Back to Journals » OncoTargets and Therapy » Volume 9
HER2 induces cell proliferation and invasion of non-small-cell lung cancer by upregulating COX-2 expression via MEK/ERK signaling pathway
Authors Chi F, Wu R, Jin X, Jiang M, Zhu X
Received 11 September 2015
Accepted for publication 28 November 2015
Published 5 May 2016 Volume 2016:9 Pages 2709—2716
DOI https://doi.org/10.2147/OTT.S96197
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Faris Farassati
Feng Chi, Rong Wu, Xueying Jin, Min Jiang, Xike Zhu
Department of Medical Oncology, Shengjing Hospital of China Medical University, Shenyang, People’s Republic of China
Abstract: HER2 positivity has been well studied in various cancers, but its importance in non-small-cell lung cancer (NSCLC) is still being explored. In this study, quantitative reverse transcription polymerase chain reaction (qRT-PCR) was performed to detect HER2 and COX-2 expression in NSCLC tissues. Then, pcDNA3.1-HER2 was used to overexpress HER2, while HER2 siRNA and COX-2 siRNA were used to silence HER2 and COX-2 expression. MTT assay and invasion assay were used to detect the effects of HER2 on cell proliferation and invasion. Our study revealed that HER2 and COX-2 expression were upregulated in NSCLC tissues and HER2 exhibited a significant positive correlation with the levels of COX-2 expression. Overexpression of HER2 evidently elevated COX-2 expression, while silencing of HER2 evidently decreased COX-2 expression. Furthermore, overexpressed HER2 induced the ERK phosphorylation, and this was abolished by the treatment with U0126, a pharmacological inhibitor of MEK, an upstream kinase of ERK. HER2-induced expression and promoter activity of COX-2 were also suppressed by U0126, suggesting that the MEK/ERK signaling pathway regulates COX-2 expression. In addition, HER2 induced activation of AKT signaling pathway, which was reversed by pretreatment with U0126 and COX-2 siRNA. MTT and invasion assays revealed that HER2 induced cell proliferation and invasion that were reversed by pretreatment with U0126 and COX-2 siRNA. In this study, our results demonstrated for the first time that HER2 elevated COX-2 expression through the activation of MEK/ERK pathway, which subsequently induced cell proliferation and invasion via AKT pathway in NSCLC tissues.
Keywords: HER2, MEK/ERK, COX-2, AKT signaling pathway, non-small-cell lung cancer
Introduction
Lung cancer is one of the most common aggressive malignancies, accounting for 1.59 million deaths worldwide in 2012.1 Non-small-cell lung cancer (NSCLC) accounts for 85% of all lung cancers and is responsible for almost 80% of lung cancer-related deaths.1,2 Athough there has been great improvement in chemotherapy and molecular-targeted therapy for the treatment of lung cancers, the outcome remains poor. The invasiveness and metastasis of tumor cells are critical challenges in the clinical management of NSCLC. Therefore, an improved understanding of the molecular mechanisms involved in the development of NSCLC is required as a basis to identify novel strategies for the treatment of lung cancer.
HER2 (ErbB2) is a member of the epidermal growth factor receptor (EGFR) family of transmembrane tyrosine kinase-type receptors. It is involved in the activation of its downstream signaling cascades, which could promote cell proliferation, metastasis, and angiogenesis in tumors.3,4 HER2 functions as an oncogene and is overexpressed in a variety of malignancies, including breast, stomach, bladder, ovarian, and lung cancers.5 Recent studies demonstrated an association between HER2 expression and survival of patients with NSCLCs.6–8 Laboratory techniques for the assessment of HER2 positivity in NSCLC include immunohistochemistry (IHC) for protein overexpression, fluorescent in situ hybridization (FISH) for gene amplification, and next generation sequencing (NGS) for gene mutations.6,8 These studies emphasize the important functional role of HER2 in the progression of NSCLC and highlight its potential as a therapeutic target. However, the molecular mechanism underlying HER2 action in NSCLC remains unclear.
COX-1 and COX-2 are the rate-limiting enzymes for the synthesis of prostaglandins from arachidonic acid.9 Generally, constitutive activation of COX-2 has been demonstrated in various tumors of the lung, including adenocarcinoma,10 atypical adenomatous hyperplasia,11 bronchiolar alveolar carcinoma,12 and squamous cell carcinoma,13 and its overexpression has been associated with poor prognosis and short survival of patients with lung cancer.14 Recently, high expression of COX-2 has been implicated in NSCLC progression and is associated with tumor invasion and metastasis.15,16 COX-2 expression is significantly increased in nodal metastasis compared to primary cancers.17 Although overexpressed COX-2 was reported to correlate with overexpressed HER2 in NSCLC,18 the intrinsic linkage has remained unclear.
In this study, our results demonstrated that HER2 elevated COX-2 expression through the activation of MEK/ERK pathway, which subsequently induced cell proliferation and invasion via AKT pathway.
