Back to Journals » International Journal of Nanomedicine » Volume 13
Hepatocellular carcinoma-targeted effect of configurations and groups of glycyrrhetinic acid by evaluation of its derivative-modified liposomes
Authors Sun Y, Dai C, Yin M, Lu J, Hu H, Chen D
Received 12 October 2017
Accepted for publication 15 January 2018
Published 16 March 2018 Volume 2018:13 Pages 1621—1632
DOI https://doi.org/10.2147/IJN.S153944
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Lei Yang
Yuqi Sun,1,2 Chunmei Dai,1 Meilin Yin,1 Jinghua Lu,1 Haiyang Hu,2 Dawei Chen2
1School of Pharmacy, Jinzhou Medical University, Jinzhou, China; 2School of Pharmacy, Shenyang Pharmaceutical University, Shenyang, China
Background: There are abundant glycyrrhetinic acid (GA) receptors on the cellular membrane of hepatocytes and hepatocellular carcinoma (HCC) cells. The receptor binding effect might be related to the structure of the guiding molecule. GA exists in two stereoisomers with C3-hydroxyl and C11-carbonyl active groups.
Purpose: The objective of this study was to investigate the relationship between the HCC-targeted effect and the configurations and groups of GA.
Methods and results: Different GA derivatives (18β-GA, 18α-GA, 3-acetyl-18β-GA [3-Ace-GA] and 11-deoxy-18β-GA [11-Deo-GA]) were used to investigate the targeting effect of GA’s configurations and groups on HCC cells. The EC50 values of competition to binding sites and the ratio of specific binding in HepG2 cells showed that 18β-GA and 3-Ace-GA demonstrated significant competitive effect with fluorescein isothiocyanate (FITC)-labeled GA. Then, the GA derivatives were distearoyl-phosphatidylethanolamine (DSPE)-PEGylated. 18β-GA-, 18α-GA-, 3-Ace-GA- and 11-Deo-GA-modified liposomes were prepared and characterized by size, zeta potential, encapsulation efficiency, loading capacity, leakage and membrane stability. Evaluation on the cellular location in vitro and tumor targeting in vivo was carried out. Compared to common long-circulation liposome (PEG-Lip), more 18β-GA- and 3-Ace-GA-modified liposomes aggregated around HepG2 cells in vitro in short time and transferred into HCC tumors in vivo for a longer time.
Conclusion: The β-configuration hydrogen atom on C18 position of GA played the most important role on the targeting effect. C11-carbonyl and C3-hydroxy groups of GA have certain and little influence on targeting action to HCC, respectively. In general, GA might be a promising targeting molecule for the research on liver diseases and hepatoma therapy.
Keywords: glycyrrhetinic acid, derivatives, liposomes, receptor competition, HCC-targeted effect
Introduction
Hepatocellular carcinoma (HCC) accounts for up to 90% of all malignant primary liver cancer worldwide and represents a major health threat.1 Currently, apart from surgery, patients with liver cancer are mainly treated by chemotherapy.2 However, chemotherapy is limited by a low response rate and severe systemic toxicity due to low specificity and resistance mechanisms of the chemotherapeutic agents toward cancer cells.3 Therefore, novel drug delivery systems are urgently needed to enhance the selective action of cytotoxic drugs to HCC and therefore minimize systemic toxicity to noncancerous tissues.
Glycyrrhetinic acid (GA), a pentacyclic triterpenoid, is widely present in the licorice plant and is the active aglycone of glycyrrhizin (GL). Abundant GA receptors have been confirmed on the cellular membrane of hepatocytes.4 The protein kinase C-α was reported as the target binding protein of GA, which had much more expression in HCC cells than in the adjacent nontumor liver cells.5 The amount of GA receptors in tumor tissue has been found to be 1.5- to 5-fold more than that in normal tissue.6 Additionally, GA showed the selective toxicity toward tumor through the downregulation of glutathione.7 Recently, some GA-modified liposomes have been developed with higher drug accumulation in the liver and better anti-HCC activity.5–9 We have confirmed GA receptors on HCC cells formerly according to the binding effect between fluorescence-labeled GA and GA receptors.10 Therefore, it could be expected that GA-modified liposomes could target selectively to HCC cells and tissues.
GA exists in two stereoisomers, including trans form 18α- and the cis form 18β-GA, lying in the spatial orientation of hydrogen atom of C18. Different configurations of GA exhibit various stabilities, solubilities11 and pharmacological effects. 18β-GA exerted protective effects against cyclophosphamide-induced hepatotoxicity.12 18β-GA also showed anti-HCC proliferation effects, which could induce the HCC cells’ apoptosis via modulation of inflammatory markers and inhibit HCC development by reversing hepatic stellate cell-mediated immunosuppression.13,14 18β-GA could reduce the amount of glucose release induced by glucagon in rat primary cultured hepatocytes, while 18α-GA did not.15 Nevertheless, 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1) is a kind of microsomal enzyme belonging to the short-chain dehydrogenase/reductase family, which is highly expressed in many glucocorticoid target tissues, such as the liver, adipose tissue, skeletal muscle and macrophages. 18α-GA selectively inhibited 11β-HSD1, but 18β-GA had no selectivity.16 18α-GA increased peroxisome proliferator-activated receptor γ expression and decreased nuclear factor-κB DNA-binding activity, inhibiting the proliferation of activated hepatic stellate cells.17 18α-GA was reported to target prostate cancer cells by downregulation of inflammation-rated genes in DU-145 cells.18
Ishida et al have proved that a carrier-mediated transport system participated in the uptake of GL into isolated rat hepatocytes and the affinity site of the transport carrier bound to GA.19 GA is a hydrolytic product of GL with the differences of hydroxyl or glycosyl group at C3. The removal of 11-carbonyl in the ring structure of GA not only eliminated pseudoaldosterone effect but also improved its anti-inflammatory, antiulcer and antiallergic activities.20 11-deoxy-18β-GA (11-Deo-GA), performing a similar action of 18α-GA, also selectively (and significantly) acted on 11-β-HSD1.21 As for anticancer properties, 11-Deo-GA induced gastric cancer cell apoptosis by upregulation of p21, downregulation of cdc2 and cyclin B1 and association with BID (a BH3 domain-only agonist) translocation from the nucleus to the mitochondria.22 These deduced that for GA’s main ring structure, the hydroxyl group at C3 and the carbonyl group at C11 had certain effect on the liver or HCC cell targeting.
Thus, in this study, we focused on the targeting effect of different GA configurations and groups to HCC cells. As shown in Figure 1, 18β-GA, 18α-GA, 3-acetyl-18β-GA (3-Ace-GA) and 11-Deo-GA were obtained, and fluorescein isothiocyanate-labeled 18β-GA (FITC-GA) was synthesized according to reported method.23 The binding site competition to HCC cells of different GA derivatives was studied. The long-circulation phospholipids with potential targeting molecular were synthesized by the GA derivatives linked with DSPE-PEG2000-NH2. Coumarin 6 (Cou6) and 1,1-dioctadecyl-3,3,3,3-tetramethylindotricarbocyanine iodide (DiR) liposomes were prepared to evaluate the targeting effect of GA’s configurations and groups in vitro and in vivo.
