Back to Journals » Journal of Hepatocellular Carcinoma » Volume 7
Hepatocellular Carcinoma Mechanisms Associated with Chronic HCV Infection and the Impact of Direct-Acting Antiviral Treatment
Authors Dash S, Aydin Y, Widmer KE, Nayak L
Received 28 June 2019
Accepted for publication 6 March 2020
Published 15 April 2020 Volume 2020:7 Pages 45—76
DOI https://doi.org/10.2147/JHC.S221187
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Ahmed Kaseb
Srikanta Dash,1– 3 Yucel Aydin,1 Kyle E Widmer,2 Leela Nayak2
1Department of Pathology and Laboratory Medicine, Tulane University Health Sciences Center, New Orleans, LA 70112, USA; 2Southeast Louisiana Veterans Health Care System, New Orleans, LA 70119, USA; 3Department of Medicine, Division of Gastroenterology, Tulane University Health Sciences Center, New Orleans, LA 70112, USA
Correspondence: Srikanta Dash
Department of Pathology and Laboratory Medicine, Tulane University Health Sciences Center, 1430 Tulane Avenue, New Orleans, LA 70112, USA
Tel +1 504-988-2519
Fax +1 504-988-7389
Email [email protected]
Abstract: Hepatitis C virus (HCV) infection is the major risk factor for liver cirrhosis and hepatocellular carcinoma (HCC). The mechanisms of HCC initiation, growth, and metastasis appear to be highly complex due to the decade-long interactions between the virus, immune system, and overlapping bystander effects of host metabolic liver disease. The lack of a readily accessible animal model system for HCV is a significant obstacle to understand the mechanisms of viral carcinogenesis. Traditionally, the primary prevention strategy of HCC has been to eliminate infection by antiviral therapy. The success of virus elimination by antiviral treatment is determined by the SVR when the HCV is no longer detectable in serum. Interferon-alpha (IFN-α) and its analogs, pegylated IFN-α (PEG-IFN-α) alone with ribavirin (RBV), have been the primary antiviral treatment of HCV for many years with a low cure rate. The cloning and sequencing of HCV have allowed the development of cell culture models, which accelerated antiviral drug discovery. It resulted in the selection of highly effective direct-acting antiviral (DAA)-based combination therapy that now offers incredible success in curing HCV infection in more than 95% of all patients, including those with cirrhosis. However, several emerging recent publications claim that patients who have liver cirrhosis at the time of DAAs treatment face the risk of HCC occurrence and recurrence after viral cure. This remains a substantial challenge while addressing the long-term benefit of antiviral medicine. The host-related mechanisms that drive the risk of HCC in the absence of the virus are unknown. This review describes the multifaceted mechanisms that create a tumorigenic environment during chronic HCV infection. In addition to the potential oncogenic programming that drives HCC after viral clearance by DAAs, the current status of a biomarker development for early prediction of cirrhosis regression and HCC detection post viral treatment is discussed. Since DAAs treatment does not provide full protection against reinfection or viral transmission to other individuals, the recent studies for a vaccine development are also reviewed.
Keywords: hepatitis C virus, HCV, hepatocellular carcinoma, HCC, interferon, IFN, direct-acting antiviral, DAA, endoplasmic reticulum stress, ER stress, autophagy
Introduction
Primary liver cancer includes hepatoblastoma (liver cancer among children), angiosarcoma (cancer derived from blood vessels of the liver), cholangiocarcinoma (cancer derived from bile duct), and HCC (cancer derived from hepatocytes). Among those, HCC remains the most common form. Although a high incidence of HCC has been reported in developing countries, the prevalence of HCC has grown tremendously over the last three decades in Western countries as well.1 Currently, HCC is the sixth most common cancer that occurs three times more in males than in females.2 Approximately 800,000 individuals are diagnosed with HCC worldwide causing more than 700,000 deaths per year.3 In the US, deaths due to HCC and cholangiocarcinoma are rising more rapidly than deaths from any other type of cancer.4 The risk factor predominantly contributing to the rise in HCC includes the high rate of HCV infection, followed by increasing rates of alcohol abuse, obesity, NAFLD, and uncontrolled type II diabetes. Autoimmune liver diseases, hemochromatosis, tyrosinemia, glycogen storage diseases, and alpha-1 antitrypsin deficiency can predispose to risk of liver cirrhosis and HCC. Among all these risk factors, HCV alone has about 5 to 20 fold higher risk for HCC development.5–10
HCV is a blood-borne pathogen that infects the liver exclusively. Most of the individuals infected with HCV fail to clear the infection naturally, leading to a stage of life-long chronic infection. The long-lasting liver inflammation due to HCV causes the onset of advanced liver diseases, such as liver fibrosis, cirrhosis, and HCC resulting in death. Approximately 71 million people are currently infected with HCV, of which only 20–30% of those develop liver cirrhosis, and 1–4% of cirrhotic patients develop HCC per year.11 In most cases, HCC develops on the background of cirrhosis.12–18 Approximately 15% of patients who developed HCC had no cirrhosis supporting the hypothesis that HCV infection can also induce HCC directly.19 HCC can also develop in non-cirrhotic liver related to NAFLD and HBV infection.20
IFN-α or PEG-IFN-α plus RBV combination antiviral therapy has been used as the standard-of-care for patients with chronic HCV infection for many years. This treatment eliminates the virus in only a little more than half of all patients, and many liver cirrhosis patients were unable to clear the infection with this regimen.21,22 The low responsiveness to IFN/RBV therapy for chronic HCV infection found to be associated with specific IL-28B genotypes.23 Because of the high association with liver cancer, HCV-related liver research remained as one of the rapidly evolving areas in Hepatology since its discovery in 1989. The development of highly effective antiviral drugs to eliminate chronic HCV infection continued to be the primary focus in many academic laboratories and pharmaceutical industries. Drug discovery efforts of HCV have been hampered for a long time due to the ongoing technical difficulties in the establishment of cell culture models. There was swift progress in many areas of basic and translational research after the cloning and sequencing of the HCV genome and development of infectious chimpanzee clones (Figure 1). Although HCV is known to replicate well in human hepatocytes, researchers have struggled for a long time to grow the virus in the laboratory using different hepatoma cell lines. It took more than ten years to develop a cell culture model for HCV minigenome (the replicon model), which was later followed by the development of an infectious cell culture system using a unique virus strain derived from a Japanese patient with fulminant hepatitis. This virus strain with a highly adapted Huh-7.5-based liver cell line allowed us to study the replication and assembly of the whole HCV genome.24 The availability of replicon and infectious cell culture models accelerated the discovery of newer DAAs. Approval of IFN-free DAAs targeting the NS3/4A protease, NS5A, and NS5B polymerase have now rapidly changed the therapeutic landscape for curing HCV infection at a very high rate.25,26 In the future, HCV cure by DAAs is expected to reduce the incidence of chronic liver disease, liver cirrhosis, HCC, which result in decreased liver-related mortality.27,28
 |
Figure 1 Progress in translational research that leads to significant breakthroughs in DAA-based antiviral drug development resulting in HCV cure. Reprinted from Semin Cell Dev Biol, Dash S, Aydin Y, Wu T. Integrated stress response in hepatitis C promotes Nrf2-related chaperone-mediated autophagy: a novel mechanism for host-microbe survival and HCC development in liver cirrhosis, Copyright (2019), with permission from Elsevier.106 |
Recently, emerging clinical studies with DAAs in patients with liver cirrhosis created a heated debate about the risk of HCC occurrence and recurrence after viral cure. Some studies support that HCV cure by DAAs benefits patients as the liver inflammation decreased, fibrosis reversed, as well as a decreasing incidence of liver transplantation and liver-related extrahepatic complications.28–39 However, some recent emerging publications from the US, Europe, and Asia indicate that DAA-induced HCV clearance in patients with liver cirrhosis decreases but does not eliminate the risk of HCC occurrence or recurrence.40–45 In light of this new development, the overall objective of this article is to review relevant pieces of literature about the long-term benefits and risks of HCC development post viral cure by DAAs as compared to previously used IFN-based antiviral therapy. This review has also discussed potential overlapping viral-induced and host-related mechanisms implicated in HCC risk after viral cure by DAAs.
The Risk of HCC Occurrence and Recurrence After HCV Cure
Chronic HCV infection is the major risk factor for HCC. A natural course of HCV based on observational model predicts that 60% of infected individuals will develop cirrhosis and 14.4% will develop HCC and remaining 37% will die of other complications related to HCV infection.46 Many earlier publications concluded that the IFN-based antiviral therapy prevented HCC risk and reduces all causes of mortality among patients with chronic HCV infection, including patients with liver cirrhosis.46–54 The overall trends of HCC occurrence and recurrence after HCV clearance by IFN-based antiviral therapy were verified through meta-analyses.55–57 Morgan et al analyzed 30 observational studies of compromising 31,528 chronic HCV patients in which 10,853 patients achieved SVR (34.4%) by IFN-based antiviral therapy, and 1742 patients developed HCC, suggesting viral cure by IFN does not eliminate the risk of HCC. When they adjusted HCC incidence per year according to the follow-up period and viral clearance, they found that HCV-cured patients develop HCC at a rate of 0.33%/year.55 Rutledge et al performed similar analysis from 31 studies involving 71,443 HCV patients treated with IFN-based regimens showing that SVR rate with IFN was 45.9%. When they adjusted HCC incidence per year in the SVR population, it was 0.7%.56 Waziry et al examined HCC occurrence and recurrence in patients treated with IFN-based treatment using an extensive electronic database. Their analysis revealed that the overall risk of HCC development is 1.13%/year. This study also reported the HCC recurrence rate was 9.2%/year when SVR was achieved.57 Amalgamation of these data is that the risk of HCC occurrence and recurrence was decreased but not eliminated by IFN-based antiviral therapy. The risk and benefit of HCV clearance by DAA therapy have not been fully established since the follow-up timing of DAA-treated patients is not long enough. However, there have been three significant developments on addressing the HCC risk after viral cure since the approval of the DAAs. These results have generated considerable debates among researchers. First, a number of investigators found that, DAA-based antiviral therapy reduced the incidence of HCC development in chronic HCV patients with pre-existing cirrhosis, but did not eradicate the risk, suggesting these patients need ongoing surveillance for HCC after viral clearance.58–67 Second, some investigators have reported that there is a high chance of HCC recurrence in cirrhotic patients who received DAAs.68–73 Third, low SVR rate was seen with DAAs in patients with active HCC associated to HCV74–80 suggests that the DAA-based HCV therapy should be carried out after HCC treatment. There are now considerable debates and disagreements among researchers all over the world on these observations. We will discuss selected publications addressing the HCC risk after HCV cure by DAAs in patients with liver cirrhosis.
