Back to Journals » International Journal of Women's Health » Volume 9
Hereditary hemorrhagic telangiectasia and pregnancy: potential adverse events and pregnancy outcomes
Received 19 February 2017
Accepted for publication 9 May 2017
Published 26 May 2017 Volume 2017:9 Pages 373—378
DOI https://doi.org/10.2147/IJWH.S131585
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Elie Al-Chaer
Omar Bari,1 Philip R Cohen2
1School of Medicine, University of California San Diego, La Jolla, CA, USA; 2Department of Dermatology, University of California San Diego, La Jolla, CA, USA
Abstract: Hereditary hemorrhagic telangiectasia (HHT) is an autosomal dominant condition with a prevalence of ~1 in 5,000 individuals. The pathophysiology of this condition centers on the lack of capillary beds between arterioles and venules, leading to direct contact between these vessels. This results in telangiectases on characteristic locations such as the face, fingers, mouth, and nasal mucosa. Visceral arteriovenous malformations (AVMs) are also observed in many patients, and these are most commonly seen in the brain, gastrointestinal tract, and lungs. Liver AVMs are present in many patients with HHT, though these individuals are usually asymptomatic; however, liver AVMs may lead to serious complications, such as high output cardiac failure. Diagnosis of HHT hinges upon fulfilling three out of four criteria: family history of the condition, mucocutaneous telangiectases, spontaneous and recurrent episodes of epistaxis, and visceral AVMs. Management is guided by international consensus guidelines and targets patients’ specific AVMs. Prognosis is good, though severe complications including hemorrhage and paradoxical emboli are possible. Novel therapeutics are being explored in clinical trials; bevacizumab and pazopanib inhibit angiogenesis, while thalidomide bolsters blood vessel maturation. Pregnancy in patients with HHT is considered high risk. While the majority of pregnancies proceed normally, severe complications have been reported in some women with HHT; these include heart failure, intracranial hemorrhage, pulmonary hemorrhage, and stroke. Such complications occur most often in the second and third trimesters when maternal changes such as peripheral vasodilation and increased cardiac output are at their maximum. Awareness of the diagnosis of HHT has been associated with improved outcomes in pregnancy. Management guidelines for pregnant patients with HHT are reviewed.
Keywords: arteriovenous, hereditary, hemorrhagic, malformations, pregnancy, telangiectasia
Introduction
Hereditary hemorrhagic telangiectasia (HHT) is an autosomal dominant condition with variable penetrance which is characterized by arteriovenous malformations (AVMs).1,2 Small AVMs, known as telangiectases, manifest on the buccal mucosa, face, fingers, gastrointestinal mucosa, lips, nasal mucosa, and tongue.1,2 Larger AVMs, which range up to several centimeters in diameter, present most commonly in the brain, lung, and liver.2
Genetics research has unveiled many mutations associated with HHT; all of them have been implicated in the transforming growth factor-beta (TGF-β) signaling pathway.1 The underlying genetic defects have led to the classification of five variants of HHT. HHT type 1 is due to mutations in the endoglin (ENG) gene, while HHT type 2 is due to mutations in the activin receptor-like kinase-1 (ALK-1) gene.3 Several hundred mutations in these two genes account for approximately 90% of cases of HHT.3 A mutation in SMAD4 accounts for an additional 2% of cases of HHT.4 HHT type 3 is linked to mutations in locus 5q31.3-q32, while HHT type 4 is associated with mutations at location 7p14.3 The specific genes associated with HHT type 3 and type 4 have not been identified.4 HHT type 5 is associated with mutations in BMP9, which is a TGF-β superfamily ligand of ALK-1.4