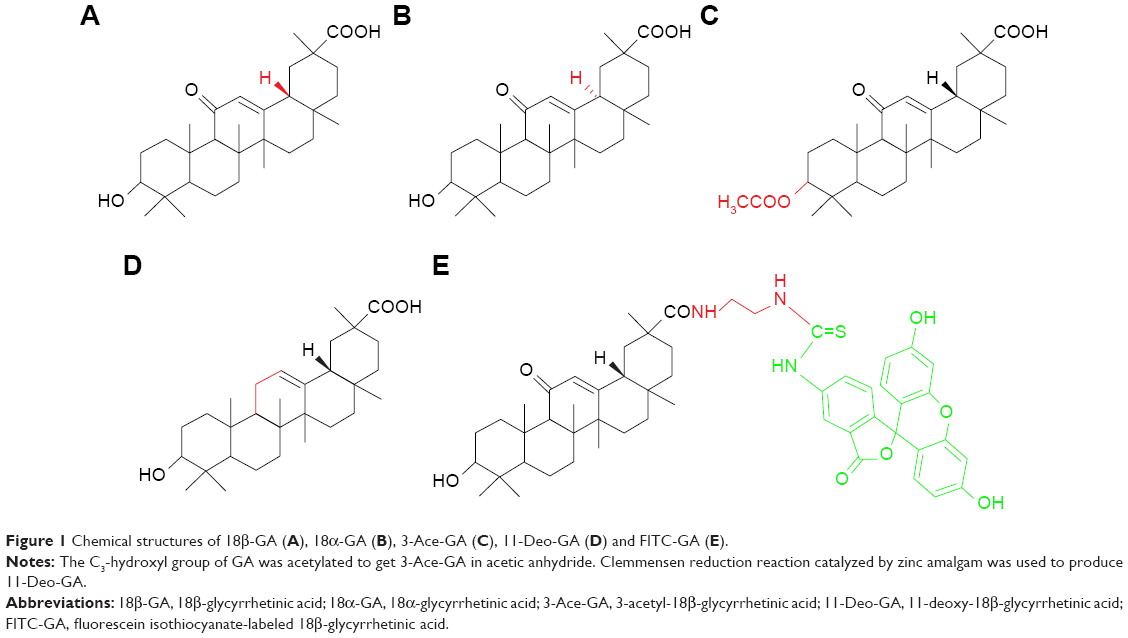 | Figure 1 Chemical structures of 18β-GA (A), 18α-GA (B), 3-Ace-GA (C), 11-Deo-GA (D) and FITC-GA (E). |
Materials and methods
Materials
18β-GA (purity 98%), 18α-GA (purity ≥98%) and Cou6 (purity 98%) were obtained from J&K Scientific Ltd. (Beijing, China). 3-Ace-GA, 11-Deo-GA and FITC-GA were synthesized and characterized in our laboratory. DSPE-PEG2000 (DSPE-PEG, purity ≥97%) and DSPE-PEG2000-NH2 (DSPE-PEG-NH2, purity ≥95%) were bought from AVT Pharmaceutical Co., Ltd (Shanghai, China). Soybean phospholipid (for injection, phosphatidylcholine ≥85%) and cholesterol (purity ≥98%) were from Tywei Pharmaceutical Co. (Shanghai, China). DiR was from AAT Bioquest Int. (Sunnyvale, CA, USA). RIPA lysis buffer, BCA protein assay kit and Hoechst 33258 were from Beyotime Institute of Biotechnology (Shanghai, China).
Human HCC (HepG2) cells and mouse ascites hepatoma (H22) cells were obtained from Cell Bank of Chinese Academy of Sciences (Shanghai, China). HepG2 cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM; Invitrogen, Carlsbad, CA, USA) with 10% fetal bovine serum (FBS; Gibco, Waltham, MA, USA). H22 cells were cultured and passaged in Kunming mouse ascites. Male BALB/c nude mice (20±2 g), supplied by the Department of Experimental Animals, Shenyang Pharmaceutical University (Shenyang, China), were acclimated under specific-pathogen-free conditions in the central animal facility of the university. All animal experiments were approved by the Ethics Committee of the Shenyang Pharmaceutical University and were carried out in accordance with the guidelines evaluated and approved by the ethics committee of Shenyang Pharmaceutical University.
Competition of binding sites
Approximately 2.5×106 HepG2 cells were seeded into 6-well culture plates. After the cells have covered the plates, the medium was removed and the cells were washed with PBS and DMEM (without FBS) successively. The cells were treated with a series concentrations of 18β-GA, 18α-GA, 3-Ace-GA, 11-Deo-GA and FITC-GA in DMEM for 2 h at 37°C. Then, FITC-GA was added to a final concentration of 100 nM. After co-incubation for 2 h at 37°C, the cells were washed with chilled PBS and lysed with 100 μL of lysis buffer in ice bath. The cell lysate was harvested, shaken and centrifuged (1.2×104 rpm for 10 min) at 4°C. The fluorescence intensity of the supernatant was measured (λex =490 nm, λem =520 nm) with the Varioskan Flash Spectral Scanning Multimode Reader (Thermo Fisher Scientific, Waltham, MA, USA). The fluorescence intensity was normalized with respect to the cells’ protein content, which was determined with a BCA protein assay kit. The fluorescence intensity of different GA derivatives with FITC-GA samples and the only FITC-GA samples normalized with the content of protein in the cells was calculated as the specific binding (B) and maximum binding (B0), respectively. The competitive curve fitting was constructed by GraphPad Prism 5.0 (GraphPad Software, La Jolla, CA, USA) software with concentration as x-axis and specific binding ratio (B/B0) as y-axis.
Synthesis and identification of DSPE-PEGylated GA derivatives
The GA derivatives and DSPE-PEG were linked together with amidation (Figure 2).24 In total, 235.5 mg 18β-GA, 235.5 mg 18α-GA, 256.2 mg 3-Ace-GA and 228.2 mg 11-Deo-GA (0.5 mMol) were dissolved in anhydrous acetone with an amount of pyridine. In total, 143.8 mg EDC·HCl (0.75 mMol), 86.3 mg NHS (0.75 mMol) and 1,100 mg DSPE-PEG were then added sequentially to the solution with stirring for 48 h under nitrogen protection at ambient temperature. The products were obtained by recrystallization from the condensed reaction solutions, which were mixed with an amount of ice-cold anhydrous ether. The precipitate was dissolved in water and dialyzed for 24 h. The DSPE-PEGylated GA derivatives were obtained by freeze-drying at last. The 1H-NMR (DMSO-d6) spectrum of DSPE-PEGylated GA derivatives was characterized at 300 Hz with an ARX-300 (600) NMR spectrometer (Bruker Optik GmbH, Ettlingen, Germany).
