Back to Journals » OncoTargets and Therapy » Volume 15
Hereditary Gynecologic Cancer Syndromes – A Narrative Review
Authors Kostov S, Watrowski R ![]() , Kornovski Y, Dzhenkov D, Slavchev S, Ivanova Y, Yordanov A
, Kornovski Y, Dzhenkov D, Slavchev S, Ivanova Y, Yordanov A
Received 17 December 2021
Accepted for publication 18 March 2022
Published 8 April 2022 Volume 2022:15 Pages 381—405
DOI https://doi.org/10.2147/OTT.S353054
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Arseniy Yuzhalin
Stoyan Kostov,1,2 Rafał Watrowski,3 Yavor Kornovski,1 Deyan Dzhenkov,4 Stanislav Slavchev,1 Yonka Ivanova,1 Angel Yordanov5
1Department of Gynecology, University Hospital “Saint Anna”, Medical University “Prof. Dr. Paraskev Stoyanov”, Varna, Bulgaria; 2Faculty of Health Care, Medical University Pleven, Pleven, Bulgaria; 3Faculty of Medicine, University of Freiburg, Freiburg, 79106, Germany; 4Department of General and Clinical Pathology, Forensic Medicine and Deontology, Division of General and Clinical Pathology, Faculty of Medicine, Medical University “Prof. Dr. Paraskev Stoyanov”, Varna, Bulgaria; 5Department of Gynecologic Oncology, Medical University Pleven, Pleven, Bulgaria
Correspondence: Angel Yordanov, Email [email protected]
Abstract: Hereditary cancer syndromes are defined as syndromes, where the genetics of cancer are the result of low penetrant polymorphisms or of a single gene disorder inherited in a mendelian fashion. During the last decade, compelling evidence has accumulated that approximately 5– 10% of all cancers could be attributed to hereditary cancer syndromes. A tremendous progress has been made over the last decade in the evaluation and management of these syndromes. However, hereditary syndromes associated with gynecologic malignancies still present significant challenge for oncogynecologists. Oncogynecologists tend to pay more attention to staging, histological type and treatment options of gynecological cancers than thinking of inherited cancers and taking a detailed family history. Moreover, physicians should also be familiar with screening strategies in patients with inherited gynecological cancers. Lynch syndrome and hereditary breast-ovarian cancer syndrome are the most common and widely discussed syndromes in medical literature. The aim of the present review article is to delineate and emphasize the majority of hereditary gynecological cancer syndromes, even these, which are rarely reported in oncogynecology. The following inherited cancers are briefly discussed: Lynch syndrome; “site-specific” ovarian cancer and hereditary breast–ovarian cancer syndrome; Cowden syndrome; Li-Fraumeni syndrome; Peutz-Jeghers syndrome; ataxia-telangiectasia; DICER1- syndrome; gonadal dysgenesis; tuberous sclerosis; multiple endocrine neoplasia type I, II; hereditary small cell carcinoma of the ovary, hypercalcemic type and hereditary undifferentiated uterine sarcoma; hereditary diffuse gastric cancer and MUTYH-associated polyposis. Epidemiology, pathogenesis, diagnosis, pathology and screening of these syndromes are discussed. General treatment recommendations are beyond the scope of this review.
Keywords: hereditary gynecologic cancer syndromes, pathogenesis, diagnosis, pathology, screening
Introduction
Hereditary cancer syndromes (HCS) are defined as syndromes, where the genetics of cancer are the result of low penetrant polymorphisms or of a single gene disorder inherited in a mendelian fashion. Hereditary syndromes associated with gynecologic malignancies present significant challenge for oncogynecologists. Physicians tend to pay more attention to staging, histological type and treatment options of gynecological cancers than taking a detailed family history.1 During the last decade, compelling evidence has accumulated that approximately 5–10% of all cancers can be attributed to hereditary cancer syndromes.2,3 Concerning these conditions facilitates the opportunity to screen and identify individuals at higher risk of developing some types of cancers.3 Moreover, identifying women with a hereditary pathogenic gene variant is essential in order to estimate the counseling strategies for cancer prevention, the future fertility, cancer surveillance and risk of transmitting pathogenic gene variants to their offspring. Additionally, other topics that should be discussed with patients are the potential initiation of hormone therapy after prophylactic surgery (bilateral salpingo-oophorectomy, mastectomy, hysterectomy) at-risk family members.1,3 The most common hereditary gynecologic cancer syndromes (HGCSs) are hereditary breast–ovarian cancer syndrome and hereditary nonpolyposis colorectal cancer (HNPCC) syndrome.4 They account approximately 10–18% of ovarian, tubal, peritoneal, and endometrial cancer cases.5 Approximately 5% of uterine cancers and 10% of ovarian cancers are due to HGCSs.6 However, other HGCSs are rarely mentioned. The aim of the present review is to describe counseling strategies, pathogenesis, pathology and screening of the following HGCSs: hereditary nonpolyposis colorectal cancer syndrome; “site-specific” ovarian cancer and hereditary breast–ovarian cancer syndrome; Cowden syndrome; Li-Fraumeni syndrome; Peutz-Jeghers syndrome; ataxia-telangiectasia; DICER1- syndrome; gonadal dysgenesis; tuberous sclerosis; multiple endocrine neoplasia type I, II; hereditary small cell carcinoma of the ovary, hypercalcemic type and hereditary undifferentiated uterine sarcoma; hereditary diffuse gastric cancer and MUTYH-associated polyposis. General treatment recommendations are beyond the scope of this review. We only mentioned risk-reducing surgery as an option and described other author’s experience, because there are many controversies and further recommendations are debatable and could cause unnecessary overtreatment.
Hereditary Gynecologic Cancer Syndromes
Hereditary Nonpolyposis Colorectal Cancer Syndrome (HNPCC) -Lynch Syndrome
Definition and Epidemiology
HNPCC syndrome, also known as Lynch syndrome (LS), is an autosomal dominant cancer predisposition characterized by increased risk and early occurrences of colorectal cancer. LS is also associated with elevated risk of cancers of the endometrium, ovary, breast, stomach, small bowel, urinary tract, biliary tract, brain (usually glioblastoma), skin, pancreas, and prostate.7,8 For patients with HNPCC syndrome, the lifetime risks of developing endometrial and ovarian cancer are approximately 30–71% and 4–24%, respectively.5,7,9 Other studies estimated that approximately 60% of women with LS present initially with cancers of the female genital tract, of which 14% are associated with ovarian and 60% of endometrial cancer, respectively.10–12 The syndrome accounts for at least 2% to 5% of all colorectal and endometrial cancers and increases the lifetime risk of colorectal cancer to as high as 60% to 85%.4,13 Some studies reported higher risk of endometrial cancer development than colorectal cancer for patient with LS.4 The average age of colorectal (mean age 41–44 years), ovarian, endometrial cancers (mean age 48–50 years) diagnosis in individuals with LS is younger than the general population – during the fourth decade.7,14,15 One study estimated an incidence of endometrial cancer in LS among Japanese endometrial cancer patients of reproductive age (≤40 years) being approximately 1.5%. However, authors also reported that 12.3% of the patients had variants of uncertain significance in MMR genes.16 The risk of second, metachronous cancer occurrence in women with LS is 25% by 10 years after diagnosis of the primary cancer and 50% by 15 years after an initial cancer diagnosis.15 Therefore, after the diagnosis of primary cancer associated with LS the patients should be adequately counseled and offered appropriate risk-reduction strategies (prophylactic surgery).4 A woman with HNPCC who survives a colon cancer has an increased risk of developing an endometrial cancer.15 The occurrence of breast cancer as part of Lynch syndrome is controversial. Some studies stated that breast cancer is not a part of LS, whereas others have suggested an epidemiologic association.7–17
Pathogenesis
HNPCC is caused by mutations in the DNA mismatch repair (MMR) genes – MLH1, MSH2, MSH6 and less commonly PMS1, PMS2. These genes are responsible for repairing single base pair mismatches and mispaired loops of DNA during the process of DNA replication.1,13 Some studies did not include mutations in the PMS1 as the cause of LS. It is reported that the cancer risk on LS depended on gene mutations and it is substantially lower for PMS2.18 Dominguez-Valentin et al observed no difference in incidence of cancer between truncating and missense or aberrant splicing pathogenic variants in MLH1 and MSH2 carriers.19 Mutation in the MMR genes causes microsatellite instability (MSI), where insertion of extra nucleotides in microsatellite repeats and leads to accumulation of gene mutations that affect proliferation and cell-cycle control.1,4,13 The MSI is the hallmark of cancers associated with LS.5 Rarely, the LS may occur due to inactivation of CHEK2 (cell cycle checkpoint kinase 2).20 It should be stressed that MMR deficiency is not an absolute feature of LS, as most of MMR deficient endometrial cancers present with somatic inactivation of the MLH1 due to hypermethylation of the promoter region.18 Cases, which are not caused by MLH1 hypermethylation or MMR gene mutation are considered sporadic via biallelic somatic MMR gene inactivation.18 Figures 1 and 2 show the risk of LS-associated cancers depending on the MMR gene mutation.1,5,21,22
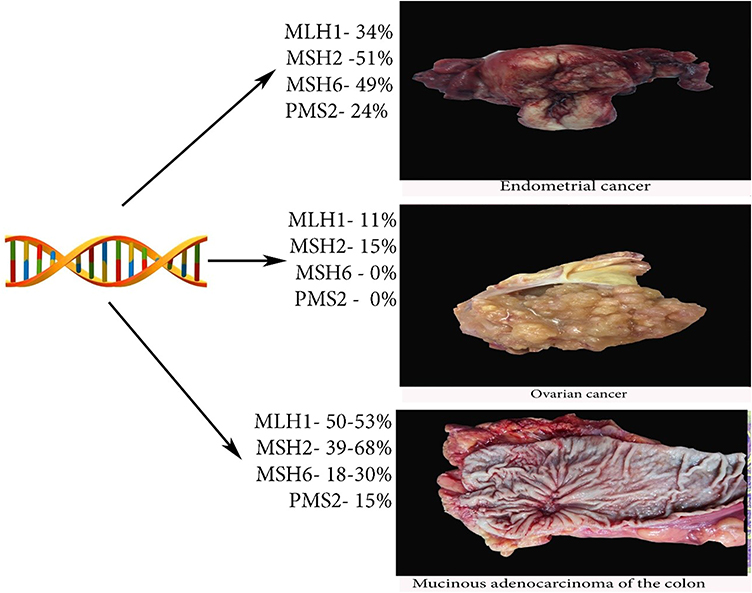 |
Figure 1 Risk of endometrial, ovarian and colon cancers in LS. |
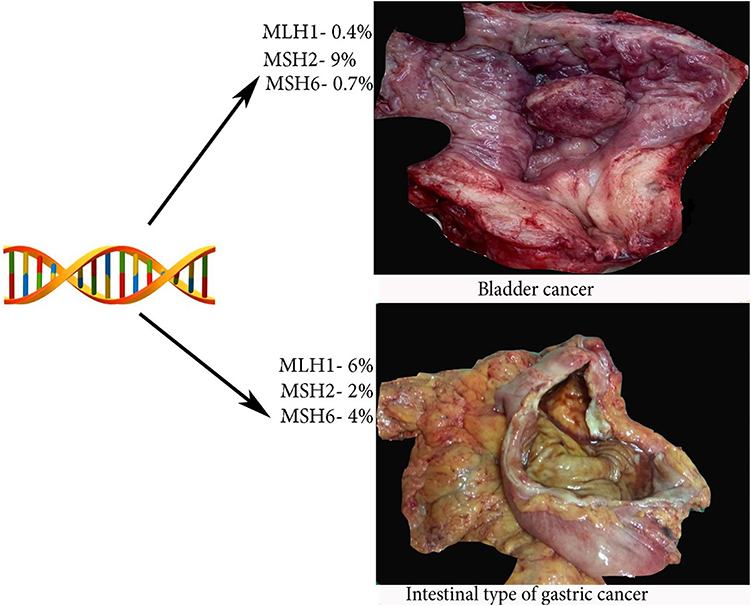 |
Figure 2 Risk of bladder and gastric cancer is LS. |
Diagnosis
The clinical criteria for the diagnosis of LS include revised Amsterdam II criteria and revised Bethesda criteria 1,4,5,13,23–25 (Table 1).