Materials and methods
Tissue samples and cell lines
Paired NSCLC and normal adjacent lung tissues were obtained, with informed consent, from 25 patients who underwent primary surgical resection of NSCLC between 2014 and 2015 at the Shengjing Hospital of China Medical University (Shenyang, People’s Republic of China). Informed consent was obtained from each patient to approve the use of their tissues for research purposes. The study protocol was approved by the Institute Research Ethics Committee at Shengjing Hospital of China Medical University. The NSCLC cell lines A549 was obtained from Shengjing Hospital and cultured in Dulbecco’s Modified Eagle’s Medium (DMEM; Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS). All cells were incubated in a humidified (37°C, 5% CO2) incubator.
Cell transfection
We constructed pcDNA3.1-HER2 and transfected as previously described.19 To knock down the expression of HER2 and COX-2 in A549 cells, transient transfection experiment with HER2 small interfering RNA (siRNA) and COX-2 siRNA was performed according to the previous publication.20 After the cells were transfected and cultured for 48 hours, they were collected for Western blot and quantitative reverse transcription polymerase chain reaction (qRT-PCR) analyses.
Luciferase assay for COX-2 transactivation
A549 cells were stably transfected with the COX-2 luciferase reporter plasmid and maintained in 5% FBS-DMEM. JB6 P+ cells carrying each reporter plasmid were seeded in 0.1% FBS-MEM for 24 hours. Cells were pretreated with (or without) U0126 for 1 hour, and transfected with pcDNA3.1-HER2 or HER2 siRNA for 24 hours. Cells were harvested using lysis buffer composed of 0.1 M potassium phosphate buffer (pH 7.8), 1% Triton X-100, 1 mM DTT, and 2 mM EDTA. Luciferase activity was measured using a luminometer (Luminoskan Ascent; Thermo Electron, Helsinki, Finland).
qRT-PCR
Total RNA was isolated from tissues and cell lines using the miRNeasy Mini Kit (Qiagen NV, Venlo, the Netherlands). The miRNA Q-PCR Detection Kit (GeneCopoeia, Rockville, MD, USA) was used for quantification of miRNA levels according to the manufacturer’s protocol. For quantification of messenger RNA (mRNA) levels, the reverse transcription reactions were conducted with the RevertAid TM H Minus First Strand cDNA Synthesis Kit (Fermentas GmbH, St Leon-Rot, Germany). PCR amplification for the quantification of HER2, COX-2, and GAPDH mRNAs was performed using an ABI PRISM 7300 Sequence Detection System (Applied Biosystems, Foster City, CA, USA) and a SYBR®Premix Ex Taq™ ii (Perfect Real Time) Kit (Takara Bio, Shiga, Japan). The primers were as follows: Homo-HER2, sense: 5′-CCATCTGCACCATTGATGTC-3′, antisense: 5′-ATGCGGGAGAATTCAGACAC-3′; COX-2, sense: 5′-CCAGCACTTCACGCATCAGT-3′, antisense: 5′-ACGCTGTCTAGCCAGAGTTTCAC-3′; and β-actin, sense: 5′-CATTAAGGAGAAGCTGTGCT-3′, antisense: 5′-GTTGAAGGTAGTTTCGTGGA-3′.
Western blot
Whole cell extracts were prepared with a cell lysis reagent (Sigma-Aldrich, St Louis, MO, USA) according to the manual, and then the protein was quantified by a bicinchoninic acid (BCA) assay (Pierce, Rockford, IL, USA). The protein samples were later separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (10 %) and detected by Western blot using the following specific antibodies: anti-HER2, COX-2, P-ERK, ERK, AKT, p-AKT, MMP-2, caspase-3, and β-actin polyclonal antibodies (Santa Cruz Biotechnology Inc., Dallas, TX, USA), the antibodies were suspended in 5% BSA. Goat anti-rabbit IgG (Pierce) secondary antibody conjugated to horseradish peroxidase and ECL detection systems (SuperSignal West Femto, Pierce) were used for detection.
Cell proliferation assay
The MTT assay was used to estimate cell viability.21 Briefly, cells were plated at a density of 1×104 cells per well in 96-well plates. After exposure to specific treatment, the cells were incubated with MTT at a final concentration of 0.5 mg/mL for 4 hours at 37°C. After the removal of the medium, 150 mM dimethyl sulfoxide (DMSO) solutions were added to dissolve the formazan crystals. The absorbance was read at 570 nm using a multiwell scanning spectrophotometer reader. Cells in the control group were considered 100% viable.
Cell invasion assay
Cells were cultivated to 80% confluence on the 12-well plates. Then, the procedures of cellular growth were observed at 24 hours. All the experiments were repeated in triplicate. The transwell invasion chambers were used to evaluate cell invasion. Then cells invading cells across the membrane were counted under a light microscope.
Statistical analysis
Each experiment was repeated at least three times. Data were shown as mean ± standard deviation (SD) and analyzed using SPSS 18.0. Statistical comparisons between groups were analyzed using Student’s t-test and a two-tailed P<0.05 was considered to indicate statistical significance.