Some studies claimed that the risk of HCC is not decreased after HCV cure by DAA therapy.58–62 These investigators demonstrated that the annual incidence of HCC development after viral treatment with DAAs is 3–5%, which is much higher than the risk observed with IFN-based therapy. In contrast, some other studies have tried to verify these observations and showed that HCV cure does reduce the risk but does not eliminate the risk of HCC development.63–67 These studies found that the annual incidence of HCC occurrence was higher in DAA-induced HCV cure as compared to IFN-induced HCV cure (2.9% vs 1.14%). On the other hand, one large study demonstrated that DAA-induced HCV clearance is associated with reduced HCC risk, similar to IFN-induced HCV clearance among veteran patients.68 Interestingly, a few reports expanded the debates claiming that HCV cure makes the liver cancer grow faster. Mainly, HCV infected patients with HCC who had a complete response to hepatic resection or local ablation subsequently developed high rates of HCC when they received DAAs.69–71 These results are consistent with two small European studies that have shown a high percentage of HCC recurrence (27.6% and 28.8%) among patients who first received treatment for HCC then received DAA within six months.59,72 Another study by Cabibbo et al reported a relatively high rate of HCC recurrence among patients who received DAAs after curative HCC therapy with occurrence rates of 12%, 26.6%, and 29.1% in 6-, 12- and 18 months.73 Moreover, a report from the US shows that 5 out of 18 patients (28%) who were transplanted for HCV-related HCC show unusually high rates of recurrent HCC within six months post DAA therapy. In contrast, only 9.5% recurrence of HCC was seen in those who did not receive HCV treatment.74 However these unexpected findings were not verified by other publications, which reported no such risk for HCC recurrence after viral cure by DAAs therapy.75–77
HCC Risk After HCV Cure Through Meta-Analysis
The risk of HCC occurrence and recurrence after HCV cure by DAAs has been determined through meta-analyses. These reports found that the risk of HCC occurrence and recurrence is almost similar between DAAs and IFN-therapy. However, the data agree with the fact that the risk of HCC occurrence persists after HCV treatment.55–57 To determine the incidence of HCC occurrence after HCV treatment with DAAs, Rutledge et al performed a more rigorous data analysis of 44 studies with a large number of patients (n=91,249). It is found that the risk of HCC development was 3.57%/year in patients who achieved SVR by DAAs and 9.83%/year in patients who did not achieve SVR.56 The study of Huang et al involving 61,334 patients who received DAAs from 16 sites showed that the risk of HCC development in SVR-achieved and SVR non-achieved groups was 3.5%/year and 9.1%/year respectively.81 Waziry et al analysis presented evidence that the overall risk for HCC occurrence is 2.96%/year and recurrence 12.16%/year when HCV infection is cleared by DAA-based therapy.57 A meta-analysis using 16 studies of IFN-based treatment of 1043 patients and 33 studies of DAA-based treatment of 4876 patients determined the risk of HCC recurrence. The recurrence rate of HCC was 16.7%/year in the DAA-based treatment group and 14.3%/year in IFN-based treatment group.56 In addition, Huang et al analysis revealed that the HCC recurrence rate is 17.4%/year following DAA-induced viral clearance.81 A comprehensive review by Singal et al summarized data of the studies that reported the risk of HCC occurrence and recurrence after DAAs treatment.82 These findings suggest that the risk of HCC occurrence and recurrence remains after viral cure by DAAs treatment. Therefore, most of the authors recommend that patients with advanced liver fibrosis (F3-4) need continuous surveillance for early HCC detection after HCV eradication by DAAs.
HCC Mechanisms Associated with Chronic HCV Infection
Hepatitis C virus infects hepatocytes, the major cell types that constitute the liver. Only 25% of individuals acutely infected with HCV can eliminate the virus naturally while the rest develop a persistent infection, and chronic liver disease, including liver fibrosis, cirrhosis, and HCC.83 Chronic HCV infection causes breakdown of immune tolerance leading to a prolonged inflammatory reaction in the liver. A wide range of non-viral agents induces cellular stress and liver injury leading to sterile chronic inflammation. During the early stage of injury, the functional and morphogenic alterations in the liver are reversible if the damage-causing agents or stress factors are removed. The persistence of chronic injury can cause irreversible harm, where the liver undergoes changes from physiological adaptation to pathological adaptation. Liver cirrhosis is a pathological adaptation to cellular stress causing structural and functional changes in the liver parenchyma to escape from injury. The nature and severity of cellular stress determine whether cellular adaptation to virus infection is reversible or irreversible. Over the years, studies using transgenic mice and cell models revealed that HCV viral proteins altered multiple cell signaling pathways implicated in cell survival, proliferation, migration, and transformation. Many of these cell-signaling pathways overlap among hepatic steatosis, alcoholic liver disease, insulin resistance, and oxidative stress, which cause inflammation, cell death, and liver cirrhosis. The evolution of HCC from cirrhosis is a pathological adaptive response to ISR. Only 1–3% of patients with chronic HCV infection develop HCC after 30 years and HCV does not infect HCC tumor cells as compared to surrounding non-tumorous hepatocytes, supporting the conclusion that HCV is not directly oncogenic. It is possible that HCV infection creates a tumorigenic environment that promote cellular transformation of uninfected hepatocytes through a bystander mechanism as seen in the case of colorectal cancer associated with Fusobacterium nucleatum and gastric cancer related to Helicobacter pylori. Based on these evidences, we propose that HCC mechanisms associated to HCV infection can be direct virus-induced cellular programming, indirect host-related inflammatory response, and an overlapping host metabolic bystander effect (Figure 2). In the following sections, we discuss the molecular basis of hepatic pathological adaption to the virus-associated stress response, indirect stress related to inflammation, and metabolic stress response of host that contributes to the persistent fibrosis and HCC. In the end, we discuss possible overlapping synergy mechanisms between viral-induced oncogenic and host-related pathways due to concomitant liver diseases that contribute to the persistent HCC risk after DAAs treatment independent of the virus.
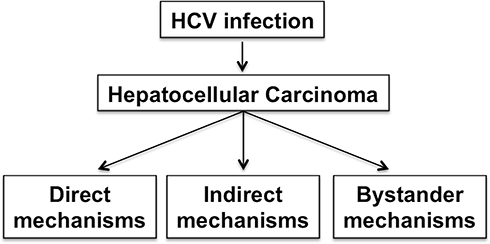 |
Figure 2 Hepatocellular carcinoma mechanisms related to chronic HCV infection are the combination of virus-mediated (direct), host-mediated (indirect), and host-related bystander effects. |
Direct Oncogenic Mechanisms
HCV Replication in Hepatocytes Generates an Integrated Stress Response
HCV is a small, enveloped, positive-stranded RNA virus belonging to the Flaviviridae family. The complete enveloped HCV particles are icosahedral with a diameter of 56–65 nm, and an embedded viral core around 45nm in size.84,85 Hepatocytes are the only cell type in the human liver that support the entire life cycle of HCV through multiple steps: attachment and entry, viral protein synthesis, virus replication, virus assembly, and release.86,87 The infection cycle involves the attachment and entry of HCV particles into hepatocytes through the interaction of its two viral envelope proteins (E1 and E2) with the cell surface receptors. Multiple host cell-surface proteins are involved in the attachment and entry of the HCV particles into hepatocytes.88,89 During the entry process, the fusion of the HCV viral envelope with the host endosomal membrane results in pH-dependent uncoating, releasing the genomic RNA into the cytoplasm. The HCV RNA genome makes viral proteins for its reproduction in a specific region of the ER called the rough ER. The positive-strand HCV RNA genome binds directly to ribosomes through an Internal ribosome entry site present in its 5ʹ untranslated region. Translation of the HCV genome leads to the production of a single large polyprotein of 3000 amino acids. This protein is subsequently cleaved by cellular and viral proteases into the structural proteins (core and envelope proteins E1 and E2), and non-structural proteins (P7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B).90,91 The viral proteins assemble in the ER membrane vesicles to reproduce more genomic HCV RNA and progeny viral RNA. The non-structural proteins are needed for genome replication.90 Multiple rounds of viral replication lead to increased accumulation of positive-strand RNA, replicative intermediates (negative-strand RNA), and viral proteins in the ER. Sustained virus replication results in the proliferation and remodeling of the ER membranes into a structure referred to as the membranous web. Structural proteins remodel the ER membranes to