Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 19
Hepatogenomics of MAFLD in Asian Population: Genetic Polymorphisms and Pathway-Based Insights
Authors Justyn M ![]() , Sutanto C, Barliana MI
, Sutanto C, Barliana MI ![]() , Heryaman H
, Heryaman H ![]() , Meiliana A
, Meiliana A ![]() , Puspitasari IM
, Puspitasari IM ![]()
Received 13 February 2026
Accepted for publication 27 April 2026
Published 10 June 2026 Volume 2026:19 603042
DOI https://doi.org/10.2147/DMSO.S603042
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Pablo Corral
Matthew Justyn,1 Cheerly Sutanto,2 Melisa Intan Barliana,3,4 Henhen Heryaman,5 Anna Meiliana,6 Irma Melyani Puspitasari4,6
1Doctoral of Pharmacy Program, Faculty of Pharmacy, Padjadjaran University, Sumedang, Indonesia; 2Faculty of Medicine, Pelita Harapan University, Tangerang, Indonesia; 3Department of Pharmaceutical Biology, Faculty of Pharmacy, Padjadjaran University, Sumedang, Indonesia; 4Pharmaceutical Care Innovation, Faculty of Pharmacy, Padjadjaran University, Sumedang, Indonesia; 5Department of Biomedical Sciences, Faculty of Medicine, Padjadjaran University, Sumedang, Indonesia; 6Department of Pharmacology and Clinical Pharmacy, Faculty of Pharmacy, Universitas Padjadjaran, Sumedang, Indonesia
Correspondence: Irma Melyani Puspitasari, Department of Pharmacology and Clinical Pharmacy, Faculty of Pharmacy, Universitas Padjadjaran, Sumedang, Indonesia, Tel +62 22 84288888, Ext.351, Email [email protected]
Abstract: Metabolic dysfunction–associated fatty liver disease (MAFLD) has emerged as a major public health concern across Asia, marked by rising prevalence, younger age at presentation, and variability in clinical course. This variability reflects a complex interplay between metabolic exposures and genetic architecture, contributing to heterogeneity in disease susceptibility and progression. While dietary patterns, sedentary lifestyle, and metabolic comorbidities remain central contributors, inherited susceptibility influences hepatic fat accumulation, progression to steatohepatitis, and fibrotic transformation. This review aims to summarize genetic polymorphisms implicated in MAFLD among Asian populations and to explore their role in identifying individuals at increased inherited risk. A pathway-oriented perspective is adopted to contextualize how these variants contribute to key biological mechanisms underlying MAFLD. A narrative review approach was employed, drawing upon genome-wide association studies, candidate gene analyses, and functional research. Genetic variants were grouped into principal pathogenic pathways, including lipid handling, insulin resistance and de novo lipogenesis, cholesterol metabolism, inflammatory signaling, and fibrogenesis. While emphasis is placed on evidence from Asian cohorts, selected variants are also discussed based on mechanistic relevance, even when direct population-based data remain limited. Differences in allele frequency and effect size between Asian and Western populations were considered to clarify ethnic variation. Among the identified variants, PNPLA3 rs738409 consistently emerges as a dominant determinant of hepatic fat accumulation and adverse histological features. Additional polymorphisms further modulate risk, with some exerting protective effects. Taken together, current evidence supports integrating genetic markers into risk stratification models for earlier recognition of genetically predisposed individuals. This pathway-based synthesis provides a framework for understanding MAFLD heterogeneity in Asian populations and may inform precision-oriented prevention and individualized management.
Keywords: MAFLD, polymorphisms, genomics, Asia
Introduction
Over the past decade, the prevalence of metabolic dysfunction-associated fatty liver disease (MAFLD) has risen significantly, becoming a major public health concern. In the Asia-Pacific region, MAFLD affects 28–40% of the population, with South Asia reporting the highest prevalence at around 34%, surpassing other subregions within Asia-Pacific.1 Alongside this growing burden, the terminology surrounding fatty liver disease has also evolved to reflect its underlying pathophysiology better. While NAFLD defined based on the exclusion of significant alcohol intake, it was redefined in 2019 as MAFLD to emphasize the central role of metabolic dysfunction. More recently, the nomenclature has continued to develop with the introduction of metabolic dysfunction-associated steatosis liver disease (MASLD).2,3 In this review, the term MAFLD used consistently, as it aligns with the metabolic focus of this discussion. Nevertheless, studies using NAFLD or related terminology included and interpreted within the same disease spectrum.
Concurrently, the prevalence of MAFLD to rise due to increasing rates of metabolic disorders, unhealthy diets, sedentary lifestyles, and low physical activity.1 Its pathogenesis is multifactorial, involving interactions among genetic, epigenetic, environmental, and metabolic factors.4 MAFLD is also associated with a higher risk of chronic macrovascular and microvascular complications.5,6 Among these contributing factors, genetic predisposition plays a pivotal role in both the development and progression of MAFLD. Specifically, single nucleotide polymorphisms (SNPs) in genes involved in hepatic lipid metabolism and remodeling have been strongly correlated with susceptibility to MAFLD7,8. Despite growing recognition of the genetic role of this disease, this area remains incompletely understood, leaving a critical gap in the identification and clinical application of genetic biomarkers for early detection and personalized management of MAFLD.
Importantly, Asian populations call for particular attention, as MAFLD in this group often presents with distinct clinical and metabolic characteristics. Compared with Western populations, individuals in Asia tend to develop hepatic fat accumulation at lower body mass index (BMI) thresholds and exhibit a higher prevalence of lean MAFLD. These differences suggest unique gene–environment interactions and highlight the need for population-specific investigation of genetic susceptibility. This phenotype underscores the importance of genetic determinants in MAFLD pathogenesis in Asian populations, where traditional markers such as BMI may underestimate disease risk.
This gap highlights the urgency of further research into genetic variants that could serve as predictive biomarkers, enabling targeted screening and risk stratification in Asian population. Despite growing recognition of the genetic basis of MAFLD, there is a lack of data on genetic variants specific to Asian populations. Most studies have focused on Western cohorts, overlooking population-specific genetic factors and gene–environment interactions in Asia. This review was developed through a structured, comprehensive literature assessment to capture current evidence on population-level genetic determinants of MAFLD in Asian populations. The primary focus was on genome-wide association studies (GWAS) and population-based genetic polymorphism studies conducted in Asian cohorts. Searches were performed in PubMed, Scopus, Web of Science, and Google Scholar using combinations of the terms “MAFLD”, “NAFLD”, and “genetic polymorphism”. Publications from 2010 to 2025 were screened. Studies conducted exclusively in non-Asian populations were not considered as primary evidence but included where relevant for mechanistic or comparative interpretation. In addition to population-based studies, mechanistic investigations exploring genetic effects on lipid metabolism and hepatic pathways were included when conducted in relevant animal models or in vitro systems. Studies conducted exclusively in non-Asian populations, case reports, conference abstracts, and reports focusing on alcoholic or viral liver disease were excluded.
The evidence was synthesized narratively, with particular attention to distinguishing findings derived from Asian cohorts from those based on non-Asian populations or mechanistic studies, to avoid overgeneralization and ensure contextual relevance to Asian MAFLD.
By integrating population-specific genetic evidence with broader mechanistic insights, this review aims to provide a more nuanced understanding of MAFLD susceptibility in Asian populations and to inform future strategies for risk stratification and precision-based management.
Variants Affect Metabolic Associated Fatty Liver Disease (MAFLD)
Numerous studies on genetic polymorphisms, particularly variants associated with MAFLD, have identified multiple genes linked to both disease progression and a reduced risk of MAFLD. Although the use of genetic data remains primarily limited to biological markers for prevention based on genetic profiles, identifying these genes is expected to support the development of future therapeutic strategies and target interventions to reduce the prevalence of MAFLD.
Genetic Variants Contribute to Increasing the Risk of MAFLD
Lipid Metabolism and Fat Accumulation
Lipid metabolism is a central aspect of the pathophysiology of MAFLD, previously known as NAFLD. The accumulation of fat in the liver is one of the hallmark features of MAFLD. Several genetic variants have been identified as contributors to this phenomenon, including the complex interplay between these variants and key metabolic pathways involved in lipid handling, insulin signaling, and hepatic fat accumulation, which contributes to the development and progression of metabolic dysfunction–associated fatty liver disease (MAFLD). Polymorphisms in genes regulating de novo lipogenesis, fatty acid uptake, triglyceride synthesis, and very-low-density lipoprotein (VLDL) secretion promote intrahepatic lipid accumulation. In parallel, genetic alterations affecting insulin sensitivity, mitochondrial function, and oxidative stress exacerbate lipid dysregulation and hepatocellular injury. These processes collectively lead to hepatic steatosis, lipotoxicity, and low-grade inflammation, thereby increasing susceptibility to MAFLD and its metabolic and cardiovascular comorbidities (Figure 1).