Results
Correlation between HER2 and COX-2 in clinical NSCLC tissues
HER2 and COX-2 expression were reported to be upregulated in NSCLC tissues.6,22 mRNA expression levels of HER2 and COX-2 were determined in 25 paired clinical NSCLC and human normal lung tissues using qRT-PCR. Compared with their noncancerous counterparts, significant upregulation of HER2 and COX-2 was observed in all the 25 NSCLC samples (Figure 1A and B). Then we assessed the correlation between HER2 and COX-2. As expected, we found that the levels of HER2 exhibited a significant positive correlation with the levels of COX-2 mRNA (Pearson’s correlation coefficient of 0.7532, P<0.01) (Figure 1C). Overall, these findings demonstrate that COX-2 and HER2 correlated significantly in terms of mRNA expression in clinical NSCLC tissues.
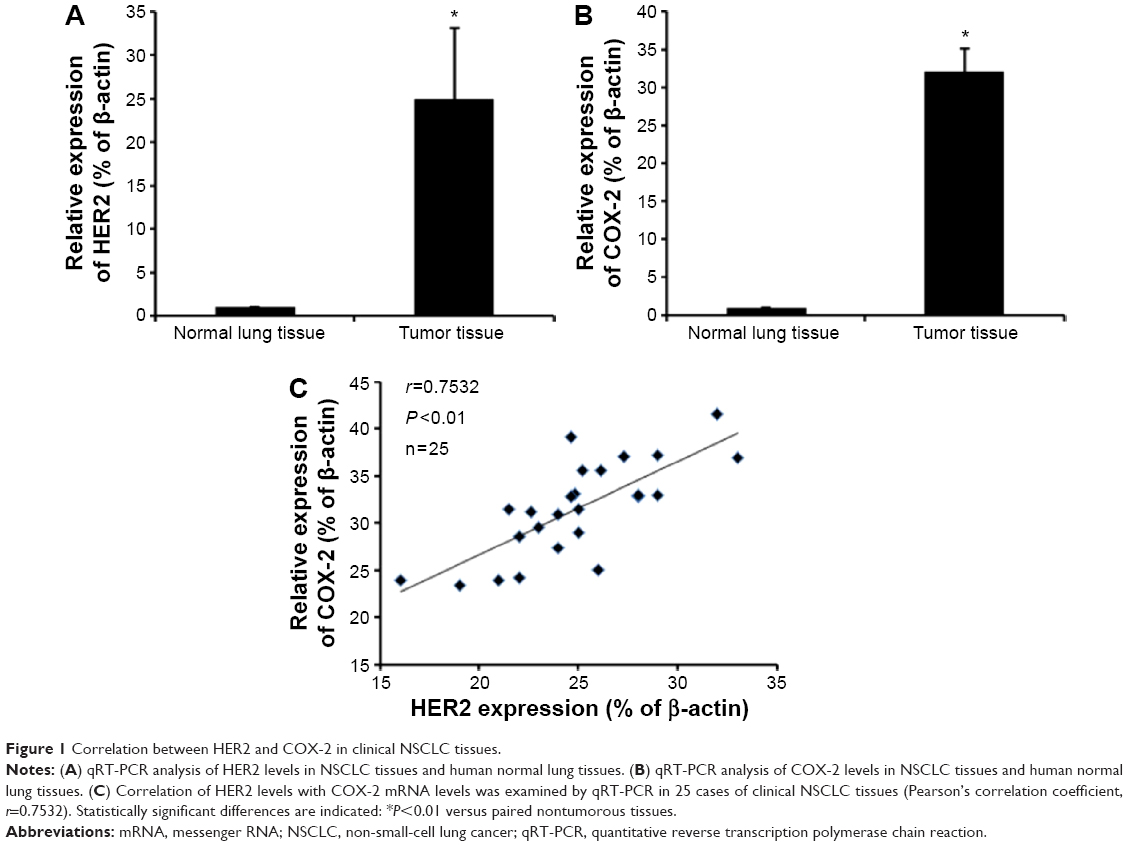 | Figure 1 Correlation between HER2 and COX-2 in clinical NSCLC tissues. |
HER2 induces COX-2 expression and activity in NSCLC
To investigate whether HER2 induces COX-2 expression, we treated cells with pcDNA3.1-HER2 and analyzed COX-2 expression by Western blot analysis. pcDNA3.1-HER2 markedly elevated COX-2 expression and HER2 siRNA markedly repressed COX-2 expression (Figure 2B–D). To determine whether HER2 upregulates COX-2 through transactivation of the COX-2 gene, we evaluated the effect of HER2 on COX-2 promoter activity using a luciferase assay. As shown in Figure 2A, HER2 significantly increased the COX-2 promoter activity.