recruit more protein components needed for virus replication, assembly, and release.92 It is not clear how the replication and assembly processes occur one after the other, but they appear to occur in lipid droplets accumulated in the ER vesicles. Intrahepatic HCV replication stimulates lipid metabolism, leading to the accumulation of lipid droplets in multilayer membrane vesicles, which facilitates virus assembly and maturation. Virus particle assembly and release occur in the membranous web, a link to very low-density lipoprotein synthesis and secretion. HCV RNA replication is catalyzed by the viral NS5B, an RNA-dependent RNA polymerase, via the negative-strand RNA. The protease and helicase domains of NS3 play essential roles during the viral genome replication. The NS5A protein is required for the formation of the membranous web that supports viral replication and assembly. Studies have shown that NS5A inhibitors prevent the formation of membranous web structures in the ER by decreasing the complex formation with phosphatidylinositol 4 kinase III alpha.93 Several viral proteins and host cellular factors essential for HCV replication could be promising targets for antiviral therapy. Among these, the most successful viral marks used in DAAs treatment are the HCV NS3 protease, NS5A, and the NS5B viral RNA polymerase (Figure 3). A recent review describes the mechanism of how different DAAs inhibit HCV replication.94 Although the causal relationship between chronic HCV infection and the development of HCC has been established, the exact HCV cancer mechanism is unknown.95,96 Hepatocytes are specialized cells in the liver with an elaborate ER membrane network. The ER membrane network transports proteins and lipids to lysosomes, mitochondria, secretory vesicles, the Golgi apparatus, endosome, and the cell membrane. The ER plays an essential role in maintaining hepatocyte function in the liver. The proteins synthesized in the ER must be correctly folded and undergo post-translational modifications such as glycosylation, disulfide bridge formation, and oligomerization. All of these processes take place in the ER. The ER stress response can develop due to alterations in protein synthesis, degradation or folding during viral infection. Due to these reasons, the stress response in the ER can activate pathways that trigger cell injury, inflammation associated with chronic liver disease, liver cirrhosis, and HCC related to both viral and non-viral causes.97
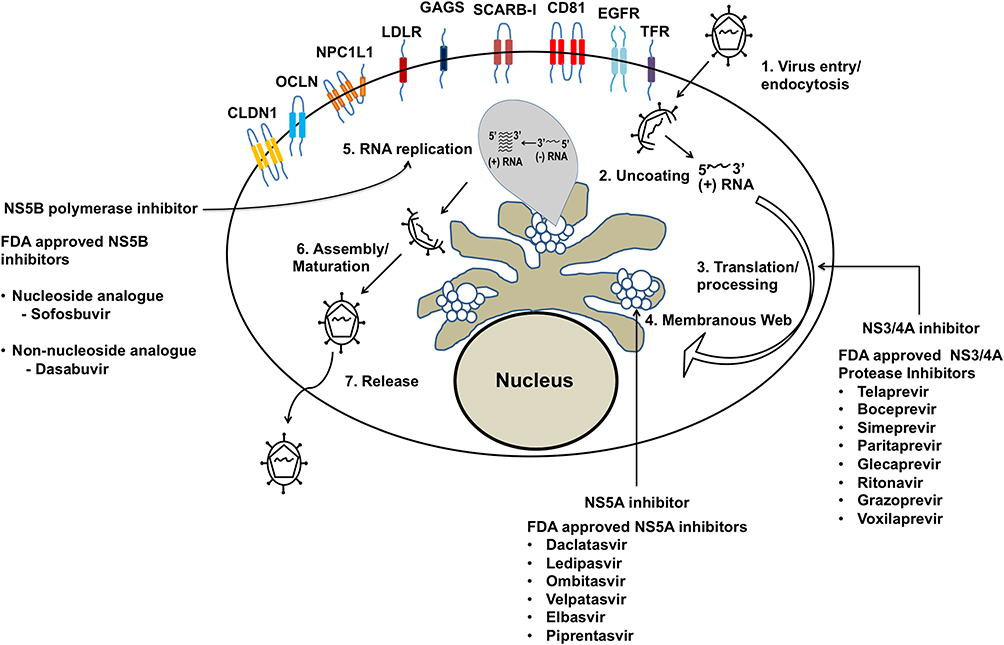 |
Figure 3 Chronic HCV replication in liver cells induces a multifaceted stress response. The infection is initiated by the attachment and entry of the virus particle through several cell surface receptors. The HCV RNA binds to the ribosome and translates a single large polyprotein, which is processed into structural and non-structural proteins. Accumulation of viral proteins induces proliferation of ER-membranes and formation of membranous web structure. HCV replication in the membranous web produces many new genomic positive-strand and negative-strand RNA. Genomic HCV positive-strand RNA packages into complete infectious virus particles that release through the secretory pathway. Arrows show the emerging new DAAs targeting the viral protein and HCV replication cycle.Note: Adapted from Chapter 8, Figure 2 of Viral Polymerases: Structures, Functions and Roles as Antiviral Drug Targets, Dash S, Aydin Y, Stephens CM. In: Gupta SP, editor. Hepatitis C Virus NS5B RNA-Dependent RNA Polymerase Inhibitor: An Integral Part of HCV Antiviral Therapy, 211-235, copyright (2019), with permission from Elsevier. 383 |
Hepatocytes in the Liver Undergo Pathological Adaptation to HCV Microbial Stress
The increase in cellular energy demand during chronic virus infection imposes all types of caloric restrictions, and low levels of ATP, amino acids, and sugar, leading to increased ER stress.98 Autophagy is activated when the ATP and amino acid levels are depleted during infection. Low intracellular sugars will cause a defect in protein glycosylation and alter ER and Golgi function leading to the stress response. Low oxygen supply (hypoxia) during viral replication creates oxidative stress that generates ROS, which can cause ER stress. Chronic HCV infection induces lipid metabolism in the liver, leading to the accumulation of fat in hepatocytes and hepatic steatosis. To study the complex lipid metabolic remodeling in infected cells, Hofmann et al determined the lipid composition of whole cells and subcellular fractions of HCV–infected culture using a bioinformatics approach. They showed that HCV infection accumulates membrane lipids, especially cholesterol and phospholipids, with a higher abundance of phosphatidylcholines and triglycerides with longer fatty acyl chains. The accumulation of longer fatty acids and cholesterol in the infected cells could inhibit the ubiquitin-dependent protein degradation pathway; therefore, increasing ER stress.99 Persistent virus replication increases cellular DNA damage response, the frequency of DNA repair, transcriptional fidelity, and genomic instability, all of which create additional cellular stress. If the viral-induced stress becomes persistent and severe, that results in irreversible injury or death of infected cells. Hepatocytes experience different levels of cellular stress during virus infection that activates various cell death cascades (apoptotic, necrotic, or autophagic) during chronic liver disease. The release of DAMP from lysed necrotic cells can produce ROS and RNS, which are known to cause lipid peroxidation.100 This process can activate the unfolded protein response and NF-κB pathway, which is the hallmark of an inflammatory response implicated in the development of liver fibrosis and HCC during chronic HCV infection.101 In some individuals, combinations of multiple non-viral insults may contribute to liver disease progression. One of the host-related factors responsible for the progression of liver disease is the degree of hepatocellular injury. Aminotransferases are a group of enzymes that synthesize and break down amino acids and convert them into energy storage molecules. Increased ALT and AST levels in the blood are a direct indication of a liver injury. Prior studies found that 1–14% of cells in the HCV–infected liver showed Ki-67 labeling, suggesting that the hepatic parenchymal mass is maintained through continuous hepatocyte regeneration and self-replication.102,103 The physiological adaptions at the level of cellular metabolic activity and cellular function, which often occur during chronic infection, are reversible. However, pathological adaptation due to severe microbial stress can result in an increase or decrease in cellular appearance and cell proliferation. Due to these reasons, hepatocyte proliferation rates drop significantly during the stage of liver cirrhosis due to pathological adaption to stress.104,105 Infected hepatocytes can manage the stress response through the induction of ISR that promotes the transcription of numerous genes for cell survival.106 The significant reduction of hepatocyte proliferation in liver cirrhosis could activate stem cell compartment through epigenetic programming.107–109 The stress signal reprograms to switch cell death to cell proliferation, and it will abet the emergence of malignancies like HCC. In a previously published series, we showed that chronic HCV infection induces ER stress and the expression of ER stress marker remains high in patients with liver cirrhosis.110 Using the HCV cell culture model, we showed that excessive ER stress activates NRF2-mediated autophagy switching to promote cell survival.111 The excessive stress contributes to HCC development in liver cirrhosis. HCC grown in the cirrhotic liver undergoes autophagy switching from a protective state characterized by high macroautophagy with low CMA to an HCC-promoting state characterized by low macroautophagy with high CMA.112,113 Our data are also consistent with previous reports suggesting that persistent activation of NRF2 through the accumulation of p62 is involved in HCC development.114,115 In the following section, we review how pathological adaptation to HCV–induced stress results in cellular oncogenic programming implicated in HCC development.
Hepatic Adaptation to HCV Microbial Stress Induces Oncogenic Cell Programing
An integrated analysis of whole-exome sequencing of HCV-related HCC tumors demonstrated many mutations in cancer driver genes associated with malignant transformations.116–125 The exome sequencing study found that tumor suppressor genes are downregulated due to promoter hypermethylation, and genomic instability in HCC due to the gains and loss in chromosomes was found in more than 80% of HCC associated with chronic HCV infection.119–125 The most significant cellular pathways that are altered in HCV–induced HCC are TERT, β-catenin, p53, Rb, chromatin remodeling/epigenetic modifications, hepatocyte differentiation, PI3K-mTOR pathway, and NRF2-kelck-like ECH-associated protein (KEAP), cancer stem cells, angiogenesis and RTKs (Figure 4). In the following section, we discuss the molecular mechanism by which HCV regulates these cancer pathways.