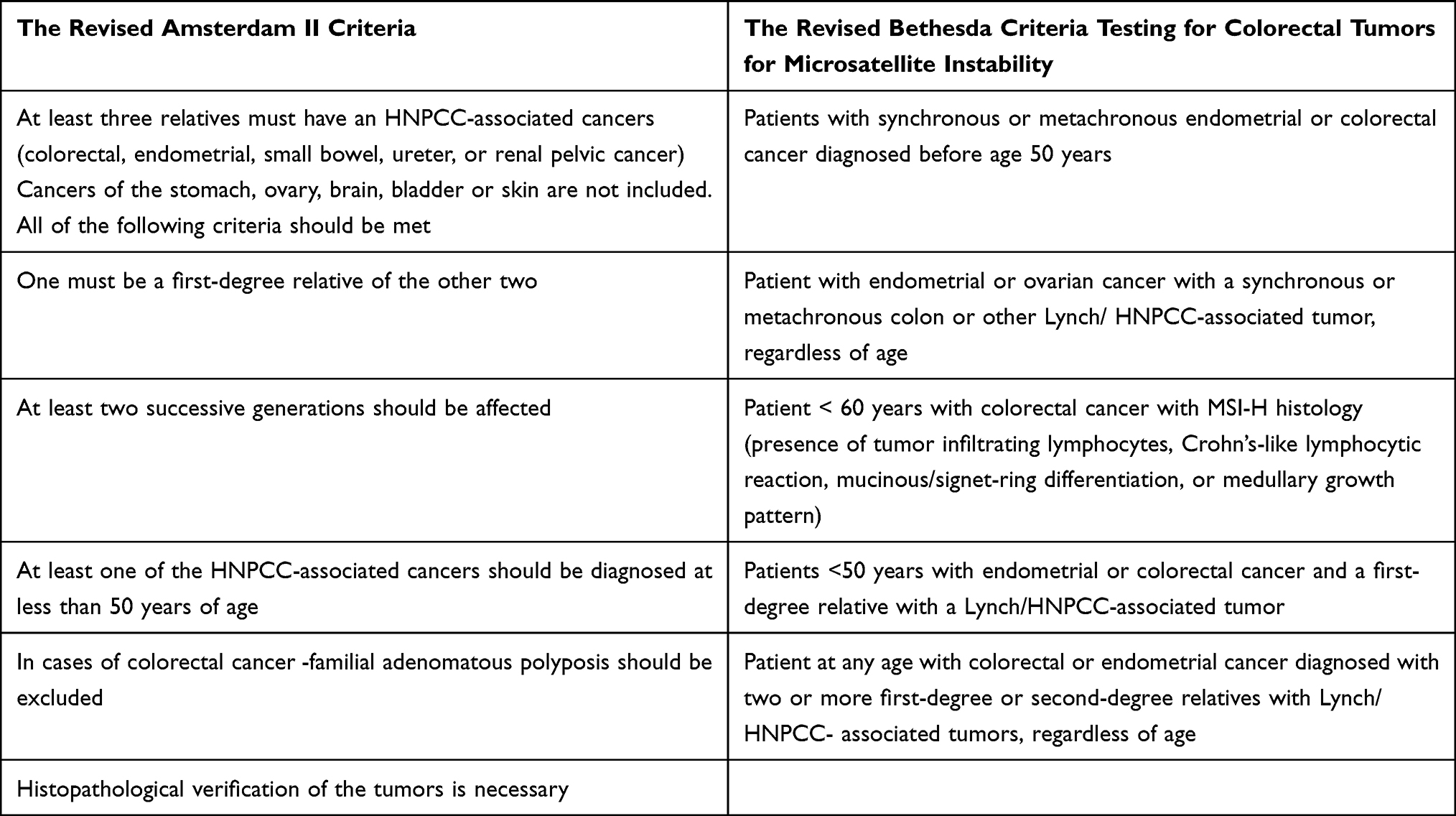 |
Table 1 The Revised Amsterdam II and Bethesda Criteria |
The purpose of the two criteria is to identify families eligible for further molecular analysis. As the specificity of the Amsterdam II criteria was high (98%), but the sensitivity low (22%), a Bethesda guideline was developed in 1997, and revised in 2004. The limitations of the Amsterdam criteria were that only 13% to 36% of families with molecularly confirmed HNPCC will meet these criteria. The revised Bethesda criteria has 82% specificity and 77% sensitivity and included endometrial cancer as sentinel cancer.4,5,26 However, it has been estimated that screening by using these criteria could miss more than one-fourth of LS cases. Therefore, it is recommended screening for all patients with colorectal cancer, either by direct germline testing for mutation, MSI testing of tumor specimens via PCR or by initial tumor testing with immunohistochemistry (IHC) for MMR genes.5,23
Although it is controversial whether immunohistochemistry or molecular testing should be performed, it is stated that detection of mutated MMR genes by IHC is reliable (especially for endometrial cancer). Loss of PMS2 staining supposes PMS2 mutation, whereas loss of MSH6 staining is suggestive of MSH6 mutation.20 Loss of MLH1 and PMS2 staining is likely to occur in MLH1 mutation, while loss of MSH2 and MSH6 staining may be observed in MSH2 mutation.20 The benefit of IHC in LS is that the presence of MLH1, MSH2, MSH6, and PMS2 proteins rules out LS and further DNA-testing for MMR gene mutation is not necessary.23 However, in cases of endometrial cancer related to LS, a MLH1 promoter methylation testing should be performed to rule out sporadic endometrial cancer, as MLH1 promoter hypermethylation is associated with sporadic cancers of the endometrium.27,28 Buchanan et al investigated 702 patients with endometrial cancer diagnosed before 60 years of age by using tumor mismatch repair immunohistochemistry and DNA MLH1 methylation testing. Authors concluded that combination such a testing combination provided the highest positive predictive value for the identification of mutation carriers.27 Other specific clinical characteristics that could be used to identify LS are the following: patients with colorectal cancer at young age, proximal colon location and the presence of synchronous or metachronous tumors.13,29
Pathology
The histopathological features of colorectal cancer related to LS differ to colorectal cancer in general. Mucinous, poorly differentiated, diploid and signet-ring cancers are common in LS. HNPCC tumors are often associated with a prominent chronic inflammatory response (Crohn’s-like reaction) (Figure 3). High numbers of tumour infiltrating lymphocytes are also observed. These features could serve as additional markers for LS recognition, although they are not specific.29,30 It is believed that LS-related colorectal carcinomas arose from adenomas, which had a more rapid progression to carcinoma compared to those arising sporadically within the population.30
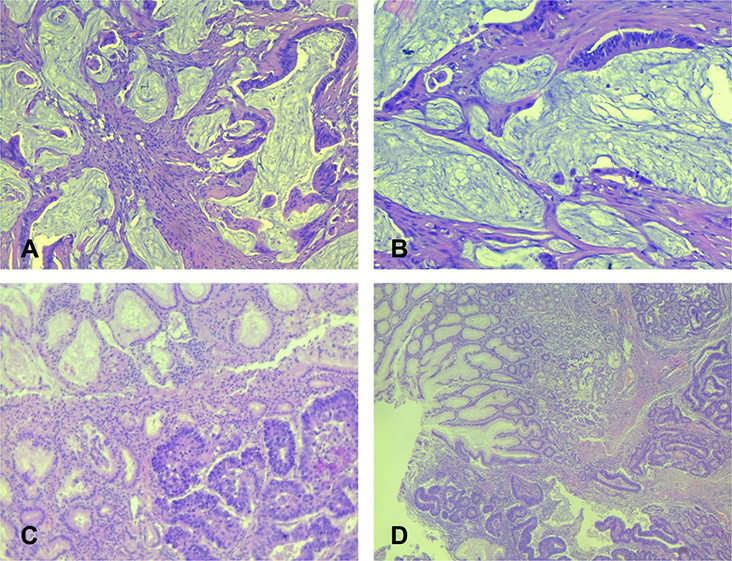 |
Figure 3 Histological features of mucinous colon adenocarcinoma and gastric carcinoma – intestinal type. (A and B) – mucinous colon adenocarcinoma. Strips of atypical tumor cells, and part of abortive glands floating in large extracellular mucin lakes comprising at least 50% of tumor area ((A) HEx200; (B) – HEx100). (C and D) – gastric cancer, intestinal type. Tumor is composed of confluent tubular structures consist of atypical cells with hyperchromatic nuclei similar to intestinal adenocarcinoma, which invade in submucosal gastric area. In upper part is antral mucosa with reactive mucinous foveolar hyperplasia ((C) - HEx100; (D) – HEx40). |
The histological features of endometrial cancer related to LS are non-specific – commonly of endometrial type, although non-endometrioid types are also observed – uterine papillary serous carcinoma, clear cell carcinoma, and uterine carcinosarcoma.2,29,31 A non-endometrioid histology in LS is often observed in patient with MSH2 mutation. Moreover, compared to the sporadic endometrial cancer, the LS-EC shows tendency to develop in the lower uterine segment.30–32 There are no differences of survival between LS-associated endometrial cancer (LS-EC) and the sporadic one.29
The histological features of ovarian carcinomas tend to be endometrioid, undifferentiated and clear cell.31,35 Ryan et al identified 53 patients (proven MMR mutations) with LS-associated ovarian cancer. Authors reported that the endometrioid ovarian adenocarcinoma was the most common histology, followed by high-grade serous, clear cell and mixed. LS-associated ovarian cancer occurred at an earlier age (59 years) compared to the sporadic ovarian cancer.36
Gastric cancer is the second most common extracolonic malignancies. It is of an intestinal type related to Helicobacter pylori-associated chronic gastritis29 (Figure 3).
The histology of LS-associated bladder and ureteral cancers do not differ from the sporadic ones.29
As mentioned above, although the presence of breast cancer in LS is controversial, Sheehan et al suggested that Lynch syndrome patients with PMS2 mutations might be at higher risk of developing breast cancer.37 Walsh et al concluded that LS-associated breast cancer was often poorly differentiated with a high mitotic index, steroid hormone receptor–negative with peritumoral lymphocytes and confluent necrosis. There is no predominance of lobular or ductal histological types, though Yee et al observed higher levels of MSI in lobular carcinomas (39%) than in infiltrating ductal cancers (13.5%).38,39
Screening
Screening recommendations are more clear for colorectal cancer than for the gynecologic cancers (GCs), as less information is available regarding GCs.4,13 Screening colonoscopy is the best screening method as it decreases the incidence of colorectal cancer and mortality in patients with Lynch syndrome.4,5 Moreover, colonoscopy is a preferable method as patients with LS have proximal colon tumors that may be missed by more limited screening tools.40 For patients with molecularly confirmed LS, colonoscopy should be performed every 1 to 2 years beginning at age 20 to 25 or 2–5 years before the earliest cancer diagnosis in a family member with a LS-associated colorectal cancer.4,5 The colonoscopy is offered annually after the age 40.4
Screening for endometrial cancer includes combination of annual/biannual transvaginal ultrasound or endometrial biopsy starting at age 25–35, as well as evaluating abnormal vaginal bleeding.5,6,8 However, evidence for endometrial cancer screening is less convincing, as ultrasound has a high false-positive rate and the efficiency of endometrial biopsy has not been evaluated.40
Although screening for ovarian cancer is controversial, pelvic examination, transvaginal ultrasound with serum Ca-125 levels should be conducted from an age of 30–35 years at 6 months or annually.23
Screening for breast cancer is the same as the population-based breast cancer screening recommendations.8 Women with LS and childbearing plans should be screened before attempting to become pregnant.8
Risk-Reducing Surgery
Risk reducing surgery is beyond the scope of this review, as there are many controversies in medical literature and further studies are needed in order to improve patients’ outcome. For instance, some authors stated that as LS cancers are associated with younger age of onset, risk-reducing surgeries are recommended between the ages of 35–45, or when childbearing is complete.3 Conversely, another study showed almost no benefit from risk-reducing surgery before 40 years of age and premenopausal bilateral salpingo-oophorectomy in MSH6 and PMS2 heterozygotes carries was not associated with increased mortality.41 Tzortzatos et al observed 41 women with LS who underwent risk-reducing surgery – 4 of them has occult complex atypical hyperplasia (2 patients) or endometrial cancer (two patients).42 Duenas et al conducted the largest study published up to the date in a single-institution. Authors concluded that risk-reducing surgeries are effective in decreasing the rate of colorectal, endometrial and ovarian cancer. They compared risk reducing gynecological surgery vs surveillance in the gynecological cohort and showed that the cumulative, incidence at 75 years was 11.2% vs 46.3% for endometrial cancer and 0% vs 12.7% for ovarian cancer. Moreover, the mortality cumulative incidence was 0% vs 52.7%.43 However, Seppala et al observed that uptake of risk-reducing gynecological surgery in LS aligned poorly regarding gene- and age-associated risk and did not correspond well with current clinical guidelines.44
“Site-Specific” Ovarian Cancer and Hereditary Breast–Ovarian Cancer Syndrome
Definition, Epidemiology and Pathogenesis
An approximately 23% of ovarian cancer is related to hereditary conditions.45 “Site-specific” ovarian syndrome is a variant of hereditary breast-ovarian cancer syndrome (HBOCS) instead of a specific syndrome, as no gene has been identified, which increased susceptibility to ovarian cancer alone.46,47 HBOCS is an autosomal dominant cancer syndrome, which is associated with an increased risk for female and male breast cancer, ovarian, tubal and primary peritoneal cancers.48,49
HBOCS is often associated with mutations in BRCA1, BRCA2 tumor suppressor genes, which are autosomal dominant with high penetrance.47 BRCA1 and BRCA2 were identified in the mid 1990s, and both are responsible for the repair of DNA double-strand breaks via homologous recombination repair to maintain genomic stability.13,50 BRCA1 and BRCA2 genes are located on chromosome 17q21 and chromosome 13q12.3, respectively.47 In the general population, the prevalence of these mutations is estimated at 1:300 to 1:500.31,48 An increased risk of carrying BRCA germline pathogenic variants was described in the Ashkenazi Jewish population, which has a carrier risk of 2.5% (1:40).13 Although these genes account for the majority of cancer cases of HBOCS (90–95%), recently other rare genes were identified such as CHEK2, RAD51C, RAD51C, BRIP1, PALB2, TP53 and BARD1.5,45 Homozygosis mutations did not occur in the BRCA1 gene, whereas rare diseases such as Fanconi’s anemia and Wilms’ tumor are related to homozygous BRCA2 mutation.47 In the general population, about 10% ovarian cancer cases and 3–5% of breast cancer cases are associated with BRCA1 or BRCA2 gene mutations.9 The risk for ovarian cancer occurrence varies between 39% and 63% for patients with BRCA1 mutation and 16.5–27% for BRCA2 mutation carriers, respectively. The risk of breast malignancy in individuals with BRCA1 and BRCA2 varies between 46–87% and 38–84%, respectively.48 According to the World Health Organization (WHO), the incidence rate for breast cancer is 46% and 52% for BRCA1 and BRCA2 carriers, respectively.31 The incidence rate for ovarian cancer is 12% and 6% for BRCA1 and BRCA2 carriers, respectively.31 The mean age at ovarian cancer diagnosis for BRCA1, 2 carriers is younger than for patients with sporadic cancer (54–63 years).46 The median age of ovarian cancer diagnosis for patients with BRCA1 carriers is 45 to 51 years and 57 to 63 years for BRCA2 carriers, respectively.13 Recently, growing evidence suggests that the fallopian tube epithelia as an etiological site for the development of high-grade serous ovarian cancer.51 Molecular and genetic data indicated that high-grade serous ovarian, tubal and peritoneal cancer had a similar origin and should be described collectively as HGSC.51 Therefore, BRCA1, 2 mutations are also responsible for tubal and peritoneal cancer. Sakurada et al estimated the presence of BRCA mutations among 40 patients with primary fallopian tube cancer. The authors found that 26.7% of patients had BRCA mutations.52
Additionally, it should be stressed that approximately 10–20% of women with HBOCS 10–20% of HBOC, which underwent germline BRCA1/2 testing harboring variants of unknown significance. These variants are defined as an alteration of a DNA nucleotide sequence, which shows uncertain outcomes – corresponding protein loss or the possible risk of developing diseases. Fanale et al concluded that verifying new potentially pathogenic BRCA1/BRCA2 could urge for their re-classification.53
Diagnosis
The diagnosis of BRCA1- and BRCA2-associated hereditary breast and ovarian cancer is estimated by finding a heterozygous germline pathogenic variant in BRCA1 or BRCA2 on molecular genetic testing.48 Molecular testing includes BRCA1 and BRCA2 gene panel and a multigene panel.48 Multigene panel is superior to only BRCA1/2 gene panel as it could estimate RAD51C, RAD51D, BRIP1, PALB2, BARD1, and the MMR gene mutations, which accounts for 4% of hereditary cancers.5 The DNA recombinase RAD51 is one of the main proteins, which involves in DNA repair by homologous recombination. RAD51 include 5 paralogs (RAD51B, RAD51C, RAD51D, XRCC2 and XRCC3). It was estimated that relative risk of ovarian and breast cancer for RAD51D mutation carriers was 6.30 and 1.32%, respectively. Meindl et al observed six monoallelic pathogenic mutations in RAD51C that conferred a high risk for ovarian and breast cancer. Rafnar et al concluded that BRIP1 behaved like a typical tumor suppressor gene in ovarian cancer.54–56 Therefore, multigene panel could be considered as a definitive diagnostic method.5 National comprehensive cancer network (NCCN) established the testing criteria for hereditary breast-ovarian cancer syndrome. In general, patients with personal history of breast or ovarian cancer at younger age or patients with family history (one or more than one close blood relatives with ovarian or breast cancer) should be tested. Moreover, patients with Ashkenazi Jewish ancestry, patients with relatives with male breast and prostatic cancer, triple negative breast cancer diagnosis at age <60 are also candidates for gene testing.57 Additionally, patients who meet the diagnostic criteria, but are negative with previous limited testing, should be considered for further testing.57 Generally, studies recommended BRCA testing for all patients with non-mucinous epithelial ovarian, tubal, and peritoneal cancers.5 The percentage of identifying BRCA1 or BRCA2 for patients with a history of primary breast cancer who subsequently develop ovarian cancer is approximately 20%.6 Sister, daughter or other relatives of women with positive BRCA mutations, should be considered for predictive testing and screening and preventive measures.6
Pathology
Ovarian cancer associated with this syndrome has a distinct histologic subtype.5 Currently, studies established that ovarian cancer associated with BRCA mutations arises subsequently in the tubal fimbria from occult serous tubal intraepithelial carcinoma.45 High-grade serous adenocarcinomas with prominent intraepithelial lymphocytes, marked nuclear atypia and abundant mitoses are typical histological features of BRCA-associated ovarian cancer.48 Some authors also include HBOCS with endometrioid ovarian adenocarcinoma.5,46 Mucinous, borderline ovarian cancers and nonepithelial tumors are uncommon for this hereditary syndrome.46
BRCA1-related breast cancers tend to be bilateral and of medullar histology with high histological nuclear grade and high percentage of negative estrogen and progesterone receptors. HER2 overexpression is not common.48,50 There is association between BRCA1 mutation and triple-negative breast cancer46–52 (Figure 4).