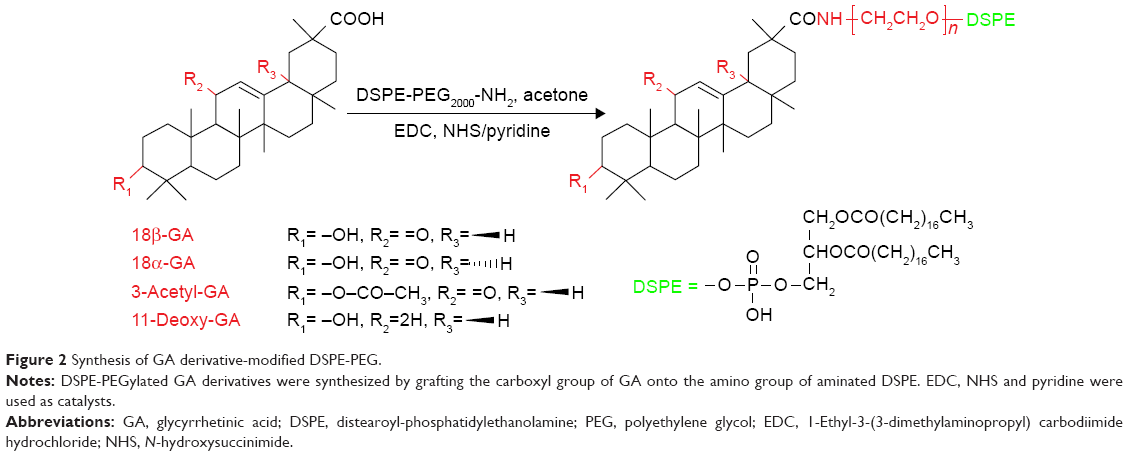 | Figure 2 Synthesis of GA derivative-modified DSPE-PEG. |
Preparation and characterization of Cou6 liposomes
The liposomes were prepared by a modified film dispersion method according to our previous report.23 Briefly, 18.2 mg DSPE-PEG, 21.2 mg 18β-GA-PEG-DSPE, 21.2 mg 18α-GA-PEG-DSPE, 21.4 mg 3-Ace-GA-PEG-DSPE or 21.0 mg 11-Deo-GA-PEG-DSPE (6.6 μM) with 50 mg phospholipid (about 1:10 molar ratio), 20 mg cholesterol and 2 mg Cou6 was dissolved in 10 mL of ethanol-dichloromethane (2:1). A thin lipid film was produced by rotary evaporation and hydrated in 10 mL of water at 60°C, followed by stirring at ambient temperature. The mixture was sonicated for 10 min at 200 W. The colloidal solution obtained was then centrifuged at 1.2×104 rpm/min for 10 min and filtered through 0.22 μm Millipore filters. All the procedures were conducted in darkness. The Cou6 liposomes mixed with DSPE-PEG (PEG-Cou6-Lip), 18β-GA-PEG-DSPE (18β-GA-Cou6-Lip), 18α-GA-PEG-DSPE (18α-GA-Cou6-Lip), 3-Acel-GA-PEG-DSPE (3-Ace-GA-Cou6-Lip) and 11-Deo-GA-PEG-DSPE (11-Deo-GA-Cou6-Lip) were prepared.
The particle sizes, polydispersity index (PDI) and zeta potentials of the liposomes were measured with a Nano Analyzer (ZS90; Malvern Instruments, Malvern, UK). The entrapment efficiency (EE), loading capacity (LC) and leakage (LK) of liposomes were determined with mini-column centrifuge-fluorescence spectroscopy (λex =497 nm, λem =523 nm) methods.
Analysis of liposomal membrane stability
To evaluate the stability of the liposomal membranes, the resistance of the liposomes against nonionic surfactant Triton X-100 was investigated.25 A series concentration (0.5%, 1%, 2%, 5%, 10%, 20%, 50%, 100%) of Triton X-100 solutions was prepared. The Triton X-100 solutions (10 μL) were added to the GA derivative-mediated liposomes (190 μL) in the 96-well plates in triplicate. The plates were shaken for 15 min at 25°C. The absorbance at 590 nm was measured by Multimode Reader. The turbidity was recorded as A/A0 (590 nm), where A is the absorbance of samples with a predetermined concentration of Triton X-100 addition and A0 is the initial absorbance of samples.
Cytotoxicity assay in vitro
Effect of the liposomes on HepG2 cells growth was assessed by MTT assay. In total, 5×103 HepG2 cells were seeded into the 96-well culture plates in quintuplicate and incubated overnight. The medium was then replaced with the GA derivative-mediated liposomes at gradient Cou6 concentrations of 2 μg/mL (low), 10 μg/mL (medium) and 50 μg/mL (high). After incubation for 6 h, the cultured medium was then discarded and refreshed with DMEM. In total, 20 μL of MTT solution (5 mg/mL in PBS) was added to each well for additional 4 h of incubation. Formazan was dissolved in 150 μL of DMSO, and the absorbance at 570 nm was measured using the Multimode Reader. The inhibition was calculated according to the absorbance.
Confocal observation
Laser scanning confocal microscopy was used to visualize the subcellular localization of different Cou6 liposomes.26 HepG2 cells were cultured on sterile microscope slides in the 6-well culture plates (2×105 cells/well) and incubated overnight. The media were replaced by the five liposomes in DMEM (without FBS), and the cells were further incubated for 0.25, 1 and 6 h. Then, the media were removed and the cells were washed 3 times with cold PBS. Hoechst 33258 (10 μg/mL, 10 min) was used to localize nuclei. Finally, the cells were fixed with 4% formaldehyde at ambient temperature and captured by laser scanning confocal microscope (Olympus FV1000-IX81; Olympus Corporation, Tokyo, Japan). The content of Cou6 in each sample was measured accordingly as we reported previously.10
In vivo targeting ability study
The DiR liposomes containing DSPE-PEG (PEG-DiR-Lip) and mediated by different GA derivatives (18β-GA-DiR-Lip, 18α-GA-DiR-Lip, 3-Ace-GA-DiR-Lip and 11-Deo-GA-DiR-Lip) were prepared according to the previously mentioned method. Male BALB/c mice were inoculated subcutaneously in the right axillary fossa with 0.2 mL of H22 cell suspension (2×106 cells/mL). Once the tumor reached a volume of 150–200 mm3, the H22 tumor-bearing mice were obtained. The mice were divided into five groups and intravenously injected with 0.2 mL of various DiR liposomes. In the period of 12 h postinjection, the mice under anesthetic state were scanned using a Kodak In Vivo Imaging System FX Pro (Carestream Health Inc., Rochester, NY, USA) at different time.
Statistical analysis
All the experiments were performed in triplicate, and the values are presented as mean ± standard deviation. The data were analyzed by applying one-way ANOVA, followed by the methods of Student’s t-test. p-value <0.05 indicated statistically significant differences.
Results and discussion
Competitive effect on binding sites
According to the differences in the configurations and in the main modified parts at C3-hydroxyl and C11-carbonyl groups of GA,27 the compounds including 18β-GA, 18α-GA, 3-Ace-GA and 11-Deo-GA were obtained. As is known, receptor competition is increasingly common to discovery of new drugs28 and screen for targeting ligands. Higher binding effect means stronger physiological activity and longer signaling pathway influence. The results on the competitive effect of GA derivatives with FITC-GA in HepG2 cells are shown in Figure 3. The reversible binding of receptors and ligands accounted for 70% of maximum binding (B0) of FITC-GA after the inhibitor (18β-GA, 18α-GA, 3-Ace-GA or 11-Deo-GA) was added. EC50 values of different GA derivative competition to binding sites in HepG2 cells were calculated. Among them, 18β-GA decreased the specific binding ratio (B/B0) significantly. The EC50 of 18β-GA was 100.1 nM. B/B0 value decreased to 10% of B/B0 when 200 nM of 18β-GA was added. The competitive curve of 18α-GA with FITC-GA was flatter and the EC50 was 563.1 nM indicating the weak binding effect of GA receptors and competitive ability with FITC-GA. It demonstrated that the 18-H configuration of GA influenced the binding effect greatly. The addition of 3-Ace-GA gave a similar competitive curve of 18β-GA and 111.7 nM of EC50, indicating a little influence of 3-OH of GA. The competitive curve of 11-Deo-GA lay between 18α-GA and 3-Ace-GA, with 121.2 nM of EC50 demonstrating the competitive effect with FITC-GA. 11-Carbonyl group influenced the targeting action of GA.
