW, Oh CS, et al. Developmental pattern of the hip in patients with hereditary multiple exostoses. BMC Musculoskelet Disord. 2015;16:54. doi:10.1186/s12891-015-0514-5
47. Vaishya R, Swami S, Vijay V, et al. Bilateral total hip arthroplasty in a young man with hereditary multiple exostoses. BMJ Case Rep. 2015;2015:bcr2014207853–bcr2014207853. doi:10.1136/bcr-2014-207853
48. Clement ND, Porter DE. Can deformity of the knee and longitudinal growth of the leg be predicted in patients with hereditary multiple exostoses? A cross-sectional study. Knee. 2014;21:299–303. doi:10.1016/j.knee.2012.10.029
49. Noonan KJ, Feinberg JR, Levenda A, et al. Natural history of multiple hereditary osteochondromatosis of the lower extremity and ankle. Pediatr Orthop. 2002;22:120–124. doi:10.1097/01241398-200201000-00025
50. Matsumoto Y, Matsumoto K, Harimaya K, et al. Scoliosis in patients with multiple hereditary exostoses. J Bone Joint Surg Am. 2009;91:1942–1948. doi:10.2106/JBJS.H.00762
51. Staal HM, Goud AL, van der Woude HJ, et al. Skeletal maturity of children with multiple ostechondromas: is diminished stature due to a systemic influence? J Child Orthop. 2015;9(5):397–402. doi:10.1007/s11832-015-0680-x
52. Rozeman LB, de Bruijn IH, Bacchini P, et al. Dedifferentiated peripheral chondrosarcomas: regulation of EXT-downstream molecules and differentiation related genes. Mod Path. 2009;22:1489–1498. doi:10.1038/modpathol.2009.120
53. Marrero Barrera PA, Marrero Ortiz PB. Ewing sarcoma superimposed on a previuos ostechondroma in multiple ostechondromatosis. Orthopedics. 2014;37(4):e403–e406. doi:10.3928/01477447-20140401-65
54. Goud A, de Lange J, Scholtes VA, et al. Pain physical and social functioning, and quality of life in individuals with multiple hereditary exostoses in the Nederlands: a national cohort study. J Bone Joint Surg Am. 2012;94:1013–1020. doi:10.2106/JBJS.K.00406
55. Kok HK, Fitzgerald L, Campbell N, et al. Multimodality imaging features of hereditary multiple exostoses. Br J Radiol. 2013;86:20130398. doi:10.1259/bjr.20130398
56. Northrup BE, Slat DF, Loomans RU, et al. The myriad of diseases that present with polyostotic bone lesions. Curr Probl Diagn Radiol. 2014;43(4):186–204. doi:10.1067/j.cpradiol.2014.01.003
57. Sonne-Holm E, Wong C, Sonne-Holm S. Multiple cartilaginous exostoses and development of chondrosarcomas – a systematic review. Dan Med J. 2014;61(9):A4895.
58. Faruqi T, Dhawan N, Bahl J, et al. Molecular, phenotypic aspects and therapeutic horizons of rare genetic bone disorders. Biomed Res Int. 2014;2014:670842. doi:10.1155/2014/670842
59. Cappuccio G, Genesio R, Ronga V, et al. Complex chromosomal rearrangements causing langer-giedion syndrome atypical phenotype: genotype-phenotype correlation and literature review. Am J Med Genet A. 2014;164A(3):753–759. doi:10.1002/ajmg.a.36326
60. Fisher TJ, Williams N, Morris L, et al. Metachondromatosis: more than just multiple osteochondromas. J Child Orthop. 2013;7(6):455–464. doi:10.1007/s11832-013-0526-3
61. Sciubba DM, Macki M, Bydon M, et al. Long-term outcomes in primary spinal osteochondroma: a multicenter study of 27 patients. J Neurosurg Spine. 2015;22(6):582–588. doi:10.3171/2014.10.SPINE14501
62. Wu Q, Xiao BO, Li LI, Feng LI. Atypical teratoid/rhabdoid tumor with hereditary multiple exostoses in an 18-year-old male : a case report. Oncol Lett. 2015;10:1561–1564. doi:10.3892/ol.2015.3389
63. Alvarez CM, De Vera MA, Heslip TR, et al. Evaluation of the anatomic burden of patients with hereditary multiple exostoses. Clin Orthop Relat Res. 2007;462:73–79. doi:10.1097/BLO.0b013e3181334b51
64. Woodside JC, Ganey T, Gaston RG. Multiple osteochondroma of the hand : initial and long-term follow-up study. Hand. 2015;10:616–620. doi:10.1007/s11552-015-9775-6
65. Darilek S, Wicklund C, Novy D, et al. Hereditary multiple exostosis and pain. J Pediatr Orthop. 2005;25(3):369–376. doi:10.1097/01.bpo.0000150813.18673.ad
66. Roach JW, Klatt JWB, Faulkner ND. Involvement of the spine in patients with multiple hereditary exostoses. J Bone Joint Surg Am. 2009;91:1942–1948. doi:10.2106/JBJS.H.00762
67. Ashraf A, Larson AN, Ferski G, Guidera KJ. Spinal stenosis frequent in children with multiple hereditary exostoses. J Child Orthop. 2013;7:183–194. doi:10.1007/s11832-013-0484-9