 |
Figure 4 Triple negative atypical medullary breast cancer associated with BRCA1 mutation. AMBC – atypical medullary breast cancer. Absence of estrogen receptor (ER), progesterone receptor (PR) and HER2 immunohistochemistry expression. |
Although some authors suggest the association between BRCA1 mutations and serous uterine cancer, more studies are needed to estimate the real risk.57
Screening
Ovarian cancer screening for BRCA mutation carriers has no adequate positive or negative predictive value. Moreover, it does not decrease morbidity and mortality and false-positive rates in premenopausal women are high.4,13 However, ovarian cancer screening for BRCA mutation carriers include transvaginal ultrasound combined with serum Ca-125 every 3 or 6 months starting either at age 35 or 5 to 10 years younger than the youngest affected member in the family.13,57
Breast cancer screening for BRCA carriers includes annual magnetic resonance imaging (MRI) with contrast starting at age 25 years.57
Risk-Reducing Surgery
The same as the LS, risk-reducing surgery in HBOCS is associated with many controversies and debates. Risk-reducing salpingo-oophorectomy (RRSO) is recommended between 35 and 40 years or after completion of child bearing. Evans et al observed three cases of occult ovarian cancer at surgery among 160 BRCA1/2 carriers, who underwent bilateral sapling oophorectomy.58 RRSO for BRCA2 mutation carriers should be performed between 40 and 45 years, as the onset of ovarian cancer for women with BRCA2 mutation is 8–10 years later than in women with BRCA1 mutation.57 Salpingectomy alone is not recommended.57 However, Liu et al concluded that there are many uncertainties, and shared decision-making should be used for risk-reducing surgery related to HBOCS. Authors also observed benefit of risk-reducing surgery for pathogenic variants in BRCA1/2 as well as BRIP1 and RAD51C/D.59 Moreover, Öfverholm et al found that overall mortality remained increased after risk-reducing surgery in BRCA1/2 carriers compared to the women in the general population.60
Cowden Syndrome
Definition, Epidemiology and Pathogenesis
Cowden syndrome (CS) is part of multiple hamartoma syndrome, which is caused by mutations in pentaerythritol tetranitrate (PTEN) gene, which has a role in the cell cycle regime. The gene is located in the long arm of the chromosome 10 and act as a tumor suppressor gene.1,5,61–67 Mutations in the PTEN gene cause chromosomal instability and uncontrolled proliferation of cells, which result in characteristic hamartomas and malignancies.67 Approximately 80% of patients with CS have mutations in the PTEN gene.65 Somatic mutations in PTEN are one of the most commonly altered genes in EC.61 Multiple hamartoma syndrome also includes Bannayan-Riley-Ruvalcaba syndrome, PTEN-related Proteus syndrome and proteus-like syndrome.66 Bannayan-Riley-Ruvalcaba syndrome and CS have overlapping phenotypic features.64 The incidence of Cowden syndrome is roughly 1 in 200,000. CS is an autosomal dominant disorder, which is associated with an increased risk of breast and endometrial cancer occurrence.62–64 Other conditions related to the syndrome include epithelial (predominantly follicular) thyroid cancer, facial trichilemmomas (hamartomas of the infundibulum of the hair follicle), fibrocystic breast disease, gastrointestinal hamartoma and macrocephaly.62 Mucocutaneous lesions (cutaneous papules, mucosal papillomas) are considered as pathognomonic finding.5 Other authors described colonic polyps and esophageal glycogenic acanthosis as pathognomic findings in CS.66 The lifetime risk of breast cancer development for PTEN carriers varies 77% to 85%, whereas endometrial cancer occurs at the ages of 30–49 years, and the lifetime risk is 13–28%.31,62 However, cases of endometrial cancer development associated with CS have been described in adolescent.63,64 Some authors suggested formal evaluation (germline PTEN mutation analysis) for CS to be considered for adolescent patients with endometrial cancer.63
Diagnosis, Testing and Screening
CS testing should be performed for individuals from a family with known PTEN mutation or with a personal history of Bannayan-Riley-Ruvalcaba syndrome.57 Additionally, lack of PTEN expression by immunochemistry may help in diagnosis.61 Early diagnosis of CS is essential in order to prevent the development of malignancies.65 NCCN guidelines include major and minor testing criteria for CS. Individuals have two or more major criteria (one must be macrocephaly), three major criteria (without macrocephaly), one major and three or more minor criteria or 4 and more minor criteria are candidates for CS testing. Criterias are available on NCCN site.57 Cases, which cover some, but not all diagnostic criteria are known as Cowden-like syndrome.31
Annual mammography or MRI with contrast should be considered for women with CS at age 30–35 years or 5–10 years before the earliest known family breast cancer.57,65 MRI with contrast is the preferable imaging modality as is it more sensitive than mammography. Moreover, it can identify invasive breast cancer earlier compared to mammography.57,65
Although endometrial cancer screening has no proven benefit and the value of surveillance is limited, it is recommended endometrial sampling and transvaginal ultrasound every 1–2 years after the age of 30–35 years or 5 years before age of earliest endometrial cancer in the family.62,65 Transvaginal ultrasound in premenopausal women is not recommended for screening.57
Pathology
Low-grade endometrioid carcinomas and rarely serous endometrioid carcinoma are associated with CS.31,68
Risk-Reducing Surgery
As there are many controversies, the risk-reducing bilateral mastectomy should be considered on an individual basis.61 NCCN recommends prophylactic hysterectomy after childbearing and after careful discussion with patients.57
Li–Fraumeni Syndrome
Definition, Pathogenesis and Epidemiology
Li-Fraumeni Syndrome (LFS), also known as sarcoma, breast, leukemia, and adrenal gland (SBLA) cancer syndrome, is an autosomal-dominant hereditary cancer caused by germline loss-of-function mutations in TP53 (missense mutations).62,69–72 The TP53 gene is located at the chromosome 17p13.1.69 Gene mutations are more likely to be inherited or can arise early in embryogenesis, but 14% of patients harbor de novo TP53 mutations.62,69 It is estimated that TP53 mutations occurred in approximately 70% of Li-Fraumeni syndrome and 40% of Li-Fraumeni–like families.69 Mutations of that gene lead to an estimated 50-fold risk over the general population of developing several types of cancer.70
The incidence of LFS is approximately one in 20,000 people with predominance in female population.70 Breast cancer is the most observed cancer and affects approximately 54% of women by the age of 70 years.62 Roughly, 3–8% of women with breast cancer and that syndrome are diagnosed under 30 years.57 Juvenile onset of cancers is often observed in LFS and more than half of cancers or soft-tissue sarcomas related to LFS occur before age 30.71 The most common malignancies associated with LFS are soft tissue sarcoma, osteosarcoma, adrenocortical carcinoma, brain tumors, premenopausal breast, leukemia, bronchoalveolar lung cancer, colorectal, and gastric cancer. Soft tissue sarcoma, brain tumors, and adrenocortical carcinoma occur in young patient and sarcomas account 36.8% of all cancers in childhood.62,70 Among them, the most common type are osteosarcomas (40.4%), sarcoma not otherwise specified (17%), rhabdomyosarcoma (16.5%), leiomyosarcoma (9.1%), and liposarcoma (4.9%).70 The risk of developing malignancy is approximately 40% by age 20, and more than 90% by age 70. More than half of these patients will develop any type of second tumor.72 Approximately 7–8% of patients will develop leiomyosarcoma of any site at a median age of 40 years.73 Although there are cases of endometrial or ovarian cancer and LFS, even in adolescent, the link to the syndrome is not definitely established.71,74
Diagnosis and Testing
Different authors had proposed many testing criteria for LFS through the years, but the most commonly used are the classic Li criteria and the Chompret criteria (Table 2).57,69,75 Individuals who meet LFS testing criteria or patients with pathogenic variant verified on tumor genomic testing should undergo germline testing.57 Additionally, an individual from a family with a known TP53 mutation should also be tested for LFS.57
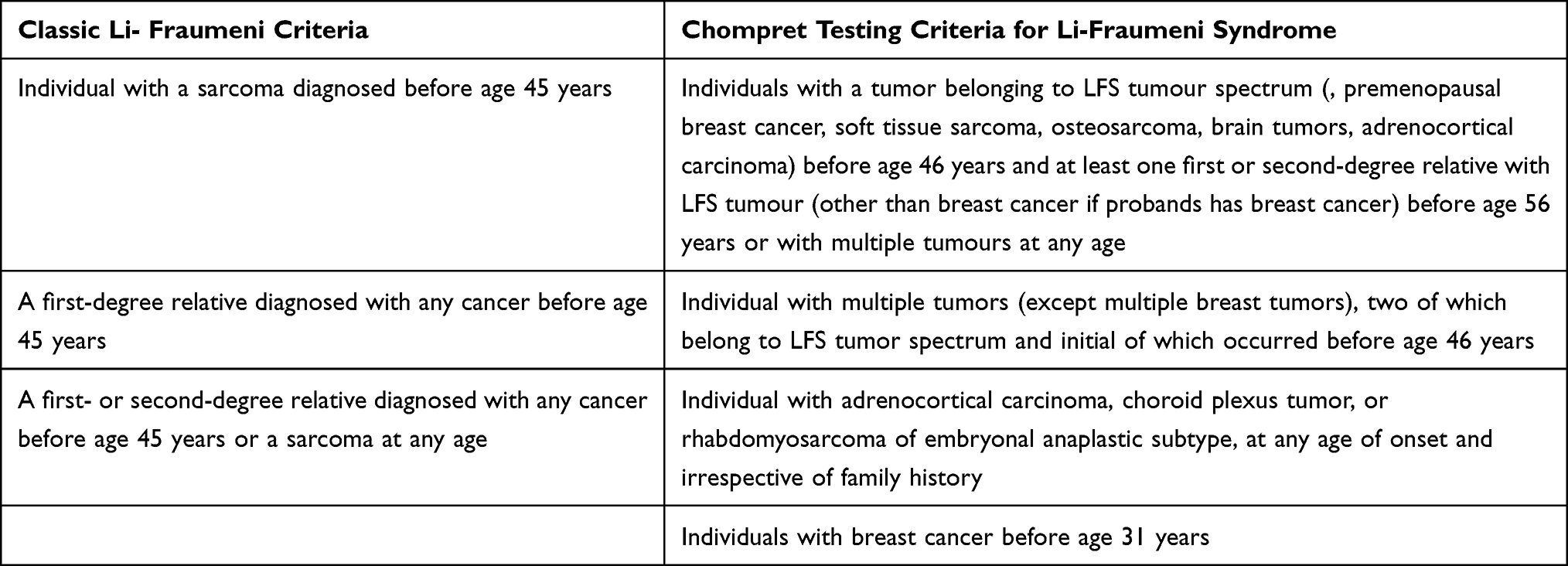 |
Table 2 Classic and Chompret Testing Criteria for Li-Fraumeni Syndrome |
Screening
Screening for LFS associated breast cancer starts after age 20 years and includes annual breast MRI with contrast and mammogram with eventual tomosynthesis.57 As preoperative diagnosis of uterine leiomyosarcoma has not been established yet, screening is not possible. However, for PT53 mutation carriers annual whole-body MRI is recommended.57
Pathology
Breast cancer associated with LFS tend to be mostly of ductal type and estrogen receptor positive with frequent HER2 co-expression. Prominent tumor infiltrating lymphocytes, nuclear atypia and high mitotic activity are observed.76 A case of small cell carcinoma of the ovary, hypercalcemic type and concurrent pleomorphic liposarcoma of the cervix associated with LFS has been described in medical literature.77
Risk-Reducing Surgery
As TP53 is frequently encountered in various cancers, it rarely has pathogenic variants in the germline.62 As it is difficult to establish the diagnosis by tumor profiling, a prophylactic bilateral mastectomy should be discussed after accurate medical and family history of each patient.62
Peutz-Jeghers Syndrome
Definition and Epidemiology
Peutz-Jeghers syndrome (PJS) is a rare autosomal dominant disease, which is characterized by gastrointestinal hamartomatous polyposis and mucocutaneous pigmentation in the oral mucosa, lips, wings of the nose, fingers and toes.78–80 Mucocutaneous pigmentation occurs in approximately 95% of patients with PFS, but tend to disappear with age.62,78 The syndrome is also associated with increased risk of malignancies of the gastrointestinal tract and extra-gastrointestinal organs with predominance of gynecologic ones (malignancies of breast, ovary, uterine cervix and endometrium).79 However, increased risk of leukemia and renal, pulmonary, bone cancer has been observed.81 The estimated incidence of the disease is 1 in 50,000–200,000 live births.79 Despite the lung carcinoma, which is more common in men, there is no significant difference of developing malignant tumors according to sex.79 However, females have earlier onset of malignant tumors than men.79 The relative risk of ovarian, cervical and uterine cancers associated with this syndrome is 18–21%, 10% and 9%, respectively.71 The estimated lifetime risk for breast cancer is approximately 24–54%.67
Pathogenesis
PJS is caused by a germline mutation in the tumor suppressor serine/threonine kinase 11 gene (STK11), which was previously known as liver kinase B1 (LKB1).67,81 The gene is located on chromosome 19p.13.3 and it is involved in cell cycle activities, mTOR (mammalian target of rapamycin) regulation, p53-dependent apoptosis and the regulation of vascular endothelial growth factor.67,80 STK11 is found in approximately 94% of affected families and 25% of newly diagnosed cases are due to sporadic de novo mutations.67
Pathology
PJS-associated gynecological cancers have characteristic histological types.71 Ovarian tumors related to PJS are mainly the sex-cord tumors with annular tubules (SCTAT) with morphologic features between granulosa cell and Sertoli cell tumors. SCTAT are observed in 21% of women with an average age of diagnosis 28 years.62 Other histological ovarian types that have been observed in this syndrome are Sertoli cell, mucinous, serous and mature teratoma.71 SCTAT associated PJS have a lower risk of malignant transformation than the sporadic ones.71 In the majority of cases, SCTAT with PJS was multifocal, bilateral, small and required microscopy to confirm the diagnosis, whereas SCATAT in general population are often unilateral and large.80
PJS-related cervical tumors include minimal deviation adenocarcinoma (MDA), lobular endocervical glandular hyperplasia (LEGH) and cervical gastric-type mucinous adenocarcinoma.31,62,82 The risk of MDA occurrence in PJS patients is estimated to be 15–30%.79 The average age of onset is 34–40 years, whereas that of MDA patients without PJS is 55 years.62,80 Actually, MDA is an adenocarcinoma of the uterine cervix, which shows gastric-type differentiation.78 LGEH has been identified adjacent to up to 50% of MDA and it is known as a precursor lesion of MDA.83 MDA is a highly aggressive tumor with a dismal prognosis. Recurrences occur in the majority of patients within 2 years of diagnosis.77 In Figure 5 is shown lobular endocervical glandular hyperplasia.