The classical features of HHT are shared amongst the different variants, though specific vascular abnormalities may be associated with particular genotypes.5 For example, HHT type 1, which appears to predominate in America and Northern Europe, is more commonly associated with pulmonary and cerebral AVMs.4,5 HHT type 2, which is classically seen in Mediterranean nations and Argentina, has more hepatic AVMs.4,5
HHT: condition features
History
HHT has its roots in the 19th century, when Rendu presented a patient with anemia, recurrent epistaxis, and skin telangiectases; he hypothesized that this condition was hereditary since the patient’s father had a history of recurrent melena.6 In 1901, Osler presented three cases with multiple vascular lesions and recurrent epistaxis; he also suspected a hereditary link due to family history in these cases.6 In 1907, Weber reported a woman with multiple mucosal and skin lesions, whose children were also affected.2 Subsequently, this disease was dubbed Osler–Weber–Rendu syndrome.6 Yet, in 1909, the term hereditary hemorrhagic telangiectasia (HHT) was established at Johns Hopkins University.6
Epidemiology
HHT prevalence is likely underestimated since the phenotype is variable even within a single family. The highest prevalence is seen in the Netherlands Antilles where 1 of 1,331 people >12 years of age receive the diagnosis.7 Worldwide prevalence is estimated to range between 1 in 5,000 to 1 in 8,000 individuals.1
Pathogenesis
The pathophysiology of HHT centers on an inability to properly connect arterioles and venules, such that they come into direct contact without an intervening capillary bed.1 It is theorized that an inability to properly recruit smooth muscle cells during angiogenesis may be a contributing factor.5,8 This hypothesis is tenable since the known mutations implicated in HHT affect the TGF-β signaling pathway associated with angiogenesis.1
Diagnostic criteria
Consensus diagnostic criteria, known as the Curaçao Criteria, were established in 2000.2 These criteria encompass four components: family history of a first degree relative with HHT, mucocutaneous telangiectases at characteristic locations such as fingers and lips, recurrent and spontaneous epistaxis, and visceral AVMs.2 The diagnosis is definite when three criteria are met, possible if two are fulfilled, and unlikely if fewer than two are seen.1,2 The criteria are best used in adults. The lack of adequate criteria to fulfill the diagnosis of HHT in a child may be misleading since the symptoms of HHT emerge gradually.2
Cutaneous manifestations
Cutaneous manifestations of HHT are variable and often present 10–30 years after initial episodes of epistaxis.9 Telangiectases may present on the face, fingers (Figure 1), tongue (Figure 2), lips (Figure 3), and nailbeds; they often present as pinpoint to pinhead-sized pink or red lesions, or as larger, sometimes raised, purple lesions.1,2 Facial lesions favor sun-exposed areas.1 Telangiectases blanch with pressure and color returns when pressure is released; this distinguishes them from petechiae and angiomas.2 Lesions may bleed at times with little trauma, due to their proximity to the skin surface and also on account of their tortuous vessels.2 Notably, telangiectases often increase in number as the patients age.1
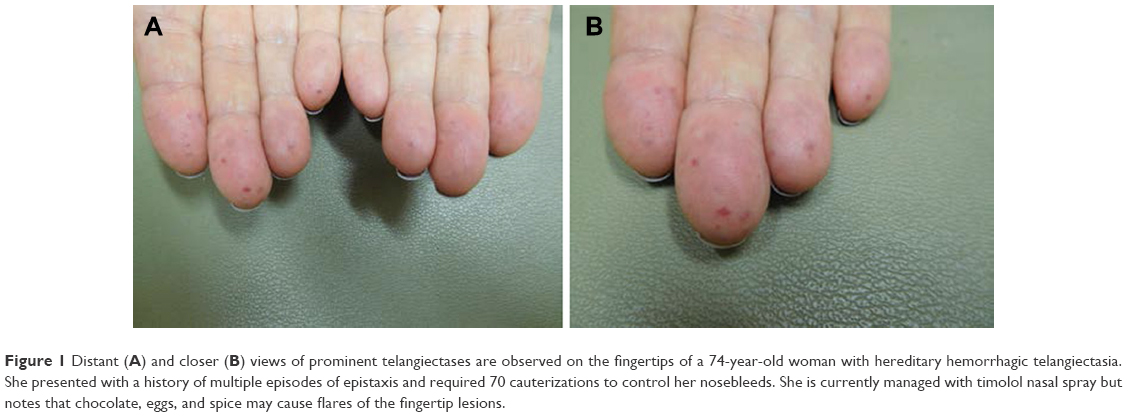 | Figure 1 Distant (A) and closer (B) views of prominent telangiectases are observed on the fingertips of a 74-year-old woman with hereditary hemorrhagic telangiectasia. She presented with a history of multiple episodes of epistaxis and required 70 cauterizations to control her nosebleeds. She is currently managed with timolol nasal spray but notes that chocolate, eggs, and spice may cause flares of the fingertip lesions. |
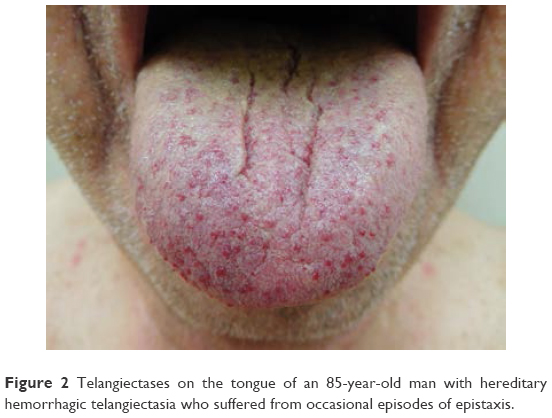 | Figure 2 Telangiectases on the tongue of an 85-year-old man with hereditary hemorrhagic telangiectasia who suffered from occasional episodes of epistaxis. |
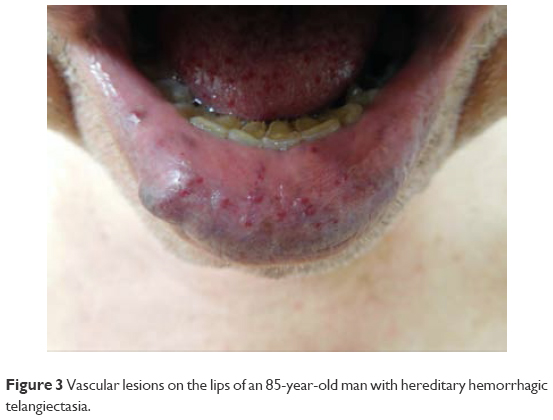 | Figure 3 Vascular lesions on the lips of an 85-year-old man with hereditary hemorrhagic telangiectasia. |
Systemic manifestations
