Back to Journals » Drug Design, Development and Therapy » Volume 19
Herb-Drug Interaction Between Sailuotong and Pitavastatin: A Systematic Pharmacokinetic Investigation and Mechanism Analysis
Authors Zhang Y, Yi H, Yang H ![]() , Song Y, Ren C, Li X, Zhang Y
, Song Y, Ren C, Li X, Zhang Y
Received 20 March 2025
Accepted for publication 31 July 2025
Published 20 August 2025 Volume 2025:19 Pages 7135—7149
DOI https://doi.org/10.2147/DDDT.S529385
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Anastasios Lymperopoulos
Ying Zhang,* Huan Yi,* Hongtian Yang, Yuchen Song, Changying Ren, Xiang Li, Ying Zhang
Beijing Key Laboratory of Pharmacology of Chinese Materia Medica, Institute of Basic Medical Sciences, Xiyuan Hospital, China Academy of Chinese Medical Sciences, Beijing, 100091, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Ying Zhang, Beijing Key Laboratory of Pharmacology of Chinese Materia Medica, Institute of Basic Medical Sciences, Xiyuan Hospital, China Academy of Chinese Medical Sciences, Beijing, 100091, People’s Republic of China, Tel +86-10-62835639, Email [email protected]
Propose: Sailuotong (SLT), a standardized Chinese herbal preparation for vascular dementia (VaD), is frequently co-administered with pitavastatin (PIV). Potential herb-drug interactions (HDIs) between these agents remain uncharacterized. Given the high likelihood of using this combination to treat VaD, this study aims to systematically evaluate the effects of SLT on PIV’s pharmacokinetics and elucidate underlying mechanisms.
Methods: Rats received single or repeated doses of SLT followed by oral or intravenous PIV. Plasma and hepatic PIV concentrations were quantified via LC-MS/MS. Biliary excretion and enterohepatic circulation interruption models assessed elimination pathways. The expression of hepatic and intestinal transporters was analyzed by RT-PCR and Western blot. Transporter functionality was validated using MDR1 substrates (digoxin and betrixaban).
Results: Single-dose SLT increased PIV’s Cmax and AUC0-9h by approximately 23.30% and 15.70%, respectively. Repeated SLT administration significantly decreased PIV’s AUC by 32.90%, and reduced its hepatic accumulation by 68.96%. Intravenous studies revealed that SLT primarily affected the later exposure phases. Multiple doses of SLT decreased PIV’s total biliary excretion by 33.10%. Mechanistically, SLT significantly induced the expression of MDR1 mRNA and proteins in the intestine and liver, and MRP2 in the liver. Additionally, SLT significantly decreased the exposure levels of digoxin and betrixaban, with betrixaban’s Cmax and AUC remarkably reduced by 86.44% and 79.74%, respectively.
Conclusion: Combining SLT and PIV can lead to HDIs, with multiple doses of SLT significantly reducing the plasma and hepatic exposure of PIV in rats. The primary mechanism appears to be the induction of the intestinal efflux transporter MDR1, resulting in decreased bioavailability of PIV.
Keywords: sailuotong, pitavastatin, herb-drug interaction, pharmacokinetics, MDR1
Graphical Abstract:

Introduction
Cerebrovascular disease has long been a major global concern, with the risk of developing this condition increasing with age. Vascular dementia (VaD), the second most common form of dementia after Alzheimer’s disease, is a type of cognitive dysfunction caused by cerebrovascular disease, primarily characterized by memory and cognitive impairments.1–3 Its pathophysiological mechanisms are complex, involving multiple factors such as chronic cerebral hypoperfusion, small vessel disease, neuroinflammation, oxidative stress, and synaptic plasticity impairment.4–7 Although current drug development targets these pathways, single-target therapies often have limited efficacy.
Natural products offer unique advantages in managing multi-factorial diseases like VaD due to their multi-target regulatory properties.8–10 Based on the traditional Chinese medicine theory of pathogenesis, which states that “qi stagnation and blood stasis” are the root cause of VaD, Sailuotong (SLT) has been developed as a standardized Chinese herbal preparation. It consists of Ginseng (Panax ginseng C. A. Meyer, root and rhizome, qi-tonifying) combined with Ginkgo leaf (Ginkgo biloba L.) and Saffron (Crocus sativus L., stigma, blood-activating and stasis-resolving). Extensive pharmacological studies confirm that SLT effectively alleviates focal cerebral ischemia/reperfusion injury and improves memory deficits in animal models, providing a scientific basis for its use in VaD and ischemic stroke recovery.11–14 Clinical evidence further solidifies its status. Trials completed in Australia demonstrated SLT’s significant improvement in working memory in healthy adults and cognitive function in VaD patients.13,14 A Phase II randomized controlled trial in China confirmed its safety and efficacy for mild to moderate VaD.15,16 In addition, SLT is already a registered drug in Singapore.15 Therefore, SLT represents an ethnopharmacologically relevant, evidence-based therapeutic choice for VaD with robust experimental and clinical validation.
VaD patients often require concomitant use of foundational medications, such as statins, to control cardiovascular risk factors.17–21 Among statins, Pitavastatin (PIV) is a key focus of this study due to its unique pharmacokinetic (PK) profile. Unlike statins extensively metabolized by CYP3A4/CYP2C9 (eg, Lovastatin, Atorvastatin, Fluvastatin), PIV exhibits well-defined and stable metabolic pathways, significantly reducing the risk of cytochrome P450 enzyme-mediated drug-drug interactions (DDIs).22,23 PIV effectively inhibits cholesterol synthesis at lower doses and offers prolonged action duration and higher bioavailability. Notably, PIV demonstrates ameliorative effects in L-methionine-induced vascular dementia models,24 highlighting its particular therapeutic relevance in the VaD patient. Despite its metabolic stability minimizing enzyme-mediated DDI risks, PIV’s hepatic uptake and disposition are highly dependent on transporter systems, similar to most statins. In the liver, uptake transporters of organic anion-transporting polypeptide 1B1 and 1B3 (OATP1B1, OATP1B3) extract statins from the portal blood into hepatocytes.25–27 In addition, breast cancer resistance protein (BCRP) and multidrug resistance protein 1 (MDR1/P-glycoprotein) are important in the efflux of metabolites or parent drugs from the hepatocyte.28–32
Given SLT’s promising clinical application for VaD and PIV’s status as a metabolically stable statin preferred for VaD patients needing to avoid CYP-mediated DDIs, the co-administration of SLT and PIV in clinical practice is highly probable. However, the potential inhibitory or inductive effects of SLT’s multi-component nature on critical drug transporters (eg, OATP1B1/1B3, BCRP, P-gp) remain unexplored. A transporter-mediated herb-drug interaction (HDI) could result in abnormally increased systemic exposure to PIV or altered tissue distribution, elevating its toxicity risks (eg, myopathy, liver injury).33 Therefore, this study focuses on the impact of SLT on the pharmacokinetics of PIV, with the primary objective of elucidating whether SLT alters the in vivo disposition of PIV by modulating main hepatic drug transporters, particularly OATP1B1/1B3. This research will provide crucial scientific evidence and potential dose-adjustment strategies for the safe and effective clinical co-administration of SLT and statins, directly contributing to the optimized treatment of VaD patients.
Materials and Methods
Materials
Ginseng extract (batch 20170429), ginkgo extract (19041521), and saffron extract (18100301) were produced under Good Manufacturing Practice (GMP) conditions at Shineway Pharmaceutical Group (Hebei, China). These three extracts were mixed in a fixed weight ratio to prepare SLT. To assess the quality of the extracts, the total active components and major constituents were quantified using ultraviolet (UV) spectroscopy and high performance liquid chromatography (HPLC)-UV methods.13 Pitavastatin calcium was purchased from Shanghai Yuanye Bio-Technology Co., Ltd (Shanghai, China). Atorvastatin calcium was obtained from Sigma-Aldrich (Livonia, America). Digoxin was acquired from National Institute for the Control of Pharmaceutical and Biological Products (Beijing, China). Betrixaban was purchased from Bide Pharmaceuticals (Shanghai, China). Formic acid (HPLC grade) was purchased from J.T. Baker (Phillipsburg, USA). Methanol and acetonitrile (HPLC grade) were purchased from Merck (Darmstadt, Germany). All other reagents were of analytical grade.