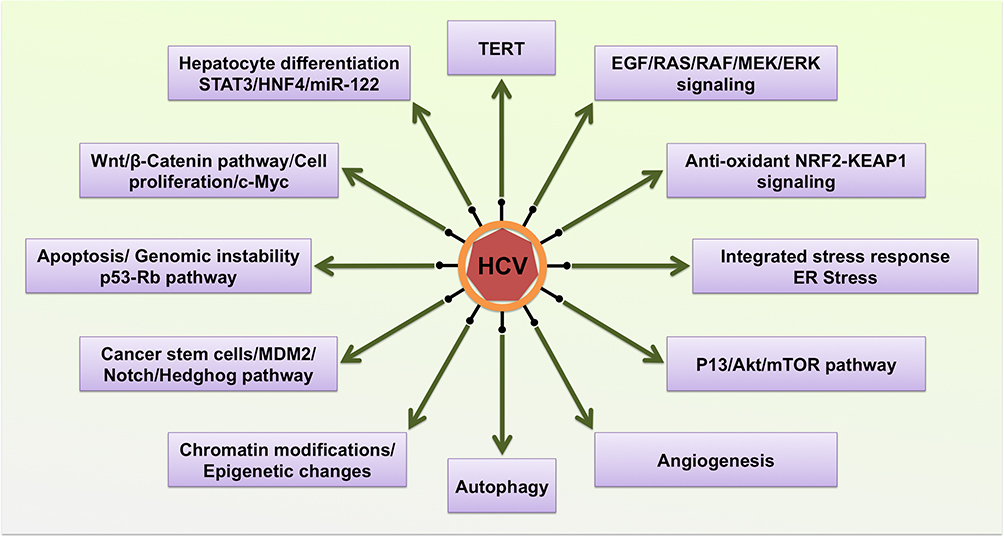 |
Figure 4 Hepatic adaptive response to chronic HCV infection activates hepatocyte cell survival programming. HCV structural and non-structural proteins activate multiple cellular pathways. |
Telomerase Reverse Transcriptase (TERT)
Telomeres are DNA-protein structures located at the end of each chromosome. They are composed of a tandem repeat of six-nucleotide sequence (5ʹ-TTAGGG-3ʹ) coated with shelterin proteins. The telomere can reach a length of 15,000 base pairs. Telomeres function by preventing chromosomes from losing base pair sequences and fusing at their ends. When the telomere length becomes too short, the cell dies by apoptosis. Telomere length is maintained by an enzyme called TERT. Telomerase is an enzyme made of protein and RNA subunits that elongates chromosomes by adding TTAGGG sequences to the end of existing chromosomes. Telomeres and telomerase play an essential role in the progression of liver disease and liver cirrhosis. Hepatocyte senescence and development of liver cirrhosis relate to the absence of telomerase activity and shortening of telomeres. Telomerase detected in human cancer cells is found to be 10–20 times more active than in healthy body cells.126 Increase telomerase provides a selective growth advantage to tumor cells. Complete HCC genome sequencing found many host spots activating mutations in the promoter regions of the TERT gene in about 85% of human tumors, including HCC.127,128 The modifications are present at nucleotides 124 (mostly G>A and rarely G>T) or 146 (G>A) before the ATG start site in the TERT promoter region.127 These mutations favor the binding of transcription factors to the promoter, causing TERT overexpression and activation. The long telomeres and increased TERT activity are found to be associated with aggressive HCC with poor prognosis.129 A previous study showed a mutation in the TERT gene among patients with liver cirrhosis of diverse etiologies: alcohol, hepatitis B, and hepatitis C infection as compared to healthy controls.128 Reduced telomerase activity could result in telomere shortening, chromosomal instability through end-to-end chromosome fusion, and HCC if the p53 and Rb are defective. Recent publications by Zhu et al suggest that HCV core and NS3 proteins can activate TERT expression and reverse transcriptase activity.130,131
The p53-Rb Pathway
The two tumor suppressors, Rb and the p53 are involved in the etiology of many cancers. The p53 protein levels are increased in response to DNA damage, hypoxia, and oncogene activation. p53 regulates gene expression that leads to cell cycle arrest or apoptosis. The Rb protein prevents cell proliferation by repressing the activation of E2F transcription factors. The interaction between Rb and E2F transcription factors are critical, preventing the S-phase of the cell cycle and mitosis.132 Many earlier studies have shown that transient expression core, NS3, and NS5A proteins of HCV alter p53 tumor suppressor function without using the infectious HCV replication model. We discuss here some of the studies that examined the impact of HCV infection on the roles of p53 and Rb tumor suppressors. Mitchell et al showed that HCV infection disrupts p53 function through activation of cellular protein kinase R.133 Stanley Lemon’s laboratory demonstrated that HCV infection negatively regulates Rb protein stability mediated by NS5B protein.134,135 Studies conducted in our laboratory showed that excessive ER stress due to HCV replication degrades p53 in the lysosomes. The degradation of p53 in HCV culture occurs independently of MDM2. We showed that CMA activation by stress degrades p14ARF, another alternative open reading frame protein (ARF), a tumor suppressor that activates p53.136 The subsequent publication showed that excessive ER stress induces MDM2-mediated Rb degradation.137 All these results are consistent with human studies that show more than 70% of HCC cases having alterations in the p53-Rb pathway that leads to mitosis, cell cycle progression, and genomic instability.
The Wnt/β-Catenin/c-Myc Pathway
The Wnt/β-catenin pathway is altered in 66% of HCC, and in 51% of these cases, activation of Wnt/β-catenin occurs due to altered gene expression.117 The Wnt/β-catenin pathway is activated when a Wnt ligand binds to the frizzled receptor, leading to activation of the transcription factor β-catenin and subsequent activation of pro-survival genes. Wang et al showed that HCV infection could activate the Wnt/β-catenin pathway. The HCV core protein was shown to activate Wnt/β-catenin signaling at multiple steps, including elevated expression of Wnt ligands, and frizzled receptor, and downregulation of the expression of low-density lipoprotein receptor-related protein 5/6. Core protein has been shown to decreases the expression of the Wnt antagonists; dickkopf and secreted frizzled-related protein by recruiting DMT1, and HDAC1 to their transcription start sites.138,139 The NS5A protein activates PI3K/Akt signaling, leading to the inactivation of GSK3β and subsequently reducing the degradation of β-catenin.140,141 The activation of c-Myc oncogene through Wnt/β-catenin pathways has been shown to promote HCC in HCV transgenic mice model.142
Receptor Tyrosine Kinases (RTKs)
The RTKs are a large superfamily of cell surface receptors representing for a wide variety of growth factors, including epidermal growth factor, nerve growth factor, PDGF, VEGF, FGF, insulin and the insulin-like growth factors. Among these, EGFR controls the cascade of oncogenic cell signaling involved in cell proliferation that contributes to hepatocarcinogenesis. The EGFR is highly expressed in the adult liver and plays an essential role in hepatocyte proliferation. The EGFR pathway is activated in 60–80% of HCC and correlates with aggressive tumors and patient survival.143–150 The receptor-mediated endocytosis and lysosomal degradation are the major negative feedback loops for EGFR signaling. We showed that HCV induces impaired autophagy response to inhibit degradation of EGFR at the level of autophagosome-lysosome fusion leading to the activation of downstream RAS/RAF/MEK/ERK signaling.111 In principle, impaired autophagy due to HCV could potentially stabilize RTK on the cell surface of infected cells by impairing their endocytosis and lysosomal degradation. Other researchers have also shown that EGFR activation favors the HCV entry process through co-internalization of an HCV-CD81-EGFR complex following binding of EGFR ligands to the receptor and subsequent endocytosis.151 The viral NS5A protein disturbs EGFR trafficking and degradation, therefore, activates EGFR signaling.152 All these data support that HCV infection activates EGFR signaling, which contributes to the HCV-associated HCC development. The EGFR pathway activation can cross-talk with Wnt/β-catenin since EGFR can phosphorylate β-catenin at residue Tyr654, therefore dissociating from the multi-receptor complex and leading to nuclear entry and gene expression.153 The EGFR stimulates PI3K/Akt and RAS/RAF/MEK/ERK cascade that can activate β-catenin through GSK3β activity. Wnt/β-catenin signaling also activates FGF signaling implicated in HCC development secondary to chronic HCV infection by inducing expression of FGF18 and FGF20.154,155
PI3K/Akt/mTOR Pathway
The activation of the mTOR pathway is associated with HCC development related to chronic viral infection.156,157 Immunohistochemical staining revealed that 33 out of 73 (45%) HCC patients showed increased expression of total S6k, which is correlated with mTOR activation and tumor size.158 In a large cohort of HCC patients, the activation of the mTOR pathway was associated with tumor differentiation, staging, vascular invasion, and expression of phosphoS6.159 The mTOR pathway can be activated by growth factors, cytokines, TLR ligands, low cellular energy (ATP/AMP ratio), hypoxia, and DNA damage. The activation of mTOR can confer many growth advantages to cancer stem cells or progenitor cells, such as promoting cell proliferation and resistance to apoptosis induced by various stress signals such as hypoxia and nutrient deficiency.160 In addition, the mTOR pathway can regulate telomerase activity in HCC since rapamycin significantly decreases telomerase activity at the protein level.161 NS5A can activate PI3K-mTOR signaling by directly binding to the p85 subunit of PI3K.162 The mTOR activation by the NS5A protein blocks apoptosis through binding to FKBP38, an immunosuppressant FK506-binding protein.163
Angiogenesis
Angiogenesis, a physiological process that generates new blood vessels from the existing vessels, is linked to HCC development. It has been shown that HCV promotes angiogenesis process during an advanced stage of liver disease.164,165 In healthy tissues, the angiogenesis process is inhibited by interactions between proangiogenic and antiangiogenic factors.165 Angiogenesis is activated when tumor tissue requires additional nutrient and oxygen supply.166 HCC cells secrete proangiogenic factors and activate endothelial cells by VEGF and FGFs. The expression levels of angiogenic growth factors, VEGF-A, angiopoietin-2, and PDGF are elevated in HCC patients.167,168 HCV infection can trigger hepatic angiogenesis and HCC growth through HIF-1α and VEGF regulation.169,170 The VEGF activation can occur through several other cell survival pathways, including PI3K/Akt, ERK1/2, NF-κB, and STAT3, which stabilize HIF-1α. It has been demonstrated that the core protein of HCV can trigger angiogenesis through a mechanism that involves cross-talk between TGF-β2, VEGF, and CD34 expression.171
Cancer Stem Cell