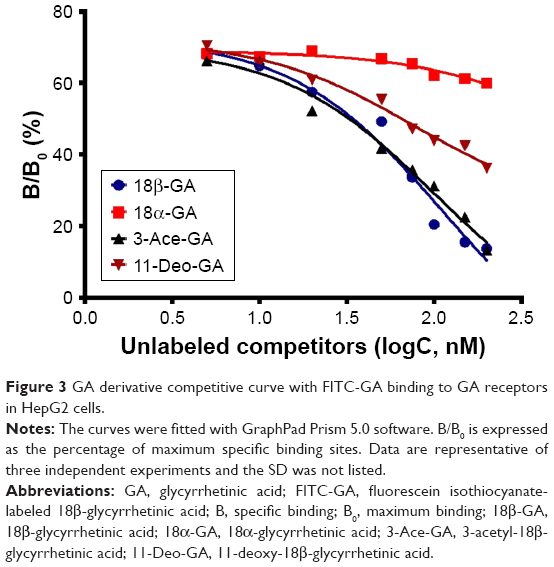 | Figure 3 GA derivative competitive curve with FITC-GA binding to GA receptors in HepG2 cells. |
Characterization of GA derivative-PEG-DSPE
The DSPE-PEGylated GA derivatives were synthesized by grafting the carboxyl group of GA onto the amino group of aminated DSPE. The 1H-NMR spectrum in Figure 4 shows that the signals at δ 0.68–1.11 ppm belonged to the methylene and the methyl groups of the pentacyclic triterpenoid structure of GA. The peaks at δ 1.30–1.40 ppm were 19-H and at δ 5.30–5.50 ppm were 12-H of the GA rings. The peaks at δ 1.20–1.35 ppm and δ 3.30–3.60 ppm mainly attributed to the −CH2 protons of stearic acid groups of DSPE and the glycol blocks of PEG2000, respectively. In the 1H-NMR spectrum, a single peak with one unit area at δ 7.40–7.80 ppm was found (18β-GA-PEG-DSPE 7.78 ppm, 18α-GA 7.63 ppm, 3-Ace-GA 7.50 ppm, 11-Deo-GA 7.47 ppm), which belonged to the −CO−NH− proton when −COOH of GA derivatives acetylated with the −NH2 of DSPE-PEG-NH2. The integration value ratio for the peak of amide group and the peak of methyl (δ 0.75–0.83) in stearyl group was about 1:6. Therefore, the long-circulation phospholipids with GA derivatives have been successfully synthesized. The yield of 18β-GA-PEG-DSPE, 18α-GA-PEG-DSPE, 3-Ace-GA-PEG-DSPE and 11-Deo-GA-PEG-DSPE was 69.3%, 53.2%, 75.0% and 66.4%, respectively.
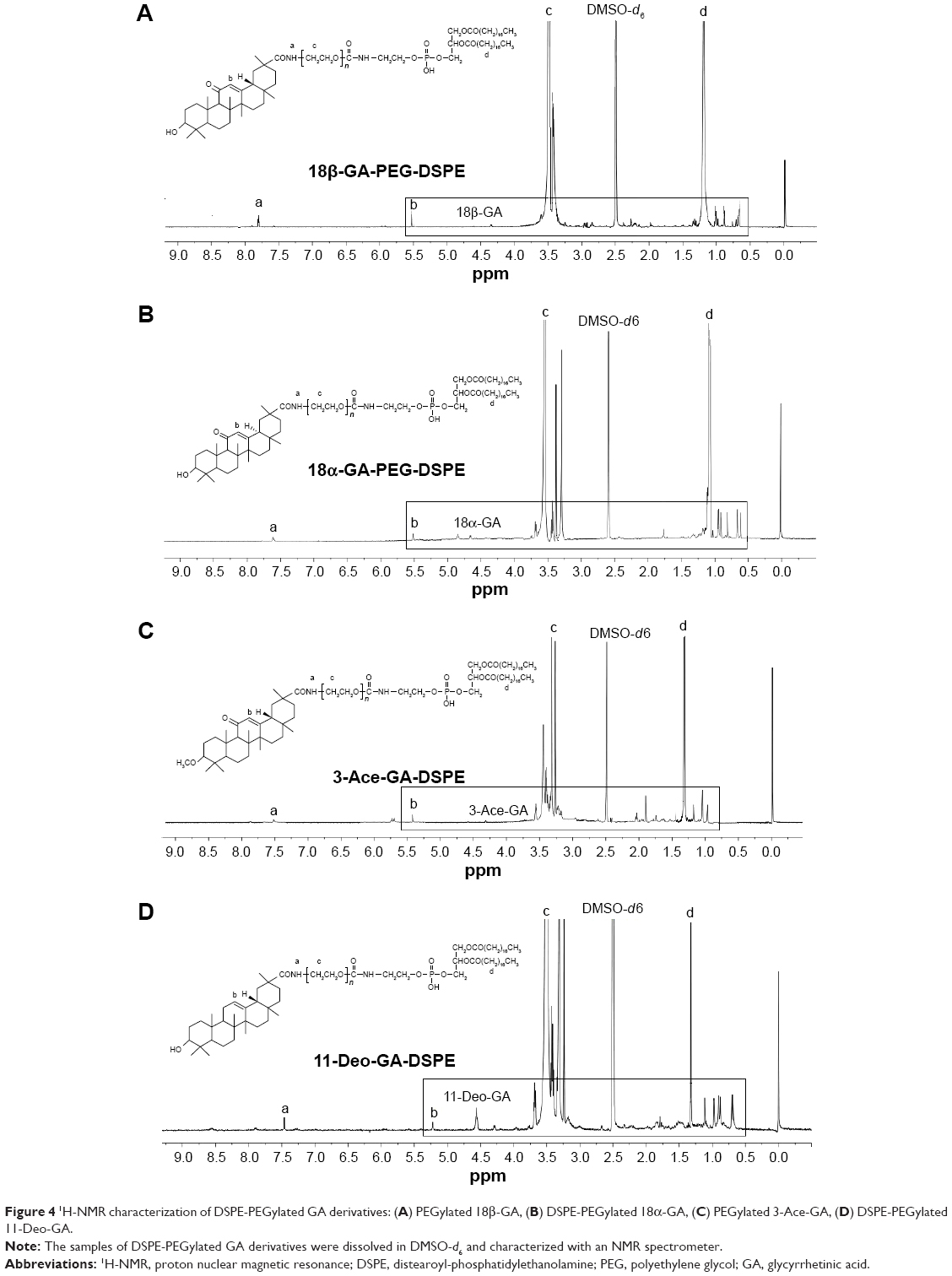 | Figure 4 1H-NMR characterization of DSPE-PEGylated GA derivatives: (A) PEGylated 18β-GA, (B) DSPE-PEGylated 18α-GA, (C) PEGylated 3-Ace-GA, (D) DSPE-PEGylated 11-Deo-GA. |
Pharmaceutical characters of liposomes
Liposome size is an important factor that can influence drug dosage, uptake, targeting, clearance and lysosomal accumulation.29 The liposomal characters are shown in Table 1. The particle sizes of the five liposomes were less than 180 nm, which were analyzed by dynamic light scattering. The sizes of Cou6 liposomes with DSPE-PEGylated GA derivatives were smaller than those of PEG-Cou6-Lip. The possible reason might be that the hydrophobicity of GA derivatives wrapped the PEG chain tightly and squeezed the particles further. The PDI (0.182–0.213) demonstrated relatively narrow distribution of the liposomes. The zeta potential of the five liposomes was negative ranging from −10 to −18 mV. With the addition of DSPE-PEGylated GA derivatives, the zeta potential was 8 mV or so lower. A greater zeta potential is necessary to inhibit liposomal aggregation.30 The LK of the Cou6 liposomes was lower (<7%, 14 d). The GA derivative-mediated liposomes showed higher stability with lower potential and lower LK. As a regulatory requirement, EE is a greatly significant parameter to control the quality of the liposomes.31 Results showed that the values of EE and the LC of the five liposomes were all higher than 65% and 17.5 mg/g. 3-Ace-GA-Cou6-Lip and 11-Deo-GA-Cou6-Lip showed the highest and lowest values of EE, respectively. The DSPE-PEGylated GA derivatives had no significant effect on the EE and LC of Cou6. Therefore, the liposomes exhibited good preparation characteristics. Because the optimum size of PEG-liposomes for prolonged circulation is 160–220 nm,32 we could expect a good long-circulation effect of these GA derivative-modified liposomes.