Animals
Healthy male Sprague-Dawley rats (6–7 weeks old, weighing 200–220 g) were purchased from Spielberg (Beijing) Biotechnology Co., Ltd. (license No. SCXK 2019-0010) and housed in the Safety Laboratory of Xiyuan hospital, China Academy of Chinese Medical Sciences (CACMS), under a 12-hour light/dark cycle at 21–24°C. All rats were acclimated for three days prior to the experiment. All animal housing, care, feeding and experimental procedures were in accordance with the National Guidelines for Animal Protection. The ethics committee of the Xiyuan Hospital approved all animal experiments (No. 2022XLC030-2).
Animal Study
Impact of SLT on the Pharmacokinetics and Hepatic Uptake of PIV After Co-Administration with PIV Orally
12 rats were randomly divided into two groups: a PIV group and a SLT+PIV group. All rats were fasted for 12 h prior to dosing and given free access to water. On the first day, the PIV group was given water, while the SLT+PIV group was given SLT by gavage. Then each group was given PIV by gavage 30 min later. 0.2 mL of blood was collected from the rats’ retro-orbital sinus into heparinized centrifuge tubes at 10, 30 min, 1, 2, 4, 6, and 9 h after dosing. Plasma samples were obtained by immediate centrifugation at 4°C and 3000 rpm for 10 min after sampling. The supernatant was stored at −30°C for analysis. The PIV group was then given water and the SLT+PIV group was given SLT once daily from day 2. On day 15, plasma samples were collected according to the same protocol as on the first day, after the last dose. After blood sampling at 6 h, the rats were anesthetized and their livers and a 10 cm section of jejunums were removed. These were washed with cold saline, dried with filter paper, and placed in liquid nitrogen for quick freezing. All samples were stored at −80°C until analysis.
Impact of SLT on the Pharmacokinetics and Hepatic Uptake of PIV After Co-Administration with PIV Intravenously
The animals were grouped and treated in the same way as in section 2.3.1, except that PIV (1 mg/kg) was administered intravenously.
Effects of SLT on Pharmacokinetics and Biliary Excretion of PIV in Bile Intubated Rats
12 rats were randomly divided into two groups: a PIV group and a SLT+PIV group. The PIV group was given water, while the SLT+PIV group was given SLT by gavage for 14 consecutive days. On the day 15, under light anesthesia with diethyl ether, bile fistulas were cannulated with PE-5 polyethylene tubes for bile collection. The rats were then allowed to recover from the anesthetic before receiving water or SLT, followed by PIV 30 min later. Bile was collected in successive vials on ice at time intervals of 0–1, 1–2, 2–4, 4–8, 8–12, 12–24, 24–30, and 30–36 h after PIV administration. Blood samples were taken from the tail vein using heparinized microcapillaries at 10, 30 min, 1, 2, 5, and 9 h after PIV administration. The samples were then centrifuged at 3000 rpm for 10 min to obtain plasma. During the bile and blood collection, the rats were kept in restraining cages with free access to water and food 2 h after administration. All bile and blood samples were stored at −30°C until analysis.
MDR1 Probe Drug Experiments
12 rats were randomly divided into two groups: digoxin/ betrixaban and SLT+ digoxin/ betrixaban. The former were given water, while the latter were given SLT by gavage once daily for 14 consecutive days. After the final gavage, each group was administered digoxin (0.2 mg/kg) or betrixaban (5 mg/kg) by gavage 30 min later. Blood samples were collected from the retro-orbital sinus of rats in heparinized centrifuge tubes at 10, 30 min, 1, 2, 4, 6, 10, 24 h after digoxin dosing, and at 30 min, 1, 1.5, 2, 3, 5, 7, 10, 24, 36, 48 h after betrixaban administration. Plasma was obtained by centrifugation at 3000 rpm for 10 min and stored at −30°C until analysis.
Determination of PIV in Bio-Samples
Sample Preparation
Each 50 μL of rat plasma was spiked with 50 μL of atorvastatin (the internal standard, 25 ng/mL in methanol) and 150 μL of acetonitrile. For bile samples, 20 μL of rat bile was spiked with 190 μL of atorvastatin (25 ng/mL in methanol) and 190 μL of 50% acetonitrile containing 0.05% formic acid. For liver samples, 0.1 g from each rat was taken and homogenized with water containing 0.033% formic acid at a ratio of 20:1 (v/w). Each 50 µL of liver homogenate was spiked with 50 μL of atorvastatin (25 ng/mL in methanol) and 150 μL of acetonitrile. All of the above mixtures were extracted by vortex shaking for 2 min and then centrifugation at 12,000 rpm for 5 min. The supernatant was then transferred to a sample vial and injected into a liquid chromatography-tandem mass spectrometry (LC-MS/MS) system for analysis.
LC-MS/MS Measurement
The concentration of PIV in bio-samples was analyzed using an LC-MS/MS method. This analysis was performed using an API 4000 QTRAP LC-MS/MS system (Applied Biosystems, USA). Chromatographic separation was performed using an Agilent ZORBAX SB-C18 column (2.1 × 50 mm, 5 μm) (Agilent Technologies, USA). The mobile phases consisted of a mixture of water, methanol and acetonitrile (9:0.5:0.5, v/v, Phase A), and a mixture of acetonitrile and methanol (1:1, v/v, Phase B), both containing 0.05% formic acid. Linear gradient elution was applied at a flow rate of 0.35 mL/min, with the percentage of B increasing from 40% to 68% (0–0.3 min), then held at 68% (0.31–1.4 min), before decreasing back to 40% for equilibration (1.5–4.5 min). The autosampler was maintained at 4°C and the injection volume was 4 μL. The electrospray ionization (ESI) source of the mass spectrometer was operated in the positive ion mode. Multiple reaction monitoring was performed for the transitions at 422.2→290.0 m/z for PIV and 559.3 → 440.2 m/z for the atorvastatin internal standard (IS). The corresponding declustering potential and the collision energy were set to 121 V and 39 eV for PIV, and 106 V and 31 eV for the IS, respectively. The optimized ion source parameters were as follows: curtain gas, 20 psi; nebulizer gas 1, 30 psi; auxiliary gas, 40 psi; ion spray voltage, 5500 V; and source temperature, 500°C. The LC-MS/MS methods used for quantification were adapted from protocols previously validated in our laboratory. The bioanalytical methods were validated in accordance with FDA guidelines. Detailed information is provided in the supplementary information.
Determination of Digoxin in Plasma
Sample Pretreatment
100 μL of rat plasma was spiked with 50 μL of NH4Cl buffer solution (pH 8.8) and 300 μL of acetonitrile containing 20 ng/mL of digitoxin (IS). After vortex-shaking for 2 min, and centrifugation at 12,000 rpm for 5 min, the 350 μL upper organic layer was transferred and evaporated to dryness under a nitrogen flow in a 50°C water bath. The residue was then reconstituted in 100 μL of 30% acetonitrile before being centrifuged at 12,000 rpm for 10 min. The resulting supernatant was transferred to a sample vial, from which10 μL was injected into an LC-MS/MS system for analysis.
LC-MS/MS Measurement
The concentration of digoxin in rat plasma was determined using the same LC-MS/MS instrumental system as for PIV. Chromatographic separation was performed using an Agilent ZORBAX XDB-C18 column (2.1 × 50 mm, 5 μm) (Agilent Technologies, USA). The mobile phases comprised a mixture of water and acetonitrile (9:1, v/v, Phase A) and acetonitrile (Phase B), both containing 0.01% formic acid. A linear gradient elution was applied at a flow rate of 0.4 mL/min, with B increased from 15% to 60% (0.1–0.5 min), held at 60% for 1.0 min, then decreased to 15% for equilibration from 2.1–6.0 min. The ESI source of the mass spectrometer was operated in negative ion mode. Multiple reaction monitoring was performed for the transitions at 779.4→649.2 m/z for digoxin and 796.3 → 633.2 m/z for IS. The corresponding declustering potential and the collision energy were set to −140 V and −46 eV for digoxin, and −115 V and −42 eV for IS, respectively. The optimized ion source parameters were as follows: curtain gas, 15 psi; nebulizer gas, 50 psi; auxiliary gas, 40 psi; ion spray voltage, −4000 V; and source temperature, 500°C.