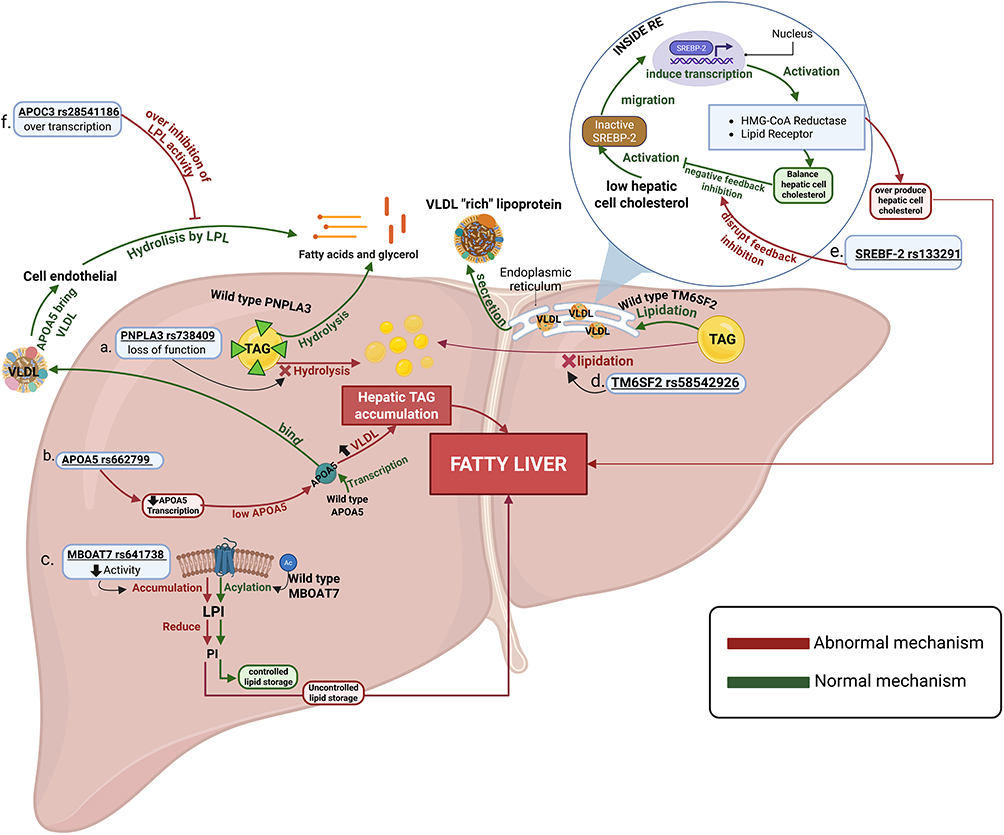 |
Figure 1 Mechanism by which Genetic polymorphisms Influence Lipid Metabolism and Hepatic Steatosis to Elevate MAFLD Risk. Created with BioRender.com. Justyn M, 2026. Available at: https://BioRender.com/wcpxngl. Abbreviations: LPI, Lysophosphatidylinositol; PI, phosphatidylinositol; TAG, triacylglycerol; VLDL, very-low density lipoprotein; RE, reticulum endoplasm. |
PNPLA3 rs738409 C>G
Hepatic lipid remodeling has appeared as a central determinant of disease severity in MAFLD, with genetic variation in lipid droplet biology playing a pivotal role. Patatin-like phospholipase domain–containing protein 3 (PNPLA3), encoded on chromosome 22q13.31, is a lipid droplet–associated enzyme expressed in hepatocytes and hepatic stellate cells, where it regulates intracellular triglyceride turnover. Under physiological conditions, PNPLA3 exhibits triacylglycerol (TAG) lipase activity, catalysing the hydrolysis of TAG stored within hepatic lipid droplets into diacylglycerol (DAG) and free fatty acids (FFA), thereby facilitating lipid mobilization and preventing excessive lipid droplet expansion in the liver.9,10 The PNPLA3 rs738409 polymorphism results in an isoleucine-to-methionine substitution at residue 148 (I148M), which profoundly impairs the TAG hydrolase activity of PNPLA3.9 This loss of function leads to defective hydrolysis of lipid droplets, resulting in progressive intracellular triglyceride retention. Moreover, the mutant PNPLA3 I148M protein accumulates on the surface of hepatic lipid droplets, where it sterically hinders access by other lipolytic enzymes, further suppressing triglyceride mobilization.10 The final effect is excessive enlargement of lipid droplets and marked hepatic fat accumulation, hallmarks of PNPLA3-driven MAFLD. Beyond simple steatosis, evidence from experimental and mechanistic studies further suggests that PNPLA3 rs738409 exerts pleiotropic effects on disease progression. In hepatic stellate cells, the I148M variant disrupts retinyl ester hydrolysis, leading to retinoid retention, stellate cell activation, and enhanced extracellular matrix deposition.11,12 This dual impairment provides a compelling biological explanation for the association of rs738409 with not only MAFLD susceptibility but also steatohepatitis, advanced fibrosis, and hepatocellular carcinoma.
From a population perspective, studies in Asian cohorts demonstrate that PNPLA3 rs738409 is highly prevalent, with reported minor allele frequencies ranging from approximately 30–45% in East Asian populations, including Chinese, Japanese, and Korean cohorts, and similarly high frequencies in Southeast Asian populations such as Indonesia.13,14 In these populations, carriers of the G allele consistently exhibit higher hepatic triglyceride content, elevated aminotransferase levels, and increased risks of MAFLD and fibrosis, even among lean individuals. This pattern is considered a major contributor to the high prevalence of lean and non-obese MAFLD phenotypes across Asia.15
Consistent association have also been reported in large-scale studies and meta-analysis conducted in broader or predominantly non-Asian populations, further supporting the role of PNPLA3 rs738409 in hepatic fat accumulation and disease progression.10,12,15 Although these findings reinforce the biological relevance of this variant, differences in genetic background and gene–environment interactions across populations suggest that extrapolation to Asian MAFLD should be interpreted with caution.
MBOAT7 rs641738 C>T
The MBOAT7 gene encodes an enzyme called membrane-bound O-acyltransferase domain–containing 7. This enzyme participates in hepatic phospholipid remodeling through the Lands’ cycle. Normally, in hepatocytes, MBOAT7 supports the acylation of lysophosphatidylinositol (LPI) with polyunsaturated acyl-CoA substrates, generating phosphatidylinositol (PI) species enriched in polyunsaturated fatty acids. This mechanism is essential for keeping cellular membrane integrity, balanced phosphoinositide signalling, and controlled inflammatory responses in the liver.
On the other hand, the MBOAT7 rs641738 C>T polymorphism is associated with reduced gene expression, leading to decreased enzymatic activity and impaired conversion of LPI to PI, resulting in the accumulation of LPI and the depletion of PI species. This disruption in phospholipid composition alters intracellular lipid signalling pathways and promotes metabolic stress in hepatocytes, thereby contributing to the pathogenesis of MAFLD.16
Evidence from mechanistic and mixed-population studies suggests that individuals carrying the rs641738 T allele exhibit increased susceptibility to hepatic lipid accumulation, not primarily through direct enhancement of FFA influx, but rather through dysregulated lipid signalling and membrane remodelling that facilitate triglyceride deposition, thereby contributing to lipotoxicity in hepatocytes.17 Excess LPI may amplify inflammatory signalling and sensitize hepatocytes and hepatic stellate cells to injury. Over time, this lipid-induced inflammatory milieu promotes progression from simple steatosis to metabolically driven steatohepatitis and fibrogenesis, thereby increasing the risk of advanced liver disease. Notably, the rs641738 T allele has been consistently associated with increased histological severity, including inflammation and fibrosis, independent of traditional metabolic risk factors.
From a population perspective, the rs641738 T allele appears to be relatively prevalent in Asian populations, with available evidence, including data from Chinese cohorts, suggesting carrier frequencies approximately 20% to 40%.18,19 This variant has been associated with increased susceptibility to hepatic steatosis and more severe histological features of liver disease.
Although much of the supporting evidence, including large meta-analyses, has been derived from mixed or non-Asian populations, these studies consistently demonstrate a similar direction of effect, particularly regarding increased hepatic fat accumulation and fibrosis risk.16,17 This consistency across populations provides supportive evidence for the biological relevance of the rs641738 variant. However, given potential differences in genetic background and gene–environment interactions, its specific impact within Asian MAFLD populations still requires further validation.
FTO rs1421085, rs8050136, and rs9939609
The FTO gene encodes the fat mass and obesity-associated protein, an mRNA demethylase that regulates energy homeostasis, adipogenesis, and appetite control. Under normal conditions, it regulates energy homeostasis, adipogenesis, and appetite control. Under normal conditions, mitochondrial activation occurs in beige adipose tissue. However, polymorphisms such as rs9939609 (T>A), rs8050136 (C>A), and especially rs1421085 (C>T) disrupt these systems. The FTO rs9939609 A allele is associated with hyperphagia and impaired satiety. Carriers exhibit increased postprandial appetite and higher caloric intake, driven by altered FTO expression within the hypothalamus.20 Meanwhile, the rs1421085 T risk allele interferes with the ARID5B repressor binding site, resulting in repression of the IRX3 and IRX5 genes, which favor white adipocyte differentiation over beige adipocyte differentiation, thereby diminishing mitochondrial thermogenesis. A recent study demonstrated that adipocyte precursors carrying the rs1421085 risk allele show exhibit down regulation of thermogenic genes, impaired mitochondrial activity, and accelerated lipid accumulation. Collectively, these variants impair metabolic flexibility and promote systemic lipid overload, raising the risk of hepatic fat deposition and progression to NAFLD/NASH.21 In Asian populations, the minor allele frequency (MAF) of rs9939609 ranges from ~12–20% in East Asian groups (eg, Chinese Han, Koreans, Japanese, Filipinos) to ~30–33% in Southeast Asian populations (Malaysia, Indians, Singapore).22 In Indonesia, population-specific data show MAF around 24% for rs9939609, and studies in Balinese adults found that AA homozygotes of rs9939609 and CC homozygotes of rs1421085 were associated with a ~1.1–1.3 kg/m2 increase in BMI (p < 0.05).23 Similarly, a 2024 study among Thai adults reported that carriers of the rs9939609 and rs1421085 risk alleles exhibited significantly higher consumption of sugar and saturated fat. This data shows that FTO polymorphism modulates behavioral patterns that exacerbate obesity risk and its hepatic consequences (ORs of 2.22 and 1.86, respectively; p < 0.05).24
TMNSF2 rs58542926 (C>T)
Transmembrane 6 superfamily member 2 (TM6SF2) encodes a multi-pass transmembrane protein localized to the endoplasmic reticulum (ER) and Golgi complex, expressed in hepatocytes and enterocytes. Under normal physiological conditions, TM6SF2 plays a pivotal role in the assembly, lipidation, and secretion of apolipoprotein B (ApoB)–containing lipoproteins, particularly VLDL. Functionally, TM6SF2 protein facilitates the transfer of triglycerides into nascent ApoB particles, stabilizes ApoB to prevent premature degradation, and supports ER-to-Golgi vesicular trafficking, ensuring proper luminal lipid composition required for VLDL maturation. Through these processes, TM6SF2 acts as a molecular checkpoint that determines the balance between hepatic triglyceride storage and systemic lipid export, thereby influencing hepatic lipid flux and circulating levels of triglycerides, LDL-cholesterol, and total cholesterol.25
The common nonsynonymous variant rs58542926 (c.449 C>T), resulting in the E167K (Glu167Lys) substitution, leads to misfolding and accelerated proteasomal degradation of the TM6SF2 protein. This loss-of-function state disrupts the normal VLDL assembly pathway by impairing triglyceride loading onto ApoB and reducing ApoB stability, diminishing VLDL-TG secretion. Consequently, hepatocytes accumulate triglyceride-rich lipid droplets, exhibit altered lipidomic signatures, and develop ER stress, collectively resulting in the characteristic features of hepatic fat retention despite paradoxically lower circulating lipids. These alterations increase susceptibility to MAFLD, promote progression to NASH, and worsen fibrosis severity, as reflected by elevated ALT/AST and histological injury markers. Evidence from meta-analyses and large-scale studies across diverse populations consistently shows that the E167K allele significantly increases the odds of MAFLD/NAFLD in both adults and children.25,26
In Asian populations, multiple cohort and case–control studies have demonstrated that the rs58542926-T allele is present with variable frequency, generally ranging from approximately 6–13% across East Asian populations, including Chinese, Korean, and Japanese cohorts.27,28 Studies conducted in Asian cohorts further show that carriers of the T allele exhibit increased susceptibility to hepatic fat accumulation and more severe histological features of liver disease. One genotype analysis conducted by Boonvisut et al in a Japanese population (n = 3013) showed that individuals carrying the T allele are more susceptible to intrahepatic lipid deposition.27 Furthermore, a study by Koo et al involving biopsy-confirmed NAFLD patients and controls from South Korea demonstrated that the TM6SF2 rs58542926 variant was associated not only with NASH (OR = 1.86, 95% CI = 1.04–3.32, p = 0.035) but also with significant fibrosis (≥F2) (OR = 1.88; 95% CI = 1.02–3.46), even after adjustment for metabolic risk factors.28
These findings in Asian populations are consistent with observations from broader multi-ethnic and non-Asian studies, reinforcing the role of TM6SF2 E167K as a key determinant of hepatic lipid retention and disease progression. Taken together, both population-specific and cross-population evidence support the functional and clinical relevance of TM6SF2 as a genetic contributor to MAFLD, with consistent effects observed across different ethnic groups.