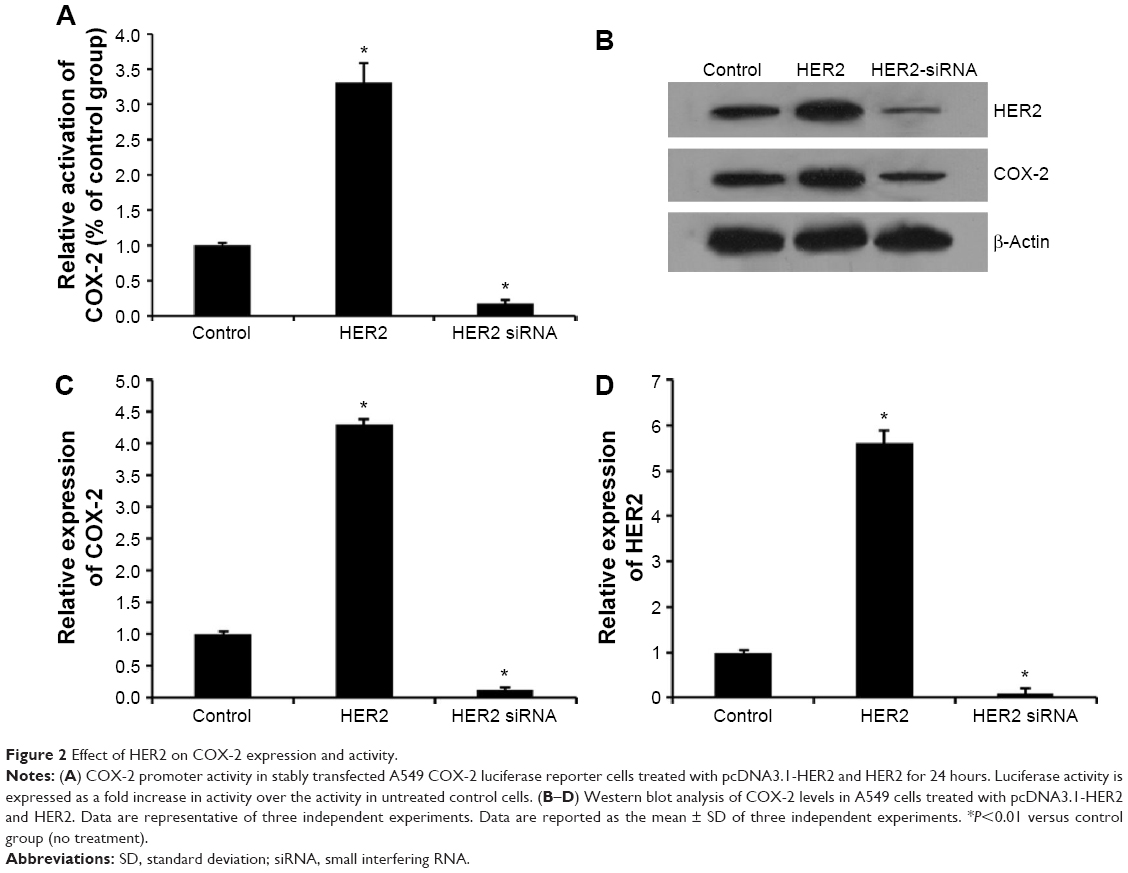 | Figure 2 Effect of HER2 on COX-2 expression and activity. |
HER2 induced COX-2 upregulation by MEK/ERK pathway and in turn influenced AKT pathway
MEK/ERK pathway is the important downstream signaling of HER2 in various tumors.8 Here, we investigated whether the ERK pathway is activated by HER2 in NSCLC. Transfection with pcDNA3.1-HER2 resulted in ERK phosphorylation compared with control cells (Figure 3A). Moreover, pretreatment with U0126, an MEK inhibitor, abolished HER2-induced ERK phosphorylation (Figure 3A), suggesting that ERK phosphorylation in the presence of HER2 is MEK-dependent. To investigate the mechanism underlying HER2-induced COX-2 expression in A549 cells, we examined the effect of MEK inhibitor U0126 on COX-2 expression and promoter activity. As shown in Figure 3A and B, U0126 pretreatment abolished HER2-induced COX-2 expression and promoter activity. In conclusion, HER2-induced COX-2 upregulation is accompanied by ERK activation and both events are MEK-dependent.