Determination of Betrixaban in Plasma
Sample Pretreatment
50 μL of rat plasma was spiked with 150 μL of acetonitrile containing rivaroxaban (IS) at a concentration of 10 ng/mL. Following vortex shaking for 2 min and centrifugation at 12,000 rpm for 5 min, the supernatant was transferred to a sample vial and analysed using an LC-MS/MS system.
LC-MS/MS Measurement
The concentration of betrixaban in rat plasma was determined using the same LC-MS/MS instrumental, analytical column, and mobile phases as for digoxin. A linear gradient elution was applied at a flow rate of 0.4 mL/min, with B increased from 15% to 60% (0.1–0.3 min), held at 60% for 0.2 min, then decreased to 15% for equilibration from 0.6–5.0 min. The ESI source of the mass spectrometer was operated in positive ion mode. Multiple reaction monitoring was performed for the transitions at 452.2→324.2 m/z for digoxin and 436.0→145.0 m/z for IS. The corresponding declustering potential and the collision energy were set to 116 V and 33 eV for betrixaban, and to 96 V and 39 eV for IS, respectively. The optimized ion source parameters were as follows: curtain gas, 15 psi; nebulizer gas, 50 psi; auxiliary gas, 50 psi; ion spray voltage, 5500 V; and source temperature, 600°C.
RT-PCR Analysis
Total RNA was isolated from the intestine and liver using TRNzol Universal Reagent (Tiangen Biochemical Technology, Beijing, China). RNA quality and concentration were then measured using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Waltham, USA). A 500 ng purified sample of total RNA was synthesized into cDNA using a ReverTra Ace® qPCR RT Master Mix (bio-toyobo, Toyobo, Japan). The qPCR primers were designed and synthesized by Sangon Biotech (Shanghai, China), and the forward and reverse sequences are provided in Table 1. RT-PCR was conducted with an initial denaturation at 95°C for 10 min, followed by 40 cycles of 95°C for 15 seconds, and 60°C for 1 minute. The final extension step was performed at 95°C for 15 seconds and 60°C for 1 min. Gene expression was analyzed using the 2−ΔΔCt method with GAPDH as the reference gene. Quantitative real-time PCR reactions were prepared using the PowerUp™ SYBR™ Green Master Mix (Thermo Fisher Scientific, Waltham, USA) and performed on a StepOne Real-Time PCR System (Thermo Fisher Scientific, Waltham, USA). All procedures were carried out following the manufacturer’s protocols.
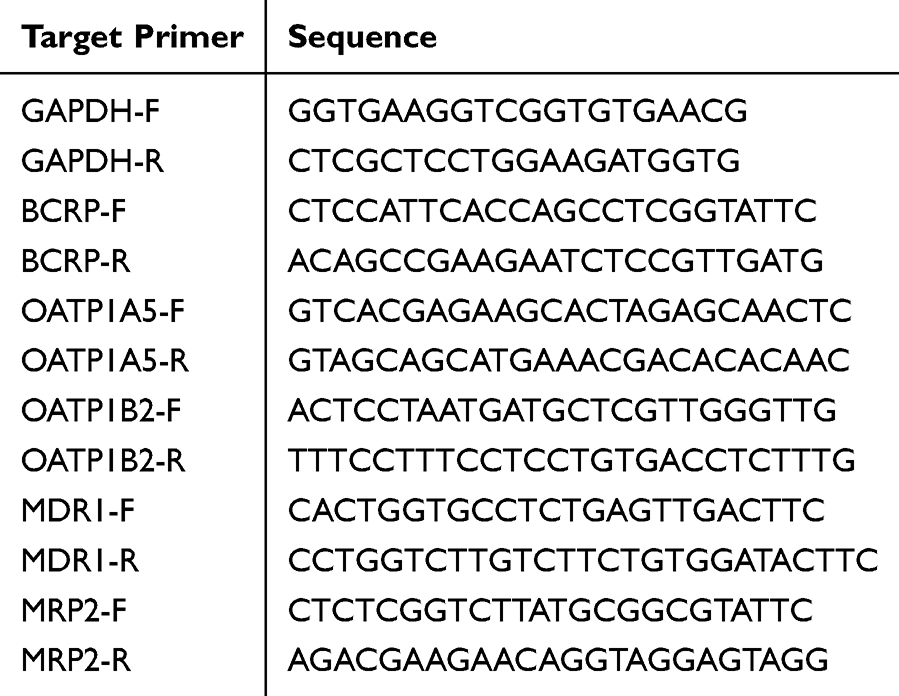 |
Table 1 The Used Primers for RT-PCR |
Western Blot Analysis
Intestine and liver samples were lysed in RIPA buffer (Beyotime Technology, Shanghai, China) containing 1 mM PMSF in an ice bath, followed by centrifugation at 12,000 rpm for 5 min. The supernatant was collected and the protein concentration determined using the Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific, Waltham, USA). The protein extracts (3 μg/μL) were then mixed with loading buffer and denatured by boiling at 100°C for 10 min. The denatured proteins were separated by SDS-PAGE and transferred to PVDF membranes (Millipore Corporation, Tullagreen, Ireland). The membranes were blocked with 5% BSA (Beyotime Technology, Shanghai, China) at room temperature for 1 h, then incubated with primary antibodies overnight at 4°C, followed by incubation with horseradish peroxidase-conjugated secondary antibodies. Bands were visualized using an ECL Western blotting substrate (Solarbio Life Sciences, Beijing, China) and detected using a ChemiDoc XRS+ imaging system (Bio-Rad, California, USA). The grey values of the bands were quantified using ImageJ and normalized to GAPDH. The antibodies and their dilutions used are listed below: anti-MRP2 (1:1000, Affinity Biosciences, USA), anti-BCRP (1:1000, Affinity Biosciences, USA), anti-P-glycoprotein (1:1000, Cell Signaling Technology, MA), and anti-GAPDH (1:50,000, Proteintech, China).
Data Analysis
The pharmacokinetic parameters of the terminal elimination half-life (T1/2), the area under the curve (AUC0-t), total body clearance (CLZ/F), and the terminal exponential volume of distribution (VZ/F) were calculated using non-compartmental analysis. These were obtained from plasma concentration-time curves using WinNonlin 6.1 (Pharsight Corporation, USA). Peak concentration (Cmax) and time to reach Cmax (Tmax) were obtained from the actual data. All pharmacokinetic parameters were assumed to follow a log-normal distribution. Comparisons between the two groups were made using an unpaired t-test in SPSS 17.0. Statistical significance was defined as P < 0.05.
Result and Discussion
PK and Hepatic Uptake of Oral PIV After Co-Administration with SLT
The plasma concentration-time curves of PIV following single and repeated administration of SLT are shown in Figure 1A and B. As can be seen from the figures, a single dose of SLT did not alter the pharmacokinetics of PIV. However, repeated SLT pretreatment markedly decreased plasma concentrations over time. The obtained pharmacokinetic parameters are presented in Table 2. Compared to the PIV group, a single dose of SLT resulted in non-significant increases in Cmax and AUC0-9h of 23.3% and 15.7%, respectively. Conversely, repeated administration of SLT significantly reduced the AUC of oral PIV by 32.9% (P < 0.05), and Cmax by 33.9%, with an unchanged T1/2.