HHT may result in AVMs in numerous organs: brain, intestine, lung, nose, and spinal cord involvement have been observed.1 Epistaxis is often the first presenting symptom, and 50%–95% of patients develop nosebleeds by 20 years of age.1 The severity of nosebleeds increases with time and may lead to anemia.1 It is important to recognize that epistaxis may be severe, and in some cases can be life threatening.10
Pulmonary AVMs develop in 15%–50% of patients. The patients are usually asymptomatic. Lung AVMs cause right-to-left shunting, which cause an array of possible symptoms including dyspnea, hypoxia, and pulmonary hypertension.1 Moreover, pulmonary AVMs can result in paradoxical septic or non-septic emboli; brain, spinal, or systemic abscesses have been observed, secondary to septic emboli, in patients with pulmonary AVMs.11
Central nervous system involvement is usually secondary to paradoxical emboli from pulmonary AVMs; the emboli can result in stroke or transient ischemic attack. However, some patients suffer from complications of cerebral AVMs.1 The risk of central nervous system AVM hemorrhage is low; one study found that the bleeding risk secondary to HHT-related cerebral vascular malformations was 0.27%–0.46% per year compared to a 2%–4% per year risk for sporadic central nervous system AVMs.12
Gastrointestinal involvement is also common. The stomach and small intestine are most commonly affected. Fifteen percent to 45% of HHT patients develop a gastrointestinal bleed during their lives, usually after 50 years of age.1 Liver involvement has been shown to be common in patients with HHT; however <10% of patients with liver AVMs are symptomatic.2 Patients with liver involvement can present with biliary dysfunction and/or portal hypertension.1 It is important to recognize that a severe complication associated with hepatic AVMs is high output cardiac failure.1 Due to reduced systemic vascular resistance, patients develop high cardiac output secondary to sodium and water metabolism, which activates the renin-angiotensin-aldosterone system. The increased cardiac output is disproportionately shunted through hepatic AVMs, eventually leading to heart faliure.13
Management
Consensus guidelines
International consensus management guidelines for HHT emerged in 2011.14 Epistaxis is common and significantly affects quality of life.15 Initially, epistaxis should be managed with measures such as humidification and lubricants.14 For refractory nosebleeds, chemical cautery, cryotherapy, laser coagulation, septodermoplasty, and sodium tetradecyl sulfate injections may be considered.1,14
All patients should be screened for pulmonary AVMs.14 Transthoracic contrast echocardiography is the preferable test for screening.1,14 If an AVM >3 mm is discovered, embolization is recommended.14
Infants with HHT should be screened for cerebral malformations, using non-contrast brain magnetic resonance imaging (MRI), within the first 6 months of life; however, this recommendation is controversial.14 Adults should also be screened for cerebral malformations; however, repeat imaging is not required if central nervous system AVMs are not seen.1 Management of central nervous system malformations should be decided on a case by case basis.1
To evaluate for gastrointestinal involvement, an annual hemoglobin is recommended for all patients >35 years of age; upper endoscopy should be considered if the patients’ anemia is out of proportion to their epistaxis.14 Liver imaging is not routine and should be done only when patients are symptomatic or if abnormal liver function tests are found.14 If a liver AVM is discovered and the patient is symptomatic, the patient should be managed medically; if patients fail to respond to therapy and progressively worsen, invasive therapeutic options such as transarterial embolization or liver transplant may be warranted.16
Drug therapy
A specific drug for the treatment of HHT has not currently been established. However, new therapeutic options are being actively explored.1 For example, thalidomide has been proposed; this drug boosts pericyte recruitment, which may enhance new blood vessel maturation.17