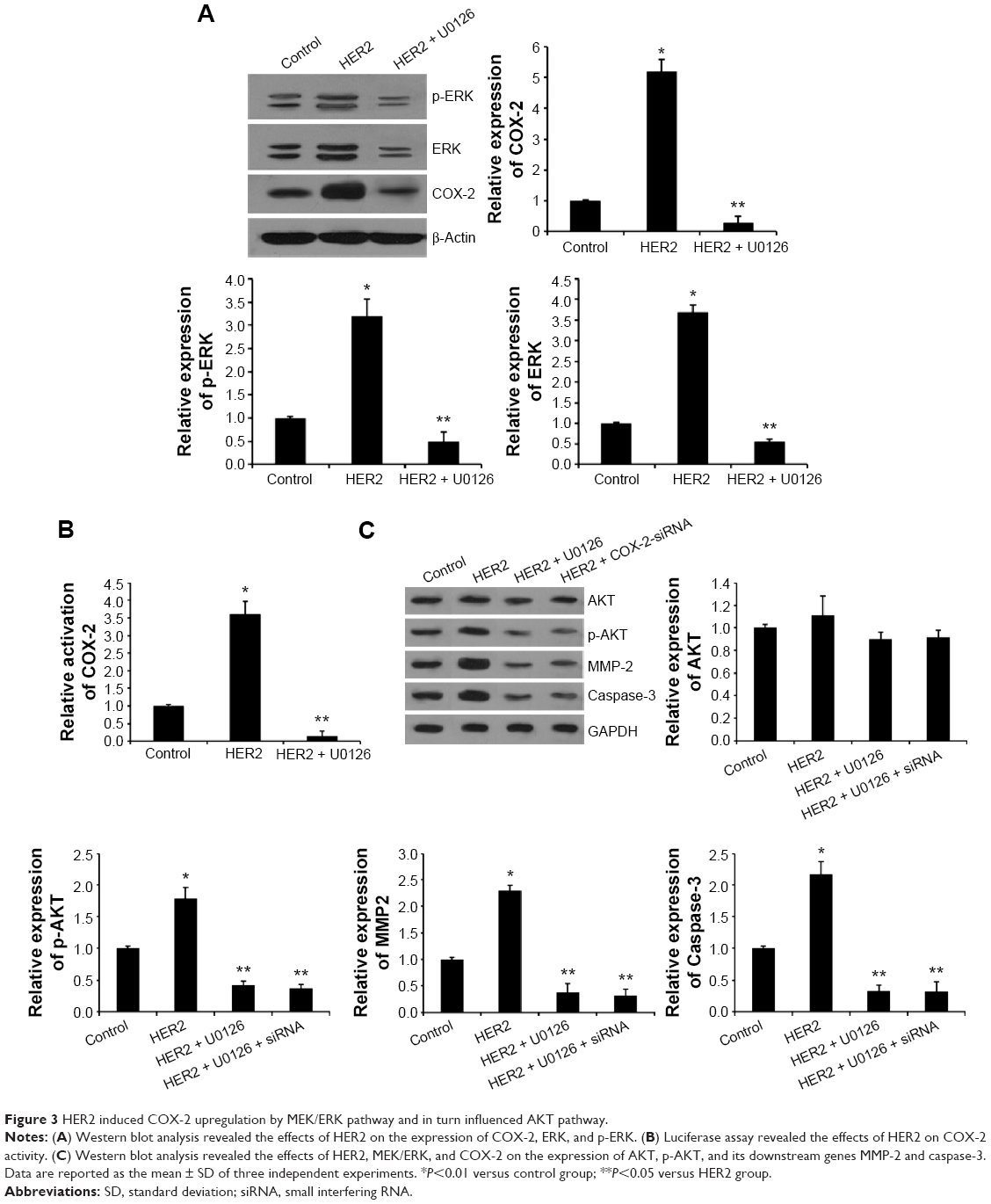 | Figure 3 HER2 induced COX-2 upregulation by MEK/ERK pathway and in turn influenced AKT pathway. |
Recently, COX-2-mediated PKB/AKT activation was reported to be involved in multiple tumor processes.23,24 Here, we determined the effects of HER2-induced COX-2 upregulation on the activation of AKT signaling pathway. HER2 elevated the expression of p-AKT and its downstream genes MMP-2 and caspase-3, while the expression of total AKT was not affected (Figure 3C). To further investigate the relationship between the AKT signal and HER2, COX-2 siRNA and U0126 were used for inhibition experiment. The results showed that COX-2 siRNA and U0126 led to the obvious decrease of p-AKT, MMP-2, and caspase-3 expression in the overexpressed HER2 A549 cells. Collectively, these findings suggest that HER2-mediated MEK/ERK regulates the expression of COX-2, which subsequently suppresses the AKT signaling pathway in NSCLC cells.
HER2-mediated MEK/ERK induced cell proliferation via COX-2 pathway
Next, the proliferation and invasion of A549 cell transfected with HER2 or pretreated with COX-2 siRNA and U0126 were detected by using MTT assay and invasion assay. As shown in Figure 4A, HER2 significantly increased cell proliferation ability, which was reversed by COX-2 siRNA or U0126. The transwell invasion assay manifested that cell invasion was greatly increased when the cells were transfected with HER2, but pretreatment with COX-2 siRNA and U0126 could reverse elevation of cell invasion mediated by pcDNA3.1-HER2 in the cells (Figure 4B). Therefore, these results indicated that HER2-mediated MEK/ERK induced cell proliferation and invasion via COX-2/AKT pathway.