APOA5 rs662799
Apolipoprotein A5 (APOA5) is a liver-derived apolipoprotein encoded on chromosome 11q23 within the APOA1–APOC3–APOA4–APOA5 gene cluster and functions as a key regulator of plasma triglyceride metabolism through its effects on triglyceride-rich lipoproteins.29 Under normal physiological conditions, APOA5 is synthesized and secreted by hepatocytes, it enhances lipoprotein lipase (LPL)–mediated hydrolysis of very low-density lipoprotein (VLDL) triglycerides, thereby promoting efficient clearance of circulating triglyceride-rich particles and limiting excessive lipid flux to the liver.29 In addition, APOA5 facilitates hepatic uptake of remnant lipoproteins through interactions with heparan sulphate proteoglycans and LDL receptor–related pathways, thereby contributing to the maintenance of triglyceride homeostasis.
The functional promoter polymorphism rs662799 (−1131T>C; A>G) reduces APOA5 transcriptional activity, resulting in decreased circulating APOA5 levels and impaired triglyceride clearance.29,30 This loss of function diminishes LPL activation, leading to hypertriglyceridemia, prolonged circulation of triglyceride-rich lipoproteins, and increased delivery of fatty acids to the liver.29,30 Consequently, hepatic triglyceride accumulation is enhanced, de novo lipogenesis is stimulated, and insulin resistance is exacerbated, creating a metabolic milieu that favours the development of fatty liver disease within the MAFLD spectrum.
Population-based studies, including those conducted in Asian cohorts, demonstrate that carriers of the rs662799 G allele exhibit higher serum triglyceride levels and increased prevalence of metabolic risk factors compared with non-carriers.30,31 In East Asian populations, particularly Chinese and Southeast Asian cohorts, the rs662799 G allele is relatively common, with reported minor allele frequencies ranging from approximately 20% to 35%, which are generally higher than those observed in European populations.29–31 While some supporting evidence comes from non-Asian populations, the consistent effect of this variant on triglyceride metabolism across ethnic groups underscores its biological relevance.
Collectively, APOA5 rs662799 contributes to MAFLD susceptibility primarily by disrupting triglyceride clearance pathways, linking dyslipidaemia, hepatic fat accumulation, and insulin resistance within a genetically susceptible metabolic context.
APOC3 rs2854116
A finely balanced regulatory system governs postprandial lipid handling, and excessive inhibition of triglyceride clearance constitutes a major pathway toward hepatic lipid accumulation. Within the APOA1–APOC3–APOA4–APOA5 gene cluster, apolipoprotein C-III (APOC3) functions as a key negative regulator of triglyceride metabolism by suppressing lipoprotein lipase (LPL) activity and delaying hepatic uptake of triglyceride-rich lipoprotein remnants.30 Under physiological conditions, controlled APOC3 expression modulates lipid flux; however, dysregulation of this mechanism predisposes to prolonged triglyceride exposure and metabolic stress.
The polymorphism rs2854116 (−455T>C) alters APOC3 transcriptional regulation, leading to increased APOC3 expression and enhanced inhibition of LPL-mediated triglyceride hydrolysis.32,33 Consequently, carriers of the risk allele exhibit elevated circulating triglyceride levels, prolonged residence time of triglyceride-rich lipoproteins, and increased fatty acid delivery to the liver.32 Elevated APOC3 levels not only impair peripheral triglyceride clearance but also counteract the triglyceride-lowering effects of APOA5, which is in the same gene cluster, thereby shifting the balance toward lipid retention. When APOC3 gain-of-function occurs concurrently with reduced APOA5 activity—either through APOA5 rs662799–associated downregulation or functional insufficiency—the combined effect may amplify hypertriglyceridemia, increase fatty acid flux to hepatocytes, and accelerate hepatic triglyceride accumulation. This metabolic perturbation promotes insulin resistance, enhances de novo lipogenesis, and creates a metabolic environment conducive to the development of fatty liver disease within the MAFLD spectrum.
Emerging evidence from Asian populations highlights the clinical relevance of the APOC3 rs2854116 variant. Nutrigenetic studies demonstrate that the metabolic impact of this variant is modulated by dietary fat intake, with high-fat dietary patterns amplifying its triglyceride-raising and steatogenic effects.32 In Chinese population studies, the rs2854116 polymorphism has been associated with an increased risk of fatty liver disease, an effect mediated through hypertriglyceridemia rather than obesity.33 Given the high prevalence of APOC3 promoter variants in East Asian populations and the frequent co-occurrence of APOA5 and APOC3 risk alleles within the same genomic locus, dysregulation of the APOA5–APOC3 axis represents an important mechanism linking dietary lipid exposure, dyslipidaemia, and MAFLD in Asian populations. Collectively, these findings support APOC3 rs2854116 as a genetic contributor to MAFLD susceptibility and highlight its potential role as a modifier of APOA5-mediated lipid regulation.
SREBF-2 rs133291
Disordered hepatic cholesterol metabolism recognized as a central pathogenic axis in MAFLD, particularly among Asian populations, in whom fatty liver often develops amid modest adiposity and subtle metabolic perturbations. Sterol regulatory element-binding factor-2 (SREBF-2) encodes sterol regulatory element-binding protein-2 (SREBP-2), a master transcriptional regulator of intracellular cholesterol homeostasis that governs hepatic cholesterol biosynthesis, uptake, intracellular trafficking, and lipoprotein remodeling through transcriptional control of targets such as HMG-CoA reductase and the LDL receptor.34 Under physiological conditions, SREBP-2 activation tightly coupled to intracellular cholesterol depletion, thereby keeping metabolic balance and preventing hepatocellular cholesterol overload.
The rs133291 C>T polymorphism disrupts this regulatory feedback loop, predisposing carriers to inappropriate activation of cholesterol biosynthetic and uptake pathways, even before overt metabolic disease becomes clinically apparent.34 This genetically mediated accumulation of cholesterol promotes oxidative stress, endoplasmic reticulum dysfunction, and inflammatory signalling within hepatocytes. These mechanisms recognized as key drivers of MAFLD progression beyond simple triglyceride storage. Importantly, cholesterol-induced lipotoxicity exerts a disproportionately strong effect on hepatocellular injury and fibrogenic activation, providing a mechanistic explanation for the association between SREBF-2 activity and steatohepatitis in metabolically susceptible individuals.
From an Asian perspective, clinical and epidemiological observations consistently indicate that MAFLD frequently develops at lower BMI thresholds and is strongly influenced by dysregulated lipid and cholesterol metabolism rather than generalized obesity.35–37 These findings highlight the central role of cholesterol-driven metabolic disturbance in Asian MAFLD and provide indirect support for the relevance of SREBF-2–mediated pathways in this population. Emerging evidence from Asian cohorts, particularly in multi-gene or polygenic risk models, has also identified SREBF-2 variants as potential contributors to disease susceptibility, although their independent effects remain less well defined.
In contrast, more direct genetic evidence is available from non-Asian populations. Evidence from longitudinal cohort studies conducted in European populations has shown that the SREBF-2 rs133291 variant independently predicted the future development of fatty liver in conjunction with emerging metabolic dysfunction, including worsening insulin resistance, β-cell dysfunction, postprandial dyslipidaemia, and endothelial impairment.34 Notably, this predictive effect observed in individuals who were initially non-obese and free of overt metabolic syndrome. This clinical phenotype closely mirrors the lean, non-obese MAFLD phenotype commonly observed in Asian populations.