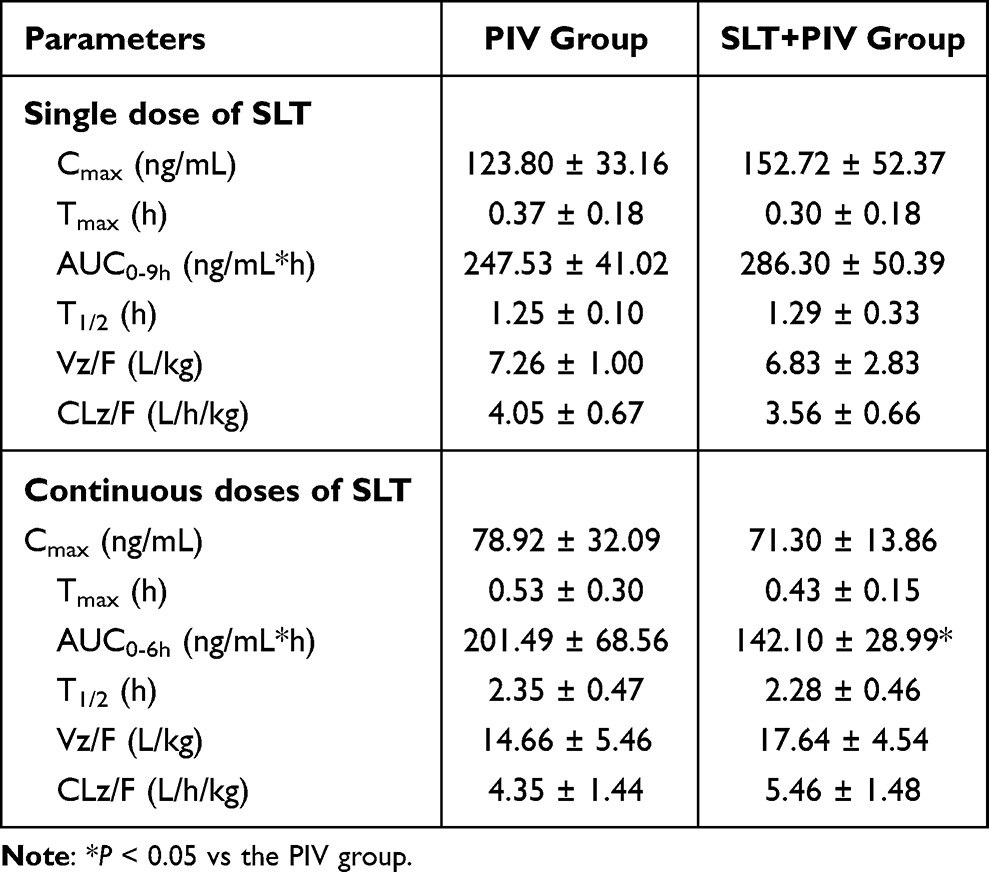 |
Table 2 Pharmacokinetic Parameters of PIV After Oral Single and Continuous Doses of SLT (Mean ± SD, n=6) |
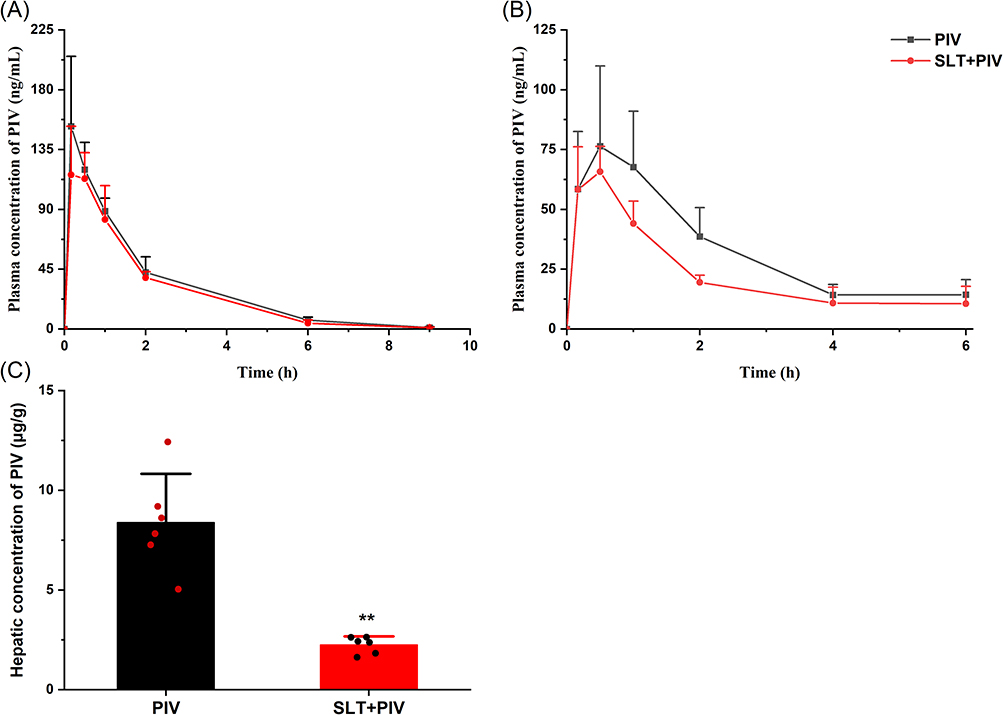 |
Figure 1 Effect of SLT on the pharmacokinetics of oral PIV (n=6). (A) Concentration-time curves of PIV after a single oral administration of SLT; (B) Concentration-time curves of PIV after continuous oral administration of SLT; (C) The concentrations of PIV in the liver after continuous administration of SLT. Data are shown as mean ± SD. **P < 0.01 vs the PIV group. |
PIV is an HMG-CoA reductase inhibitor and its pharmacological target tissue is the liver, hepatotoxicity is also associated with hepatic drug concentrations.34 Although changes in plasma exposure levels often reflect changes in drug exposure in target organs, studies have shown that inhibiting hepatic uptake transporters for PIV can significantly increase plasma exposure levels while decreasing hepatic concentrations.35 Therefore, analyzing hepatic concentrations may better reflect the potential pharmacodynamic and safety changes caused by DDIs. This study analyzed hepatic PIV levels after 2 weeks of SLT pretreatment, with the results shown in Figure 1C. The hepatic concentration of PIV was found to be significantly reduced by 68.96% at 6 hours post-dose compared to the PIV group.
The dose of SLT (66 mg/kg) administered to the rats in the study was based on the high dose used in the clinical trials to investigate drug interactions. The results indicate that continuous SLT administration can significantly reduce both PIV plasma and hepatic concentrations, indicating the occurrence of HDIs. In vivo exposure to orally administered drugs is related to their absorption and elimination rates, and a decrease in absorption or an increase in elimination could lead to a reduction in AUC. The observed reduction in both plasma and hepatic exposure suggests that decreased absorption of PIV induced by SLT may be the underlying cause of HDIs. Further pharmacokinetic experiments were conducted by intravenous administration to explore the reasons leading to the decrease in PIV exposure.
PK and Hepatic Uptake of PIV in Means of Intravenous PIV Administration After Co-Administration with SLT
The effects of SLT on the pharmacokinetics of intravenous PIV are shown in Figure 2A and B. Following the co-administration of a single SLT dose, the plasma concentration of PIV increased, with greater changes observed over time. Table 3 shows that a single dose of SLT caused a 25% increase in the AUC0-9h of PIV and a 52.3% increase in the AUC2-9h. Although T1/2 was slightly prolonged and clearance (CL) decreased non-significantly, repeated SLT administration reduced the total AUC by 30.15%, with a pronounced 45.61% decrease in post-2h exposure. CL increased by 39.1% with no change in T1/2. Hepatic PIV concentrations at 6 h showed a non-significant reduction (Figure 2C).