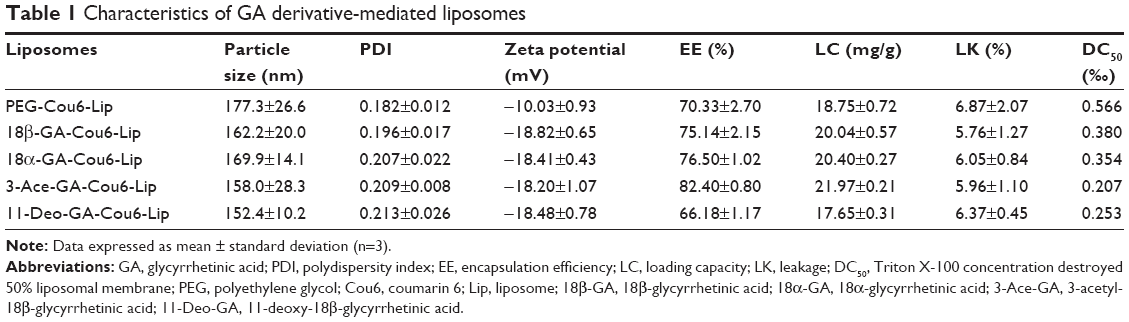 | Table 1 Characteristics of GA derivative-mediated liposomes |
Liposomal membrane stability
Triton X-100, a polyoxyethylene nonionic surfactant, has been used to determine the membrane stability of different Cou6 liposomes. Three relevant transitions would take place when Triton X-100 was added into the liposomal suspension. First, lower concentration led to a slight increase in the size of liposome, which might be attributed to the incorporation of surfactant monomers in the lipid bilayer. Second, increasing surfactant amounts led to a progressive decrease in intensity for mixed liposomes, which corresponded to the saturation of Triton X-100 in bilayer and the formation of lipid micelles. The size of micelles was far less than that of liposomal particles. Finally, higher concentration made a slight fall in the particle size, which might be attributed to the progressive enrichment in Triton X-100 of the mixed micelles formed.33
The variations of relative turbidity (RTU) for the suspension vs Triton X-100 concentration were plotted in logarithmic coordinates (Figure 5). The spots showed the inverted S-type distribution when a series concentration of Triton X-100 was added into the liposomal suspensions. The RTU expressed horizontal distribution when lower (<0.15‰) and higher (>0.60‰) concentrations of Triton X-100 were added because of the less membrane action and thorough membrane destroy, respectively. When a medium concentration of Triton X-100 was added, the liposomal membrane was destroyed in a concentration-dependent manner and the RTU decreased sharply; a straight line (linear regression) was fitted for the relationship of RTU to logarithm concentration of Triton X-100. When half of the liposomal membrane was destroyed (RTU =0.5), the destruction concentration of Triton X-100 (DC50) was calculated (Table 1) to evaluate the stability of the membrane. The results showed that the membrane of PEG-Cou6-Lip was extremely stable and the membrane stability of GA derivative-modified liposomes decreased slightly.
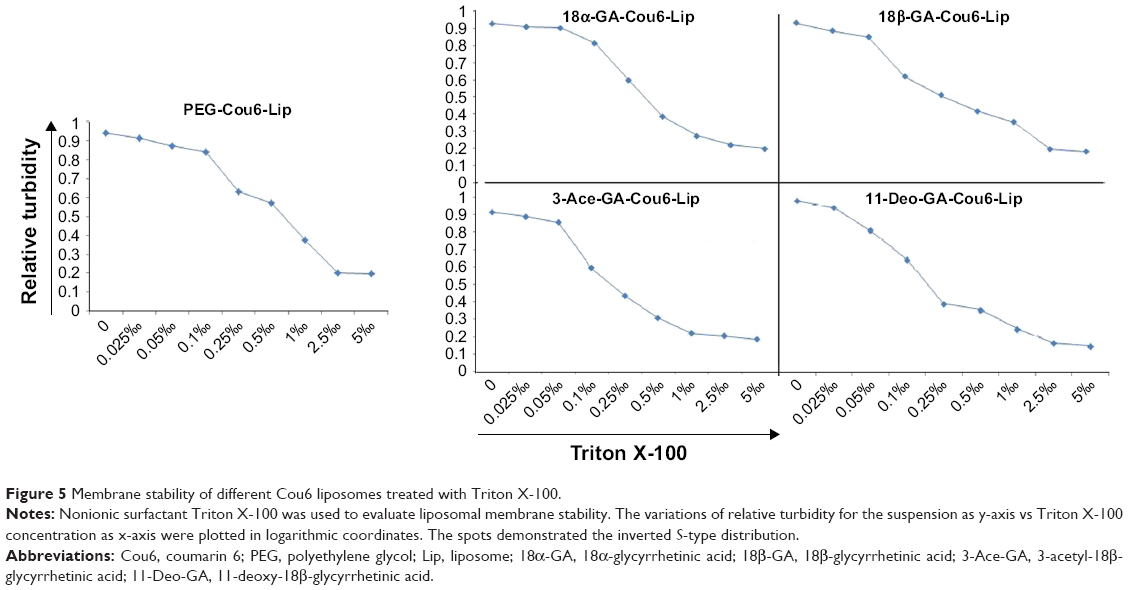 | Figure 5 Membrane stability of different Cou6 liposomes treated with Triton X-100. |
Cytotoxicity assay
The cytotoxic effects of PEG-Cou6-Lip, 18β-GA-Cou6-Lip, 18α-GA-Cou6-Lip, 3-Ace-GA-Cou6-Lip and 11-Deo-GA-Cou6-Lip on HepG2 cells in 6 h were investigated by MTT assay. As shown in Figure 6, different concentrations (2, 10, 50 μg/mL) of Cou6 used in different liposomes showed certain cytotoxicity against HCC cells. No apparent cytotoxicity was observed at low concentration (IR <2.0%). Compared to PEG-Cou6-Lip, there were no significant differences in cytotoxicity at 10 μg/mL of Cou6 in four GA derivative-mediated liposomes. High concentration action showed significant differences in cell viability. As a result, 10 μg/mL of Cou6 in liposomes was chosen as the optimal concentration in the further trial and 6 h was taken as the longest action time.