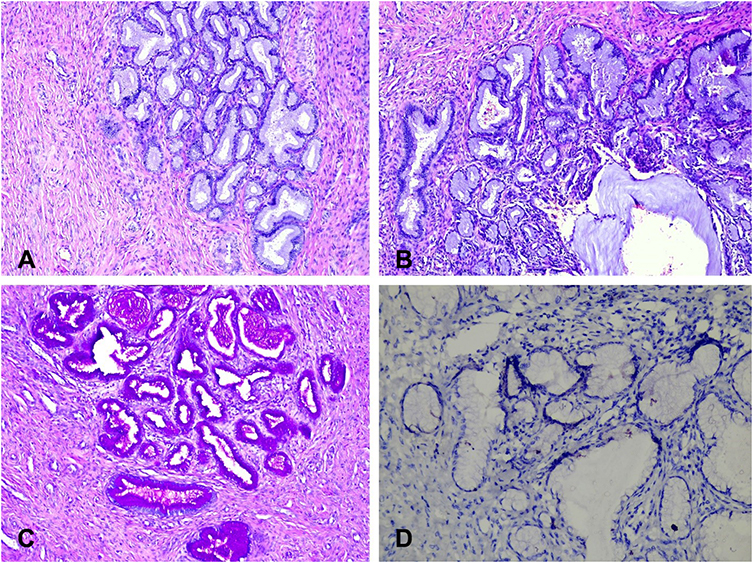 |
Figure 5 Histology of lobular endocervical glandular hyperplasia. (A) – Lobular endocervical glandular hyperplasia (HE x100). (B) – Glandular hyperplasia with lobular/acinar architecture composed of a central gland with cystic dilation, surrounded by smaller glands and cysts arranged in a floret-like pattern (HE x200).(C) – Lobular endocervical glandular hyperplasia (PAS reaction x100). PAS+ neutral mucin is observed in the cytoplasm of glandular cells. (D) – Lobular endocervical glandular hyperplasia (IHC-CEA x200). |
Screening
It is possible for cervical cancer associated with PJS syndrome and include Pap smear and pelvic examination annually after 18–20 years of age. Screening for breast cancer related to PJS includes mammography and MRI annually after 30 years of age. Adolescent with suspected PJS should undergo annual physical examination for precocious puberty after 8 years of age.57
Risk-Reducing Surgery
The benefits of RRS for gynecological cancers associated with Peutz-Jeghers syndrome have not been estimated.
Ataxia-Telangiectasia
Definition and Epidemiology
Ataxia-telangiectasia (AT) is an autosomal recessive hereditary disease, which include progressive cerebellar ataxia, oculomotor apraxia, choreoathetosis, sinopulmonary infections and oculocutaneous telangiectasia. The syndrome is also associated with radiosensitivity, primary immunodeficiency, sterility and high incidence of malignancies.31,84–86 The overall incidence of the AT syndrome is estimated to be 1 case per 40,000–100,000 live births.31,87
Pathogenesis
AT is caused by a punctual mutation in ATM gene localized in chromosome 11q22.3-23.1. This gene encodes a serine/threonine protein kinase that regulates the cell cycle and repair of DNA double-stranded breaks. Absence of ATM leads to genomic instability or DNA damage response, leading to carcinogenesis.31,84–86
AT and Gynecological Malignancies
Patients with AT syndrome have an increased risk of developing leukemias and lymphomas.81 According to the WHO, the ovary is the only gynecological site with neoplasms.31 However, different studies showed association between uterine, breast cancer and AT syndrome.88,89 Veiga- Fernandez et al reported for an increased risk (20–30%) for ATM gene carriers for developing breast cancer.88 Authors concluded that ATM carriers have a moderate-high risk of developing breast cancer.88
Ovarian tumors in AT syndrome patients are from non-epithelial origin – often dysgerminomas, and rarely gonadoblastomas and yolk sac tumors (Figures 6 and 7).31,84–86
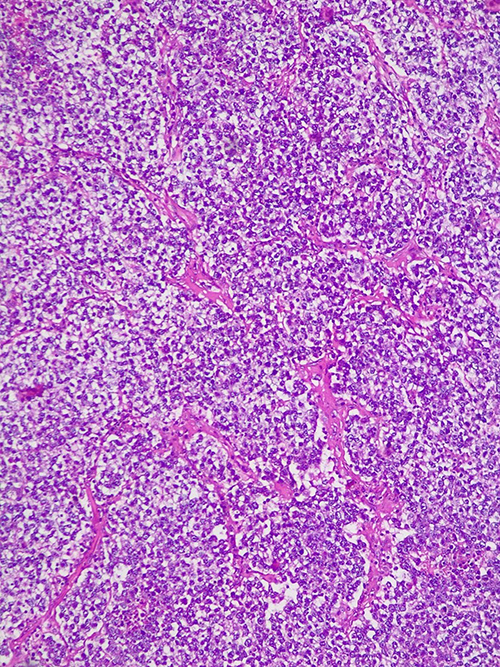 |
Figure 6 Histology features of ovarian dysgerminoma. Sheets and nests of monotonous tumor cells separated by thin fibrous septa. Cells are polygonal with well-defined cell borders and abundant clear cytoplasm (HE x 10). |
 |
Figure 7 Histological features of ovarian yolk sac tumor. Mesenchyme-like pattern – tumor cells with variable atypia scattered in edematous and myxomatous connective tissue (HE x 10). |
Risk-Reducing Surgery
It is not reported as patients with AT syndrome have an average life expectancy of 25 years.31,87
DICER1 Syndrome
Definition and Epidemiology
DICER1 syndrome, first described in pulmonary blastoma, is a rare genetic autosomal dominant condition, which is associated with increased risk of neoplasms – pleuropulmonary blastoma, Wilms tumor, renal sarcoma, pineoblastoma, pituitary blastoma, thyroid cancers, embryonal rhabdomyosarcoma of the uterine cervix, gynandroblastomas and Sertoli-Leydig ovarian cell tumor.32,90–96 It is estimated that approximately 1:2529–1:10,600 of people carry a pathogenic or likely pathogenic DICER1 variant.93,94
Pathogenesis
The syndrome is associated with mutation in the DICER1 gene, which is located on chromosome 14q32.13.90 The gene encodes an RNA endonuclease (Dicer), which is essential for processing microRNAs in the RNA interference pathway.31,90 The children of DICER1 carries have a 50% chance of inheriting the mutation as it is inherited as an autosomal dominant condition with decreased penetrance.93 Mosaicism for missense variants in the five hotspot codons in the RNase IIIb domain (E1705, D1709, G1809, D1810 and E1813) have been found. That explains the possible presence of tumors in adolescence without a DICER 1 germline mutation.91,92
Testing
Genetic counseling and testing is recommended for individuals with a personal history of at least one major or two minor indications for testing. Additionally, individuals, which have one minor indication and a family history of a major or minor indication, are also candidates for testing. Some of the major and minor indications are shown in Table 3. The majority of indications for DICER1 genetic counseling and testing surveillance strategies are well described by Schultz et al.93
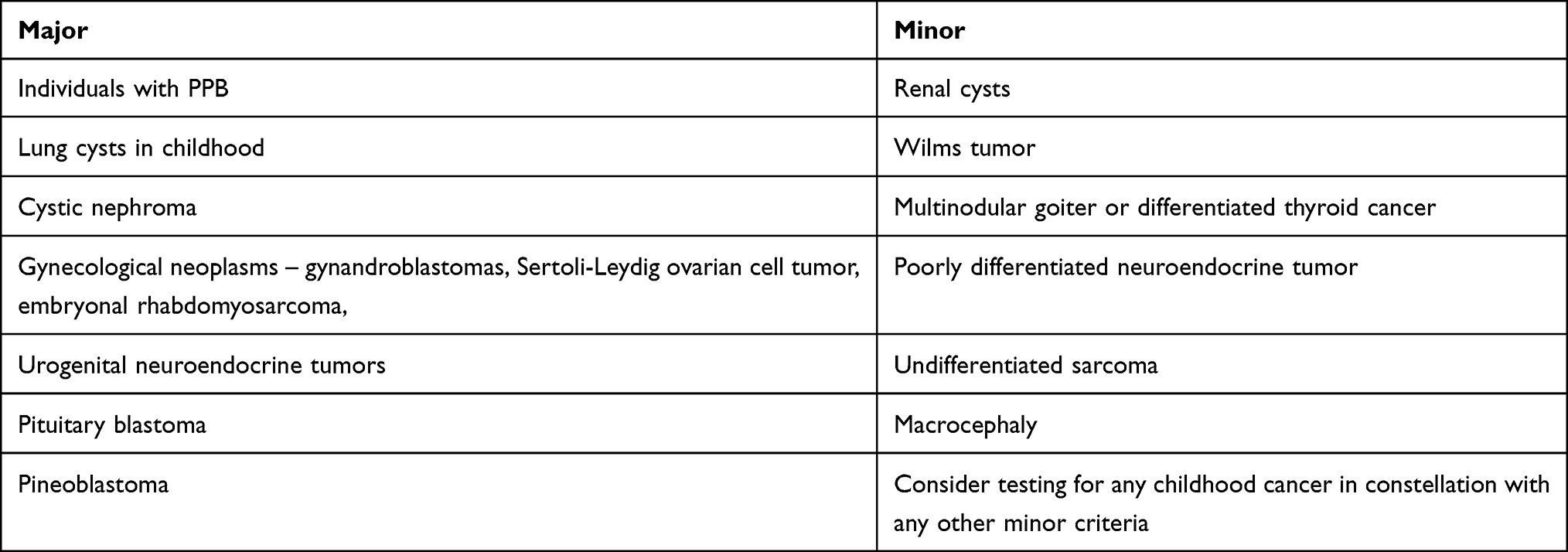 |
Table 3 Some of the Major and Minor Indications for DICER1 Genetic Counseling |
DICER1 Syndrome and Gynecological Malignancies
As mentioned above, the gynecological neoplasms associated with DICER1 syndrome are embryonal rhabdomyosarcoma of the uterine cervix, gynandroblastomas and Sertoli-Leydig ovarian cell tumor.90–97 Ovarian carcinoid tumor in a DICER1 carrier has also been described.97 Most of Sertoli-Leydig ovarian tumors are unilateral and diagnosed at early stage (Stage I).98 The majority of these tumors occur in adolescence. Young patients with gynandroblastomas and Sertoli-Leydig ovarian cell tumors often presented with virilization or amenorrhea.97 Therefore, gynecologists should be aware of a DICER1 syndrome and such symptoms in adolescence.