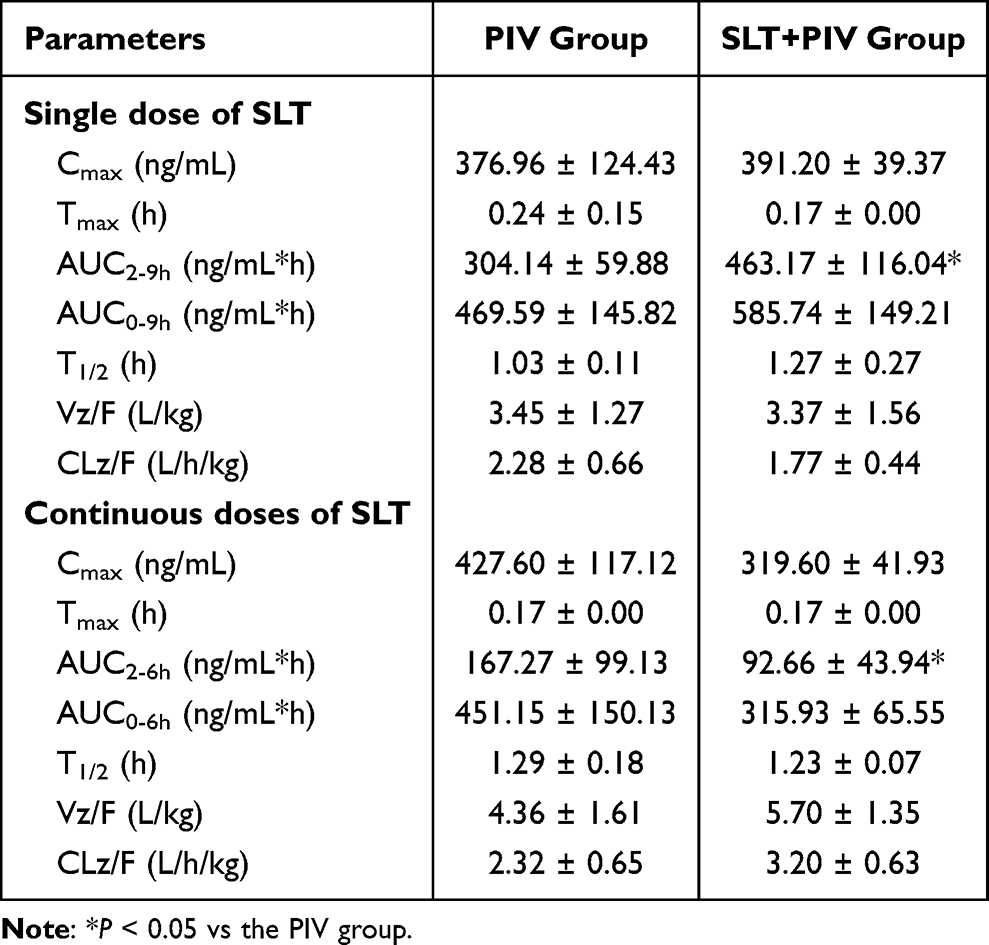 |
Table 3 Effect of Single and Continuous Administration of SLT on the Pharmacokinetics of Intravenous PIV (Mean ± SD, n=6) |
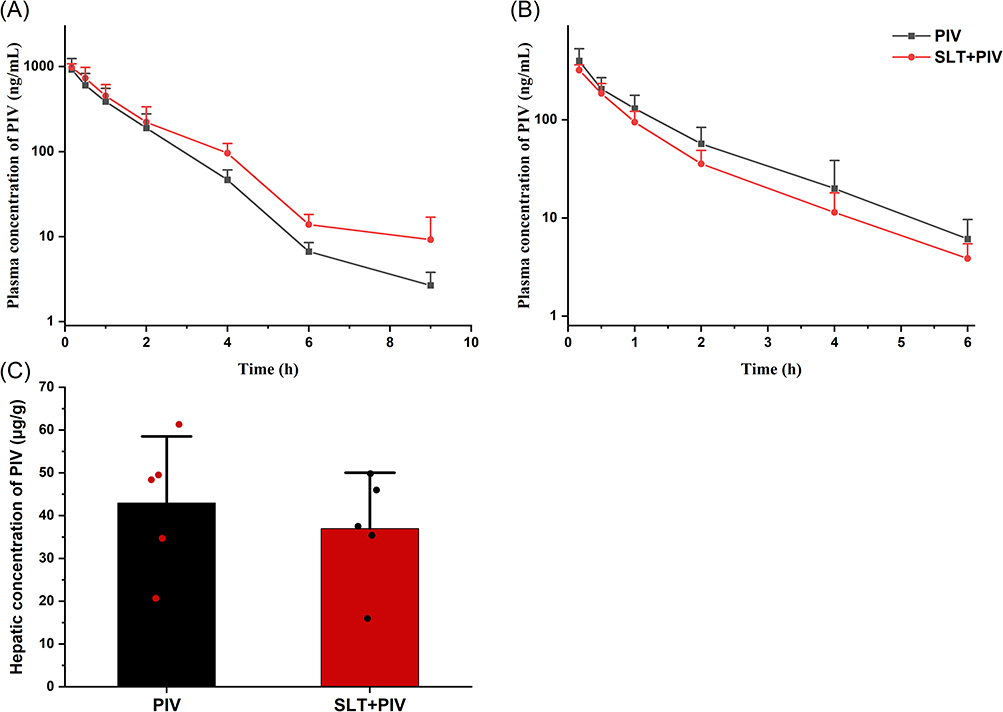 |
Figure 2 Effect of SLT on the pharmacokinetics of intravenous PIV (n=6). (A) Concentration-time curves of PIV after a single dose of SLT; (B) Concentration-time curves of PIV after continuous administration of SLT; (C) The concentrations of PIV in the liver after continuous doses of SLT. Data are shown as mean ± SD. |
Intravenous data confirmed the dominance of intestinal route in SLT-PIV interactions. Repeated SLT reduced oral PIV exposure more severely (32.9% reduction in the AUC) than intravenous exposure (30.15% reduction), indicating that the primary interaction site is intestinal absorption. This is consistent with the elimination profile of PIV (> 90% biliary excretion) and its enterohepatic circulation dynamics.36,37 As enterohepatic circulation occurs later than direct gastrointestinal absorption, its impact is more pronounced in the later phase of the plasma concentration-time curve. This is consistent with the significant changes in PIV exposure levels observed after 2 hours in this study (Figure 2B). Furthermore, SLT did not affect PIV’s T1/2 or CL. It has been speculated that a single dose of SLT may promote the enterohepatic reabsorption of PIV to some extent, thereby increasing its exposure level. However, repeated dosing induces efflux transporters that override this effect, ultimately leading to an overall reduction in PIV exposure levels.
Given that SLT is typically used in long-term clinical treatment, investigating the drug interactions caused by continuous administration is important. Differences in the effects of SLT on the pharmacokinetics of PIV via oral and intravenous administration suggest that SLT is likely to reduce systemic PIV exposure after continuous administration by reducing intestinal absorption. PIV is metabolically stable and minimally eliminated via the kidneys. Biliary excretion of the parent drug following hepatic uptake is the predominant elimination pathway, meaning that PIV excreted in the bile reflects the total amount of drug absorbed. Further studies using a bile duct cannulation model were conducted to confirm that the reduction in PIV exposure caused by SLT was due to its effects on the absorption pathway and to verify the role of SLT in PIV intestinal absorption and its impact on enterohepatic circulation.
Effects of Continuous SLT Administration on Biliary Excretion of PIV and Its Pharmacokinetics After Enterohepatic Circulation Blockage
In order to quantify the impact of SLT on PIV absorption, rats with a cannulated bile duct that had been pretreated with repeated SLT were given an oral dose of PIV. Figure 3A shows that cumulative biliary excretion results in rapid PIV elimination (> 80% within the first 4 h), with total recovery reaching 90% of the initial dose in PIV group. This confirms that biliary excretion is the primary elimination route. SLT pretreatment significantly reduced cumulative biliary excretion by 33.17% over 0–36 h compared to the PIV group (P < 0.05), which directly demonstrates impaired intestinal absorption.