Taken together, while direct genetic evidence in Asian MAFLD still limited, the convergence of metabolic, epidemiological, and cross-population genetic findings supports a biologically plausible role for SREBF-2 in disease pathogenesis. Nevertheless, further population-specific studies required to clarify its contribution as a genetic susceptibility locus in Asian populations.
Insulin Resistance and Lipogenesis
Insulin resistance (IR) is a key feature of metabolic dysfunction and is deeply entwined with the pathogenesis of MAFLD. Insulin resistance leads to higher circulating insulin levels and hyperglycemia, which exacerbate lipogenesis (the synthesis of fat) and impair hepatic fat utilization. Several genetic variants contribute to this process by influencing insulin signaling and lipid production in the liver.
IRS1 rs1801278 (G>A)
Insulin receptor substrate 1(IRS1) gene, which is located at chromosome 2q36, is a member of the IRS protein family. It also encodes IRS-1 protein, which plays an important role in signal transmission between the insulin receptor and Phosphoinositide 3-kinase (PI3K). The rs1801278 polymorphism (G>A) in IRS1 results in an amino acid change from Glycine (G) to Arginine (A) at position 972 (Gly972Arg). This polymorphism significantly impacts insulin sensitivity, leading to insulin resistance (IR), which causes hyperglycemia, a key factor in the pathogenesis of MAFLD.38,39
The Gly972Arg variant in IRS1 impairs insulin sensitivity, thereby promoting the progression of insulin resistance in liver cells. In individuals with this variant, the liver becomes less responsive to insulin regulatory effects.26 Elevated insulin levels subsequently stimulate the liver to produce more triglycerides, leading to their accumulation in hepatocytes, a hallmark feature of hepatic steatosis. Over time, this dysfunction can trigger liver inflammation and progress to NASH and fibrosis, which are severe stages of MAFLD.40 In vivo studies involving IRS1 knockout (KO) mice have demonstrated that the absence of IRS1 leads to impaired insulin signaling. Insulin-induced phosphorylation of the protein kinase B signaling pathway was significantly suppressed in KO mice, resulting in hepatic fat accumulation, glucose dysregulation, and lipid metabolic disorders, resembling the features of MAFLD seen in humans. These findings underscore the pivotal role of IRS1 in regulating lipid metabolism in the liver.41
The Gly972Arg variant is particularly prevalent in populations with high rates of insulin resistance and metabolic dysfunction, making it a significant risk factor for MAFLD. Research in Asian populations, such as in Indonesia and India, has shown a strong association between the A allele of rs1801278 and increased liver fat content. Studies have also highlighted its prevalence among individuals with obesity, type 2 diabetes, and dyslipidemia, all of which worsen the progression of MAFLD.42
GCKR rs1260326 (C>T) and rs780094 (C>T)
The GCKR gene encodes glucokinase regulatory protein (GKRP), which plays a central role in regulating glucokinase activity in the liver and pancreas.43 Glucokinase (GK) is essential for hepatic glucose metabolism, catalyzing the phosphorylation of glucose to glucose-6-phosphate as the first step in glycolysis.44,45 Under physiological conditions, GKRP binds to GK in the nucleus during fasting, maintaining it in an inactive state. When glucose levels rise, GK is released into the cytoplasm to facilitate glucose utilization.
The rs1260326 C>T polymorphism in the GCKR gene alters this regulatory interaction by reducing the binding affinity between GKRP and GK, resulting in constitutively increased glucokinase activity. This leads to enhanced hepatic glucose uptake and glycolysis, which may lower fasting glucose levels while simultaneously promoting de novo lipogenesis and increasing triglyceride synthesis. As a result, carriers of the T allele exhibit a characteristic metabolic profile characterized by improved glycemic parameters but increased lipid accumulation, which contributes to a higher risk of hepatic steatosis, particularly in individuals with insulin resistance or metabolic syndrome. A similar metabolic effect has been observed for the rs780094 C>T polymorphism, which is in strong linkage disequilibrium with rs1260326 and influences hepatic glucokinase regulation in a comparable manner.43,45,46
Population-based studies, including those conducted in Asian cohorts such as Chinese Han populations, have demonstrated associations between GCKR variants and components of metabolic syndrome, including hypertriglyceridemia and altered glucose metabolism.46 However, direct evidence linking these variants specifically to MAFLD in Asian populations remains relatively limited, and much of the current understanding is extrapolated from metabolic and diabetes-related studies. While these variants are present across different populations, their contribution to MAFLD risk may be modulated by dietary patterns, insulin resistance, and other environmental factors prevalent in specific regions.
Supporting mechanistic evidence from in vitro and in vivo studies further reinforces the role of GCKR in metabolic regulation. Experimental models, including GCKRP-deficient mice, demonstrate significant alterations in hepatic glucose production and lipid metabolism, particularly increased de novo lipogenesis, thereby recapitulating key features of fatty liver disease.47 Taken together, GCKR polymorphisms illustrate a metabolically complex genetic pathway in which improved glucose handling occurs at the expense of increased hepatic lipid accumulation, highlighting their relevance in the intersection between insulin resistance and MAFLD pathogenesis.
TCF7L2 rs7903146
Transcription factor 7-like 2 (TCF7L2) is a high-mobility group (HMG) box transcription factor located on chromosome 10q25.3. It serves as a key nuclear effector of the canonical Wnt/β-catenin signaling pathway, regulating genes involved in glucose metabolism, pancreatic β-cell proliferation, and incretin hormone production.48 Under normal physiological conditions, Wnt ligand activation stabilizes cytoplasmic β-catenin, enabling its translocation to the nucleus, where it complexes with TCF7L2 to promote transcription of genes critical for insulin secretion, proglucagon processing, GLP-1 synthesis, and enteroendocrine cell function.48 In addition, TCF7L2 also modulates hepatic glucose output, insulin biosynthesis, and adipocyte lipid signaling, making it a central regulator of systemic insulin sensitivity and metabolic homeostasis.
The rs7903146 C>T polymorphism affects TCF7L2 transcriptional activity by altering enhancer function, resulting in impaired incretin-stimulated insulin secretion, reduced β-cell function, increased hepatic glucose production, and heightened insulin resistance.48,49 These metabolic disturbances promote hepatic lipid influx, enhance de novo lipogenesis, and contribute to the development of fatty liver disease within the MAFLD spectrum, particularly in insulin-resistant individuals.49
In Asian populations, the association between rs7903146 and MAFLD appears to be heterogeneous. Studies in South Asian populations, particularly among Asian Indians, have reported a positive association between the T allele and increased risk of NAFLD, with carriers demonstrating higher BMI, HOMA-IR, and transaminase levels.50 In contrast, studies in Chinese Han populations have reported no overall association, although subgroup analyses suggest a possible protective effect of the T allele in non-obese individuals.49 This variability may reflect differences in allele frequency, underlying metabolic phenotype, and gene–environment interactions across Asian populations. Notably, the minor allele frequency of rs7903146 in East Asians is low (~3–5%), which is substantially lower than in South Asians and European populations, and may partly explain the inconsistent association signals. Collectively, TCF7L2 rs7903146 influences multiple metabolic pathways that intersect with hepatic lipid accumulation; however, its phenotypic impact in MAFLD appears to be population-dependent and strongly modified by the degree of insulin resistance and obesity.
Inflammation and Fibrosis
Inflammation is a critical component of MAFLD pathogenesis. As hepatic fat accumulates, it triggers an inflammatory response in the liver that can progress to fibrosis and, eventually, cirrhosis. Several genetic variants influence these inflammatory pathways, making them key contributors to the progression of MAFLD.
STAT3 rs744166 (G>A)
The STAT3 gene encodes signal transducer and activator of transcription 3, a transcription factor and mitochondrial regulator integral to energy metabolism, lipid homeostasis, and inflammatory signaling. Under normal conditions, STAT3 maintains metabolic balance by modulating transcription of genes involved in fatty acid synthesis, β-oxidation, and glucose homeostasis, while its mitochondrial form supports oxidative phosphorylation and ATP production.51 The rs744166 (G > A) polymorphism, located within an intronic enhancer region, alters STAT3 expression and signaling dynamics. Specifically, the A allele has been associated with increased STAT3 activity in peripheral blood mononuclear cells, skewing metabolism toward adipogenesis and reducing lipid clearance, which are central processes in the development and progression of MAFLD.51,52
In vivo studies in animal models have shown that STAT3 deficiency or mutations that impair STAT3 signaling lead to increased hepatic inflammation, oxidative stress, and fibrosis, all of which are key features of MAFLD progression. Mice with hepatocyte-specific STAT3 knockout exhibit accelerated hepatic steatosis accompanied by a more pronounced inflammatory response, recapitulating the clinical features observed in human MAFLD.52
Oxidative Stress and Mitochondrial Dysfunction
Oxidative stress and mitochondrial dysfunction are intricately linked and represent central pathogenic mechanisms in metabolic dysfunction–associated fatty liver disease (MAFLD). Metabolic overload and insulin resistance increase mitochondrial electron transport chain activity, resulting in excessive reactive oxygen species (ROS) generation. In genetically susceptible individuals, variants in genes regulating mitochondrial function, antioxidant defenses, and redox balance may impair ROS detoxification and mitochondrial homeostasis, leading to oxidative damage of mitochondrial components and reduced oxidative phosphorylation efficiency.53
ALDH rs671
Oxidative stress and mitochondrial injury represent central drivers of hepatic steatosis progression and hepatocellular damage in metabolic-associated fatty liver disease, particularly in populations with impaired aldehyde detoxification capacity. In this context, aldehyde dehydrogenase 2 (ALDH2) functions as a key mitochondrial defense enzyme by catalyzing the clearance of reactive aldehydes generated from ethanol metabolism and lipid peroxidation, thereby limiting aldehyde-induced cytotoxicity, preserving mitochondrial function, and maintaining intracellular redox balance.53 Within hepatocytes, intact ALDH2 activity mitigates oxidative injury, suppresses inflammatory signaling, and protects against mitochondrial dysfunction that would otherwise exacerbate lipid-induced cellular stress.