Surveillance Recommendations for Gynecological Malignancies
Patients and their parents should be familiar with possible gynecological syndromes due to DICER1-related tumours – hormonal symptoms including virilization, recurrent abdominal pain, and abdominal mass. That will provide an immediate imaging evaluation if clinically indicated. Schultz et al recommended pelvic and abdominal sonography every 6–12 months. Moreover, they stated that pelvic US should continue throughout adulthood. Surveillance imaging should continue until at least 40 years of age, although the ultimate time of imaging remains undetermined.93
Gonadal Dysgenesis
Definition and Epidemiology
Gonadal dysgenesis (GD) comprises a variety of sexual differentiation disorders with different somatic or genetic features.77,78 The syndrome includes the following subtypes – 46, XY pure (complete) GD (Swyer syndrome); 46, XX complete GD; 45, X complete GD (Turner syndrome), partial GD and mixed GD (asymmetric or atypical GD – 45, X/46, XY mosaicism).23,77,78 The syndrome affects one in 4500–5000 births.79 GD is associated with an increased risk of gonadal tumor formation.77
Gonadal Dysgenesis and Gynecological Cancers
Patients with GD are at increased risk of pure or mixed germ cell tumors.99–102 The incidence of gonadal tumors in Swyer syndrome is approximately 30%, of which the majority are gonadoblastomas. Gonadoblastomas occur in 96% of patients with GD and tend to be bilateral. Gonadoblastomas are considered to be in situ germ cell neoplasms, which are associated with up to 50–60% risk of developing dysgerminoma.99,103 Other germ cell tumors such as immature teratoma, seminomas, yolk sac tumors, embryonal carcinomas and choriocarcinoma can be encountered in patients with GD.99–103 Approximately 6% of patients with Turner syndrome have a mosaic karyotype (partial fragments of the Y chromosome).99 The majority of patients with Turner syndrome, who develop gonadal tumors are those with mosaic karyotype. In conclusion, gonadal tumors often occur in females with GD and Y-chromosome or Y-derived sequences in their genome.103 GD-related tumors have the same histological features as their non-syndromic counterparts.31
Pathogenesis
Genes DDX3Y and TSPY, which are localized on the proximal part of the short and long arms of the Y chromosome, are found in the majority of patients with gonadal dysgenesis and gonadoblastomas or germinoma.31,104
Diagnosis
Preoperative diagnosis of GD and ovarian masses is mandatory as it can help surgeons to evaluate the necessity of gonadectomy before surgery.102 Therefore, karyotyping or genetics testing should be evaluated in patients with amenorrhea, delayed pubertal development and pelvic masses.102,105 Capito et al stated that in cases of ovarian tumour, delayed pubertal development, moderate βHCG level and elevated FSH level, Swyer syndrome and dysgerminoma should be suspected.105 Authors concluded that preoperative karyotype analysis should be performed in such cases.105
Risk-Reducing Surgery
The risk of GD-associated malignancies occurs in adolescence and increases with age in patients with Y chromosome or Y-derived sequences. The optimal protocol in the management of DSD is controversial.106 According to some authors, prophylactic gonadectomy should be performed when the diagnosis of GD is established.99 Currently, the majority of authors stated that the optimal timing of gonadectomy should be decided by multiple factors including the subgroup of disorder (gonadal dysgenesis, androgen insensitivity syndrome, androgen synthesis defects, etc), age and development of secondary sex characteristics.103 Lui et al concluded that the optimal time for prophylactic gonadectomy is around age 16–18 when secondary sexual characteristics are developed. However, this step requires an accurate diagnostic evaluation in order to exclude premalignancy.103 Abaci et al stated that safe and well-accepted guidelines for prophylactic gonadectomy in patients with GD are needed, as there are only a few prospective studies.106
Tuberous Sclerosis
Tuberous sclerosis (TS) is an autosomal dominant neurocutaneous disorder, which is associated with the occurrence of benign congenital tumors in multiple organs. Neoplasms such as perivascular epithelioid cell differentiation (PEComa) including angiomyolipoma and lymphangioleiomyomatosis, can occur in association with TS. PEComas are an unusual group of rare mesenchymal neoplasms, composed of cells, which coexpress smooth muscle and melanocytic markers. These cells are also known as “perivascular epithelioid cells”. In the female genital tract, the most common location of PEComa is the uterine corpus, although other genital locations have been described (Figure 8).107–109 Although association between TS and PEComa is controversial, many case reports described PEComa in TS (Figure 8).107–110 Further larger studies are needed to prove the described association.
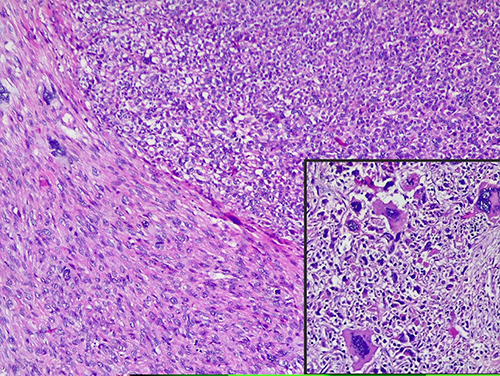 |
Figure 8 Histological features of uterine PEComa. Upper right - solid nodules of epithelioid cells with eosinophilic-to-pale cytoplasm, well-defined cellular membranes and mild-to-moderate nuclear pleomorphism. Some cells displayed large bizarre nuclei /insertion/. Lower left - spindle-cell component (HE x 10, insertion HE X 40). |
Multiple Endocrine Neoplasia Type I and Type II
Multiple endocrine neoplasia Type I (MEN I) is an autosomal dominant inherited syndrome, which is associated with parathyroid, anterior pituitary and enteropancreatic endocrine cell tumors.111,112 The MEN I gene is a tumor suppressor gene, which is located on chromosome 11q13. The MEN I gene encodes the protein menin.113,114 Tumor occurrence in MEN I is explained by the “two hits” hypothesis where women affected by familial MEN I inherit one wild-type and one mutant allele. Tumor development is associated with later mutation of the remaining wild-type allele.113 MEN II is an autosomal dominant inherited syndrome characterized by medullary thyroid carcinoma, with or without pheochromocytoma and hyperparathyroidism.111,112 MEN II is divided into three subtypes: MEN IIA (Sipple’s syndrome), MEN IIB and familial medullary thyroid carcinoma. MEN II is caused by the rearranged during transfection (RET) gene mutations.115 Numerous case studies reported the association between MEN I/II and ovarian carcinoids (Figure 9). Tamsen and Mazur reported a case of a woman with ovarian stromal carcinoid and MEN IIA. Authors concluded that MEN could be associated with ovarian carcinoids.116 Spaulding et al described a case of ependymoma and carcinoid tumor arising from a mature cyst teratoma in a patient with MEN I. Authors discussed that MEN I could alert the pathogenesis of ovarian teratomas as they undergo new neoplastic transformation.113 Recently, case reports of ovarian carcinoids and genetically confirmed cases of MEN I have been described.111,117 However, large prospective studies are needed to prove the association and to establish adequate diagnosis, screening and management of such patients.
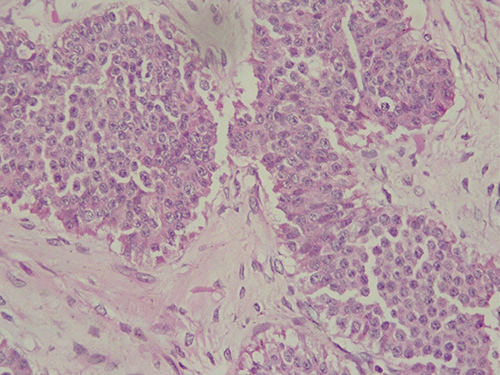 |
Figure 9 Histological features of ovarian carcinoid. Round to oval tumor cells with pink cytoplasm and centrally located nuclei with salt-and-pepper chromatin, arranged in solid nests (HE x 10). |
Rhabdoid Tumour Predisposition Syndrome
Rhabdoid tumour predisposition syndrome is characterized by germline mutations involving the SMARCB1 gene or the SMARCA4 gene. SMARCB1 gene mutations are associated with renal rhabdoid tumor, whereas SMARCA4 gene mutations are related with manifestation of rare gynecological neoplasm.10
Small cell carcinoma of the ovary, hypercalcemic type (SCCOHT) and undifferentiated uterine sarcoma (UUS) are rare and aggressive tumors of the female genital tract with a dismal prognosis.118,119 The median age of diagnosis of SCCOHT is 24 years. The majority of patients with SCCHOT presented at advanced stage of the disease, but even if the tumor is diagnosed at stage I, the 5-year OS is 30–55%.120–124 It has been reported that these tumors are oncogenically driven by somatic and/or germline mutations in SMARCA4.118,119 That gene encodes the BRG1 protein, which participates in SWI/SNF chromatin remodeling and functions as a gene activator or repressor.120–124 Connor et al described a case of a single family with an inherited germline SMARCA4 mutation. SCCOHT and UUS occurred in two different family members (a daughter and her mother).118 Authors also made a literature review and found SMARCA4 inactivating genomic alterations in 48 cases of SCCOHT and 17 cases of UUS. Germline mutations of patients with SCCOHT and UUS were 38% and 11.8%, respectively.118 Studies concluded that SCCOHT and USS are morphologically and genetically related and a germline link between these tumors is further established.118,120,121 Witkowski et al and Pejovic et al described cases of SCCOHT in families affected by germline SMARCA4.122,124 Therefore, genetic consultation should be offered for all patients with suspected SCCOHT and USS. If a germline SMARCA4 mutation is identified, genetic testing is recommended for all risk family members.118
There is no effective screening program for SMARCA4 carriers. Pejovic et al stated that serial pelvic ultrasound and serum calcium level measurements could be useful for patient with SCCOHT.124 Additionally, there are limited data about risk-reducing surgery.118,122,123 In medical literature, there are two case reports of risk-reducing surgery in patients diagnosed with SCCOHT and SMARCA4 mutation.122,124 Pejovic et al reported a case of 13-year-old pubertal Caucasian girl with a family history of SCCOHT. Her mother and maternal aunt were diagnosed with SCCOHT prior to the identification of the SMARCA4 mutations and died soon after the diagnosis. The variant was maternally inherited as the patient was found to carry the same mutation. The patient underwent bilateral salpingo-oophorectomy at the age 13 after patient family consultation.123 Berchuck et al described a case of a 33-year-old multipara patient, which had three female relative previously diagnosed with SCCOHT. After careful explanation and consulting with the patient, she underwent total laparoscopic hysterectomy with bilateral-salpingo-oophorectomy.123 Authors concluded that prophylactic oophorectomy could prevent SCCOHT in carriers of germline SMARCA4 mutations.123 Berchuck et al stated that patients who desire children might be best served by oocyte cryopreservation prior to prophylactic oophorectomy.123 Further studies are needed to provide adequate risk reducing surgery guidelines.