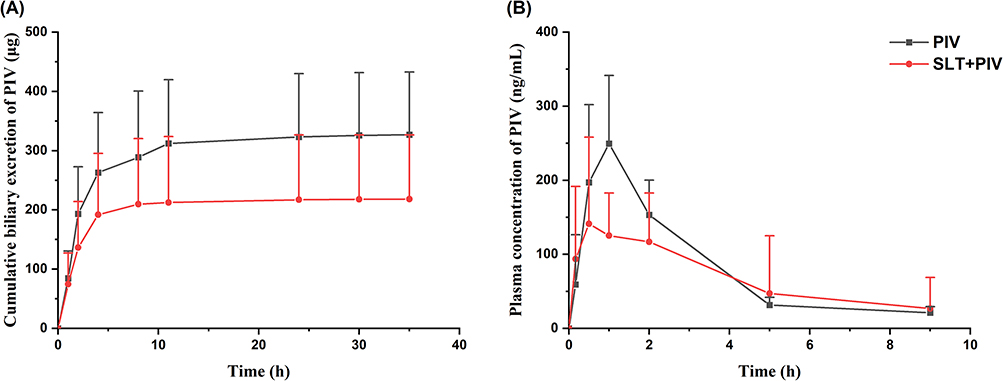 |
Figure 3 Effect of continuous administration of SLT on biliary excretion of PIV and pharmacokinetics after hepatic-intestinal circulation blockade (n=6). (A) Cumulative biliary excretion of PIV after multiple doses of SLT; (B) Concentration-time curves of PIV after multiple doses of SLT on biliary intubation with concomitant oral administration of PIV. Data are shown as mean ± SD. |
When enterohepatic circulation was blocked by cannulation, plasma profiles (Figure 3B) revealed significantly lower PIV concentrations in the SLT+PIV group during the first 2 hours post-dosing (the absorption phase), but comparable levels thereafter. This contrasts with the data for intact circulation (Figure 1B, sustained reduction), indicating that SLT affects both initial absorption and enterohepatic reabsorption. Pharmacokinetic parameters (Table 4) showed non-significant reductions in Cmax and AUC0-9h for the SLT+PIV group, with unchanged T1/2. Taken together, these data prove that the effects of continuous SLT on orally administered PIV are due to its combined action on intestinal absorption and reabsorption via enterohepatic circulation.
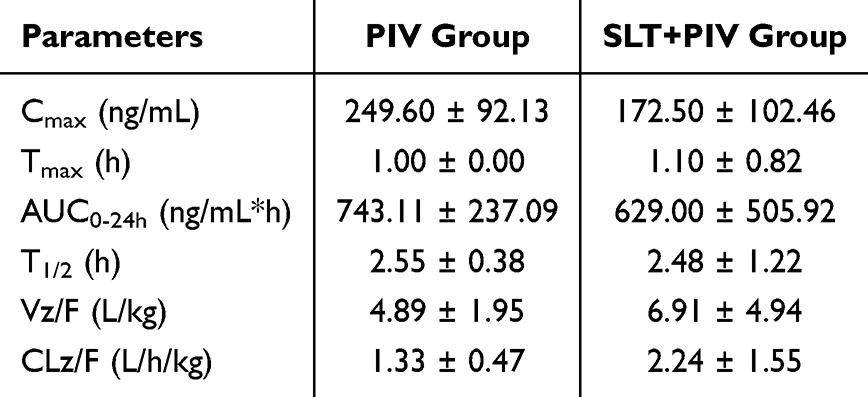 |
Table 4 Effect of Continuous SLT on Pharmacokinetic Parameters of Biliary Intubation with Concomitant Oral PIV (Mean ± SD, n=6) |
Effects of SLT on Rat Hepatic and Intestinal Transporters
RT-PCR analysis revealed that repeated SLT administration increased the levels of mRNA for key transporters in the intestine and liver (Figure 4), with significant increases in intestinal and hepatic MDR1 and hepatic MRP2 compared to the PIV group (P < 0.05). Western blot analysis (Figure 5) confirmed the increase in corresponding proteins, particularly intestinal MDR1 and hepatic MRP2 (P < 0.05).
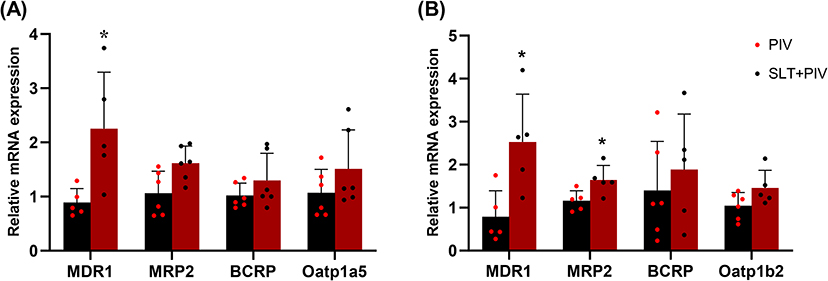 |
Figure 4 Effect of continuous SLT administration on mRNA levels of transporters by RT-PCR (n=6). (A) The mRNA expression of MDR1, MRP2, BCRP and Oatp1a5 in the intestine; (B) The expression of MDR1, MRP2, BCRP and Oatp1b2 in the liver. Data are shown as mean ± SD. *P < 0.05 vs the PIV group. |
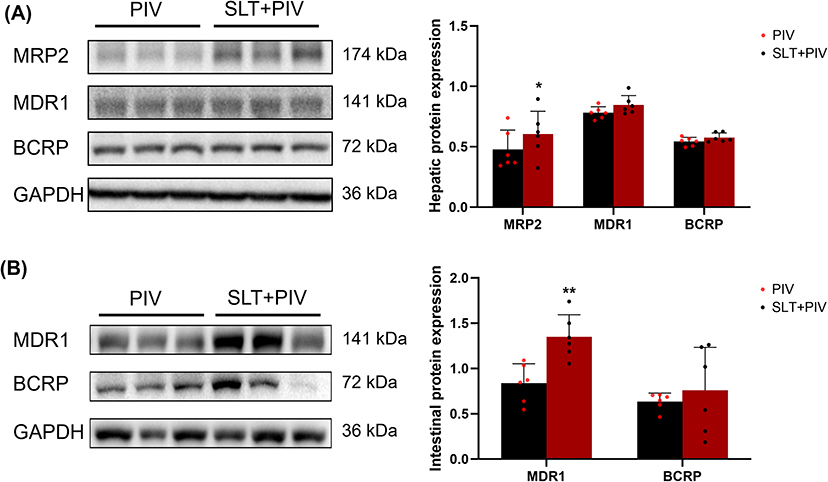 |
Figure 5 Effect on transporter protein levels with continuous SLT administration by WB (n=6). (A) The expression of major transporters in the liver; (B) The expression of major transporters in the intestine. Data are shown as mean ± SD. **P < 0.01, *P < 0.05 vs the PIV group. |
As PIV is an MDR1/BCRP substrate,38 its absorption depends on the intestinal OATP1A5-MDR1 balance.39 The significant upregulation of MDR1 in the intestine directly impairs PIV absorption by enhancing efflux. In the liver, human OATP1B1/1B3 and rat Oatp1b2 mediate the uptake,40 while MDR1, BCRP, and MRP2 mediate its biliary excretion,41 with MRP2 playing a lesser role.42 The experimental results demonstrate that multiple doses of SLT induce the up-regulation of various efflux and uptake transporters involved in PIV transport in both the liver and intestine, at mRNA and protein levels. Among these, the induction of MDR1 mRNA and protein expression was most pronounced in the rat intestine and liver. While the up-regulation of hepatic MDR1 can promote the biliary excretion of PIV, the increased intestinal MDR1 efflux caused by SLT treatment may reduce the amount of the drug that is directly absorbed and reabsorbed via enterohepatic circulation. This results in a significant decrease in PIV’s overall bioavailability.