The rs671 polymorphism (Glu504Lys; E504K) profoundly disrupts ALDH2 enzymatic activity by impairing tetramer stability and catalytic efficiency, resulting in markedly reduced aldehyde detoxification capacity.53 This loss of function leads to intracellular accumulation of toxic aldehydes, amplifying oxidative stress, promoting mitochondrial damage, and accelerating lipid peroxidation. Experimental and clinical evidence demonstrate that ALDH2 deficiency removes a critical cytoprotective barrier, thereby facilitating hepatocellular injury, inflammatory activation, and enhanced vulnerability to fatty liver disease progression.53 Rather than directly initiating steatosis, ALDH2 rs671 primarily acts as a disease modifier, intensifying hepatic injury once lipid accumulation has occurred.
Beyond its hepatic effects, ALDH2 rs671 contributes to a broader cardiometabolic phenotype that may further aggravate MAFLD risk. Population-based studies in East Asian cohorts demonstrate associations between the rs671 variant and elevated blood pressure, as well as adverse lipid profiles characterized by increased triglyceride and LDL cholesterol levels.54 These metabolic alterations may increase hepatic lipid delivery and oxidative burden, thereby reinforcing insulin resistance and mitochondrial dysfunction. Importantly, the rs671 variant is highly prevalent in East Asian populations, with allele frequencies approaching 30–50%, highlighting its role as a population-specific genetic modifier of oxidative stress–related liver injury. Collectively, ALDH2 rs671 represents a biologically plausible link between defective aldehyde detoxification, mitochondrial dysfunction, dyslipidemia, and progression within the MAFLD spectrum in Asian populations.
Table 1 summarizes key SNPs associated with increased susceptibility to MAFLD in Asian populations, encompassing pathways related to hepatic lipid metabolism, insulin resistance, de novo lipogenesis, energy balance, cholesterol homeostasis, and oxidative stress–related mechanisms.
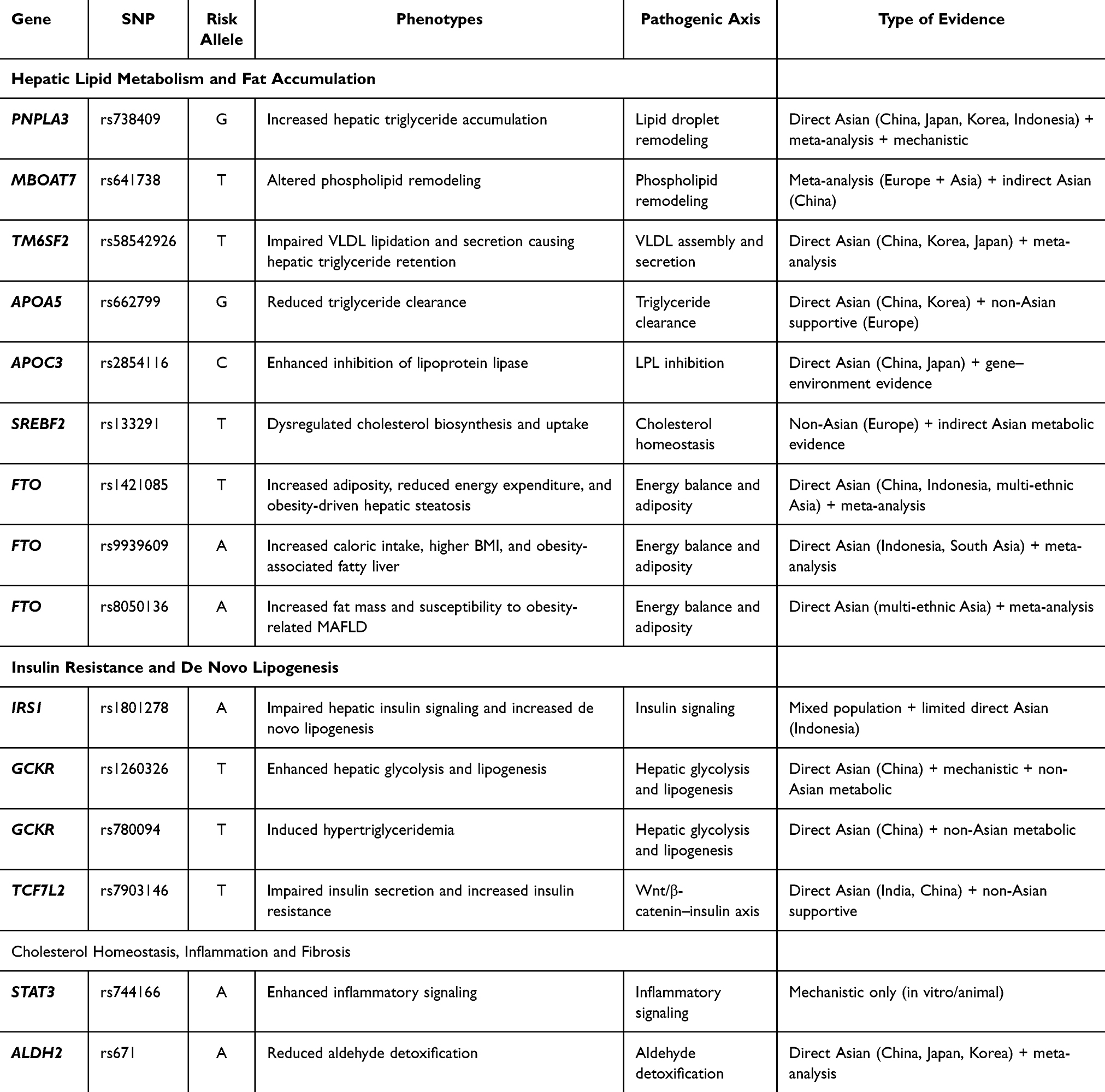 |
Table 1 Genetic Polymorphism Associated with Increased Risk of MAFLD in Asian Population |
Protective Genetic Variants and Liver Protection
While many genetic variants contribute to the risk of developing MAFLD, some variants confer protective effects by maintaining lipid homeostasis, reducing inflammation, and limiting liver fibrosis.
Lipid Homeostasis and Liver Protection
Emerging evidence indicates that specific genetic variants exert protective effects against MAFLD by promoting hepatic lipid homeostasis and preserving liver integrity. Genes involved in lipid handling and export, lipid droplet regulation, and fatty acid partitioning can limit hepatocellular lipid accumulation and reduce lipotoxic stress. In parallel, variants affecting hepatocellular stress responses and cytoprotective pathways may attenuate inflammation and cellular injury. Collectively, these protective genetic determinants highlight compensatory mechanisms that mitigate steatosis and modulate MAFLD susceptibility and progression.
HSD17B13 rs72613567
The HSD17B13 gene encodes 17-beta-hydroxysteroid dehydrogenase type 13 (17β-HSD13), a liver-enriched enzyme associated with hepatic lipid droplets and involved in lipid remodeling processes.55 This protein is expressed in hepatocytes and has been implicated in lipid droplet expansion and lipogenic activity. Notably, HSD17B13 expression is upregulated in individuals with fatty liver disease within the MAFLD spectrum, suggesting a role in disease progression. However, certain genetic variants of HSD17B13, particularly loss-of-function mutations, have been shown to confer protection against liver inflammation and fibrosis.55
The rs72613567 (T>TA) insertion variant results in a truncated, loss-of-function protein and was initially identified in association with lower serum alanine aminotransferase (ALT) levels.56 Subsequent studies demonstrated that this variant is associated with a reduced risk of chronic liver disease, including approximately 30% lower risk of NAFLD and reduced susceptibility to alcoholic liver disease among homozygous carriers.56 Rather than directly influencing hepatic fat accumulation, this variant attenuates hepatocellular injury and inflammatory responses, thereby limiting disease progression.
From a population perspective, the frequency of the rs72613567 variant varies across ethnic groups, with high prevalence observed in East Asian populations (~34%), followed by European populations (~24%), and lower frequencies in African populations (~5%).55 In Asian cohorts, multiple studies provide supporting evidence for its protective role. For instance, Seko et al demonstrated that the HSD17B13 variant attenuates the fibrogenic effect of the PNPLA3 risk allele in Japanese patients with fatty liver disease.57 Similarly, studies in Chinese Han and Pakistani populations have reported that this variant is associated with reduced risk of alcohol-related liver disease and other chronic liver conditions.58,59 These findings suggest that HSD17B13 functions as a genetic modifier that mitigates liver injury across different etiologies, including those within the MAFLD spectrum. Collectively, HSD17B13 rs72613567 represents a key protective genetic factor that modulates disease severity rather than disease initiation, highlighting its potential relevance for risk stratification and therapeutic targeting.
MARC1 rs2642438
The MARC1 gene encodes mitochondrial amidoxime reducing component-1, an enzyme involved in hepatic redox reactions and detoxification processes. The rs2642438 polymorphism (G>A) results in an A165T amino acid substitution, in which alanine replaced by threonine at position 165, altering protein structure and stability. This substitution has been associated with reduced MARC1 enzymatic activity and favorable hepatic phenotypes, including lower levels of liver enzymes such as ALT and AST.60 Mechanistically, reduced MARC1 activity thought to influence hepatic lipid handling, potentially enhancing triglyceride export and limiting intracellular lipid retention, thereby contributing to a protective effect against hepatic steatosis and disease progression within the MAFLD spectrum.
Supporting this, mechanistic and experimental studies, including in vitro models using hepatocyte-like cells, suggest that the A165T variant may alter protein localization and stability, which in turn are associated with reduced intracellular lipid accumulation and increased triglyceride secretion. In addition, Mendelian randomization analyses conducted in European populations have demonstrated that carriers of the A allele show a lower risk of cirrhosis and liver-related mortality.58 The A165T variant is common in European populations, with a minor allele frequency of approximately 25%, but is less prevalent in East Asian and African populations.