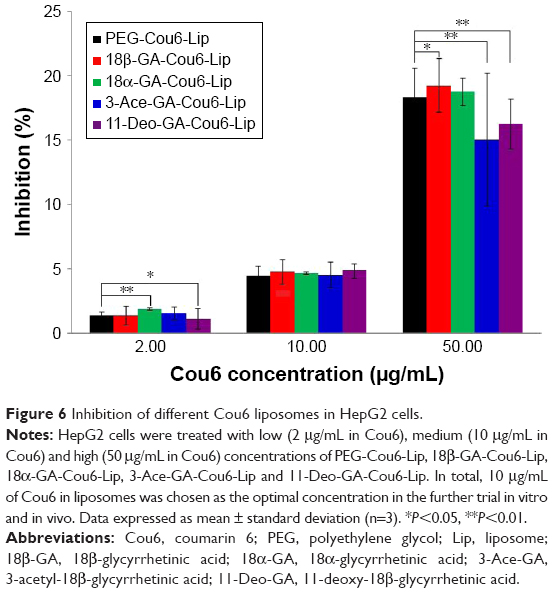 | Figure 6 Inhibition of different Cou6 liposomes in HepG2 cells. |
Cellular localization
Cou6 with high quantum yield has been widely used in cellular localization of nano-drug delivery system. Most cells have no background influence in the fluorescence spectrum range of Cou6. In addition, raw Cou6 cannot be directly internalized by the cells.34 In other words, the fluorescence measured in the uptake samples reflects the liposomes taken up by the cells but not the released Cou6. Figure 7 shows the confocal laser scanning microscopy (CLSM) images and quantitative analysis of different liposomes loading Cou6 after 15 min, 2 h and 6 h in HepG2 cells. For 18β-GA-Cou6-Lip and 3-Ace-GA-Cou6-Lip, much more green fluorescence intensity was aggregated around the HepG2 cells after incubation for 15 min, demonstrating a good affinity to the cytomembrane. Meanwhile, weak fluorescence was observed near and in the cells when the other three liposomes were added. After co-incubation for 2 h, the green fluorescence was internalized and gathered in the cells. The HCC cells treated with 18β-GA-Cou6-Lip and 3-Ace-GA-Cou6-Lip showed distinct cell outlines with strong fluorescence intensity. However, the cells treated with PEG-Cou6-Lip, 18α-GA-Cou6-Lip and 11-Deo-GA-Cou6-Lip showed obscure outlines. The green fluorescence intensity was enhanced with increasing the incubation time for all the liposomes. Furthermore, after co-incubation for 6 h, most fluorescence was gathered in the cells and all the liposomes were internalized by cells. It was difficult to distinguish the binding sites of the cells. The results indicated that GA-modified nano-carriers showed liver and HCC targeting effects. The C3-hydroxyl group of GA had little influence on the targeting ability, which was consistent with the reports of Tian et al.35 The spatial configuration and C11-carbonyl group affected the HCC cell targeting of GA.
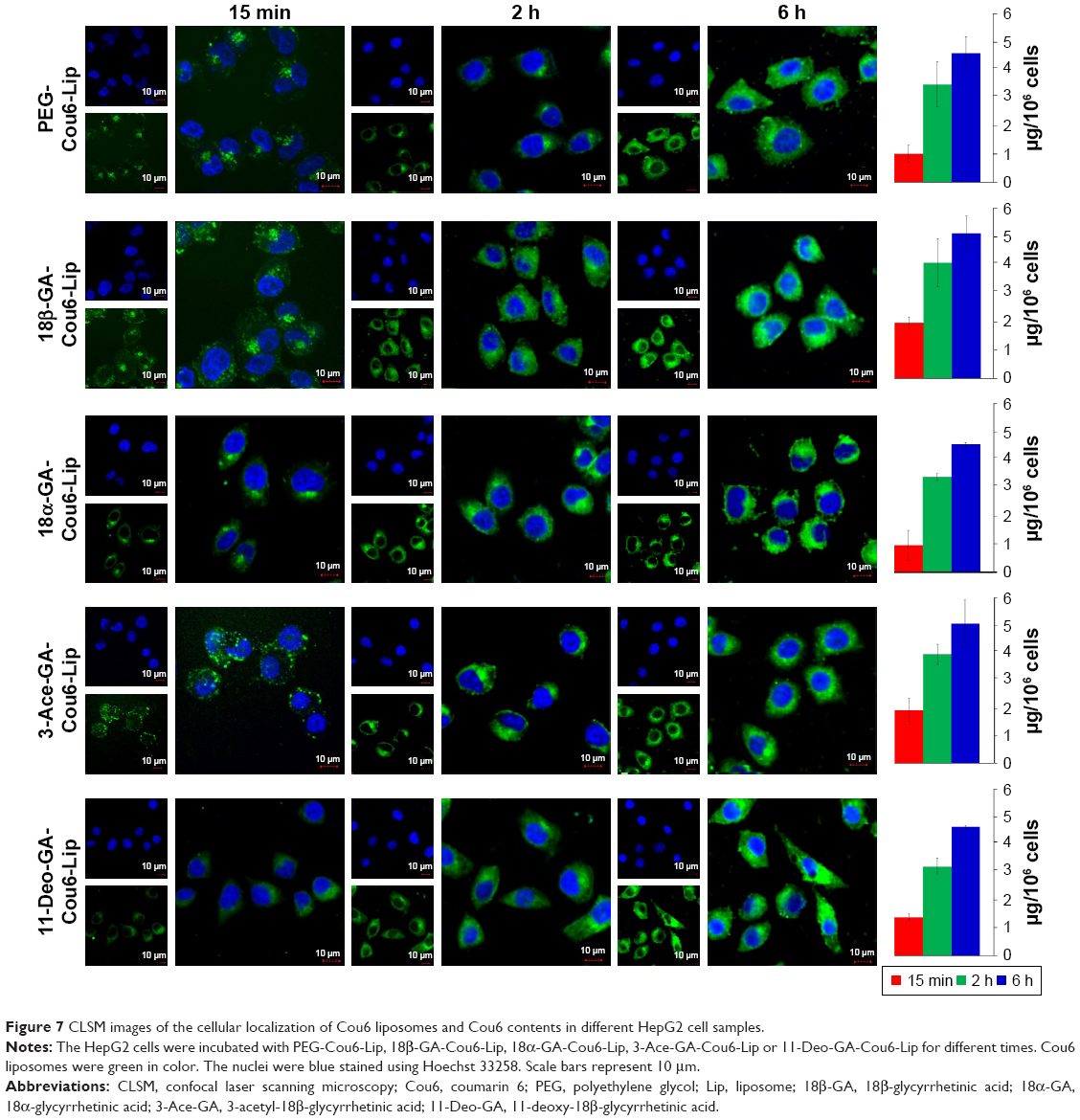 | Figure 7 CLSM images of the cellular localization of Cou6 liposomes and Cou6 contents in different HepG2 cell samples. |
Tumor targeting in vivo
The in vivo biodistribution and targeting ability of the liposomes in mice were evaluated using a near-infrared fluorescence image system. DiR was chosen as a fluorescent marker, which is a type of near-infrared fluorescent dye with an excitation spectrum of 750 nm and an emission spectrum of 782 nm. As a lipophilic tracer, DiR allowed the noninvasive tracking of cells for several days in vivo.36 Figure 8 shows the real-time images in H22 tumor-bearing nude mice at 0.5, 2 and 12 h after intravenous (caudal-vein) injection of PEG-DiR-Lip, 18β-GA-DiR-Lip, 18α-GA-DiR-Lip, 3-Ace-GA-DiR-Lip and 11-Deo-GA-DiR-Lip. All the liposomes had a time-dependent liver and tumor accumulation in the mice. Most DiR accumulated in the liver at 0.5 h postadministration of the liposomes, and then there was a decrease in the fluorescence intensity from 2 to 12 h. With the prolongation of time, the fluorescence intensity in the tumor region was increased. 18β-GA- and 3-Ace-GA-modified liposomes revealed distinct uptake, which were transported more quickly to the HCC tumors after 2 h of injection. The highest accumulation in an H22 xenograft tumor model after 12 h of administration was found, which was consistent with CLSM results of the two liposomes in HCC cells. As to PEG-DiR-Lip and 18α-GA-DiR-Lip, only a little fluorescence was measured in the tumor tissues. However, the fluorescence intensity in the liver decreased significantly. The targeting effect of 11-Deo-GA-DiR-Lip differed from 18β-GA-DiR-Lip and 3-Ace-GA-DiR-Lip, as well as PEG-DiR-Lip and 18α-GA-DiR-Lip. Part of liposome was transported and accumulated in the HCC tumors. There was no obvious fluorescent signal appearing in kidney tissue. This suggested that the metabolism in the liver was the main elimination route of GA derivative-modified liposomes. The tumor-targeting results indicated that the liposomes were transferred quickly in the liver. There were no significant differences whether GA derivatives modified or not. Different GA derivative ligands showed different HCC tumor-targeting effects. The configuration and groups of GA influenced different degrees on the targeting effects.