Confirmation of the Transporter Mechanism of SLT-PIV Interaction
To validate SLT-induced MDR1 upregulation, the pharmacokinetics of two MDR1 substrates (digoxin and betrixaban) were assessed following repeated SLT dosing. The Cmax of digoxin in the SLT+PIV group decreased by 34.78%, and its AUC0-24h decreased by 13.2%, compared with the PIV group (Figure 6A and Table 5). In contrast, SLT significantly reduced betrixaban’s Cmax and AUC0-24h by 86.40% and 79.70%, respectively. Furthermore, Vz/F and CLz/F increased markedly by 396.2% and 367.4%, respectively (Figure 6B).
 |
Table 5 Effect of SLT on Pharmacokinetic Parameters of Digoxin and Betrixaban (Mean ± SD, n=6) |
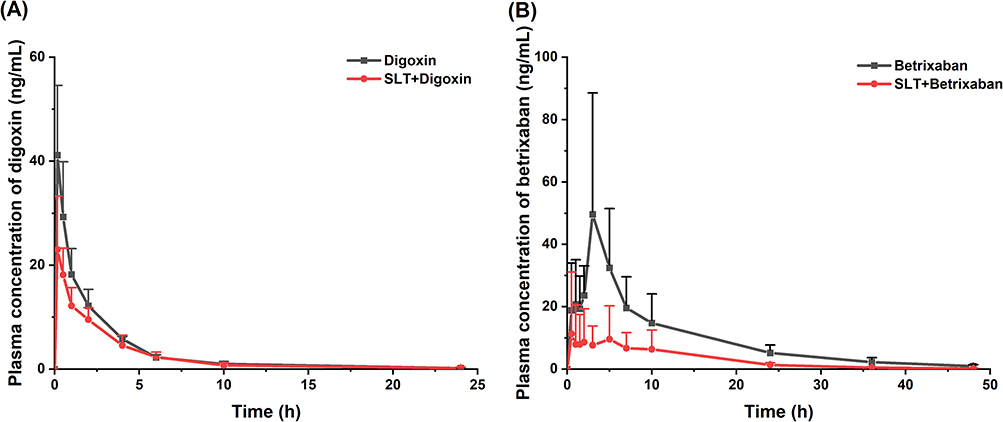 |
Figure 6 Effect of continuous administration of SLT on the pharmacokinetics of digoxin and betrixaban in rats (n=6). (A) Concentration-time curves of digoxin; (B) Concentration-time curves of betrixaban. Data are shown as mean ± SD. |
Both sets of results suggest that SLT impacts the pharmacokinetics of the two MDR1 substrates, digoxin and betrixaban, in a similar way to its effect on PIV. However, the extent of these effects differed significantly between the two probes. MDR1 expression in the intestine of rodents increases progressively from the duodenum to the colon,43 enhancing the efflux function of MDR1 for drugs undergoing enterohepatic recirculation, as confirmed in a previous study.44 Although digoxin undergoes partial biliary excretion, its primary elimination route is renal,45 and its relatively low enterohepatic recirculation rate may explain why SLT caused a weaker reduction in digoxin’s overall exposure levels. Betrixaban, a novel oral anticoagulant that directly inhibits factor Xa, has a T1/2 of 17–19 hours, ensuring a consistent pharmacological effect within a 24-hour period.46 The results demonstrated that continuous SLT administration significantly reduced betrixaban’s plasma concentrations and systemic exposure. As a substrate of the efflux transporter MDR1, betrixaban is not metabolized by CYP450. Approximately 85% of the oral dose is excreted via the bile,46 which is similar to the in vivo processing of PIV. Previous studies have shown that the AUC of orally administered betrixaban is 25 times higher in normal rats than in MDR1-knockout rats, whereas digoxin shows only a 1.6-fold difference,47 indicating that betrixaban is a more sensitive and specific substrate for MDR1. This makes betrixaban an ideal drug with which to evaluate the impact of continuous SLT administration on efflux function in rats.
Conclusion
Repeated administration of SLT reduces systemic and hepatic exposure of PIV by 32.9% and 69.0%, respectively, through intestinal MDR1 induction. This transporter-mediated mechanism diminishes drug absorption and enterohepatic recirculation, lowering PIV bioavailability. Clinicians should monitor efficacy of MDR1 substrate drugs (eg, statins) when co-administered with SLT.
Abbreviations
AUC, area under the curve; BCRP, breast cancer resistance protein; CLz/F, total body clearance; Cmax, peak concentration; CYP3A4, cytochrome P450 3A4; ESI, electrospray ionization; HPLC, high performance liquid chromatography; IS, internal standard; LC-MS/MS, liquid chromatography-tandem mass spectrometry; MDR1, multidrug resistance protein 1; MRP2, multi-drug resistance protein 2; OATP1B1, organic anion-transporting polypeptide 1B1; PIV, pitavastatin; SLT, sailuotong; T1/2, half-life; Tmax, time to reach Cmax; UV, ultraviolet; VaD, vascular dementia; Vz/F, terminal exponential volume of distribution.
Data Sharing Statement
The data in this study are available from the corresponding author upon reasonable request.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This research was funded by the National Natural Scientific Foundation of China (grant number: 81873179) and Scientific and Technological Innovation Project of China Academy of Chinese Medical Sciences (grant number: CI2021A04906).
Disclosure
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
1. O’Brien JT, Thomas A. Vascular dementia. Lancet. 2015;386(10004):1698–1706. doi:10.1016/s0140-6736(15)00463-8
2. Bir SC, Khan MW, Javalkar V, et al. Emerging concepts in vascular dementia: a review. J Stroke Cerebrovasc Dis. 2021;30(8):105864. doi:10.1016/j.jstrokecerebrovasdis.2021.105864
3. Morgan AE, Mc Auley MT. Vascular dementia: from pathobiology to emerging perspectives. Ageing Res Rev. 2024;96:102278. doi:10.1016/j.arr.2024.102278
4. Du SQ, Wang XR, Xiao LY, et al. Molecular mechanisms of vascular dementia: what can be learned from animal models of chronic cerebral hypoperfusion? Mol Neurobiol. 2017;54(5):3670–3682. doi:10.1007/s12035-016-9915-1
5. Iadecola C. The pathobiology of vascular dementia. Neuron. 2013;80(4):844–866. doi:10.1016/j.neuron.2013.10.008
6. Kalaria RN. The pathology and pathophysiology of vascular dementia. Neuropharmacology. 2018;134:226–239. doi:10.1016/j.neuropharm.2017.12.030
7. Iadecola C, Duering M, Hachinski V, et al. Vascular cognitive impairment and dementia: JACC scientific expert panel. J Am Coll Cardiol. 2019;73(25):3326–3344. doi:10.1016/j.jacc.2019.04.034
8. Paine MF, Shen DD, McCune JS. Recommended approaches for pharmacokinetic natural product-drug interaction research: a NaPDI Center Commentary. Drug Metab Dispos. 2018;46(7):1041–1045. doi:10.1124/dmd.117.079962
9. Liu Y, Jiang YR, Zhang JC, et al. Integrative Chinese and western medicine on atherosclerosis of coronary heart disease: what are the new control strategies? Chin Sci Bull. 2014;59(11):1091–1096. doi:10.1007/s11434-014-0114-z
10. Liu P, Kong MW, Yuan SH, et al. History and experience: a survey of traditional Chinese medicine treatment for Alzheimer’s disease. Evid Based Complement Alternat Med. 2014;2014:642128. doi:10.1155/2014/642128
11. Hosseini A, Razavi BM, Hosseinzadeh H. Pharmacokinetic properties of Saffron and its active components. Eur J Drug Metab Pharmacokinet. 2018;43(4):383–390. doi:10.1007/s13318-017-0449-3
12. Steiner GZ, Seto SW, Kwan YW, et al. Complementary medicine for the modification of risk factors for cognitive impairment. Evid Based Complement Alternat Med. 2017;2017:9472859. doi:10.1155/2017/9472859
13. Zhang Y, Miao L, Lin L, et al. Repeated administration of Sailuotong, a fixed combination of Panax ginseng, Ginkgo biloba, and Crocus sativus extracts for vascular dementia, alters CYP450 activities in rats. Phytomedicine. 2018;38:125–134. doi:10.1016/j.phymed.2017.02.007
14. Fan XD, Yao MJ, Yang B, et al. Chinese herbal preparation SaiLuoTong alleviates brain ischemia via Nrf2 antioxidation pathway–dependent cerebral microvascular protection. Front Pharmacol. 2021;12:16. doi:10.3389/fphar.2021.748568
15. Jia J, Wei C, Chen S, et al. Efficacy and safety of the compound Chinese medicine SaiLuoTong in vascular dementia: a randomized clinical trial. Alzheimers Dement. 2018;4(1):108–117. doi:10.1016/j.trci.2018.02.004
16. Liang J, Li F, Wei C, et al. Rationale and design of a multicenter, phase 2 clinical trial to investigate the efficacy of traditional Chinese medicine SaiLuoTong in vascular dementia. J Stroke Cerebrovascular Dis. 2014;23(10):2626–2634. doi:10.1016/j.jstrokecerebrovasdis.2014.06.005
17. Aoki T, Nishimura H, Nakagawa S, et al. Pharmacological profile of a novel synthetic inhibitor of 3-hydroxy-3-methylglutaryl-coenzyme A reductase. Arzneimittelforschung. 1997;47(8):904–909.