Hereditary Diffuse Gastric Cancer
Hereditary diffuse gastric cancer (HDGC) is an autosomal dominant cancer syndrome, which is characterized by susceptibility to diffuse gastric cancer (DGC) and lobular breast cancer (LBC).125–127 Its incidence is approximately 5–10 per 100,000 births.125 HDGC is caused by inactivating germline mutations in the CDH1 tumour suppressor gene. It is the coding gene for E-cadherin and it is located on chromosome 16q22.1.125–127 The majority of cases of DGC and LBC in CDH1 carriers occur before age 40 years.127 The estimated cumulative risk of gastric and breast cancers in females by age 80 years is 56% and 42%, respectively.127 CDH1 carriers with HDGC have lower 1- and 5-years survival rates compared to HDGC patients without germline CDH1 mutations. LBC could be the first manifestation of HDGC.126,128 Benusiglio et al even recommended a revision of the definition of HDGC and proposed for consideration the term ‘Hereditary Diffuse Gastric and Lobular Breast Cancer’.128 Corso et al observed that CDH1 germline mutations are mutually exclusive with BRCA1/2 alterations.129
Testing
Blair et al proposed a panel of 3 family criteria and 9 individual criteria for genetic testing (Table 4).125
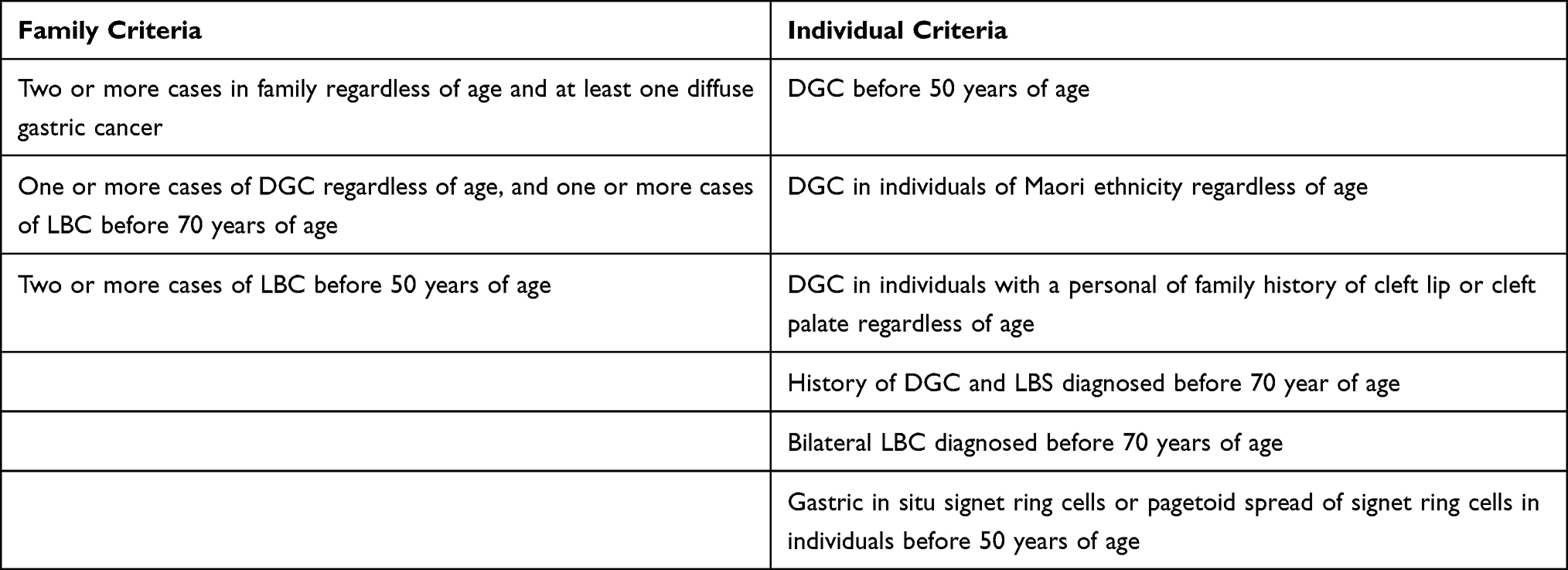 |
Table 4 Genetic Testing Criteria for HGC |
Screening and Risk Reducing Surgery
Although prophylactic gastrectomy carries a 3–6% mortality rate and a 100% morbidity rate, it is recommended for CDH1 carriers in their 20 or 30s. If gastrectomy is contraindicated or the patient refuses surgery, an endoscopic surveillance should be considered as an option. Prophylactic gastrectomy is not recommended after 70 years of age.125–127 Screening recommendation for breast cancer includes breast self-examinations after 20 years of age, clinical breast examinations every six month after 30 years of age and MRI breast examination annually starting at age 30. MRI is the preferable imaging modalities as the sensitivity of detecting lobular breast cancer by mammography is low.126,127 Prophylactic bilateral mastectomy is not routinely recommended as there are very limited number of CDH1 mutation carriers who underwent prophylactic mastectomy.126,127 However, after genetic counseling, this surgical procedure could be offered accordingly with the patient.129 In cases of LBC in CDH1mutation carriers, contralateral mastectomy could be considered.126,130
MUTYH-Associated Polyposis
MUTYH-associated polyposis is an autosomal recessive condition, which is characterized by germline mutations in the mutY homolog (MUTYH) base excision repair gene. The gene is located on the short arm of chromosome 1 and encodes MYH glycosylase enzyme, which is associated with DNA damage repair. The cumulative risk of colorectal cancer is approximately 63% by age 60. Both monoallelic and biallelic MUTYH carriers are at increased risks of other cancer development, despite its autosomal recessive pattern.131 Biallelic MUTYH mutations are inherited from both parents and occur in 0.01–0.04% of the Caucasian population. The estimated risk of ovarian cancer for biallelic MUTHYH mutation carriers is 14%.132 Win et al estimated that biallelic MUTYH mutation carriers had a 17-fold increased risk of ovarian cancer, compared with the general population.132 Monoallelic carriers do not demonstrate an elevated risk of ovarian cancer, but according to some authors there is an increased risk of endometrial and breast cancer.131,132 However, further studies are warranted to prove the association. The MUTYH mutated ovarian tumors are often of serous histology, but the MUTYH mutation and mechanism for oncogenesis demonstrated that multiple ovarian cancer histologies could be related to such a mutation.131–134 More studies are needed to determine the optimal screening and risk-reducing surgery protocols.
The most important information of hereditary gynecologic cancer syndromes in the present article is summarized in Table 5.4–10,20–47,135–139
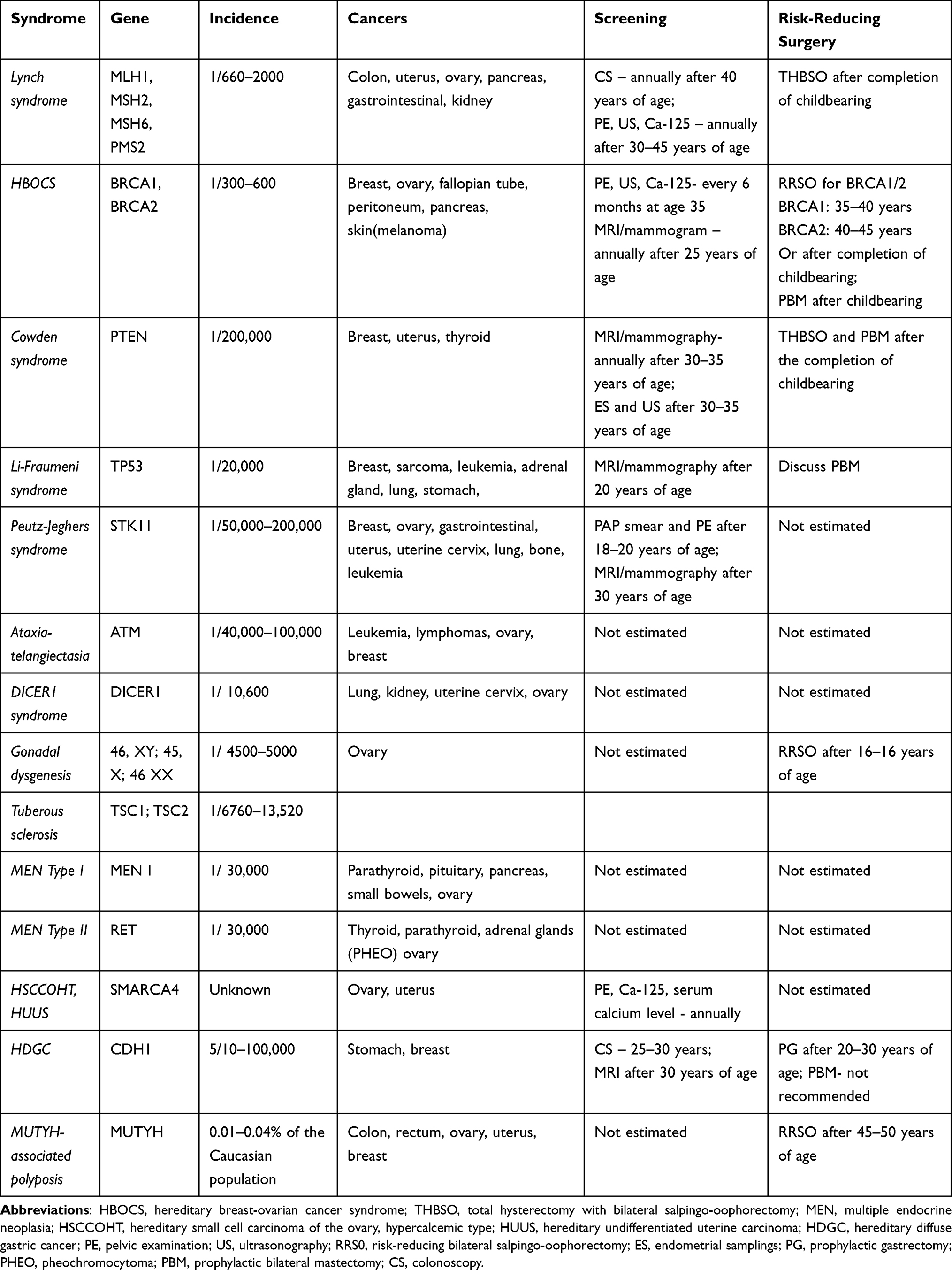 |
Table 5 Hereditary Gynecologic Cancer Syndromes |
Conclusion
Hereditary gynecologic cancer syndromes are a huge group of syndromes, where a tremendous progress has been made over the last decade in the evaluation and management. The availability and improvement of clinical genetic testing allows us to identify and consult patients and their families, which are at high risk of gynecologic cancer occurrence. Moreover, recommendations regarding epidemiology, pathology, screening and risk-reducing surgery have been developed for some gynecologic cancer syndromes. However, there also are many other questions that still need to be addressed regarding screening and risk-reducing surgery for some of the rarest inherited gynecologic cancers. Additionally, advances of molecular biology and genetic testing may improve treatment of these syndromes. Oncogynecologists should look beyond the horizon and start playing a greater role in the management of these high-risk patients. It must be underlined the importance of multidisciplinary management for such patients in order to achieve improvement of the quality of care and life.
Data Sharing Statement
The authors declare that all related data are available concerning researchers by the corresponding author’s email.
Acknowledgments
The authors wish to thank Dr Dimitar Metodiev and Dr Margarita Nikolova for their technical support.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This research received no external funding.
Disclosure
The authors declare no conflicts of interest for this work.
References
1. Devlin LA, Morrison PJ. Inherited gynaecological cancer syndromes. Obstetr Gynaecolog. 2008;10(1):9–15. doi:10.1576/toag.10.1.009.27371
2. Daniels MS, Lu KH. Genetic predisposition in gynecologic cancers. Semin Oncol. 2016;43(5):543–547. doi:10.1053/j.seminoncol.2016.08.005
3. Chen L, Blank SV, Burton E, Glass K, Penick E, Woodard T. Reproductive and hormonal considerations in women at increased risk for hereditary gynecologic cancers: Society of Gynecologic Oncology and American Society for reproductive medicine evidence-based review. Gynecol Oncol. 2019;155:508–514. doi:10.1016/j.ygyno.2019.06.017
4. Kehoe SM, Kauff ND. Screening and prevention of hereditary gynecologic cancers. Semin Oncol. 2007;34(5):406–410. doi:10.1053/j.seminoncol.2007.07.004
5. Randall LM, Pothuri B. The genetic prediction of risk for gynecologic cancers. Gynecol Oncol. 2016;141(1):10–16. doi:10.1016/j.ygyno.2016.03.007
6. Lu KH. Hereditary gynecologic cancers: differential diagnosis, surveillance, management and surgical prophylaxis. Fam Cancer. 2007;7(1):53–58. doi:10.1007/s10689-007-9144-x
7. Brown GJE, St. John DJB, Macrae FA, Aittomäki K. Cancer risk in young women at risk of hereditary nonpolyposis colorectal cancer: implications for gynecologic surveillance. Gynecol Oncol. 2001;80(3):346–349. doi:10.1006/gyno.2000.6065
8. Idos G, Valle L. Lynch syndrome. In: Adam MP, Ardinger HH, Pagon RA, editors. GeneReviews®. Seattle: University of Washington, Seattle; 2004:1993–2021.
9. Mutch D, Denny L, Quinn M. Hereditary gynecologic cancers. Int Jurnal of Gynecol Obstetr. 2013;124(3):189–192. doi:10.1016/j.ijgo.2013.12.001
10. Shanbhogue KP, Prasad AS, Ucisik-Keser FE, Katabathina VS, Morani AC. Hereditary ovarian tumour syndromes: current update on genetics and imaging. Clin Radiol. 2021;76:
11. Nakamura K, Banno K, Yanokura M, et al. Features of ovarian cancer in Lynch syndrome (Review). Mol Clin Oncol. 2014;2(6):909–916. doi:10.3892/mco.2014.397
12. Bartosch C, Clarke B, Bosse T. Gynaecological neoplasms in common familial syndromes (Lynch and HBOC). Pathology. 2018;50(2):222–237. doi:10.1016/j.pathol.2017.10.010
13. Davidson SA. Hereditary gynecologic cancer syndromes. Postgrad Obstetr Gynecol. 2010;30(1):1–7. doi:10.1097/01.pgo.0000364891.73439.70
14. Goetsch AL, Kimelman D, Woodruff TK. Hereditary gynecologic cancer predisposition syndromes. In: Fertility Preservation and Restoration for Patients with Complex Medical Conditions. Springer; 2017:7–18. doi:10.1007/978-3-319-52316-3_2
15. Lu KH, Dinh M, Kohlmann W, et al. Gynecologic cancer as a “sentinel cancer” for women with hereditary nonpolyposis colorectal cancer syndrome. Obstet Gynecol. 2005;105(3):569–574. doi:10.1097/01.aog.0000154885.44002.ae
16. Makabe T, Yamagami W, Hirasawa A, et al. Incidence of germline variants in lynch syndrome-related genes among Japanese endometrial cancer patients aged 40 years or younger. Int J Clin Oncol. 2021;26:1767–1774. doi:10.1007/s10147-021-01953-5
17. Cohen SA, Pritchard CC, Jarvik GP. Lynch syndrome: from screening to diagnosis to treatment in the era of modern molecular oncology. Annu Rev Genomics Hum Genet. 2019;20(1):293–307. doi:10.1146/annurev-genom-083118-015406
18. Post CCB, Stelloo E, Smit VT, et al. Prevalence and prognosis of lynch syndrome and sporadic mismatch repair deficiency in endometrial cancer. J Natl Cancer Inst. 2021;113(9):1212–1220. doi:10.1093/jnci/djab029
19. Dominguez-Valentin M, Plazzer J-P, Sampson JR, et al. No difference in penetrance between truncating and missense/aberrant splicing pathogenic variants in MLH1 and MSH2: a prospective lynch syndrome database study. J Clin Med. 2021;10:2856. doi:10.3390/jcm10132856
20. Findeis-Hosey J, Gonzalez RS. Lynch syndrome. Available from: https://www.pathologyoutlines.com/topic/colontumorlynch.html.