Currently, direct evidence from Asian MAFLD cohorts remains limited. However, available data from multiethnic and mechanistic studies suggest that the protective effects of this variant may extend across populations, particularly in individuals with metabolic risk factors such as obesity or insulin resistance.61 These findings should interpret with caution in the Asian context, and further population-specific studies to clarify the role of MARC1 in Asian MAFLD.
Table 2 summarizes two SNPs that are directly and indirectly associated with the protective effect through MAFLD risk progression in the Asian population.
 |
Table 2 Genetic Polymorphism Associated with Reduced Risk of MAFLD in the Asian Population |
Synergistic Genetic Interplay and Its Contribution to MAFLD Progression
Understanding the genetic architecture of MAFLD extends beyond identifying individual risk alleles to determining the interconnections and cumulative contributions of genetic polymorphisms to liver-related mortality. Various studies have demonstrated that the progression of MAFLD, particularly in Asian populations, is not driven by a single gene variant but rather by a complex hierarchy of genes, a phenomenon known as polygenicity. Furthermore, interactions between genes, environmental factors, comorbidities, and lifestyle also play a significant role in increasing mortality.
Importantly, the relevance of this polygenic framework is particularly pronounced in Asian populations, where MAFLD frequently develops at lower BMI thresholds and in the absence of overt obesity (lean MAFLD). Epidemiological and genetic studies in Asian cohorts demonstrate that lean individuals can exhibit distinct genetic susceptibility profiles, with variants such as PNPLA3 exerting a stronger effect on hepatic fat accumulation compared with overweight or obese individuals.62 Moreover, population-based studies have identified differential distribution and metabolic impact of polymorphisms such as GCKR and FTO in lean versus obese NAFLD, further supporting a genotype-dependent disease phenotype.63 Given that lean MAFLD is highly prevalent in Asia and defined using lower BMI thresholds (eg, <23 kg/m2), these findings suggest that genetic susceptibility may play a proportionally greater role compared with Western populations, where excess adiposity is a more dominant driver.64,65
Among all variants associated with MAFLD, the PNPLA3 rs738409 variant consistently contributes to MAFLD progression. This is because this variant directly disrupts intrahepatic lipid metabolism, leading to triglyceride deposition in hepatocytes. This variant’s susceptibility to MAFLD is significant, especially in carriers with metabolic syndrome. Research conducted by Xia et al showed that the PNPLA3 rs738409 C>G polymorphism, both homogeneous and heterogeneous mutations, increases liver fat content (LFC). Increased LFC has been shown to lead to liver-specific mortality, with a two-fold increased risk in individuals with a BMI ≥ 24 kg/m2 or other metabolic syndrome conditions such as hypertension, diabetes, and hyperlipidemia.66
In addition to PNPLA3, variants in the TM6SF2 and MBOAT7 genes contribute more downstream, acting after lipid metabolism or formation in the liver. The TM6SF2 rs58542926 polymorphism disrupts VLDL secretion. This functional mechanism was demonstrated by in vitro studies using the human liver cell line (Huh-7). The researchers analyzed the differences between wild-type (C) cells and mutant (T) cells at the transcriptome and proteome levels to determine the duration of the mutation’s effect. The comparative study results show that the TM6SF2 gene at the transcriptome level, that is, the expression level of RNA, is basically the same in wild-type cells and mutant cells; However, at the proteome level, the amount of TM6SF2 protein in mutant cells was significantly reduced, reaching only 46% of the levels observed in wild-type cells.67 Through this cell-level study, these data suggest that the TM6SF2 E167K variant does not alter transcriptional activity; instead, it induces structural changes that accelerate protein degradation, reducing overall protein stability and abundance. Therefore, the TM6SF2 E167K mutation is also considered a loss-of-function mutation that inactivates the TM6SF2 protein and impairs VLDL secretion, thereby promoting hepatic lipid deposition.68,69 This accumulation remains subclinical or undetectable on standard serum lipid panels, as circulating biomarkers often fail to reflect the extent of intrahepatic lipid burden accurately.
Therefore, understanding genetic predisposition or performing imaging studies to assess liver morphology is crucial. Furthermore, this mutation also affects the expression of genes involved in triglyceride metabolism, such as PNPLA3. Further studies have shown that the E167K mutation at position rs58542926 of the TM6SF2 gene alters the expression of genes involved in triglyceride metabolism, such as PNPLA3.26 Although its allele frequency is lower than that of PNPLA3 in Asian populations, the functional impact of this gene on endoplasmic reticulum stress, lipoprotein metabolism, and fibrogenic signaling confers an increased clinical risk for MAFLD, particularly in more advanced stages of the disease. Similarly, MBOAT7 rs641738 contributes to MAFLD through impaired phospholipid remodeling and altered inflammatory lipid signaling, rather than through direct triglyceride overload.
While PNPLA3 modulates lipid remodeling and TM6SF2 facilitates intracellular VLDL secretion, the APOA5 and APOC3 variants exert the opposite effect on systemic lipid profiles. Both genes play a role in the circulation of triglyceride-rich lipoproteins. Mutations in these genes are known to disrupt lipoprotein lipase activity, thereby dysregulating lipid homeostasis and increasing the risk of intrahepatic lipid accumulation. Overall, these findings support a multilevel model of genetic contribution to MAFLD in Asian populations, with PNPLA3 rs738409 acting as a core driver of liver fat accumulation, followed by moderately impactful modulators of lipid handling and phospholipid remodeling, population-level regulators of triglyceride flux (Figure 2).
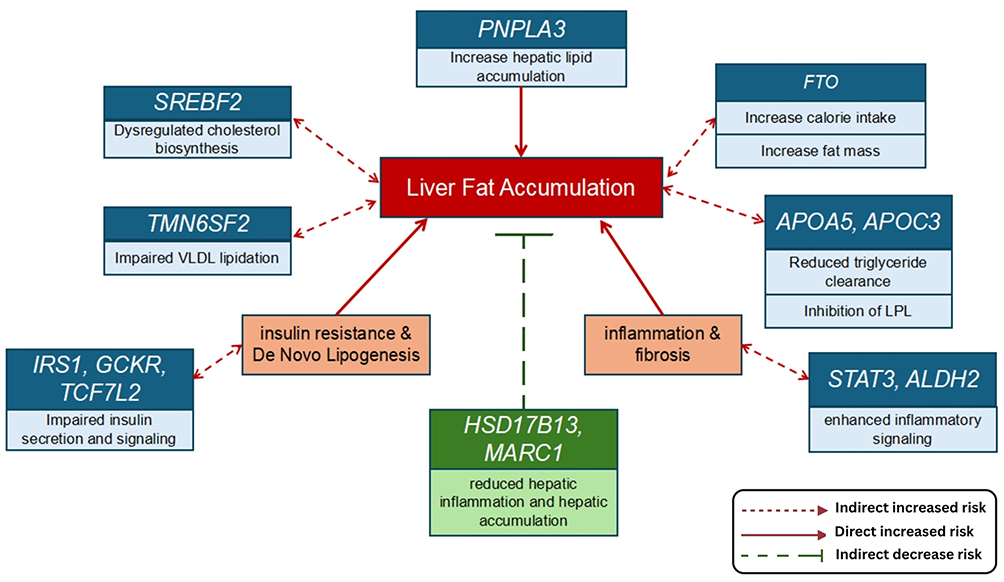 |
Figure 2 Genetic contribution to MAFLD in Asian population. Created in BioRender. Justyn, (M) (2026) https://BioRender.com/t84pgse. |
Clinical Implications and Polymorphism Profiling for Risk Prediction
The growing burden of MAFLD in Asian populations, particularly among lean and non-obese individuals, underscores the need for approaches that extend beyond traditional metabolic risk assessment. The genetic variants discussed in this review provide clinically relevant insights into interindividual susceptibility and disease progression that are not captured by conventional biomarkers.
High-frequency variants such as PNPLA3 rs738409,TM6SF2 rs58542926,MBOAT7 rs641738, and polymorphisms within the APOA5–APOC3 gene cluster may help identify individuals at increased risk of hepatic steatosis, steatohepatitis, and fibrosis despite modest adiposity or metabolic abnormalities (Figure 2). Incorporating these variants into risk stratification models may improve early detection of MAFLD, particularly in Asian populations where fatty liver often develops regardless of BMI.
Furthermore, the identification of protective variants (HSD17B13 and MARC1) highlights the potential for genetically informed prognostic assessment. Individuals carrying protective alleles may exhibit attenuated disease progression despite exposure to metabolic risk factors, suggesting a role for hepatogenomic profiling in refining surveillance intensity, lifestyle intervention strategies, and therapeutic prioritization. Collectively, these findings support the integration of population-specific genetic information into precision hepatology frameworks for MAFLD management in Asia.
Future Direction
Future research should focus on translating hepatogenomic discoveries into clinically actionable tools for MAFLD. Large-scale, multiethnic cohort studies across Asian populations are needed to validate the cumulative and interactive effects of MAFLD-associated variants and to define robust polygenic risk scores (PRS) tailored to Asian genetic architectures.
Integrative approaches that combine genetic data with metabolic, lifestyle, and environmental factors, such as dietary patterns and physical activity, will be essential for elucidating gene–environment interactions that modulate MAFLD risk and progression. Nutrigenomic studies may clarify how variants in lipid-handling genes (eg, APOA5, APOC3, GCKR) influence individual responses to dietary fat and carbohydrate intake, enabling personalized lifestyle interventions.
From a therapeutic perspective, functional characterization of protective variants may uncover novel molecular targets for disease modification. Additionally, advances in single-cell transcriptomics, liver organoid models, and CRISPR-based functional genomics offer promising avenues for dissecting cell-type–specific effects of MAFLD-associated polymorphisms and accelerating the development of genotype-informed therapies.
Strength and Limitation
A major strength of this review lies in its population-specific focus, synthesizing genetic evidence relevant to Asian populations underrepresented in the existing MAFLD literature. By integrating mechanistic insights spanning lipid metabolism, insulin resistance, inflammation, and mitochondrial dysfunction, this review moves beyond descriptive gene lists to propose a hierarchical, interconnected model of genetic contributions to MAFLD progression.