18. Goldstein LB, Toth PP, Dearborn-Tomazos JL, et al. Aggressive LDL-C lowering and the brain: impact on risk for dementia and hemorrhagic stroke: a scientific statement from the American Heart Association. Arterioscler Thromb Vasc Biol. 2023;43(10):e404–e442. doi:10.1161/atv.0000000000000164
19. Biffi A, Devan WJ, Anderson CD, et al. Statin treatment and functional outcome after ischemic stroke: case-control and meta-analysis. Stroke. 2011;42(5):1314–1319. doi:10.1161/strokeaha.110.605923
20. Climent E, Benaiges D, Pedro-Botet J. Lipid-lowering treatment in secondary prevention of ischaemic cerebrovascular disease. Clin Investig Arterioscler. 2020;32(4):175–182. doi:10.1016/j.arteri.2019.12.002
21. Perreault S, Ellia L, Dragomir A, et al. Effect of statin adherence on cerebrovascular disease in primary prevention. Am J Med. 2009;122(7):647–655. doi:10.1016/j.amjmed.2009.01.032
22. Miida T, Takahashi A, Ikeuchi T. Prevention of stroke and dementia by statin therapy: experimental and clinical evidence of their pleiotropic effects. Pharmacol Ther. 2007;113(2):378–393. doi:10.1016/j.pharmthera.2006.09.003
23. Sharma B, Singh N, Singh M. Modulation of celecoxib- and streptozotocin-induced experimental dementia of Alzheimer’s disease by pitavastatin and donepezil. J Psychopharmacol. 2008;22(2):162–171. doi:10.1177/0269881107081553
24. Koladiya RU, Jaggi AS, Singh N, et al. Ameliorative role of Atorvastatin and Pitavastatin in L-Methionine induced vascular dementia in rats. BMC Pharmacol. 2008;8(1):14. doi:10.1186/1471-2210-8-14
25. Hua WJ, Hua WX, Fang HJ. The role of OATP1B1 and BCRP in pharmacokinetics and DDI of novel statins. Cardiovasc Ther. 2012;30(5):e234–e241. doi:10.1111/j.1755-5922.2011.00290.x
26. Niemi M. Role of OATP transporters in the disposition of drugs. Pharmacogenomics. 2007;8(7):787–802. doi:10.2217/14622416.8.7.787
27. Kalliokoski A, Niemi M. Impact of OATP transporters on pharmacokinetics. Br J Pharmacol. 2009;158(3):693–705. doi:10.1111/j.1476-5381.2009.00430.x
28. Szakács G, Váradi A, Özvegy-Laczka C, et al. The role of ABC transporters in drug absorption, distribution, metabolism, excretion and toxicity (ADME-Tox). Drug Discov Today. 2008;13(9–10):379–393. doi:10.1016/j.drudis.2007.12.010
29. Bodó A, Bakos E, Szeri F, et al. The role of multidrug, transporters in drug availability, metabolism and toxicity. Toxicol Lett. 2003;140:133–143. doi:10.1016/s0378-4274(02)00497-6
30. Benes LB, Bassi NS, Davidson MH. The risk of hepatotoxicity, new onset diabetes and rhabdomyolysis in the era of high-intensity statin therapy: does statin type matter? Prog Cardiovasc Dis. 2016;59(2):145–152. doi:10.1016/j.pcad.2016.08.001
31. Lopez JL, Tayek JA. Voriconazole-induced hepatitis via Simvastatin- and Lansoprazole-mediated drug interactions: a case report and review of the literature. Drug Metab Dispos. 2016;44(1):124–126. doi:10.1124/dmd.115.066878
32. Groll AH, Kolve H, Ehlert K, et al. Pharmacokinetic interaction between voriconazole and ciclosporin A following allogeneic bone marrow transplantation. J Antimicrob Chemother. 2004;53(1):113–114. doi:10.1093/jac/dkh022
33. du Souich P, Roederer G, Dufour R. Myotoxicity of statins: mechanism of action. Pharmacol Ther. 2017;175:1–16. doi:10.1016/j.pharmthera.2017.02.029
34. Schierwagen R, Uschner FE, Magdaleno F, et al. Rationale for the use of statins in liver disease. Am J Physiol Gastrointest Liver Physiol. 2017;312(5):G407–g412. doi:10.1152/ajpgi.00441.2016
35. Wang ZJ, Yang HY, Xu J, et al. Prediction of atorvastatin pharmacokinetics in high-fat diet and low-dose streptozotocin-induced diabetic rats using a semiphysiologically based pharmacokinetic model involving both enzymes and transporters. Drug Metab Dispos. 2019;47(10):1066–1079. doi:10.1124/dmd.118.085902
36. Mukhtar RY, Reid J, Reckless JP. Pitavastatin. Int J Clin Pract. 2005;59(2):239–252. doi:10.1111/j.1742-1241.2005.00461.x
37. Kajinami K, Mabuchi H, Saito Y. NK-104: a novel synthetic HMG-CoA reductase inhibitor. Expert Opin Investig Drugs. 2000;9(11):2653–2661. doi:10.1517/13543784.9.11.2653
38. Deng F, Tuomi S-K, Neuvonen M, et al. Comparative hepatic and intestinal efflux transport of statins. Drug Metab Dispos. 2021;49(9):750–759. doi:10.1124/dmd.121.000430
39. Shirasaka Y, Suzuki K, Shichiri M, et al. Intestinal absorption of HMG-CoA reductase inhibitor pitavastatin mediated by organic anion transporting polypeptide and P-glycoprotein/multidrug resistance 1. Drug Metab Pharmacokinet. 2011;26(2):171–179. doi:10.2133/dmpk.dmpk-10-rg-073
40. Izat N, Kaplan O, Celebier M, et al. An isolated perfused rat liver model: simultaneous LC-MS quantification of Pitavastatin, Coproporphyrin I, and Coproporphyrin III levels in the rat liver and bile. ACS Omega. 2024;9(17):19250–19260. doi:10.1021/acsomega.4c00109
41. Hirano M, Maeda K, Matsushima S, et al. Involvement of BCRP (ABCG2) in the biliary excretion of pitavastatin. Mol Pharmacol. 2005;68(3):800–807. doi:10.1124/mol.105.014019
42. Abe K, Bridges AS, Yue W, et al. In vitro biliary clearance of angiotensin II receptor blockers and 3-hydroxy-3-methylglutaryl-coenzyme A reductase inhibitors in sandwich-cultured rat hepatocytes: comparison with in vivo biliary clearance. J Pharmacol Exp Ther. 2008;326(3):983–990. doi:10.1124/jpet.108.138073
43. Wada S, Kano T, Mita S, et al. The role of inter-segmental differences in P-glycoprotein expression and activity along the rat small intestine in causing the double-peak phenomenon of substrate plasma concentration. Drug Metab Pharmacokinet. 2013;28(2):98–103. doi:10.2133/dmpk.DMPK-12-RG-005
44. Murakami T, Takano M. Intestinal efflux transporters and drug absorption. Expert Opin Drug Metab Toxicol. 2008;4(7):923–939. doi:10.1517/17425255.4.7.923
45. Tsujimoto M, Dan Y, Hirata S, et al. Influence of SLCO1B3 gene polymorphism on the pharmacokinetics of digoxin in terminal renal failure. Drug Metab Pharmacokinet. 2008;23(6):406–411. doi:10.2133/dmpk.23.406
46. Huisman MV, Klok FA. Pharmacological properties of betrixaban. Eur Heart J Suppl. 2018;20(E):E12–E15. doi:10.1093/eurheartj/suy016
47. Miyake T, Tsutsui H, Haraya K, et al. Quantitative prediction of P-glycoprotein-mediated drug-drug interactions and intestinal absorption using humanized mice. Br J Pharmacol. 2021;178(21):4335–4351. doi:10.1111/bph.15612
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.