21. Concin N, Matias-Guiu X, Vergote I, et al. ESGO/ESTRO/ESP guidelines for the management of patients with endometrial carcinoma. Int J Gynecol Cancer. 2020. doi:10.1136/ijgc-2020-002230
22. Møller P, Seppälä T, Bernstein I, et al. Cancer incidence and survival in Lynch syndrome patients receiving colonoscopic and gynaecological surveillance: first report from the prospective Lynch syndrome database. Gut. 2017;66:464–472. doi:10.1136/gutjnl-2015-309675
23. Tanakaya K. Current clinical topics of Lynch syndrome. Int J Clin Oncol. 2018;24:1013–1019. doi:10.1007/s10147-018-1282-7
24. Vasen H, Watson P, Mecklin J, Lynch H. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative Group on HNPCC☆. Gastroenterology. 1999;116(6):1453–1456. doi:10.1016/s0016-5085(99)
25. Umar A, Boland CR, Terdiman JP, et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst. 2004;96(4):261–268. doi:10.1093/jnci/djh034
26. Duraturo F, Liccardo R, De Rosa M, Izzo P. Genetics, diagnosis and treatment of Lynch syndrome: old lessons and current challenges (Review). Oncol Lett. 2019. doi:10.3892/ol.2019.9945
27. Buchanan DD, Tan YY, Walsh MD, et al. Tumor mismatch repair immunohistochemistry and DNA MLH1 methylation testing of patients with endometrial cancer diagnosed at age younger than 60 years optimizes triage for population-level germline mismatch repair gene mutation testing. J Clin Oncol. 2014;32:90–100. 45. doi:10.1200/JCO.2013.51.2129
28. Mills AM, Liou S, Ford JM, Berek JS, Pai RK, Longacre TA. Lynch syndrome screening should be considered for all patients with newly diagnosed endometrial cancer. Am J Surg Pathol. 2014;38:1501–1509. doi:10.1097/PAS.0000000000000321
29. Aarnio M. Clinicopathological features and management of cancers in lynch syndrome. Patholog Res Int. 2012;2012:1–6. doi:10.1155/2012/350309
30. Walsh S. The pathology of lynch syndrome. Diagn Histopathol. 2015;21(4):161–164. doi:10.1016/j.mpdhp.2015.04.007
31. WHO. Female Genital Tumors, World Health Organization Classification of Tumours.
32. Zhao S, Chen L, Zang Y, et al. Endometrial cancer in Lynch syndrome. Int J Cancer. 2021. doi:10.1002/ijc.33763
33. Broaddus RR, Lynch HT, Chen LM, et al. Pathologic features of endometrial carcinoma associated with HNPCC: a comparison with sporadic endometrial carcinoma. Cancer. 2006;106:87–94. doi:10.1002/cncr.21560
34. Westin SN, Lacour RA, Urbauer DL, et al. Carcinoma of the lower uterine segment: a newly described association with Lynch syndrome. J Clin Oncol. 2008;26:5965–5971. doi:10.1200/JCO.2008.18.6296
35. Jensen KC, Mariappan MR, Putcha GV, et al. Microsatellite instability and mismatch repair protein defects in ovarian epithelial neoplasms in patients 50 years of age and younger. Am J Surg Pathol. 2008;32(7):1029–1037. doi:10.1097/PAS.0b013e31816380c4
36. Ryan NAJ, Evans DG, Green K, Crosbie EJ. Pathological features and clinical behavior of Lynch syndrome-associated ovarian cancer. Gynecol Oncol. 2017;144(3):491–495. doi:10.1016/j.ygyno.2017.01.005
37. Sheehan M, Heald B, Yanda C, et al. Investigating the link between lynch syndrome and breast cancer. Eur J Breast Health. 2020;16(2):106–109. doi:10.5152/ejbh.2020.5198
38. Walsh MD, Buchanan DD, Cummings MC, et al. Lynch syndrome-associated breast cancers: clinicopathologic characteristics of a case series from the colon cancer family registry. Clin Cancer Res. 2010;16(7):2214–2224. doi:10.1158/1078-0432.CCR-09-3058
39. Yee CJ, Roodi N, Verrier CS, Parl FF. Microsatellite instability and loss of heterozygosity in breast cancer. Cancer Res. 1994;54:1641–1644.
40. Meyer LA, Broaddus RR, Lu KH. Endometrial cancer and Lynch syndrome: clinical and pathologic considerations. Cancer Control. 2009;16(1):14–22. doi:10.1177/107327480901600103
41. Dominguez-Valentin M, Crosbie EJ, Engel C, et al. Risk-reducing hysterectomy and bilateral salpingo-oophorectomy in female heterozygotes of pathogenic mismatch repair variants: a prospective lynch syndrome database report. Genet Med. 2021;23:705–712. doi:10.1038/s41436-020-01029-1
42. Tzortzatos G, Andersson E, Soller M, et al. The gynecological surveillance of women with Lynch syndrome in Sweden. Gynecol Oncol. 2015;138(3):717–722. doi:10.1016/j.ygyno.2015.07.016
43. Dueñas N, Navarro M, Teulé À, et al. Assessing effectiveness of colonic and gynecological risk reducing surgery in lynch syndrome individuals. Cancers. 2020;12(11):3419. doi:10.3390/cancers12113419
44. Seppälä TT, Dominguez-Valentin M, Crosbie EJ, et al. Uptake of hysterectomy and bilateral salpingo-oophorectomy in carriers of pathogenic mismatch repair variants: a prospective lynch syndrome database report. Eur J Cancer. 2021;148:124–133. doi:10.1016/j.ejca.2021.02.022
45. Toss A, Tomasello C, Razzaboni E, et al. Hereditary ovarian cancer: not OnlyBRCA1 and 2 genes. Biomed Res Int. 2015;2015:1–11. doi:10.1155/2015/341723
46. Prat J, Ribé A, Gallardo A. Hereditary ovarian cancer. Hum Pathol. 2005;36(8):861–870. doi:10.1016/j.humpath.2005.06.006
47. Russo A, Calò V, Bruno L, Rizzo S, Bazan V, Di Fede G. Hereditary ovarian cancer. Crit Rev Oncol/ Hematol. 2009;69(1):28–44. doi:10.1016/j.critrevonc.2008.06.003
48. Petrucelli N, Daly MB, Pal T, et al. BRCA1- and BRCA2-associated hereditary breast and ovarian cancer. In: Adam MP, Ardinger HH, Pagon RA, editors. GeneReviews®. Seattle: University of Washington, Seattle; 1998:1993–2021.
49. Nakonechny QB, Gilks CB. Ovarian cancer in hereditary cancer susceptibility syndromes. Surg Pathol Clin. 2016;9(2):189–199. doi:10.1016/j.path.2016.01.003
50. Yoshida R. Hereditary breast and ovarian cancer (HBOC): review of its molecular characteristics, screening, treatment, and prognosis. Breast Cancer. 2020. doi:10.1007/s12282-020-01148-2
51. George SH, Garcia R, Slomovitz BM. Ovarian cancer: the fallopian tube as the site of origin and opportunities for prevention. Front Oncol. 2016;6:108. doi:10.3389/fonc.2016.00108
52. Sakurada S, Watanabe Y, Tokunaga H, et al. Clinicopathologic features and BRCA mutations in primary fallopian tube cancer in Japanese women. Jpn J Clin Oncol. 2018;48(9):794–798. doi:10.1093/jjco/hyy095
53. Fanale D, Pivetti A, Cancelliere D, et al. BRCA1/2 variants of unknown significance in hereditary breast and ovarian cancer (HBOC) syndrome: looking for the hidden meaning. Crit Rev Oncol Hematol. 2022;172:103626. doi:10.1016/j.critrevonc.2022.103626
54. Loveday C, Turnbull C, Ramsay E, Hughes D, Ruark E, Rahman N. Germline mutations in RAD51D confer susceptibility to ovarian cancer. Nat Genet. 2011;43(9):879–882. doi:10.1038/ng.893
55. Meindl A, Hellebrand H, Wiek C, et al. Germline mutations in breast and ovarian cancer pedigrees establish RAD51C as a human cancer susceptibility gene. Nat Genet. 2010;42(5):410–414. doi:10.1038/ng.569
56. Rafnar T, Gudbjartsson DF, Sulem P, et al. Mutations in BRIP1 confer high risk of ovarian cancer. Nat Genet. 2011;43(11):1104–1107. doi:10.1038/ng.955
57. National Comprehensive Cancer Network. Log in page. Available from: https://www.nccn.org/professionals/physician_gls/pdf/genetics_bop.pdf.
58. Evans DG, Clayton R, Donnai P, Shenton A, Lalloo F. Risk-reducing surgery for ovarian cancer: outcomes in 300 surgeries suggest a low peritoneal primary risk. Eur J Human Genet. 2009;17(11):1381–1385. doi:10.1038/ejhg.2009.60
59. Liu YL, Breen K, Catchings A, et al. Risk-reducing bilateral salpingo-oophorectomy for ovarian cancer: a review and clinical guide for hereditary predisposition genes [published online ahead of print, 2021 Sep 28]. JCO Oncol Pract. 2021;OP2100382. doi:10.1200/OP.21.00382
60. Öfverholm A, Einbeigi Z, Wigermo A, Holmberg E, Karsson P. Increased overall mortality even after risk reducing surgery for BRCA-positive women in Western Sweden. Genes. 2019;10(12):1046. doi:10.3390/genes10121046
61. Manning-Geist BL, Gatius S, Liu Y, et al. Diagnosis and management of an endometrial cancer patient with Cowden syndrome. Gynecol Oncol. 2021;163:14–21. doi:10.1016/j.ygyno.2021.08.00
62. Ueki A, Hirasawa A. Molecular features and clinical management of hereditary gynecological cancers. Int J Mol Sci. 2020;21(24):9504. doi:10.3390/ijms21249504
63. Baker WD, Soisson AP, Dodson MK. Endometrial cancer in a 14-year-old girl with Cowden syndrome: a case report. J Obstetr Gynaecol Res. 2012;39(4):876–878. doi:10.1111/j.1447-0756.2012.02052.x
64. Schmeler KM, Daniels MS, Brandt AC, Lu KH. Endometrial cancer in an adolescent: a possible manifestation of Cowden syndrome. Obstet Gynecol. 2009;114(2 Pt 2):477–479. doi:10.1097/AOG.0b013e31819dade8
65. Heaney RM, Farrell M, Stokes M, Gorey T, Murray D. Cowden syndrome: serendipitous diagnosis in patients with significant breast disease. Case series and literature review. Breast J. 2016;23(1):90–94. doi:10.1111/tbj.12691
66. El-Halaby A, Gonzalez R. Cowden syndrome. Available from: https://www.pathologyoutlines.com/topic/colontumorcowden.html.
67. Katabathina VS, Menias CO, Khanna L, et al. Hereditary gastrointestinal cancer syndromes: role of imaging in screening, diagnosis, and management. RadioGraphics. 2019;180185. doi:10.1148/rg.2019180185
68. Mahdi H, Mester JL, Nizialek EA, Ngeow J, Michener C, Eng C. Germline PTEN, SDHB-D, and KLLN alterations in endometrial cancer patients with Cowden and Cowden-like syndromes: an international, multicenter, prospective study. Cancer. 2015;121(5):688–696. doi:10.1002/cncr.29106
69. George A, Gheena S. Li- Fraumeni syndrome- a review. Int J Sci Eng Dev Res. 2017;2(8):154–159.
70. Farid M, Ngeow J. Sarcomas associated with genetic cancer predisposition syndromes: a review. Oncologist. 2016;21(8):1002–1013. doi:10.1634/theoncologist.2016-0079
71. Neto N, Cunha TM. Do hereditary syndrome-related gynecologic cancers have any specific features? Insights Imaging. 2015;6(5):545–552. doi:10.1007/s13244-015-0425-x
72. Gupta A, Malkin D. Sarcomas and cancer predisposition syndromes. Available from: http://sarcomahelp.org/articles/sarcoma-predisposition-syndromes.html.
73. George S, Serrano C, Hensley ML, Ray-Coquard I. Soft tissue and uterine leiomyosarcoma. J Clin Oncol. 2018;36(2):144–150. doi:10.1200/JCO.2017.75.9845
74. Clark MB, Menderes G, Azodi M, Finberg K, Canosa S, Parkash V. Endometrial carcinoma as the presenting malignancy in an 18-year-old patient with Li-Fraumeni syndrome. Gynecol Oncol. 2016;141:183–184. doi:10.1016/j.ygyno.2016.04.473
75. Bougeard G, Renaux-Petel M, Flaman J-M, et al. Revisiting Li-Fraumeni syndrome from TP53 mutation carriers. J Clin Oncol. 2015;33(21):2345–2352. doi:10.1200/jco.2014.59.5728
76. Kuba MG, Lester SC, Bowman T, et al. Histopathologic features of breast cancer in Li–Fraumeni syndrome. Mod Pathol. 2020. doi:10.1038/s41379-020-0610-4
77. Andon B, Hagemann IS, Maluf HM, Pfeifer JD, Al-Kateb H. Association of Li-Fraumeni syndrome with small cell carcinoma of the ovary, hypercalcemic type and concurrent pleomorphic liposarcoma of the cervix. Int J Gynecol Pathol. 2017;36(6):593–599. doi:10.1097/PGP.0000000000000365
78. Meserve EEK, Nucci MR. Peutz-Jeghers Syndrome. Surg Pathol Clin. 2016;9(2):243–268. doi:10.1016/j.path.2016.01.006
79. Ishida H, Tajima Y, Gonda T, Kumamoto K, Ishibashi K, Iwama T. Update on our investigation of malignant tumors associated with Peutz–Jeghers syndrome in Japan. Surg Today. 2016;46(11):1231–1242. doi:10.1007/s00595-015-1296-y
80. Banno K, Kisu I, Yanokura M, et al. Hereditary gynecological tumors associated with Peutz-Jeghers syndrome (Review). Oncol Lett. 2013;6(5):1184–1188. doi:10.3892/ol.2013.1527
81. Chen H-Y, Jin X-W, Li B-R, et al. Cancer risk in patients with Peutz–Jeghers syndrome: a retrospective cohort study of 336 cases. Tumor Biol. 2017;39(6):101042831770513. doi:10.1177/1010428317705131
82. Kim Y, Kim EY, Kim TJ, et al. A rare case of gastric-type mucinous adenocarcinoma in a woman with Peutz-Jeghers syndrome. Obstetr Gynecol Sci. 2019;62(6):474–477. doi:10.5468/ogs.2019.62.6.474
83. Turashvili G. Gastric type adenocarcinoma. PathologyOutlines.com website. Available from: https://www.pathologyoutlines.com/topic/cervixGAS.html.