Pathological adaptive response to microbial stress results in altered gene expression through epigenetic alterations without changing the DNA nucleotide sequences.172,173 The epigenetic modifications of chromatin are involved in the maintenance of viral latency and reactivation of human viruses such as HIV, cytomegalovirus, Epstein-Barr virus and many others.174 The potential role of epigenetic modification in cell differentiation, tissue homeostasis, and regeneration has been recently understood through stem cell research. The pluripotent embryonic stem cells are capable of giving rise to different cell types in embryo that serve as a valuable model to study epigenetic mechanisms involved in tissue development and tissue-specific gene expression.175 Disruption of epigenetic processes can lead to altered gene expression and malignant transformation.176 The chromatins that package chromosomes are the macromolecular complex of DNA and histone proteins. The basic functional unit of chromatin includes a nucleosome that contains 147 base pairs of DNA, which wrapped around a histone octamer that contains two of each histone H2A, H2B, H3, and H4. Condensed chromatin called heterochromatin that leads to gene silencing, whereas open chromatin called euchromatin leads to active gene transcription.177 The epigenetic modifications occur through DNA methylation, histone modifications, nucleosome positioning, and ncRNAs. The interplay between these chromatin modification mechanisms creates an epigenetic landscape of mammalian gene expression. DNA methylation of CpG-rich DNA sequences called CpG island leads to gene silencing. Histone proteins N-terminal tails undergo a variety of post-translational modifications, including methylation, acetylation, ubiquitination, sumoylation, and phosphorylation. Histone modification can lead to either gene activation or repression, depending upon which histone residues are modified. For example, lysine acetylation correlates with transcriptional activation, whereas lysine methylation leads to transcriptional activation or repression depending on which residue is modified and the degree of methylation. The trimethylation of lysine four on histone H3 (H3K4Me3) is enriched at the promoter that is transcriptionally active, whereas trimethylation of H3K9 (H3K9Me3) and H3K27 (H3K27Me3) of the promoter are transcriptionally inactive.176 For example, embryonic stem cells possess bivalent domains with both active (H3K4Me3), and repressive (H3K27Me3) marks at the promoters of the developmental genes that regulate multiple cell types in a tissue. The activity of such bivalent domains is regulated by two counteracting groups of chromatin proteins called the trithorax group and polycomb group proteins.178,179 In mammals, the polycomb group proteins are an epigenetic modifier that promotes H3K27 trimethylation and condenses chromatin resulting in gene silencing. This interaction favors the binding of the trithorax group of proteins to the activating H3K4 trimethylation mark that leads to open chromatin and active gene transcription. This bivalency hypothesis has been found to contribute to phenotypic plasticity and initiation of cancer stem cells. This condition allows differentiated cells to lose this bivalency through rigid chromatin structure, whereas the maintenance of active chromatin states favors tumorigenesis, stem cell renewal, and proliferation. Some reviews describe the importance of epigenetic modification on cell fate and cancer initiation.179–181 The polycomb repressive protein complexes (PRC1 and PRC2) are involved in CSCs to establish dynamic epigenetic changes needed for the embryonic stem cell gene signature.180–182 These advances have led to the discovery of epigenetic drugs that have been now FDA-approved for the treatment of cancer, and viral infection including DMT inhibitors (azacytidine, decitabine) and HDAC inhibitors (vorinostat, romidepsin, belinostat, panobinostat).183 A study published by Ali et al shows that HCV replication in cultured cells can induce expression of cancer stem markers including, doublecortin and CaM kinase-like-1, LGR5, CD133, AFP, cytokeratin-19, LIN28, and c-Myc. They showed that curing HCV replication of these cells results in diminished expression of these factors.184 Another report showed HCV NS5A transgenic mice fed with alcohol developed HCC with stem cell regulator Nanog expression through the TLR4 signaling.185 Human genome microarray study using a Huh-7.5 cell line stably-replicating HCV sub-genomic RNA revealed the upregulation of cancer stem cell markers: octamer-binding protein 3, SOX2 and suppressor of zeste 12 homolog.186,187 The authors showed that the expression of enhancer of zeste homolog 2, a member of PRC protein was overexpressed in HCC as compared to the normal liver.186 Notch signaling is involved in the maintenance of CSCs.188 Studies found that NS3 protein can activate notch signaling through binding Snf2-related CBP activator protein and enhances hairy enhancer of split-1 promoter activity that leads to increased hairy enhancer of split-1 expression, a transcriptional repressor of cell differentiation, thus providing a mechanism on how HCV infection promotes cancer stemness.189–191 The Hedgehog pathway is also important for the maintenance of stem cell homeostasis is activated during HCV infection.192,193 One study found that the Hedgehog pathway plays a role in HCV replication and viral permissiveness.194 We showed that HCV–induced cellular ER stress could activate MDM2 that degrades Rb tumor suppressor independent of p53.136 A recent report shows that MDM2 can associate with the PRC2 and enhances stemness through chromatin modification independent of p53.195 This evidence suggests there are many oncogenic pathways that HCV replication probably induces epigenetic cell programing that induces CSCs leading to HCC.
Epigenetic Modifications
Epigenetic modifications are frequently seen in human HCC with hypermethylation of tumor suppressor genes, hypomethylation of oncogenes, and methylation of repetitive elements. Stefanska et al reported that approximately 3700 promoters that are hypomethylated in HCC using a combination of methylated DNA immunoprecipitation and hybridization. The demethylated genes are mainly involved in cell growth, cell adhesion and communication, signal transduction, mobility, and invasion, functions that are essential for cancer progression and metastasis.196 A study by Nishida et al indicates that the number of tumor suppressor genes is hypermethylated in human HCC.197 Some recent publications have examined genome-wide epigenetic changes during the progression of HCV-associated cirrhosis to HCC, and their findings showed a potential prognostic value of DNA methylation of some specific gene promoters, CpG islands and CpG island shores.198,199 Alterations in histone modifications also have been observed in HCC development.200 Increased expression of HDACs that are aberrantly expressed in HCC, which provides a rationale for HCC treatment using a novel pan-HDAC inhibitor (panobinostat).201 Numerous small RNA and ncRNAs are also involved in the epigenetic mechanisms involved in HCV–induced HCC development. Some of them have been used as biomarkers for early detection of HCC.202,203 A recent report showed that long ncRNAs are highly expressed in HCC, and they promote liver cancer stem cell growth through epigenetic regulation.204 Cheng et al study showed that liver-specific miR-122 is epigenetically suppressed by HOTAIR, leading to activation of Cyclin G1 and HCC growth.205 Recent research from our laboratory shows that hepatic adaptive response to HCV–induced ER stress and oxidative stress can silence miR-122 through induction of the STAT3-HNF4α-miR-24 inflammatory feedback loop.206 In this study, we found that miR-122 levels severely depleted among patients who developed cirrhosis. These data suggest that pathological adaptive response to HCV microbial stress epigenetically silence HNF4α and miR-122.
Indirect Oncogenic Mechanisms
The liver is the most immune-privileged organ in the human body. It filters most of the microbial pathogens that enter through the hepatic artery and portal vein blood. The liver is also regularly exposed to the gut microbiota, toxic metabolites, food allergens derived from the gastrointestinal organs through the portal vein. A durable protective innate immune response through the activation of immune and non-immune cells eliminates pathogens to prevent liver injury. In this section, we describe basic molecular mechanisms of how persistent HCV replication breakdown immune tolerance in the liver that leads to the chronic infection and liver disease progression. The liver is composed of primary hepatocytes (80%), and the remaining non-parenchymal cells are KCs, stellate cells, sinusoidal endothelial cells that are essential for innate immune surveillance.207 The liver contains many other cell types that constitute the innate and adaptive immune cells of lymphoid origin, such as T cells, NK cells, NKT cells, MAIT cells, and B cells.208 The liver houses abundant APCs, such as MDCs, PDCs, KCs, sinusoidal endothelial cells, monocytes/macrophages, neutrophils, stellate cells.209,210 During HCV infection, a both innate and adaptive immune response is activated through multiple mechanisms. For example, hepatocytes and other antigen-presenting cells quickly sense the conserved PAMP receptors present in HCV (HCV genomic RNA, structural and non-structural proteins) by different pattern recognition receptors such as RIG-1, TLRs, The acute inflammatory response can also be generated by the DAMP released in response to cell death (called sterile agents) during integrated cellular stress and cell damage. The innate immune signaling pathways are amplified through the production of IFN (type 1 and type III), ISGs, and proinflammatory cytokines to eliminate virus. In the case of HCV, a very few individuals are resolved infection; naturally, the majority of cases disease becomes chronic. It has been demonstrated that several HCV proteins impair the cytotoxic and immunoregulatory activities of immune cells such as APCs, NK cells, CD4 T cells, and CD8 T cells, therefore, overcomes the host innate and adaptive immune response leading to a stage of chronic infection.211–214 We review different immune cells that involved in innate and adaptive immune response and basic mechanisms of how the rapid expansion of early T cell exhaustion or depletion leads to chronic HCV infection (Figure 5).