68. Kivioja A, Ervasti H, Kinnunen J, et al. Chondrosarcoma in a family with multiple hereditary ̈exostoses. J Bone Joint Surg B. 2000;82(2):261–266. doi:10.1302/0301-620X.82B2.10139
69. Huegel J, Enomoto-Iwamoto M, Sgariglia F, et al. Heparanase stimulates chondrogenesis and is up-regulated in human ectopic cartilage. a mechanism possibly involved in hereditary multiple exostoses. Am J Pathol. 2015;185:1676–1685. doi:10.1016/j.ajpath.2015.02.014
70. Huegel J, Mundy C, Sgariglia F, et al. Perichondrium phenotype and border function are regulated by Ext1 and heparan sulfate in developing long bones: a mechanism likely deranged in hereditary multiple exostoses. Dev Biol. 2013;377(1):100–112. doi:10.1016/j.ydbio.2013.02.008
71. Sinha S, Mundy C, Bechtold T, et al. Unsuspected osteochondroma-like outgrowths in the cranial base of hereditary multiple exostoses patients and modeling and treatment with a BMP antagonist in mice. PLoS Genet. 2017;13(4):e1006742. doi:10.1371/journal.pgen.1006742
72. Inubushi T, Lemire I, Irie F, et al. Palovarotene inhibits osteochondroma formation in a mouse model of multiple hereditary exostoses. J Bone Miner Res. 2018;33(4):658–666. doi:10.1002/jbmr.3341
73. Shimono K, Tung WE, Macolino C, et al. Potent inhibition of heterotopic ossification by nuclear retinoic acid receptor γ agonists. Nat Med. 2011;17(4):454–460. doi:10.1038/nm.2334
74. D’Ambrosi RD, Barbato A, Caldarini C, Biancardi E, Facchini RM, Ambrosi RD. Gradual ulnar lengthening in children with multiple exostoses and radial head dislocation : results at skeletal maturity. J Child Orthop. 2016;10(2):127–133. doi:10.1007/s11832-016-0718-8
75. Jager M, Westhoff B, Portier S, et al. Clinical outcome and genotype in patients with hereditary multiple exostoses. Clinical outcome and genotype in patients with hereditary multiple exostoses. J Orthop Res. 2007;25:1541–1551. doi:10.1002/(ISSN)1554-527X
76. Wirganowicz PZ, Watts HG. Surgical risk for elective excision of benign exostoses. J Pediatr Orthop. 1997;17:455–459. doi:10.1097/01241398-199707000-00008
77. Passanise AM, Mehlman CT, Wall EJ, et al. Radiographic evidence of regression of a solitary osteochondroma: a report of 4 cases and a literature review. J Pediatr Orthop. 2011;31:312–316. doi:10.1097/BPO.0b013e31820fc676
78. Bassett GS, Cowell HR. Metachondromatatosis. J Bone Joint Surg Am. 1985;67:811–814. doi:10.2106/00004623-198567050-00022
79. Kang S, Kim JY, Park SS. Outcomes of hemiepiphyseal stapling for genu valgum deformities in patients with multiple hereditary exostoses: a comparative study of patients with deformities of idiopathic cause. J Pediatr Orthop. 2017;37:265–271. doi:10.1097/BPO.0000000000000628
80. Kelly JP, James MA. Radiographic outcomes of hemiepiphyseal stapling for distal radius deformity due to multiple hereditary exostoses. J Pediatr Orthop. 2016;36:42–47. doi:10.1097/BPO.0000000000000394
81. Lee DY, Kim JI, Song MH, et al. Fibular lengthening for the management of translational talus instability in hereditary multiple exostoses patients. J Pediatr Orthop. 2014;34:726–732. doi:10.1097/BPO.0000000000000181
82. Guindani N, Eberhardt O, Wirth T, et al. Surgical dislocation for pediatric and adolescent hip deformity: clinical and radiographical results at 3 years follow-up. Arch Orthop Trauma Surg. 2017;137:471–479. doi:10.1007/s00402-017-2644-8
83. Pacifici M. Hereditary multiple exostoses: new insights into pathogenesis, clinical complications and potential treatments. Curr Osteoporos Rep. 2017;15(3):142–152. doi:10.1007/s11914-017-0355-2
84. Beltrami G, Ristori G, Scoccianti G, Tamburini A, Capanna R. Heriditary multiple exostoses: a review of clinical appearance and metabolic pattern. Clin Cases Miner Bone Metab. 2016;13(2):110–118. doi:10.11138/ccmbm/2016.13.2.110
85. Koyoma E, Young B, Nagayama M, et al. Conditional Kif3a ablation causes abnormal hedgehog signaling topography, growth plate dysfunction, and excessive bone and cartilage formation during mouse skeletogenesis. Development. 2007;134:2159–2169. doi:10.1242/dev.001586
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.