84. Gálvez-Cuitiva EA, Ridaura-Sanz C, Yamazaki-Nakashimada MA, et al. Germ cell ovarian tumor in an adolescent with ataxia-telangiectasia. Acta Pediatr Mex. 2015;36(6):464–472. doi:10.18233/APM36No6pp464-472
85. Pecorelli S, Sartori E, Favalli G, Ugazio AG, Gastaldi A. Ataxia-telangiectasia and endodermal sinus tumor of the ovary: report of a case. Gynecol Oncol. 1988;29(2):240–244. doi:10.1016/0090-8258(88)90219-3
86. Cao J, Tan RYC, Li S-T, et al. Identifying ataxia-telangiectasia in cancer patients: novel insights from an interesting case and review of literature. Clin Case Rep. 2021;9:995–1009. doi:10.1002/ccr3.3543
87. Rothblum-Oviatt C, Wright J, Lefton-Greif MA, McGrath-Morrow SA, Crawford TO, Lederman HM. Ataxia telangiectasia: a review. Orphanet J Rare Dis. 2016;11(1):159. doi:10.1186/s13023-016-0543-7
88. Veiga-Fernández A, Perdigón MD, Claverol MB, et al. 194 Ataxia-teleangiectasia followed up in a hereditary gynaecological cancer unit of a tertiary hospitalInternational. J Gynecolog Cancer. 2020;30:A91–A92.
89. Gatti RA, Nieberg R, Boder E. Uterine tumors in ataxia-telangiectasia. Gynecol Oncol. 1989;32(2):257–260. doi:10.1016/s0090-8258(89)
90. Caroleo AM, De Ioris MA, Boccuto L, et al. DICER1 syndrome and cancer predisposition: from a rare pediatric tumor to lifetime risk. Front Oncol. 2021;10:614541. doi:10.3389/fonc.2020.614541
91. Brenneman M, Field A, Yang J, et al. Temporal order of RNase IIIb and loss-of-function mutations during development determines phenotype in pleuropulmonary blastoma/ DICER1 syndrome: a unique variant of the two-hit tumor suppression model. F1000Res. 2015;4:214. doi:10.12688/f1000research.6746.1
92. González IA, Stewart DR, Schultz KAP, et al. DICER1 tumor predisposition syndrome: an evolving story initiated with the pleuropulmonary blastoma. Mod Pathol. 2022;35:4–22. doi:10.1038/s41379-021-00905-8
93. Schultz KAP, Williams GM, Kamihara J, et al. DICER1 and associated conditions: identification of at-risk individuals and recommended surveillance strategies. Clin Cancer Res. 2018;24(10):2251–2261. doi:10.1158/1078-0432.CCR-17-3089
94. Kim J, Field A, Schultz KAP, Hill DA, Stewart DR. The prevalence of DICER1 pathogenic variation in population databases. Int J Cancer. 2017;141:2030–2036. doi:10.1002/ijc.30907
95. Hayes K, French A. Pediatric gynecologic malignancy and DICER1. Obstet Gynecol. 2020;135:90S. doi:10.1097/01.aog.0000664076.39
96. Ni Y, Zhou X, Wu L, et al. Ovarian sertoli-leydig cell tumor, multinodular goiter, cystic nephromas and DICER1 mutations: case report and literature review. Pharmgenomics Pers Med. 2021;14:947–953. doi:10.2147/PGPM.S317153
97. Merideth MA, Harney LA, Vyas N, et al. Gynecologic and reproductive health in patients with pathogenic germline variants in DICER1. Gynecol Oncol. 2020;156:647–653. doi:10.1016/j.ygyno.2019.12.037
98. Stewart CJR, Charles A, Foulkes WD. Gynecologic manifestations of the DICER1 syndrome. Surg Pathol Clin. 2016;9(2):227–241. doi:10.1016/j.path.2016.01.002
99. Jonson AL, Geller MA, Dickson EL. Gonadal dysgenesis and gynecologic cancer. Obstet Gynecol. 2010;116(Supplement):550–552. doi:10.1097/aog.0b013e3181e4bfe
100. Piazza MJ, Urbanetz AA. Germ cell tumors in dysgenetic gonads. Clinics. 2019;74:e408. doi:10.6061/clinics/2019/e408
101. Dicken BJ, Billmire DF, Krailo M, et al. Gonadal dysgenesis is associated with worse outcomes in patients with ovarian nondysgerminomatous tumors: a report of the Children’s Oncology Group AGCT 0132 study. Pediatr Blood Cancer. 2017;65(4):e26913. doi:10.1002/pbc.26913
102. Yang W, Wu L, He Q, Zhang Y, Zhang Y, Tian Y. A case series of patients with gonadal dysgenesis-associated mixed malignant ovarian germ cell tumor. Gynecol Endocrinol. 2020;1–4. doi:10.1080/09513590.2020.1775194
103. Liu A-X, Shi H-Y, Cai Z-J, et al. Increased risk of gonadal malignancy and prophylactic gonadectomy: a study of 102 phenotypic female patients with Y chromosome or Y-derived sequences. Human Reprod. 2014;29(7):1413–1419. doi:10.1093/humrep/deu109
104. Vogt PH, Besikoglu B, Bettendorf M, et al. Gonadoblastoma Y locus genes expressed in germ cells of individuals with dysgenetic gonads and a Y chromosome in their karyotypes include DDX3Y and TSPY. Human Reprod. 2019;34(4):770–779. doi:10.1093/humrep/dez004
105. Capito C, Arnaud A, Hameury F, et al. Dysgerminoma and gonadal dysgenesis: the need for a new diagnosis tree for suspected ovarian tumours. J Pediatr Urol. 2011;7(3):367–372. doi:10.1016/j.jpurol.2011.02.021
106. Abacı A, Çatlı G, Berberoğlu M. Gonadal malignancy risk and prophylactic gonadectomy in disorders of sexual development. J Pediatr Endocrinol Metabol. 2015;28:9–10. doi:10.1515/jpem-2014-0522
107. Lim GSD, Oliva E. The morphologic spectrum of uterine PEC-cell associated tumors in a patient with tuberous sclerosis. Int J Gynecol Pathol. 2011;121–128. doi:10.1097/pgp.0b013e3181fa5a9
108. Liang SX, Pearl M, Liu J, Hwang S, Tornos C. “Malignant” uterine perivascular epithelioid cell tumor, pelvic lymph node lymphangioleiomyomatosis, and gynecological pecomatosis in a patient with tuberous sclerosis. Int J Gynecol Pathol. 2008;27(1):86–90. doi:10.1097/pgp.0b013e318150df37
109. Nishio N, Kido A, Minamiguchi S, et al. MR findings of uterine PEComa in patients with tuberous sclerosis: report of two cases. Abdomin Radiol. 2019;44:1256–1260. doi:10.1007/s00261-019-01918-3
110. Gyure KA, Hart WR, Kennedy AW. Lymphangiomyomatosis of the uterus associated with tuberous sclerosis and malignant neoplasia of the female genital tract: a report of two cases. Int J Gynecol Pathol. 1995;14:344–351. doi:10.1097/00004347-199510000-00010
111. Jhawar S, Lakhotia R, Suzuki M, et al. Clinical presentation and management of primary ovarian neuroendocrine tumor in multiple endocrine neoplasia type 1. Endocrinol Diabetes Metabol Case Rep. 2019;19–0040. doi:10.1530/EDM-19-0040
112. Almeida MQ, Stratakis CA. Solid tumors associated with multiple endocrine neoplasias. Cancer Genet Cytogenet. 2010;203(1):30–36. doi:10.1016/j.cancergencyto.2010.09.006
113. Spaulding R, Alatassi H, Stewart Metzinger D, Moghadamfalahi M. Ependymoma and carcinoid tumor associated with ovarian mature cystic teratoma in a patient with multiple endocrine neoplasia I. Case Rep Obstet Gynecol. 2014;2014:1–4. doi:10.1155/2014/712657
114. Mele C, Mencarelli M, Caputo M, et al. Phenotypes associated with MEN1 syndrome: a focus on genotype-phenotype correlations. Front Endocrinol (Lausanne). 2020;11:591501. doi:10.3389/fendo.2020.591501
115. Liu Q, Tong D, Yuan W, et al. Different RET gene mutation-induced multiple endocrine neoplasia type 2A in 3 Chinese families. Medicine. 2017;96(3):e5967. doi:10.1097/MD.0000000000005967
116. Tamsen A, Mazur MT. Ovarian strumal carcinoid in association with multiple endocrine neoplasia, type IIA. Arch Pathol Lab Med. 1992;116(2):200–203.
117. Lou L, Zhou L, Wang W, Li H, Li Y. Atypical ovarian carcinoid tumor with widespread skeletal metastases: a case report of multiple endocrine neoplasia type 1 in a young woman. BMC Cancer. 2019;19(1). doi:10.1186/s12885-019-6332-7
118. Connor YD, Miao D, Lin DI, et al. Germline mutations of SMARCA4 in small cell carcinoma of the ovary, hypercalcemic type and in SMARCA4-deficient undifferentiated uterine sarcoma: clinical features of a single family and comparison of large cohorts. Gynecol Oncol. 2020;159:131. doi:10.1016/j.ygyno.2019.10.031
119. Ramos P, Karnezis AN, Craig DW, et al. Small cell carcinoma of the ovary, hypercalcemic type, displays frequent inactivating germline and somatic mutations in SMARCA4. Nat Genet. 2014;46(5):427–429. doi:10.1038/ng.2928
120. Lin DI, Allen JM, Hecht JL, et al. SMARCA4 inactivation defines a subset of undifferentiated uterine sarcomas with rhabdoid and small cell features and germline mutation association. Mod Pathol. 2019;32:1675–1687. doi:10.1038/s41379-019-0303-z
121. Foulkes WD, Clarke BA, Hasselblatt M, Majewski J, Albrecht S, McCluggage WG. No small Surprise - small cell carcinoma of the ovary, hypercalcaemic type, is a malignant rhabdoid tumour. J Pathol. 2014;233(3):209–214. doi:10.1002/path.4362
122. Witkowski L, Carrot-Zhang J, Albrecht S, et al. Germline and somatic SMARCA4 mutations characterize small cell carcinoma of the ovary, hypercalcemic type. Nat Genet. 2014;46(5):438–443. doi:10.1038/ng.2931
123. Berchuck A, Witkowski L, Hasselblatt M, Foulkes WD. Prophylactic oophorectomy for hereditary small cell carcinoma of the ovary, hypercalcemic type. Gynecol Oncol Rep. 2015;12:20–22. doi:10.1016/j.gore.2015.02.002
124. Pejovic T, McCluggage WG, Krieg AJ, et al. The dilemma of early preventive oophorectomy in familial small cell carcinoma of the ovary of hypercalcemic type. Gynecol Oncol Rep. 2019;28:47–49. doi:10.1016/j.gore.2019.02.002
125. Blair VR, McLeod M, Carneiro F, et al. Hereditary diffuse gastric cancer: updated clinical practice guidelines. Lancet Oncol. 2020;21(8):e386– e397. doi:10.1016/s1470-2045(20)30219-9
126. Luo W, Fedda F, Lynch P, Tan D. CDH1 gene and hereditary diffuse gastric cancer syndrome: molecular and histological alterations and implications for diagnosis and treatment. Front Pharmacol. 2018;9. doi:10.3389/fphar.2018.01421
127. Kaurah P, Huntsman DG, et al. Hereditary diffuse gastric cancer. In: Adam MP, Ardinger HH, Pagon RA, editors. GeneReviews®. Seattle: University of Washington, Seattle; 2002:1993–2021.
128. Benusiglio PR, Malka D, Rouleau E, et al. CDH1 germline mutations and the hereditary diffuse gastric and lobular breast cancer syndrome: a multicentre study. J Med Genet. 2013;50(7):486–489. doi:10.1136/jmedgenet-2012-101472
129. Corso G, Intra M, Trentin C, Veronesi P, Galimberti V. CDH1 germline mutations and hereditary lobular breast cancer. Fam Cancer. 2016;15(2):215–219. doi:10.1007/s10689-016-9869-5
130. Boughey JC, Attai DJ, Chen SL, et al. Contralateral prophylactic mastectomy consensus statement from the American Society of Breast Surgeons: additional considerations and a framework for shared decision making. Ann Surg Oncol. 2016;23(10):3106–3111. doi:10.1245/s10434-016-5408-8
131. Hutchcraft ML, Gallion HH, Kolesar JM. MUTYH as an emerging predictive biomarker in ovarian cancer. Diagnostics. 2021;11(1):84. doi:10.3390/diagnostics11010084
132. Win AK, Reece JC, Dowty JG, et al. Risk of extracolonic cancers for people with biallelic and monoallelic mutations in MUTYH. Int J Cancer. 2016;139(7):1557–1563. doi:10.1002/ijc.30197
133. Catalogue of Somatic Mutations in Cancer (COSMIC): MUTYH gene. Available from: https://cancer.sanger.ac.uk/cosmic/gene/analysis?ln=MUTYH.
134. Vogt S, Jones N, Christian D, et al. Expanded extracolonic tumor spectrum in MUTYH-associated polyposis. Gastroenterology. 2009;137(6):1976–1985.e10. doi:10.1053/j.gastro.2009.08.052
135. Marini F, Falchetti A, Del Monte F, et al. Multiple endocrine neoplasia type 2. Orphanet J Rare Dis. 2006;1:45. doi:10.1186/1750-1172-1-45
136. Marini F, Falchetti A, Monte FD, et al. Multiple endocrine neoplasia type 1. Orphanet J Rare Dis. 2006;1:38. doi:10.1186/1750-1172-1-38
137. Mirshahi UL, Kim J, Best AF, et al. A genome-first approach to characterize DICER1 pathogenic variant prevalence, penetrance, and phenotype. JAMA Netw Open. 2021;4(2):e210112. doi:10.1001/jamanetworkopen.2021.0112
138. Wataya-Kaneda M, Tanaka M, Hamasaki T, Katayama I. Trends in the prevalence of tuberous sclerosis complex manifestations: an epidemiological study of 166 Japanese patients. PLoS One. 2013;8(5):e63910. doi:10.1371/journal.pone.0063910
139. Ebrahimi-Fakhari D, Mann LL, Poryo M, et al. Incidence of tuberous sclerosis and age at first diagnosis: new data and emerging trends from a national, prospective surveillance study. Orphanet J Rare Dis. 2018;13:117. doi:10.1186/s13023-018-0870-y
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.