However, several limitations should be acknowledged. Most genetic association studies included in this review are observational, limiting causal inference. Additionally, heterogeneity in study design, diagnostic criteria, and ethnic subgroups across Asia may contribute to variability in reported associations. Functional validation data are still limited to several variants, particularly in liver-specific and population-relevant experimental models.
Despite these limitations, the convergence of genetic, mechanistic, and epidemiological evidence presented here provides a coherent framework for understanding MAFLD susceptibility in Asian populations and highlights key priorities for future translational research.
Conclusion
This review summarizes current evidence on genetic polymorphisms associated with MAFLD in Asian populations, highlighting a complex and heterogeneous genetic architecture. PNPLA3 rs738409 remains the most consistent determinant of hepatic fat accumulation, with additional contributions from TM6SF2, MBOAT7, and variants affecting triglyceride metabolism, insulin signaling, and lipid regulation.
Collectively, these findings support a polygenic and pathway-based model of MAFLD, which is particularly relevant in Asian populations where disease often occurs at lower BMI and may be more strongly influenced by genetic susceptibility.
However, the current evidence is largely observational and heterogeneous, and the clinical application of genetic markers for risk prediction should be interpreted with caution. Further large-scale and longitudinal studies in Asian populations are needed to validate these associations and support their translation into clinical practice.
Data Sharing Statement
Data sharing does not apply to this article as no data was created or analyzed in this study.
Author Contributions
Matthew Justyn: Conceptualization, Methodology, Writing – Original Draft, Resources, Formal analysis, Visualization. Cheerly Sutanto: Investigation, Software, Data Curation, Writing – Review & Editing. Melisa Intan Berliana: Validation, Writing – Review & Editing. Henhen Heryaman: Validation, Writing – Review & Editing. Anna Meiliana: Supervision, Conceptualization, Validation, Writing – Review & Editing. Irma Melyani Puspitasari: Supervision, Conceptualization, Validation, Funding acquisition, Writing – Review & Editing.
All authors have given final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This publication charge is funded by Unpad through the Indonesian Endowment Fund for Education (LPDP) on behalf of the Indonesian Ministry of Higher Education, Science and Technology, and managed under the EQUITY Program (Contract No. 4303/B3/DT.03.08/2025 and 3927/UN6. RKT/HK.07.00/2025).
Disclosure
The authors declare that they have no competing interests related to this study.
References
1. Asian Pacific Association for the Study of the Liver. Clinical practice guidelines for the diagnosis and management of metabolic dysfunction-associated fatty liver disease.
2. Fouad Y, Alboraie M, Shiha G. Epidemiology and diagnosis of metabolic dysfunction-associated fatty liver disease. Hepatol Int. 2024;18:827–18.
3. Zhu R, Xu C, Jiang S, et al. Risk factor analysis and predictive model construction of lean MAFLD: a cross-sectional study of a health check-up population in China. Eur J Med Res. 2025;30:137. doi:10.1186/s40001-025-02373-1
4. Lee GH, Phyo WW, Loo WM, et al. Validation of genetic variants associated with metabolic dysfunction-associated fatty liver disease in an ethnic Chinese population. World J Hepatol. 2020;12(12):1228–1238. doi:10.4254/wjh.v12.i12.1228
5. Sakurai Y, Kubota N, Yamauchi T, Kadowaki T. Role of Insulin Resistance in MAFLD. Int J Mol Sci. 2021;22(8):4156. doi:10.3390/ijms22084156
6. Cól JP D, de Lima EP, Pompeu FM, et al. Underlying mechanisms behind the brain-gut-liver axis and Metabolic-Associated Fatty Liver Disease (MAFLD): an update. Int J Mol Sci. 2024;25(7):3694. doi:10.3390/ijms25073694
7. Liao S, An K, Liu Z, et al. Genetic variants associated with metabolic dysfunction-associated fatty liver disease in western China. J Clin Lab Anal. 2022;36(9):e24626. doi:10.1002/jcla.24626
8. Meroni M, Longo M, Rametta R, Dongiovanni P. MBOAT7 down-regulation by genetic and environmental factors predisposes to MAFLD. EBioMedicine. 2020;57:102866. doi:10.1016/j.ebiom.2020.102866
9. Li G, Tang LJ, Zhu PW, et al. PNPLA3 rs738409 C>G variant influences the association between visceral fat and significant fibrosis in biopsy-proven nonalcoholic fatty liver disease. J Clin transl hepatol. 2022;10:439–448. doi:10.14218/JCTH.2021.00286
10. Zhao Y, Zhao W, Ma J, Toshiyoshi M, Zhao Y. Patatin-like phospholipase domain-containing 3 gene (PNPLA3) polymorphic (rs738409) single nucleotide polymorphisms and susceptibility to nonalcoholic fatty liver disease: a meta-analysis of twenty studies. Medicine. 2023;102:e33110.
11. Park J, Zhao Y, Zhang X, et al. Modeling PNPLA3-associated NAFLD using human-induced pluripotent stem cells. Hepatology. 2021;74(6):2998–3017. doi:10.1002/hep.32063
12. Dai G, Zhang X, Zhang X, et al. Association between PNPLA3 rs738409 polymorphism and nonalcoholic fatty liver disease (NAFLD) susceptibility and severity: a meta-analysis. Medicine. 2019;98(7):e14324. doi:10.1097/MD.0000000000014324
13. Hotta K, Yoneda M, Hyogo H, et al. Association of the rs738409 polymorphism in PNPLA3 with liver damage and the development of nonalcoholic fatty liver disease. BMC Med Genet. 2010;11:172. doi:10.1186/1471-2350-11-172
14. Meagratia RA, Cayami FK, Bahrudin U, et al. Adiponutrin and adiponectin gene variants in indonesian patients with non-alcoholic fatty liver disease: a preliminary study. J Biomed Transl Res. 2021;7(2):86–91. doi:10.14710/jbtr.v7i2.11777
15. Kozlitina J, Sookoian S. Global epidemiological impact of PNPLA3 I148M on liver disease. Liver Int. 2025;45(3):e16123. doi:10.1111/liv.16123
16. Teo K, Abeysekera KWM, Adams L, et al. rs641738C>T near MBOAT7 is associated with liver fat, ALT and fibrosis in NAFLD: a meta-analysis. J Hepatol. 2021;74(1):20–30. doi:10.1016/j.jhep.2020.08.027
17. Mu T, Peng L, Xie X, et al. Single nucleotide polymorphism of genes associated with metabolic fatty liver disease. J Oncol. 2022;2022:9282557. doi:10.1155/2022/9282557
18. Eslam M, Sanyal AJ, George J; International Consensus Panel. MAFLD: a consensus-driven proposed nomenclature for metabolic associated fatty liver disease. J Hepatol. 2020;73(1):202–209. doi:10.1016/j.jhep.2020.03.039
19. Xu X, Xu H, Liu X, et al. MBOAT7 rs641738 (C>T) is associated with NAFLD progression in men and decreased ASCVD risk in elder Chinese population. Front Endocrinol. 2023;14:1199429. doi:10.3389/fendo.2023.1199429