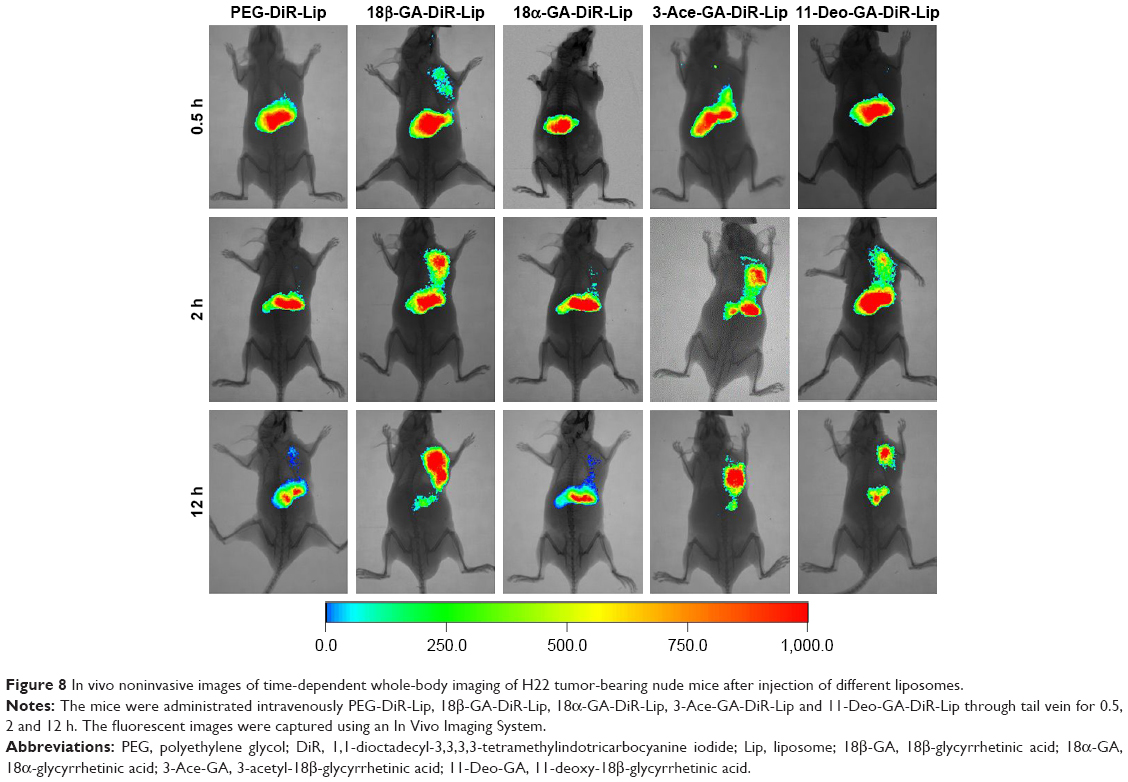 | Figure 8 In vivo noninvasive images of time-dependent whole-body imaging of H22 tumor-bearing nude mice after injection of different liposomes. |
Conclusion
GA receptor has been deemed to be a promising receptor to target HCC attributing to its safety and pharmacological potential. Focusing on the differences of structure configurations and active groups, four GA derivatives (18β-GA, 18α-GA, 3-Ace-GA and 11-Deo-GA) and mediated liposomes (18β-GA-Lip, 18α-GA-Lip, 3-Ace-GA-Lip and 11-Deo-GA-Lip) were developed to show different abilities to target the HCC cells and HCC tumors. 18β-GA and 3-Ace-GA showed significantly competitive effects with FITC-GA on HepG2 cells. The effect decreased in turn for 11-Deo-GA and 18α-GA-Lip. The liposomes modified with GA derivatives showed higher loading ability and better stability. Compared to common long-circulation liposome (PEG-Lip), more 18β-GA- and 3-Ace-GA-modified liposomes were aggregated around HepG2 cells in vitro in short time and were transferred into HCC tumors in vivo for a longer time. There were no significant differences between 18α-GA-Lip and PEG-Lip on HCC targeting. In addition, 11-Deo-GA-Lip showed certain targeting effect. Therefore, it can be concluded that GA shows the targeting action to HCC cells and HCC tumors. The β-configuration hydrogen atom at C18 position of GA contributes the most targeting effect. C11-carbonyl and C3-hydroxy groups of GA have certain and little influences on the targeting action to HCC, respectively.
Acknowledgment
This work was supported by the grants from the Science and Technology Department of Liaoning Province (2013022064, 201602312).
Disclosure
The authors report no conflicts of interest in this work.