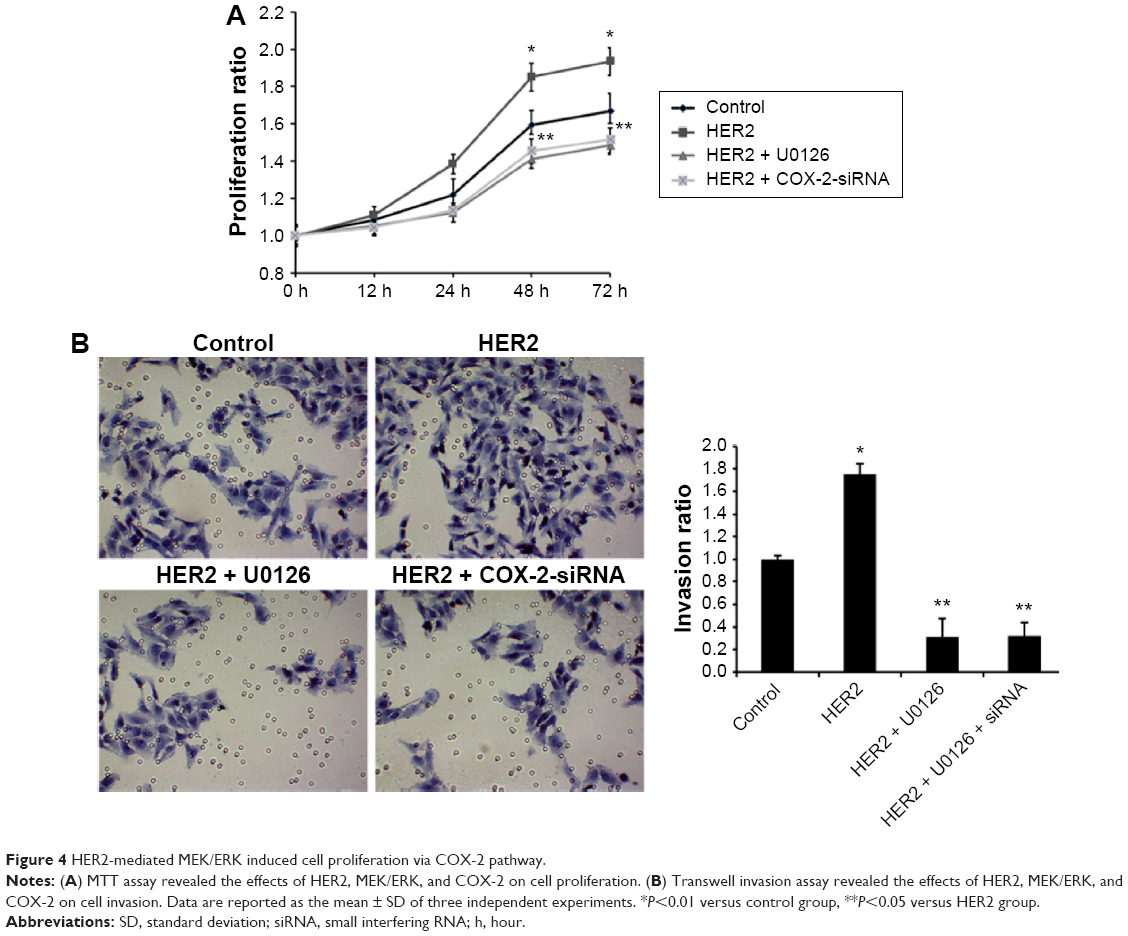 | Figure 4 HER2-mediated MEK/ERK induced cell proliferation via COX-2 pathway. |
Discussion
HER2, an important member of the EGFR family, overexpression typically occurs in the placenta, embryonic epithelial tissue, and various tumors.5 Tan et al demonstrated that HER2/neu overexpression was significantly associated with histologic subtype of NSCLC and statistical significance was observed between HER2/neu expression and tumor differentiation, with strong positive expression observed more frequently in poorly differentiated tumors.7 COX-2 is reported to be overexpressed in HER2/neu-positive breast cancer.25 COX-2 expression correlated with HER2 expression in NSCLC.18 However, the potential molecular mechanism remains unclear. Here, we applied qRT-PCR to detect the expression of HER2 and COX-2 in NSCLC tissue. Consistent with previous studies,18 HER2 and COX-2 were significantly upregulated in NSCLC tissues and HER2 exhibited a significant positive correlation with the levels of COX-2 mRNA. Furthermore, HER2 markedly elevated COX-2 expression and COX-2 promoter activity, which further confirmed the positive correlation between HER2 and COX-2.