 |
Figure 5 Hepatic adaptive response to inflammatory stress progresses to cirrhosis. Blood supply to the liver by both the hepatic artery and the portal vein brings potential pathogens, including HCV. Kupffer cells and sinusoidal endothelial cells recognize HCV-derived pathogen-associated molecular pattern (PAMP). The activation of the innate immune response through Kupffer cells recruit new adaptive immune cells (CD4 and CD8 T cells) and B cells. Sustained inflammation can lead to liver fibrosis, cirrhosis, and HCC. Chronic hepatitis is a physiological adaptation that can be reversible after HCV cure. Pathological adaptation of the inflamed liver can lead to cirrhosis and HCC. |
Antigen-Presenting Cells
KCs are the primary tissue-resident macrophages located in the liver sinusoids play an essential role in the intrahepatic innate antiviral response during HCV infection. These cells first encounter microbial pathogens that enter the liver through the natural antiviral program.215 A number of researchers have shown that HCV core and NS3 proteins can activate KCs via TLR to produce inflammatory cytokines (IFN-β, IL-1β, IL-6, TNF-α) and these inflammatory cytokines suppress HCV replication.216–220 Numbers and expression of KCs specific markers (CD163 and CD33) were found increased during chronic HCV infection.221,222 Chronic HCV patients have higher levels of IL-1β as compared to healthy controls. Some researchers showed that the NLRP3/caspase-1 inflammasome activation by hepatic macrophages during HCV infection produces IL-1β leading to proinflammatory cytokine production implicated in the liver disease progression.223 Although these reports support that HCV generates a strong innate immune response, several studies demonstrated that HCV infection is able to neutralize the proinflammatory innate immune signaling (TLR and RIG-1) in KCs and hepatocytes. Some researchers have shown that HCV is able to neutralize the inflammatory activity of peripheral mononuclear cells and hepatocytes by interfering with TLR and RIG-1 signaling.224–231 The liver also contains some populations of DCs and macrophages. Liver DCs can be two categories: MDCs and PDCs. The myeloid-derived mononuclear cells (monocytes) are the primary antigen-presenting cells that prime the innate and adaptive immunity during HCV infection. These cells support T cell activation leading to the generation of Th1 response with the production of IL-12 and TNF-α. The PDCs in the liver express PD-L1, a molecule that induces antigen-specific tolerance.232
Natural Killer Cells (NK Cells)
About 30–50% of total intrahepatic lymphocytes are NK cells in humans. Activated NK cells can kill target cells by releasing perforin and granzyme from cytotoxic granules or by engagement of death receptors on target cells. During acute infection, NK cell activation leads to increased production of IFN-γ and cytotoxicity. NK cells secrete TNF-α and IFN-γ that inhibit HCV replication as well as cytolytic enzymes that destroy HCV–infectedcells.233 The role of NK cells in HCV infection is supported by an earlier publication showing that gene encoding NK cell receptor KIR2DL3 and its ligand HLA-C1 was associated with spontaneous resolution of infection.234 The explanation for this genetic association was that the NK cells from HLA-C1 homozygous individuals show increased production of IFN-γ and greater degranulation than those from nonhomozygous individuals.235 The NK cell phenotypes and functional changes were found among patients with chronic HCV infection. Increased expressions of activating NK cell receptors such as NKp30, NKp44, NKp46, and NKG2D were reported to be increased during chronic HCV infection.236–238 Some researchers published data showing the variation of NK cell effector function, with increased cytotoxicity with reduced production of IFN-γ and TNF-α.237–239 This dichotomy of NK cell function was found to be more pronounced among patients who were receiving IFN-α antiviral therapy.240 The mechanistic insight into this observation was found to be due to the preferential activation of STAT1 expression over STAT4 by IFN-α treatment.241 At present, one report claim that DAAs-induced HCV clearance corrects the altered NK cell phenotypes and reduces STAT1 expression and phosphorylation.242 Another recent study claims that HCV–induced NK cell functional imprinting is not reversible.243 NK T cells are another group of innate cells, which comprise 26% of intrahepatic lymphocytes and secrete IFN-γ, TNF-α, and IL-2.244,245 Though its precise role in chronic infection is yet unclear, there are indications that NKT cells may influence the balance of Th1 versus Th2 responses to an HCV infection.246
CD4 and CD8 T Lymphocytes
Adaptive immune response in acute infection is activated when HCV antigen-specific T cells are primed by APCs in the lymphoid organs, where they proliferate and then migrate to the liver to execute their effector functions.247 HCV-specific CD8 T cells can kill HCV–infected hepatocytes via the perforin/granzyme lytic pathway, but also by secretion of Fas ligand (CD178 or CD95L) and inflammatory cytokines, mainly IFN-γ to clear HCV infection. The success of the adaptive immune response to HCV requires proper interactions between CD4 T cells and CD8 T cells. In addition to acute resolving infection, the CD4 T cells also contribute to the maturation of memory CD8 T cells that prevent reinfection.248,249 However, impairment of CD8 T cells response is associated with chronic HCV infection.250 The expansion of T-reg, expression of multiple co-stimulatory molecules, impaired proliferation capacity, and cytokine production are some of the mechanisms of impaired CD8 T cell response.251–255 During the chronic stage of HCV infection, CD4 T cells exhibit different phenotypes with a high expression of inhibitory receptors (TIM3, PD-1, and CTLA-4) decreasing the production of IL-21.256,257 In chronic HCV infection, PD-1 expression was associated with impaired function of CD8 T cells with studies showing that PD-1 inhibition reactivates CD8 T cells function, proliferation, IFN-γ production, and viral clearance.258–261 Tacke et al study show that HCV infection accumulates CD33+ MDSCs in human peripheral blood that suppresses CD8 T cell response through ROS.262 Lack of resolution of HCV infection due to insufficient CD8 T cell response during chronic HCV infection relate to the fibrogenic response in the liver.263
B Cell Response
The role of neutralizing antibodies and B cells in the progression of HCV–induced chronic liver disease is unclear. B cells may contribute more towards to humoral immunity and antibody-dependent cellular cytotoxicity of HCV–infected cells in the liver. Increasing evidence suggests that neutralizing antibodies are associated with spontaneous clearance of HCV infection and reinfection.264–267 HCV accounts for 85–95% of mixed cryoglobulinemia, a common extrahepatic manifestation of HCV. The cryoglobulinemia occurs by HCV antigen-driven B cell clonal proliferation leading to the deposition of circulating immune complexes in small vessels of the skin, nerves, kidney, liver, and joints. About 50% of patients with chronic HCV virus infection have circulating cryoglobulins, only about 10–15% show clinical disease of palpable purpura, arthralgias, neuropathy, and glomerulopathy.268
Pathological Adaption to Chronic Inflammation Leads to Liver Cirrhosis
The immune activation cross-talks with the metabolic pathways of immune and non-immune cells to allocate more nutrients (ATPs, amino acids, sugar, and lipids) therefore creating integrated immune stress response. However, chronic inflammation results in repeated cell death, cell injury, DNA damage leading to liver injury. Inflammatory cells also produce chemokines, metabolites, and growth factors. Production of inflammatory cytokines activates cell signaling, thus creates ISR pathways.269 Prolonged inflammation and immune activation in the liver lead to pathological adaptation. Due to this mechanism, chronic inflammation promotes liver regeneration, fibrosis, and aberrant accumulation of collagenous connective tissue leading to liver fibrosis. Liver cirrhosis represents the pathological adaptation to a long-lasting inflammation secondary to HCV infection (Figure 5). Chronic inflammation can increase the risk of HCC. The impaired immune regulation due to KCs, NK cells, CD4 T cells, CD8 T cells, CD4 T-reg cells, and inflammasome activation are associated with HCC development. A full discussion of the mechanism of chronic inflammation, liver cirrhosis, and HCC falls outside the scope of this review but can be found elsewhere.270,271
Inflammation and HCC Connection
Inflammation is a critical component of liver cirrhosis and HCC development. It is unclear whether the HCV–induced inflammation prevents HCC development (tumor suppressor) or promotes HCC (oncogenic). For example, the presence of life long chronic inflammation with repeated cycles of hepatocyte death and regeneration could be a driver of liver cancer in chronic HCV infection. The inflammatory cells promote HCC by releasing ROS, RNS, lipid peroxidation, and aberrant expression of cytotoxic cytokines. Many inflammatory cytokines released during chronic inflammation such as TNF-α, IL-1β, IL-23, IL-6 are associated with HCC development.272–274 This hypothesis is supported with the study by Ramzan et al, which showed the presence of high-level liver-infiltrating T and B cells in the cirrhotic liver with HCC as compared to cirrhotic livers without HCC. In this case, the presence of CD8 T cells in the cirrhotic liver promotes HCC development supporting the pro-carcinogenic role of inflammation.275 On the other hand, lymphocytes infiltrating the liver can be a host response to the virus when the liver failed to clear the virus leading to chronic disease. The sustained accumulation of lymphocytes in the liver can be an unsuccessful host inflammatory response to HCC as well. In this case, the virus-targeted sustained immune activation, and cell death are an anti-tumorigenic response or tumor-suppressive inflammation. The removal of virus-associated immune surveillance by DAAs therapy promotes HCC development, suggesting that HCV-associated inflammation may be tumor suppressive. The presence of low CD8 T cells and a high population of suppressive T-reg cells correlate with HCC recurrence, suggesting that antitumor surveillance role of inflammatory cells inhibited by T-reg cells. In this case, the cirrhotic microenvironment favors recruitment and expansion of committed T-reg cells, thus establishing a state of immune tolerance, ultimately contributing to the evolution of cirrhosis into cancer. A decreased number of innate immunity members, NK and NKT cells, and increased accumulation of T-reg cells supporting the hypothesis that suppressive antitumor immunity correlates with HCC development in chronic HCV infection.276 The changes in the immune landscape after HCV cure lead to the reactivation of HBV and herpes virus infection, suggesting that HCV induced immune response may also suppress the growth of other microbial contamination.277,278 The contribution of inflammation in the development of HCC is unclear. Lack of an animal model remains a significant challenge to study the function of different immune components against HCC development related to chronic HCV infection.
Bystander Oncogenic Mechanisms
Overlapping Host-Related Non-Sterile Inflammation
The concomitant liver disease related to non-viral etiologies accelerates HCV–induced liver disease, cirrhosis development, and HCC.279–287 Obesity is associated with metabolic syndromes such as insulin resistance, type 2 diabetes, and NAFLD. A recent study using more than 900,000 US adults showed that the risk of death related to HCC was 2-fold higher in men with BMI of 30–34.9 and 4.5 times higher with BMI greater than 35.279 Obesity accelerates liver disease progression and HCC risk among patients after HCV cure.280 Individuals who have metabolic syndrome with obesity, plus diabetes with HCV contributes to increased risk for HCC development. Diabetes with HCV infection has a 2 to 3-fold increase in HCC risk.281–284 The researchers also showed that HCC development increased due to the combined effect of alcohol and chronic HCV infection.285–287 In addition to non-viral agents, co-infection with the hepatitis B virus or human immunodeficiency virus increases the risk of HCC after HCV cure. Patients who have active HBV replication have double risk of HCC development and increased mortality as compared to single virus infection.288 Likewise, patients with HIV infection have an increased risk of developing liver cirrhosis and HCC.289–291 The mechanism of chronic inflammation, liver cirrhosis, and HCC development in the non-viral insults are not different than HCV. ER stress regardless of the etiology of liver disease leads to increased inflammation, cell death, tissue-regeneration, fibrosis, and HCC (Figure 6).