Another approach for the management of HHT is to target angiogenesis. Bevacizumab is an angiogenesis inhibitor which blocks vascular endothelial growth factor (VEGF), which is currently only indicated as a chemotherapy agent.18 A recent randomized controlled trial assessed the effect of bevacizumab nasal spray on epistaxis duration in HHT patients; the study failed to show a reduction in monthly epistaxis duration during the 3 months following the end of treatment when compared to placebo.19 Similarly, another randomized controlled trial did not reveal significant differences in epistaxis frequency among patients treated with topical intranasal bevacizumab, estriol, tranexamic acid, or placebo.20
Pazopanib is another VEGF inhibitor. It is also being explored for the treatment of HHT. The drug is currently in a Phase II clinical trial to determine if there is any reduction in epistaxis and anemia when compared to placebo.1
Two additional drugs, both selective estrogen receptor modulators, are used for treatment of HHT, particularly outside the United States. Raloxifene was designated an orphan drug in 2010 by both the European Medicines Agency and the United States Food and Drug Administration. Bazedoxifene was established as an orphan drug for HHT in 2014 by the European Medicines Agency alone.4 Both of these medications are thought to decrease bleeding by helping compensate for haploinsufficiency of proteins implicated in the TGF-β signaling pathway in endothelial cells.4
HHT and pregnancy
Pregnancy in patients with HHT is considered high risk. This is due to the possibility of life threatening complications. However, most pregnancies proceed without issue.21
Complication vulnerability in later trimesters
Women with HHT appear to be most vulnerable to complications in the second and third trimesters due to physiologic adaptations which occur in pregnancy. During the latter trimesters, peripheral vascular resistance decreases. In addition, cardiac output increases nearly 50% by the end of the second trimester.22 These physiologic changes may exacerbate existing shunting lesions. For example, high output heart failure secondary to hepatic AVMs could occur.23
Pregnancy outcome studies in HHT
To date, four studies have assessed pregnancy outcomes in HHT.
Study 1
The first study was from a retrospective evaluation published in 1967. Forty American women with HHT from an otolaryngology clinic were compared to 80 controls; the subjects were matched for age, race, religion, sex, and source of medical care.24 The rates of miscarriage and abnormal outcomes showed no statistically significant difference between the two groups.24
Study 2
The second study, which was published in 1995, was a retrospective series of 161 pregnancies in 47 British women with HHT.25 Similar to the 1967 study, no increase in miscarriage was observed when the rate of miscarriage in the study group was compared to the rate of miscarriage in the United Kingdom overall.25 However, patients with pulmonary AVMs had increased risk of complications; there were six cases of pulmonary AVM shunt deterioration, three strokes, and two maternal deaths from pulmonary AVM hemorrhage.25
Study 3
The third study followed 262 pregnancies in 111 British women with HHT along with 222 pregnancies in 86 first-degree relatives affected by HHT between 1999 and 2005. This was a cohort study with prospective and retrospective components. Seventy-four percent of the patients did not have an established diagnosis of HHT at the time of their pregnancy.21 While the majority of pregnancies proceeded normally, 13 patients suffered life-threatening events during pregnancy: one myocardial infarction, six pulmonary AVM hemorrhages, and six strokes were noted.21 Although all of the women had been in good health prior to pregnancy, five of them died.21 Prior awareness by the patients that they had a diagnosis of HHT was associated with increased survival.