References
Llovet JM, Villanueva A, Lachenmayer A, Finn RS. Advances in targeted therapies for hepatocellular carcinoma in the genomic era. Nat Rev Clin Oncol. 2015;12(7):408–424. | ||
Li L, Wang H. Heterogeneity of liver cancer and personalized therapy. Cancer Lett. 2016;379(2):191–197. | ||
Zhu X, Tsend-Ayush A, Yuan Z, et al. Glycyrrhetinic acid-modified TPGS polymeric micelles for hepatocellular carcinoma-targeted therapy. Int J Pharm. 2017;529(1–2):451–464. | ||
Negishi M, Irie A, Nagata N, Ichikawa A. Specific binding of glycyrrhetinic acid to the rat liver membrane. Biochim Biophys Acta. 1991;1066(1):77–82. | ||
He ZY, Zheng X, Wu XH, et al. Development of glycyrrhetinic acid-modified stealth cationic liposomes for gene delivery. Int J Pharm. 2010;397(1–2):147–154. | ||
Chen J, Jiang H, Wu Y, Li Y, Gao Y. A novel glycyrrhetinic acid-modified oxaliplatin liposome for liver-targeting and in vitro/vivo evaluation. Drug Des Devel Ther. 2015;9:2265–2275. | ||
Yamaguchi H, Yu T, Kidachi Y, et al. Selective toxicity of glycyrrhetinic acid against tumorigenic r/m HM-SFME-1 cells is potentially attributed to downregulation of glutathione. Biochimie. 2011;93(7):1172–1178. | ||
Chen Q, Ding H, Zhou J, et al. Novel glycyrrhetinic acid conjugated pH-sensitive liposomes for the delivery of doxorubicin and its antitumor activities. RSC Advance. 2016;6(22):17782–17791. | ||
Tian J, Wang L, Wang L, Ke X. A wogonin-loaded glycyrrhetinic acid-modified liposome for hepatic targeting with anti-tumor effects. Drug Deliv. 2014;21(7):553–559. | ||
Sun YQ, Dai CM, Zheng Y, Shi SD, Hu HY, Chen DW. Binding effect of fluorescence labeled glycyrrhetinic acid with GA receptors in hepatocellular carcinoma cells. Life Sci. 2017;188:186–191. | ||
Langer D, Czarczynska-Goslinska B, Goslinski T. Glycyrrhetinic acid and its derivatives in infectious diseases. Curr Issues Pharm Med Sci. 2016;29(3):118–123. | ||
Mahmoud AM, Al Dera HS. 18β-Glycyrrhetinic acid exerts protective effects against cyclophosphamide-induced hepatotoxicity: potential role of PPARγ and Nrf2 upregulation. Genes Nutr. 2015;10(6):41. | ||
Hasan SK, Siddiqi A, Nafees S, et al. Chemopreventive effect of 18β-glycyrrhetinic acid via modulation of inflammatory markers and induction of apoptosis in human hepatoma cell line (HepG2). Mol Cell Biochem. 2016;416(1–2):169–177. | ||
Kuang P, Zhao W, Su W, et al. 18β-Glycyrrhetinic acid inhibits hepatocellular carcinoma development by reversing hepatic stellate cell-mediated immunosuppression in mice. Int J Cancer. 2013;132(8):1831–1841. | ||
Nose M, Ito M, Koide T, Terawaki K, Ogihara Y. The effects of 18 alpha, beta-glycyrrhetinic acid on the hormonal induction of tyrosine aminotransferase in rat primary cultured hepatocytes. Planta Med. 1996;62(5):410–412. | ||
Classen-Houben D, Schuster D, Da Cunha T, et al. Selective inhibition of 11β-hydroxysteroid dehydrogenase 1 by 18α-glycyrrhetinic acid but not 18β-glycyrrhetinic acid. J Steroid Biochem Mol Biol. 2009;113(3–5):248–252. | ||
Zong L, Qu Y, Xu MY, Dong YW, Lu LG. 18α-glycyrrhetinic acid extracted from Glycyrrhiza radix inhibits proliferation and promotes apoptosis of the hepatic stellate cell line. J Digest Dis. 2013;14(6):328–336. | ||
Shetty AV, Thirugnanam S, Dakshinamoorthy G, et al. 18α-glycyrrhetinic acid targets prostate cancer cells by down-regulating inflammation-related genes. Int J Oncol. 2011;39(3):635–640. | ||
Ishida S, Sakiya Y, Ichikawa T, Taira Z. Uptake of glycyrrhizin by isolated rat hepatocytes. Biol Pharm Bull. 1993;16(3):293–297. | ||
Song H, Sun Y, Xu G, Hou B, Ao G. Synthesis and biological evaluation of novel hydrogen sulfide releasing glycyrrhetic acid derivatives. J Enzyme Inhib Med Chem. 2016;31(6):1457–1463. | ||
Su X, Vicker N, Lawrence H, et al. Inhibition of human and rat 11beta-hydroxysteroid dehydrogenase type 1 by 18beta-glycyrrhetinic acid derivatives. J Steroid Biochem Mol Biol. 2007;104(3–5):312–320. | ||
Lin D, Zhong W, Li J, Zhang B, Song G, Hu T. Involvement of BID translocation in glycyrrhetinic acid and 11-deoxy glycyrrhetinic acid-induced attenuation of gastric cancer growth. Nutr Cancer. 2014;66(3):463–473. | ||
Sun Y, Lu J, Yan D, Shen L, Hu H, Chen D. Cellular uptake mechanism and clearance kinetics of fluorescence-labeled glycyrrhetinic acid and glycyrrhetinic acid–modified liposome in hepatocellular carcinoma cells. Environ Toxicol Pharmacol. 2017;53:46–56. | ||
Chu Y, Li D, Luo YF, He XJ, Jiang MY. Preparation and in vitro evaluation of glycyrrhetinic acid-modified curcumin-loaded nanostructured lipid carriers. Molecules. 2014;19(2):2445–2457. | ||
Tan C, Xia S, Xue J, Xie J, Feng B, Zhang X. Liposomes as vehicles for lutein: preparation, stability, liposomal membrane dynamics, and structure. J Agric Food Chem. 2013;61(34):8175–8184. | ||
Zhang J, Chang D, Yang Y, et al. Systematic investigation on the intracellular trafficking network of polymeric nanoparticles. Nanoscale. 2017;9(9):3269–3282. | ||
Roohbakhsh A, Iranshahy M, Iranshahi M. Glycyrrhetinic acid and its derivatives: anti-cancer and cancer chemopreventive properties, mechanisms of action and structure-cytotoxic activity relationship. Curr Med Chem. 2016;23(5):498–517. | ||
Mould R, Brown J, Marshall FH, Langmead CJ. Binding kinetics differentiates functional antagonism of orexin-2 receptor ligands. Br J Pharmacol. 2014;171(2):351–363. | ||
Andar AU, Hood RR, Vreeland WN, Devoe DL, Swaan PW. Microfluidic preparation of liposomes to determine particle size influence on cellular uptake mechanisms. Pharm Res. 2014;31(2):401–413. | ||
Ochi MM, Amoabediny G, Rezayat SM, Akbarzadeh A, Ebrahimi B. In vitro co-delivery evaluation of novel pegylated nano-liposomal herbal drugs of silibinin and glycyrrhizic acid (nano-phytosome) to hepatocellular carcinoma cells. Cell J. 2016;18(2):135–148. | ||
Ran C, Chen D, Xu M, Du C, Li Q, Jiang Y. A study on characteristic of different sample pretreatment methods to evaluate the entrapment efficiency of liposomes. J Chromatogr B Analyt Technol Biomed Life Sci. 2016;1028:56–62. | ||
Awasthi VD, Garcia D, Goins BA, Phillips WT. Circulation and biodistribution profiles of long-circulating PEG-liposomes of various sizes in rabbits. Int J Pharm. 2003;253(1–2):121–132. | ||
López O, de la Maza A, Coderch L, López-Iglesias C, Wehrli E, Parra JL. Direct formation of mixed micelles in the solubilization of phospholipid liposomes by Triton X 100. FEBS Lett. 1998;426(3):314–318. | ||
Win KY, Feng SS. Effects of particle size and surface coating on cellular uptake of polymeric nanoparticles for oral delivery of anticancer drugs. Biomaterials. 2005;26(15):2713–2722. | ||
Tian Q, Wang X, Wang W, Zhang C, Liu Y, Yuan Z. Insight into glycyrrhetinic acid: the role of the hydroxyl group on liver targeting. Int J Pharm. 2010;400(1–2):153–157. | ||
Shan L. Near-infrared fluorescence 1,1-dioctadecyl-3,3,3,3-tetramethylindotricarbocyanine iodide (DiR)-labeled macrophages for cell imaging. Molecular Imaging and Contrast Agent Database (MICAD) [Internet]. Bethesda, MD: National Center for Biotechnology Information (US); 2004–2013, 2009. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.