 |
Figure 6 Sterile inflammation associated with host-related non-viral factors accelerates HCC progression during chronic HCV infection. Multiple host-related factors induce hepatic stress (ER stress) and low-grade inflammation in the liver. The most common causes of hepatic stress and inflammation include metabolic syndrome, type 2 diabetes, NAFLD associated with obesity and high-calorie diet, alcohol, gut microbiota, and autoimmune diseases. The bystander effect of inflammation associated with these non-viral causes can accelerate liver damage, persistent fibrosis, and HCC risk. |
Bystander HCC mechanisms through non-sterile inflammation:
The activation of oncogenes through point mutation and loss of tumor suppressor was thought to be a dominant mechanism for viral-induced cancer. The tumor suppressor loss and the oncogene activation are not consistent with data generated from the whole-genome sequencing of HCC tumors. This tumor centric-view is changing because HCC still develops in the absence of HCV after DAA treatment. The viable alternative hypothesis is that the cirrhotic microenvironment promotes HCC through bystander mechanisms. The recent development of microbiota studies suggesting that microbe alters the microenvironment that transform the surrounding cells through a bystander mechanism. Studies found that the metabolites derived from gut bacteria-infected cells enter the surrounding cells, induces DNA damage, and chromosomal instability that causes colon cancer.292–294 This new information has generated considerable interest in understanding whether HCV infection-associated cellular stress can promote cancer by promoting the survival of nearby uninfected cells through a bystander mechanism. A study by Kofahi et al has shown that HCV infection of cultured cells induces apoptosis and pyroptosis in both infected and uninfected bystander cells.295 The HCV–induced bystander apoptosis occurring in neighboring cells is cell-cell contact-dependent. Intracellular communication between cell-cell occurs through adhesion complexes, including adherens junctions, tight junctions, and gap junctions. Among those, intracellular communication mediated by gap junctions is vital for cell survival in various tissues.296 This observation suggests that GJIC plays an essential role in tissue homeostasis, and its downregulation causes many human diseases. In the liver, normal hepatocytes express Cx26 and Cx32, forming gap junction at the cell-cell contact areas.297,298 Altered expressions of these proteins occur in HCC tumors. One study shows that Cx26 expression abolished in HCC,299 and other studies show that Cx32 remained cytoplasm instead of the plasma membrane.300 Subsequent research indicates that the cytoplasmic Cx32 is critical for the expansion and self-renewal of cancer stem cells in hepatocellular carcinomas.301 The cytoplasmic expression of Cx32 is an integral part of cancer stem cells and high-grade malignancy.302 The radiation therapy is the most widely cited inducer of the bystander effect in cancer cells. Molecules such as ROS, RNS, protein factors, and DNA molecules can also utilize GJIP to spread from damaged cells to the surrounding healthy cells. The exosomal transfer of cargoes is implicated in the bystander cancer mechanism. The number of molecules, including TNF-α, TGF-β1, IL-6, IL-8, and nitric oxide are involved in cell-cell communications.303,304 Earlier studies showed that the overexpression of Cx32 reduces HCC growth.305 Cx32 knockout mice show increased susceptibility to chemical hepatocarcinogenesis.306–308 A similar mechanism triggered by the overlapping metabolic liver disease promoting HCC after viral cure needs to be determined.
Mechanisms of HCC Development After Viral Cure by DAAs
In this section, we review literature showing the impact of antiviral medicine on the long-term healing of liver inflammation and hepatic parenchymal injuries associated with chronic infection. We discuss some of the recent publications that have examined the benefits of viral cure at the level of cirrhosis regression, virus-induced cell programming, and indirect effect of a component of innate and adaptive response after HCV cured by DAAs.
Reversal of Cirrhosis After DAAs Treatment
Patients who have not developed cirrhosis at the time of DAAs treatment have a significantly lower risk of developing HCC as compared to those who have established cirrhosis.309 The status of cirrhosis after viral treatment is an essential predictor of HCC development. Cirrhosis is also a risk factor for increased mortality due to liver failure. The survival of patients with cirrhosis varies considerably based on the stage of the disease. For instance, one-year mortality for stage 1 fibrosis is 1%, stage 2 is 3%, stage 3 is 20%, and stage 4 is nearly 57%.310 Not all patients show cirrhosis regression after viral cure by IFN-based antiviral therapy. Previous studies have shown that SVR is associated with cirrhosis regression in nearly 55% of patients. A meta-analysis of a large number of multicenter clinical studies demonstrated that SVR by IFN-based antiviral therapy have a three-fold increased chance of cirrhosis regression that those who did not show SVR.310–316 Data on cirrhosis regression after HCV cure by DAAs is emerging based on the assessment of liver stiffness assessment by transient elastography.317–322 The available data suggest that viral cure by DAAs display significant benefits and cirrhosis regression. A study reported by Fehily et al summarizes that the DAA-induced HCV treatment improves clinical outcome and mortality associated with cirrhosis.323 Tacke et al study examined the baseline risk factors related to the success of DAAs treatment using 4946 chronic HCV patients. They found that obesity, diabetes, cirrhosis, and alcohol consumption are associated with persistent liver enzyme elevation post HCV cure.324 These data suggest that overlapping liver injury related to non-viral etiologies also contributes to persistent cirrhosis after HCV cure.
Reversal of Direct Mechanisms After DAAs Treatment
To understand the reason for increased risk of HCC after DAA-based antiviral treatment, the number of investigators started examining the cancer pathways either in liver and tumor samples after viral eradication. As discussed earlier, epigenetic changes can contribute to open chromatin, increase expression of the cancer-specific gene that leads to HCC development. Two studies have used chromatin immunoprecipitation and DNA sequence analysis from chronic HCV patients with or without DAAs treatment.325,326 Hamdane et al showed that the expression levels of 2 genes: sphingosine kinase 1 and SOX-2, a transcription factor, did not change after HCV cure. The expression levels of these two genes were examined in the samples of patients before and after HCV cure as well as HCV–induced HCC samples. These two genes are important in HCC development since the functional knockout of these two genes has been shown to inhibit HCC growth.325 A recent publication supports the data showing that SOX gene signatures are associated with the expression of liver cancer dedifferentiation markers.327 The development of an oncogenic signature relates to SOX expression. Perez et al also found that epigenetic signature was maintained after viral cure by DAAs, not by IFN treatment, therefore explaining as to why HCC is more frequent in DAA-treated group as compared to IFN. They identified Wnt10A, JUNB, FOSL2, MYCN, TNFAIP3, KLF4, and EDNA1 genes that are not reversed after DAAs. The study identified numerous genes related to cytoskeleton remodeling, endocytosis, virus release, and host cell cycle.326 Exosomes are major modulators of tumor microenvironment in cirrhosis as they carry numerous miRs implicated in cancer. Santangelo et al examined the impact of DAAs on exosomal miRs in plasma samples of chronic HCV patients. They have looked at miR-122 since it is involved in HCV replication, and miR-122 loss is associated with HCC development. They showed that miR-122-5p, miR-222-3p, miR-146-5p, miR-150-5p, miR-30C-5p, miR-378a-3p, miR-20a-5p were enriched in exosomes derived from HCV–infected cells. They found liver-specific miR-122 levels showed a significant decrease after DAAs therapy. They also found decreased expression of all these miRs mentioned above after DAAs.328 Koberle et al study also demonstrated that serum miR-122 levels remained low among patients who had SVR after DAAs.329 Villani et al study tested the hypothesis that DAAs treatment-induced VEGF expression promotes liver cancer angiogenesis. They performed an observational study using 117 cirrhotic patients who have been treated with DAAs to determine whether increased serum VEGF levels correlate with HCC development. They found serum VEGF levels increased after four weeks and remained elevated up to the end of treatment.330 Fallaci et al verified a similar hypothesis on HCC occurrence and recurrence among cirrhotic patients after DAAs. They showed that the VEGF expression was significantly related to serum angiopoietin-2 levels. They also showed that angiopoietin-2 expression in HCC and cirrhotic tissue before DAAs treatment relates to the risk of HCC recurrence and occurrence.331 Recently, we reported that HCC developed in the cirrhotic liver when the hepatic adaptive response reprogrammed to switch from pro-death to the pro-survival state through autophagy switching. In addition, our laboratory illustrated that virus-associated ER stress and tumor suppressor loss (p53 and Rb, miR-122 levels) were restored more by IFN-induced HCV clearance than DAAs in cell culture models.136,137,206 Based on these data, we propose that understanding the impact of viral cure on the resolution of direct hepatic parenchymal injuries and indirect immune restoration may explain as to why specific individuals remain at risk of HCC occurrence and recurrence.
DAAs Treatment on Innate Immunity
In the following section, we will review data correlating with the impact of HCV cure on the reversal of immune mechanism that could explain the possible reason of HCC recurrence after HCV cure. Meissner et al examined the impact of HCV clearance by DAAs on endogenous intrahepatic IFNs. They compared the expression level of type I, type II, and Type III IFN and ISGs before and after HCV treatment. The study showed that HCV clearance resulted in decreased expression of type II and type III IFNs, their receptors, and ISG in the liver. Unexpectedly, HCV cure by DAAs did not reduce the appearance of type I IFN signaling, IFNA2 gene expression remained high among those who achieved SVR. The study concluded that activation of type I IFN signaling is important for HCV elimination.332 A study by Alao et al showed that patients who achieved SVR at 12 weeks after treatment displayed higher ISG expression levels in baseline liver biopsies and a higher frequency of pSTAT1 and TRAIL-expressing, degranulating NK cells in baseline blood samples than those who experienced a virological breakthrough. The downregulation of ISGs was rapid after HCV RNA suppression by DAAs.333 Another study by Sung et al showed that the expression levels of IFN-β, IFN-induced protein 44, and CXCL10 cytokine levels decreased and normalized after DAAs treatment in the peripheral blood mononuclear cells. The conclusion of this study was that DAAs treatment normalizes type I IFN response.334 Debes et al study measured serum cytokines levels among 13 patients who developed HCC after DAAs. Among 22 different cytokines, nine of them (MIG, IL-22, TRAIL, APRIL, VEGF, IL-3, TWEAK, SCF, and IL-21) presented significantly higher in serum before treatment, which eventually developed HCC.335 These studies now provide information regarding that DAA-induced HCV clearance does not restore the innate immune surveillance completely. NK cells are an essential component of the innate immune response in HCV infection. People who clear HCV infection naturally show increased expression of the activating receptors (NKp44 and NKp46) and decreased expression of the inhibitory receptors NKG2A associated with strong T cell response.336 Golden-Mason et al reported that rapid viral clearance by DAAs results in normalization of NK cell function, reduced cytotoxicity and downregulation of cytotoxic NK cell signaling by TRAIL, NKp30, and NKp46.337 Spaan et al showed downregulation in TRAIL-mediated killing by NK cells during DAAs therapy.338 A report by Stevenson et al demonstrated that DAAs therapy normalized NK cell function, and decreased expression of CXCL-10, CXCL-11 levels associated with NK cell activation and function suggesting functional restoration of NK cells after viral cure by DAAs.339 However, some researchers claim that rapid decrease or normalized immunosurveillance causes early HCC occurrence after DAAs. The activating receptor NKG2D and its ligands play a crucial role in the immune response to HCC. Two studies found that reduced NKG2D and ligand expression in HCC correlates with HCC recurrence and early occurrence.340,341 MICA, which is one of the ligands for the NKG2D and their interaction, is vital for NK-cell cytotoxic effect for HCC cells. HCC sheds membrane-bound MICA as soluble MICA and downregulates the expression of NKG2D on the NK cell surface, explaining a mechanism of how HCC escape NK-cell mediated immune surveillance.342,343