Study 4
The most recent study was a retrospective case series of 244 pregnancies in 87 Canadian women with HHT between 1997 and 2007.26 Major complications occurred in 13% of the women and included heart failure in one patient, hemothorax in five patients, deep venous thrombosis in one patient, pulmonary embolism in one patient, myocardial infarction in one patient, myocardial ischemia in one patient, and intracranial hemorrhage in one patient.26 All of these complications were in patients who had not been screened or treated for AVMs prior to pregnancy.26 This study also noted that patients experienced more frequent episodes of epistaxis and the development of new telangiectases during pregnancy.26
Comparison of studies
The published studies assessing pregnancy outcomes in HHT were performed in North America and the United Kingdom. All four studies were retrospective, though the third study had a prospective component. The first two studies observed that miscarriages were not increased in women with HHT, implying that the fetus fares normally in pregnant women with HHT. However, possible complications from the disease appear to pose a risk to the mother. The three more recent studies emphasize that pregnant women with HHT could suffer from life-threatening complications or death. The third study, which included the largest number of pregnancies, noted that awareness of the diagnosis of HHT was important since it was associated with increased survival. The fourth study showed that severe complications only occurred in those women who were not screened or treated for underlying AVMs. All of these studies were helpful in establishing the recommendations for management of pregnancy in patients with HHT.
Management recommendations
Shovlin et al offer recommendations for management of HHT pregnancies.21 All women with HHT considering pregnancy should be screened for pulmonary AVMs with a thoracic computerized tomography scan to reduce complications such as stroke and hemorrhage; if pulmonary AVMs are found, these women should be treated prior to conception. However, if women are already pregnant and found to have asymptomatic pulmonary AVMs, they should not be treated during pregnancy. All women should also have an MRI of the spine to rule out spinal AVM, since this would enable regional anesthesia. If women have family history of cerebral hemorrhage or central nervous system symptoms, an MRI of the head is recommended. If any pregnant woman with HHT develops hemoptysis or sudden dyspnea, then urgent hospitalization is encouraged. For all pregnant women with HHT, antibiotic prophylaxis during delivery is recommended. Also, avoidance of a prolonged stage of labor in HHT patients in whom brain AVM has not been excluded is advised.21
Summary
The majority of pregnancies in patients with HHT have no complications. However, pregnancy is high risk due to the possibility of severe, life threatening complications. Thus, women with HHT should be appropriately counseled on the small but serious risks. In addition, it is important for these patients to be appropriately managed since prior awareness of HHT has been associated with enhanced survival for the mother.21
Conclusion
HHT is a rare autosomal dominant condition that can be difficult to diagnose. It presents with telangiectases in characteristic locations as well as with larger AVMs in visceral organs. Astute clinicians should be aware of this disease and its appropriate management.
HHT should be managed per international consensus guidelines. Women with HHT planning pregnancy should be advised that risks are low but serious. However, they should be reassured that close obstetric care and prior awareness of the diagnosis of HHT is usually associated with good pregnancy outcomes.
Disclosure
The authors report no conflicts of interest in this work.
References
Chung MG. Hereditary hemorrhagic telangiectasia. Handb Clin Neurol. 2015;132:185–197. | ||
McDonald J, Bayrak-Toydemir P, Pyeritz RE. Hereditary hemorrhagic telangiectasia: an overview of diagnosis, management, and pathogenesis. Genet Med. 2011;13(7):607–616. | ||
Garg N, Khunger M, Gupta A, Kumar N. Optimal management of hereditary hemorrhagic telangiectasia. J Blood Med. 2014;5:191–206. | ||