20. Osto M, Gutzwiller A. Fructose consumption and NAFLD: a multi-organ pathology. Dig Dis. 2023;41(6):440–447.
21. Zhang Z, Chen N, Yin N, et al. The rs1421085 variant within FTO promotes brown fat thermogenesis. Nat Metab. 2023;5(8):1337–1351. doi:10.1038/s42255-023-00847-2
22. Pratiwi D, Sidartha M, Wiyarta E, et al. Comparison of the risk of obesity in the FTO rs9939609 genotype in a multiethnic group in Asia: systematic review and meta-analysis. Front Med. 2025;12:1522318. doi:10.3389/fmed.2025.1522318
23. Priliani L, Oktavianthi S, Hasnita R, et al. Obesity in the Balinese is associated with FTO rs9939609 and rs1421085 single nucleotide polymorphisms. PeerJ. 2020;8:e8327. doi:10.7717/peerj.8327
24. Poosri S, Boonyuen U, Chupeerach C, Moolsuwan K, Panichsillaphakit S, Kwanbunjan K. Association of FTO variants rs9939609 and rs1421085 with elevated sugar and fat consumption in adult obesity. Sci Rep. 2024;14:25618. doi:10.1038/s41598-024-77004-6
25. Li XY, Liu Z, Li L, Wang HJ, Wang H. TM6SF2 rs58542926 is related to hepatic steatosis, fibrosis and serum lipids both in adults and children: a meta-analysis. Front Endocrinol. 2022;13:1026901. doi:10.3389/fendo.2022.1026901
26. Xue WY, Zhang L, Liu CM, et al. Research progress on the relationship between TM6SF2 rs58542926 polymorphism and non-alcoholic fatty liver disease. Expert Rev Gastroenterol Hepatol. 2022;16(4):289–301. doi:10.1080/17474124.2022.2032661
27. Boonvisut S, Nakayama K, Makishima S, et al. Replication analysis of genetic association of the NCAN-CILP2 region with plasma lipid levels and non-alcoholic fatty liver disease in Asian and Pacific ethnic groups. Lipids Health Dis. 2016;15(1). doi:10.1186/s12944-016-0181-z
28. Koo BK, Joo SK, Kim D, et al. Additive effects of PNPLA3 and TM6SF2 on the histological severity of non-alcoholic fatty liver disease. J Gastroenterol Hepatol. 2018;33(6):1277–1285. doi:10.1111/jgh.14056
29. Kim J, Baek Y, Lee S. Consumption of dietary fiber and APOA5 genetic variants in metabolic syndrome: baseline data from the Korean Medicine Daejeon Citizen Cohort Study. Nutr Metab. 2024;21:19. doi:10.1186/s12986-024-00793-0
30. You Y, Wu Y-H, Zhang Y, et al. Effects of polymorphisms in APOA5 on the plasma levels of triglycerides and risk of coronary heart disease in Jilin, northeast China: a case–control study. BMJ Open. 2018;8(6):e020016. doi:10.1136/bmjopen-2017-020016
31. de Luis DA, Izaola O, Primo D. APOA-5 genetic variant rs662799: role in lipid changes and insulin resistance after a mediterranean diet in caucasian obese subjects. Dis. Markers. 2021;2021:125–145. doi:10.1155/2021/1257145
32. Yamamoto R, Takeshita Y, Tsujiguchi H, et al. Nutrigenetic interaction between apolipoprotein C3 polymorphism and fat intake in people with nonalcoholic fatty liver disease. Curr Dev Nutr. 2023;7(4):100051. doi:10.1016/j.cdnut.2023.100051
33. Zhang Y, Liu J, Wang X, et al. Associations between APOC3 and ANGPTL8 gene polymorphisms with MASLD risk and the mediation effect of triglyceride on MASLD in the Chinese population. J Cell Mol Med. 2025;29:e70542. doi:10.1111/jcmm.70542
34. Musso G, Cassader M, Bo S, De Michieli F, Gambino R. Sterol regulatory element-binding factor-2 predicts 7-year fatty liver incidence and severity of liver disease and lipoprotein and glucose dysmetabolism. Diabetes. 2013;62:1109–1120. doi:10.2337/db12-0858
35. Patel AH, Peddu D, Amin S, et al. Fatty liver disease in lean and non-obese individuals: metabolic mechanisms and clinical implications. J Clin Transl Hepatol. 2023;11:502–515. doi:10.14218/JCTH.2022.00204
36. Liu Z, Que S, Zhou L, Zheng S. Dose-response relationship of serum cholesterol levels and risk of nonalcoholic fatty liver disease: a meta-analysis. Lipids Health Dis. 2020;19:218. doi:10.1186/s12944-020-01393-6
37. Wong VW, Wong GL, Yeung DK, et al. Incidence of non-alcoholic fatty liver disease in Hong Kong: a population study. Gut. 2015;64(4):698–704. doi:10.1136/gutjnl-2014-308175
38. Shen L, Liu J, Zhao X, Wang A, Hu X. Association between insulin receptor substrate 1 gene polymorphism rs1801278 and gestational diabetes mellitus: an updated meta-analysis. Diabetol Metab Syndr. 2024;16:62. doi:10.1186/s13098-024-01289-w
39. Bedair RN, Magour GM, Ooda SA, et al. Insulin receptor substrate-1 G972R single nucleotide polymorphism in Egyptian patients with chronic hepatitis C virus infection and type 2 diabetes mellitus. Egypt Liver J. 2021;11:2. doi:10.1186/s43066-020-00069-1
40. Duarte GBS, Pascoal GFL, Rogero MM. Polymorphisms involved in insulin resistance and metabolic inflammation: influence of nutrients and dietary interventions. Metabolites. 2025;15(4):245. doi:10.3390/metabo15040245
41. Toyoshima Y, Nakamura K, Taguchi Y, et al. Deletion of IRS-1 leads to growth failure and insulin resistance with downregulation of liver and muscle insulin signaling in rats. Sci Rep. 2025;15:649. doi:10.1038/s41598-024-84234-1
42. Sukmawan R, Akip HM, Wulandari P, et al. Gender differences of Gly972Arg polymorphism of the IRS-1 gene related to cardiovascular disease risk factors among Indonesians. Acta Med Indones. 2023;55(3):255–260.
43. Zhang Z, Ji G, Li M. Glucokinase regulatory protein: a balancing act between glucose and lipid metabolism in NAFLD. Front Endocrinol. 2023;14:1247611. doi:10.3389/fendo.2023.1247611
44. Chaudhry R, Varacallo MA. Biochemistry, Glycolysis. In: StatPearls. Treasure Island (FL): StatPearls Publishing; 2025.
45. Liu YY, Wan Q. Relationship between GCKR gene rs780094 polymorphism and type 2 diabetes with albuminuria. World J Diabetes. 2023;14(12):1803–1812. doi:10.4239/wjd.v14.i12.1803
46. Yin YW, Sun Q, Zhang BB, et al. Association between common variants of the glucokinase regulatory protein gene and metabolic syndrome in a Chinese Han population. Diabetol Metab Syndr. 2021;13(1):20. doi:10.1186/s13098-021-00637-4
47. Shen H, Pollin TI, Damcott CM, McLenithan JC, Mitchell BD, Shuldiner AR. Glucokinase regulatory protein gene polymorphism affects postprandial lipemic response in a dietary intervention study. Hum Genet. 2009;126(4):567–574. doi:10.1007/s00439-009-0700-3
48. Lin Y, Sun Z. Current views on type 2 diabetes, non-alcoholic fatty liver disease and cardiovascular disease: a comprehensive review. J Diabetes Investig. 2016;7(2):193–211.
49. Li Y, Xu F, Zheng Y, Li D, Li L. Association of TCF7L2 rs7903146 gene polymorphism with the risk of NAFLD and CAD in the Chinese Han population. J Cardiovasc Dev Dis. 2022;9(10):331. doi:10.3390/jcdd9100331
50. Bhatt SP, Nigam P, Misra A, Guleria R, Luthra K, Jain SK. Genetic variation in TCF7L2 rs7903146 is associated with non-alcoholic fatty liver disease and insulin resistance in Asian Indians. Diabetes Metab Syndr Obes. 2020;13:239–247.
51. Zhao J, Qi YF, Yu YR. STAT3: a key regulator in liver fibrosis. Ann Hepatol. 2021;21:100628. doi:10.1016/j.aohep.2020.06.010
52. Jiao J, Sanchez JI, Saldarriaga OA, et al. Spatial molecular and cellular determinants of STAT3 activation in liver fibrosis progression in non-alcoholic fatty liver disease. JHEP Rep. 2022;5(2):100628. doi:10.1016/j.jhepr.2022.100628
53. Wang Q, Chang B, Li X, Zou Z. Role of ALDH2 in hepatic disorders: gene polymorphism and disease pathogenesis. J Clin Transl Hepatol. 2021;9(1):90–98. doi:10.14218/JCTH.2020.00104
54. Liu R, Peng M, Zhang J, Qiu K, Zeng T, Chen L. The ALDH2 gene rs671 polymorphism is associated with cardiometabolic risk factors in East Asian population: an updated meta-analysis. Front Endocrinol. 2024;15:1333595. doi:10.3389/fendo.2024.1333595
55. Motomura T, Amirneni S, Diaz-Aragon R, et al. Is HSD17B13 genetic variant a protector for liver dysfunction? Future perspective as a potential therapeutic target. J Pers Med. 2021;11:619. doi:10.3390/jpm11070619
56. Abul-Husn NS, Cheng X, Li AH, et al. AProtein-TRUNCATINGHSD17B13VARIANT and protection from chronic liver disease. New Engl J Med. 2018;378:1096–1106. doi:10.1056/NEJMoa1712191
57. Seko Y, Yamaguchi K, Tochiki N, et al. Attenuated effect of PNPLA3 on hepatic fibrosis by HSD17B13 in Japanese patients with non-alcoholic fatty liver disease. Liver Int. 2020;40:1686–1692. doi:10.1111/liv.14495
58. Chen H, Zhang Y, Guo T, et al. Genetic variant rs72613567 ofHSD17B13gene reduces alcohol-related liver disease risk in Chinese Han population. Liver Int. 2020;40:2194–2202. doi:10.1111/liv.14616
59. Raja AM, Ciociola E, Ahmad IN, et al. Genetic susceptibility to chronic liver disease in individuals from Pakistan. Int J Mol Sci. 2020;21:3558. doi:10.3390/ijms21103558
60. Emdin CA, Haas ME, Khera AV, et al. A missense variant in mitochondrial amidoxime reducing component 1 gene and protection against liver disease. PLoS Genet. 2020;16(4):e1008629. doi:10.1371/journal.pgen.1008629
61. Smagris E, Shihanian LM, Mintah IJ, et al. Divergent role of mitochondrial amidoxime reducing component 1 (MARC1) in human and mouse. PLoS Genet. 2024;20(3):e1011179. doi:10.1371/journal.pgen.1011179
62. Luukkonen PK, Zhou Y, Sädevirta S, et al. Hepatic lipid composition and insulin resistance in patients with non-alcoholic fatty liver disease: the role of PNPLA3 genotype. Hepatology. 2019;69(1):132–145.
63. Zhou M, Zhu L, Cui X, et al. Genetic variants in GCKR and FTO are associated with lean NAFLD in a Chinese population. BMC Endocr Disord. 2022;22(1):12. doi:10.1186/s12902-021-00923-2
64. Wei JL, Leung JC, Loong TC, et al. Prevalence and severity of nonalcoholic fatty liver disease in non-obese patients: a population study using proton-magnetic resonance spectroscopy. Gut. 2015;64(3):476–482.
65. Younossi ZM, Golabi P, Paik JM, Henry L, Van Dongen C, Henry L. The global epidemiology of NAFLD and NASH in patients with lean phenotype: a systematic review. Hepatology. 2022;75(1):204–223.
66. Xia M, Ma S, Huang Q, et al. NAFLD-related gene polymorphisms and all-cause and cause-specific mortality in an Asian population: the Shanghai Changfeng Study. Aliment Pharmacol Ther. 2022;55(6):705–721. doi:10.1111/apt.16772
67. Younossi Z, Anstee QM, Marietti M, et al. Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol. 2018;15(1):11–20. doi:10.1038/nrgastro.2017.109
68. Holmen OL, Zhang H, Fan Y, et al. Systematic evaluation of coding variation identifies a candidate causal variant in TM6SF2 influencing total cholesterol and myocardial infarction risk. Nat Genet. 2014;46(4):345–351. doi:10.1038/ng.2926
69. Sookoian S, Castano GO, Scian R, et al. Genetic variation in transmembrane 6 superfamily member 2 and the risk of nonalcoholic fatty liver disease and histological disease severity. Hepatology. 2015;61(2):515–525. doi:10.1002/hep.27556
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.