MEK/ERK pathway is the important downstream signaling of HER2. HER2 receptor dimerization catalytically activates downstream signaling through MEK/ERK pathways, involved in cellular proliferation, differentiation, and migration.26 It is known that overexpression of HER2 in breast cancer patients lead to aberrant signaling of the MEK/ERK pathway.27,28 Lapatinib, a selective small-molecule inhibitor of EGFR and HER2 tyrosine kinases, quickly disables EGFR and HER2 signaling, resulting in the inhibition of the PI3K/AKT and MEK/ERK pathways.29 Consistent with these studies, we found that HER2 regulated MEK/ERK pathway in NSCLC. We further found that U0126, an MEK inhibitor, abolished HER2-induced COX-2 expression in A549 cells, indicating that HER2 induced COX-2 upregulation by MEK/ERK pathway. Similar results were also reported in a previous study, which showed that myricetin inhibits acrylamide-induced COX-2 expression by blocking activation of the MEK/ERK pathway.30
Recently, the mechanisms involved in COX-2-mediated tumor progression have been widely explored, especially the interplay between COX-2 and PI3K/AKT signaling. For example, wogonin, a COX-2 inhibitor, increased the sensitivity of hepatocellular carcinoma cells to chemotherapeutic agents partially through regulation of PI3K/AKT signaling.31 COX-2 was reported to positively regulate AKT signaling via suppression of PTEN activity, which is the upstream of AKT and mediates its phosphorylation.24 Another recent study showed that prostaglandin E2 (PGE2), the production of COX-2, was also involved in the activation of PI3K/AKT pathway.32 Here, we have identified that transfection with HER2 elevated the expression of p-AKT and its downstream genes, MMP-2 and caspase-3, which was reversed by U0126 and COX-2 siRNA. However, HER2, U0126, and COX-2 siRNA did not influence the total AKT expression. These results suggested that HER2-mediated MEK/ERK regulates the expression of COX-2, which subsequently suppresses the AKT signaling pathway in NSCLC cells. Furthermore, MTT assay and invasion results indicated that HER2-mediated MEK/ERK induced cell proliferation and invasion via COX-2/AKT pathway.
Conclusion
In conclusion, our results establish a functional link between HER2 and COX-2 expression in NSCLC, demonstrating that COX-2 is mediated by HER2-mediated MEK/ERK, which subsequently affect cell proliferation and invasion via AKT signaling pathway. Collectively, this finding helps us understand the molecular mechanism of NSCLC carcinogenesis and also provides potential therapeutic target for NSCLC.
Author contributions
FC, RW, and XJ designed the study, carried out the experiments, and drafted the manuscript; FC, MJ, and XZ participated in the experiments and data analysis. All authors read and approved the final manuscript. All authors contributed toward data analysis, drafting and critically revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA CancerJ Clin. 2015;65(2):87–108. | ||
Buyukcelik A, Yalcin B, Utkan G. Multidisciplinary management of lung cancer. N Engl J Med. 2004;350(19):2008–2010; author reply 2008–2010. | ||
Epis MR, Giles KM, Barker A, Kendrick TS, Leedman PJ. miR-331-3p regulates ERBB-2 expression and androgen receptor signaling in prostate cancer. J Biol Chem. 2009;284(37):24696–24704. | ||
Scott GK, Goga A, Bhaumik D, Berger CE, Sullivan CS, Benz CC. Coordinate suppression of ERBB2 and ERBB3 by enforced expression of micro-RNA miR-125a or miR-125b. J Biol Chem. 2007;282(2):1479–1486. | ||
Agus DB, Bunn PA Jr, Franklin W, Garcia M, Ozols RF. HER-2/neu as a therapeutic target in non-small cell lung cancer, prostate cancer, and ovarian cancer. Semin Oncol. 2000;27(6 Suppl 11):53–63; discussion 92–100. | ||
Aleric I, Razumovic JJ, Koprivica B. HER-2/neu oncogene and estrogen receptor expression in non small cell lung cancer patients. Med Pregl. 2012;65(5–6):210–215. | ||
Tan D, Deeb G, Wang J, et al. HER-2/neu protein expression and gene alteration in stage I–IIIA non-small-cell lung cancer: a study of 140 cases using a combination of high throughput tissue microarray, immunohistochemistry, and fluorescent in situ hybridization. Diagn Mol Pathol. 2003;12(4):201–211. | ||
Mar N, Vredenburgh JJ, Wasser JS. Targeting HER2 in the treatment of non-small cell lung cancer. Lung Cancer. 2015;87(3):220–225. | ||
Smith WL, DeWitt DL, Garavito RM. Cyclooxygenases: structural, cellular, and molecular biology. Annu Rev Biochem. 2000;69:145–182. | ||