Zarrabeitia R, Ojeda-Fernandez L, Recio L, et al. Bazedoxifene, a new orphan drug for the treatment of bleeding in hereditary haemorrhagic telangiectasia. Thromb Haemost. 2016;115(6):1167–1177. | ||
Shovlin CL. Hereditary haemorrhagic telangiectasia: pathophysiology, diagnosis and treatment. Blood Rev. 2010;24(6):203–219. | ||
Gibbs DD. Rendu-Osler-Weber disease: a triple eponymous title lives on. J R Soc Med. 1986;79(12):742–743. | ||
Westermann CJ, Rosina AF, De Vries V, de Coteau PA. The prevalence and manifestations of hereditary hemorrhagic telangiectasia in the Afro-Caribbean population of the Netherlands Antilles: a family screening. Am J Med Genet A. 2003;116A(4):324–328. | ||
Mancini ML, Terzic A, Conley BA, Oxburgh LH, Nicola T, Vary CP. Endoglin plays distinct roles in vascular smooth muscle cell recruitment and regulation of arteriovenous identity during angiogenesis. Dev Dyn. 2009;238(10):2479–2493. | ||
Plauchu H, de Chadarévian JP, Bideau A, Robert JM. Age-related clinical profile of hereditary hemorrhagic telangiectasia in an epidemiologically recruited population. Am J Med Genet. 1989;32(3):291–297. | ||
Rebeiz EE, Bryan DJ, Ehrlichman RJ, Shapshay SM. Surgical management of life-threatening epistaxis in Osler-Weber-Rendu disease. Ann Plast Surg. 1995;35(2):208–213. | ||
Moradi M, Adeli M. Brain abscess as the first manifestation of pulmonary arteriovenous malformation: a case report. Adv Biomed Res. 2014;3:28. | ||
Willemse RB, Mager JJ, Westermann CJ, Overtoom TT, Mauser H, Wolbers JG. Bleeding risk of cerebrovascular malformations in hereditary hemorrhagic telangiectasia. J Neurosurg. 2000;92(5):779–784. | ||
Cho D, Kim S, Kim M, et al. Two cases of high output heart failure caused by hereditary hemorrhagic telangiectasia. Korean Circ J. 2012;42(12):861–865. | ||
Faughnan ME, Palda VA, Garcia-Tsao G, et al. International guidelines for the diagnosis and management of hereditary haemorrhagic telangiectasia. J Med Genet. 2011;48(2):73–87. | ||
Pasculli G, Resta F, Guastamacchia E, Di Gennaro L, Suppressa P, Sabbà C. Health-related quality of life in a rare disease: hereditary hemorrhagic telangiectasia (HHT) or Rendu-Osler-Weber disease. Qual Life Res. 2004;13(10):1715–1723. | ||
Buscarini E, Leandro G, Conte D, et al. Natural history and outcome of hepatic vascular malformations in a large cohort of patients with hereditary hemorrhagic teleangiectasia. Dig Dis Sci. 2011;56(7):2166–2178. | ||
Thalgott J, Dos-Santos-Luis D, Lebrin F. Pericytes as targets in hereditary hemorrhagic telangiectasia. Front Genet. 2015;6:37. | ||
Dupuis-Girod S, Ginon I, Saurin JC, et al. Bevacizumab in patients with hereditary hemorrhagic telangiectasia and severe hepatic vascular malformations and high cardiac output. JAMA. 2012;307(9):948–955. | ||
Dupuis-Girod S, Ambrun A, Decullier E, et al. Effect of bevacizumab nasal spray on epistaxis duration in hereditary hemorrhagic telangectasia: a randomized clinical trial. JAMA. 2016;316(9):934–942. | ||
Whitehead KJ, Sautter NB, McWilliams JP, et al. Effect of topical intranasal therapy on epistaxis frequency in patients with hereditary hemorrhagic telangiectasia: a randomized clinical trial. JAMA. 2016;316(9):943–951. | ||
Shovlin CL, Sodhi V, McCarthy A, Lasjaunias P, Jackson JE, Sheppard MN. Estimates of maternal risks of pregnancy for women with hereditary haemorrhagic telangiectasia (Osler-Weber-Rendu syndrome): suggested approach for obstetric services. BJOG. 2008;115(9):1108–1115. | ||
Yeomans ER, Gilstrap LC 3rd. Physiologic changes in pregnancy and their impact on critical care. Crit Care Med. 2005;33(10 Suppl):S256–S258. | ||
Goussous T, Haynes A, Najarian K, Daccarett M, David S. Hereditary hemorrhagic telangiectasia presenting as high output cardiac failure during pregnancy. Cardiol Res Pract. 2009;2009:437237. | ||
Goodman RM, Gresham GE, Roberts PL. Outcome of pregnancy in patients with hereditary hemorrhagic telangiectasia. A retrospective study of 40 patients and 80 matched controls. Fertil Steril. 1967;18(2):272–277. | ||
Shovlin CL, Winstock AR, Peters AM, Jackson JE, Hughes JM. Medical complications of pregnancy in hereditary haemorrhagic telangiectasia. QJM. 1995;88(12):879–887. | ||
de Gussem EM, Lausman AY, Beder AJ, et al. Outcomes of pregnancy in women with hereditary hemorrhagic telangiectasia. Obstet Gynecol. 2014;123(3):514–520. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.