Wolff H, Saukkonen K, Anttila S, Karjalainen A, Vainio H, Ristimaki A. Expression of cyclooxygenase-2 in human lung carcinoma. Cancer Res. 1998;58(22):4997–5001. | ||
Hosomi Y, Yokose T, Hirose Y, et al. Increased cyclooxygenase 2 (COX-2) expression occurs frequently in precursor lesions of human adenocarcinoma of the lung. Lung Cancer. 2000;30(2):73–81. | ||
Diperna CA, Bart RD, Sievers EM, Ma Y, Starnes VA, Bremner RM. Cyclooxygenase-2 inhibition decreases primary and metastatic tumor burden in a murine model of orthotopic lung adenocarcinoma. J Thorac Cardiovasc Surg. 2003;126(4):1129–1133. | ||
Hida T, Yatabe Y, Achiwa H, et al. Increased expression of cyclooxygenase 2 occurs frequently in human lung cancers, specifically in adenocarcinomas. Cancer Res. 1998;58(17):3761–3764. | ||
Grimminger PP, Stohlmacher J, Vallbohmer D, et al. Prognostic significance and clinicopathological associations of COX-2 SNP in patients with nonsmall cell lung cancer. J Oncol. 2009;2009:139590. | ||
Lim BJ, Jung SS, Choi SY, Lee CS. Expression of metastasis-associated molecules in non-small cell lung cancer and their prognostic significance. Mol Med Rep. 2010;3(1):43–49. | ||
Lee JM, Mao JT, Krysan K, Dubinett SM. Significance of cyclooxygenase-2 in prognosis, targeted therapy and chemoprevention of NSCLC. Future Oncol. 2007;3(2):149–153. | ||
Jang TJ, Jeon KH, Jung KH. Cyclooxygenase-2 expression is related to the epithelial-to-mesenchymal transition in human colon cancers. Yonsei Med J. 2009;50(6):818–824. | ||
Brattstrom D, Wester K, Bergqvist M, et al. HER-2, EGFR, COX-2 expression status correlated to microvessel density and survival in resected non-small cell lung cancer. Acta Oncol. 2004;43(1):80–86. | ||
Li S, Ma X, Ma L, Wang C, He Y, Yu Z. Effects of ectopic HER-2/neu gene expression on the COX-2/PGE2/P450arom signaling pathway in endometrial carcinoma cells: HER-2/neu gene expression in endometrial carcinoma cells. J Exp Clin Cancer Res. 2013;32:11. | ||
Faltus T, Yuan J, Zimmer B, et al. Silencing of the HER2/neu gene by siRNA inhibits proliferation and induces apoptosis in HER2/neu-overexpressing breast cancer cells. Neoplasia. 2004;6(6):786–795. | ||
Liu B, Che W, Xue J, et al. SIRT4 prevents hypoxia-induced apoptosis in H9c2 cardiomyoblast cells. Cell Physiol Biochem. 2013;32(3):655–662. | ||
Bhat IA, Rasool R, Qasim I, et al. COX-2 overexpression and -8473 T/C polymorphism in 3′ UTR in non-small cell lung cancer. Tumour Biol. 2014;35(11):11209–11218. | ||
Cheng Y, Li Y, Liu D, Zhang R, Zhang J. miR-137 effects on gastric carcinogenesis are mediated by targeting Cox-2-activated PI3K/AKT signaling pathway. FEBS Lett. 2014;588(17):3274–3281. | ||
Li CJ, Chang JK, Wang GJ, Ho ML. Constitutively expressed COX-2 in osteoblasts positively regulates Akt signal transduction via suppression of PTEN activity. Bone. 2011;48(2):286–297. | ||
Subbaramaiah K, Norton L, Gerald W, Dannenberg AJ. Cyclooxygenase-2 is overexpressed in HER-2/neu-positive breast cancer: evidence for involvement of AP-1 and PEA3. J Biol Chem. 2002;277(21):18649–18657. | ||
Spector NL, Blackwell KL. Understanding the mechanisms behind trastuzumab therapy for human epidermal growth factor receptor 2-positive breast cancer. J Clin Onco. 2009;27(34):5838–5847. | ||
Arteaga CL, Engelman JA. ERBB receptors: from oncogene discovery to basic science to mechanism-based cancer therapeutics. Cancer Cell. 2014;25(3):282–303. | ||
Tai W, Mahato R, Cheng K. The role of HER2 in cancer therapy and targeted drug delivery. J Control Release. 2010;146(3):264–275. | ||
Konecny GE, Pegram MD, Venkatesan N, et al. Activity of the dual kinase inhibitor lapatinib (GW572016) against HER-2-overexpressing and trastuzumab-treated breast cancer cells. Cancer Res. 2006;66(3):1630–1639. | ||
Lim TG, Lee BK, Kwon JY, Jung SK, Lee KW. Acrylamide up-regulates cyclooxygenase-2 expression through the MEK/ERK signaling pathway in mouse epidermal cells. Food Chem Toxicol. 2011;49(6):1249–1254. | ||
Zhao L, Sha YY, Zhao Q, et al. Enhanced 5-fluorouracil cytotoxicity in high COX-2 expressing hepatocellular carcinoma cells by wogonin via the PI3K/Akt pathway. Biochem Cell Biol. 2013;91(4):221–229. | ||
Vo BT, Morton D Jr, Komaragiri S, Millena AC, Leath C, Khan SA. TGF-beta effects on prostate cancer cell migration and invasion are mediated by PGE2 through activation of PI3K/AKT/mTOR pathway. Endocrinology. 2013;154(5):1768–1779. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.