DCs play a critical role in sensing virus infection through pattern recognition receptors. This process coordinates the innate and adaptive immune response to HCV infection. Several reports claim that the impairment of MDC function and production of cytokines (IL-12, TNF-α, IFN-α/IFN-β) in chronic HCV infection.344–346 Laursen et al examined soluble CD163 released from activated liver macrophages in chronic HCV, and histological activity after DAAs. They found that serum CD163 levels decline rapidly after successful DAAs therapy and are associated with histological inflammatory activity and fibrosis.347 Kostadinova et al evaluated the serum level of ALT with the plasma level of sCD14, sCD163, autotaxin, and Mac2BP with HCV treatment response to DAAs. They found levels of these immune activation markers declined and normalized after DAAs. AST level was well correlated with sCD163, which is a KC activation marker.348 MAIT cells are innate-like lymphocytes that are activated in chronic HCV infection due to the synergistic action of IL-18, IL-12, IL-15, and IFN-α/IFN-β that trigger granzyme B release. MAIT cell number is significantly decreased and inversely correlates with liver inflammation and fibrosis.349 One report claims that DAA-induced HCV clearance does not restore MAIT cell defects, which seems different than IFN-based antiviral therapy.350 Cannizzo et al also found that MAIT cell function is not restored after DAAs in HIV/HCV co-infected patients.351
DAAs Treatment and Adaptive Immunity
Cytotoxic T cell response is restored when someone recovers from HCV infection naturally, which is characterized by the appearance of HCV-specific T cells expressing IL-7 receptor alpha (CD127), a marker of memory T cells and anti-apoptotic molecule Bcl-2.352 Chronic HCV infection results in T cell exhaustion and the loss of effector T cell function with the eventual disappearance of the majority of HCV-specific CD8 T cells from the blood and liver. Burchill et al examined the composition of CD8 T cells in the peripheral blood from nineteen HCV patients treated with DAAs. They found that HCV clearance leads to a rapid increase in the frequency of HCV-specific CD8 T cells. In contrast, a significant reduction of the PD-1 on HCV-specific T cells was observed, suggesting the partial restoration in the HCV-specific CD8 T cell exhaustion state.353 Weiland et al reported that TCF1+CD127+PD-1+ HCV specific T cells expressing both exhaustion and memory markers were found in subjects with chronic HCV infection, and those T cell subsets levels were found to remain unchanged during and after DAAs treatment. This subset was increased in one patient who relapsed and differentiated into terminally exhausted HCV-specific CD8 T cells. These cells express higher levels of eomes and produce low IFN-γ and TNF-α upon antigen challenge. These CD8 T cells were distinct from memory CD8 T cells (CD127+ PD-1-) that appear during spontaneous resolution.354 Aregay et al studied whether HCV cure by DAAs could restore the functionality of exhausted HCV-specific CD8T cell response. Their data showed that the exhausted HCV-specific CD8 T cells remain functionally impaired after HCV cure in all 40 patients treated with DAAs.355 Vranjkovic et al evaluated the function of circulating CD8 T cells in HCV-related cirrhotic patients. They found a progressive shift in CD8 T cells subset distribution in all cirrhotic patients even after HCV cure. The authors concluded that the severity of liver fibrosis in these patients relates to the presence of hyperfunction CD8 T cells in the liver.356 All these results suggest DAA-induced HCV cure does not restore the exhausted phenotype of HCV-specific CD8 T cell response.
CD4 helper T cells are essential for mounting appropriate HCV-specific CD8 T cell response and are needed for natural clearance of infection.357,358 Intrahepatic regulatory CD4 T cells and CD4(+)CD25(+)FoxP3(+) T-reg cells are expanded in the blood and liver of patients with chronic HCV infection, and they are not decreased after viral cure by IFN-based or DAA-based therapy.359,360 Langhans et al analyzed T-reg cells before and after DAAs at 12 and 24 weeks. They found that DAAs do not normalize the activation status of T-reg cells, and these cells persist for a long period after viral elimination.361 One recent study showed that gamma delta T cell dysfunction in chronic HCV infection is not reversed after HCV cure by DAAs, indicating this functional defect may have implications for susceptibility to HCC.362 Burchill et al showed that DAAs therapy results in (i) the reconstitution of lymphocyte populations in the peripheral blood; (ii) re‐differentiation of memory T cells towards an effector phenotype; (iii) a reduction in the expression of some, but not all inhibitory molecules on bulk and viral‐specific CD8 T cell populations; and (iv) a reduction in the activation status of circulating NK cells. B cells are the source of humoral anti-HCV antibodies that are important in resolving HCV infection.353 Although the human immune response is not the sole contributor to HCV resolution, activation of B cell response is associated with B cell-related abnormalities such as mixed cryoglobulins among 40–60% patients and CV in 5–10% patients. Comarmond et al tested the impact of DAAs treatment on HCV-related CV of 27 patients. They reported that about 88.9% of patients show the complete clinical response of CV after DAAs and restored disturbances in peripheral B cell homeostasis.363 Saadoun et al examined the remission of vasculitis after the DAAs treatment of 41 patients, and they reported 37 patients (90.2%) showed complete clinical response, which was associated with virological response.364 A study by Chang et al demonstrated that resolution of HCV infection with DAAs leads to a marked reduction in the frequency of T-bet+ B cells. Re-exposure of convalescent (cured) B cells to viremic plasma and recombinant HCV E2 protein led to the re-expression of T-bet. This study posited that the antigen-driven T-bet expression, a critical suppressor of B cell activation in chronic HCV infection, is reversed after DAAs therapy.365
Development of Accurate Biomarker for Cirrhosis Regression and Early HCC Detection
The current DAAs therapy is inducing an incredibly high rate of HCV clearance in most patients, including those with liver cirrhosis. It is expected that the cured patient population will increase in the future, and many of these cured patients will have overlapping non-viral liver diseases. There is substantial evidence showing liver injury persists after HCV cure among cirrhotic patients. These patients remain at risk of persistent cirrhosis and HCC development. The regression of liver fibrosis is a slow process after HCV treatment. Some cirrhotic patients who have underlying liver disease related to alcohol, type 2 diabetes, and obesity are still at risk of HCC development. Screening for HCC is recommended among patients who have cirrhosis at the time of DAAs treatment. There remains a clinical challenge to develop accurate tests for persistent risk of HCC in the early stratification of these patients. Liver biopsies are no longer performed to diagnose cirrhosis. The assessment of liver cirrhosis is now performed by noninvasive approaches, including liver imaging (hepatic elastography/fibroscan) and serum biomarkers.366 Two serum fibrosis scores, namely fibrosis 4 (FIB-4) and the AST to platelet ratio index (APRI), have been widely used in staging liver fibrosis.367–369 The only currently accepted HCC tumor biomarker, AFP is a 70-kDa-serum glycoprotein produced by fetal liver during development. The serum levels of AFP are increased during HCC. AFP is present in three different forms based on the lectin-binding pattern: AFP-L1 AFP-L2 and AFP-L3. Serum levels of AFP-L1 increased during chronic hepatitis and cirrhosis whereas AFP-L3 levels increased during HCC. The AFP generally has suboptimal sensitivity and specificity for HCC surveillance.370 This protein can be detected in ovarian, pancreatic, stomach and testicular cancers as well. Des-γ-carboxyprothrombin is an abnormal form of prothrombin induced by vitamin K absence-II that serum levels are elevated in patients with HCC. In addition to these serum proteins, ultrasound-based imaging is also recommended for HCC screening. Recently, computed tomography scanning and magnetic resonance imaging have shown to have better sensitivity for HCC screening. As discussed in this review, there are many potential targets that can be explored in the development of a serum biomarker for early HCC. Since the current method still lacks sensitivity for early prediction of neoplastic growth in liver, its is necessary to develop a novel biomarker that accurately measures the hepatic adaptive response to virus-associated cellular stress, immune cell activation, and liver injury.
Development of Protective Vaccine
Although the DAA-based antiviral treatment is inducing a very high rate of viral cure, the number of new HCV infection rates is increasing worldwide. Most of the new infections remain undiagnosed because both acute and chronic HCV infections are primarily asymptomatic. In the US, only 50% of infected people are aware of their HCV status.371 The risk of reinfection after DAAs treatment remains a major problem especially among intravenous drug users.372,373 The current DAA-induced viral clearance does not provide protection for reinfection or reversal of liver disease-related complications. A prophylactic vaccine is needed since DAAs treatment is unaffordable in many developing nations. Protective immunity to HCV reinfection requires expansion of a broad and polyclonal memory of CD4 T and CD8T cell response374,375 as well as generation of neutralizing antibodies.376 There are a number of challenges that exist for the development of a multivalent vaccine for HCV, including the extraordinary diversity of the virus, seven genotypes with more than 80 subtypes, viral variants associated with resistance to T cell and antibody response. At present, there are two main vaccine candidates moving for clinical trials. The first vaccine was based on purified recombinant proteins targeting the viral envelope glycoproteins called E1E2 mixed with MF59 adjuvant (an oil-in-water emulsion). The initial study shows that this formulation induced neutralizing antibody responses in chimpanzee.377,378 Preclinical evaluation of this vaccine in human volunteers showed that it generates a neutralizing antibody response as well as CD4 T cell response against viral envelope proteins E1 and E2.378,379 A second vaccine was developed based on the use of recombinant replication-defective adenovirus containing the entire non-structural proteins.380,381 The second generation of this vaccine approach includes heterologous priming first with the Chimpanzee Adenovirus 3 Nonstructural (AdCh3NS), followed by boosting with another Modified Vaccinia Ankara NS (MVA NS) vaccine. This approach appears to be generating a broad and strong T cell response. A recent study in human volunteers shows that optimal priming and boosting of these vaccine generate high frequencies of polyfunctional, elicited broad CD8 T cells response, IFN-γ production, memory cell development, and durable maintenance of HCV-specific T cell response.382 A Phase II clinical trial of this vaccine is underway in a cohort of HCV–infected intravenous drug users (clinicaltrails.gov, NCT01436357). Vaccines are available for hepatitis A and hepatitis B. Availability of vaccine for HCV will be a step forward for global immunization strategies for hepatitis viruses.
Summary and Conclusions
HCV infection is one of the major causes of cirrhosis and HCC worldwide and is associated with high mortality. It is expected that DAA-based antiviral treatment will reduce the progressive liver disease and liver-related mortality associated with HCV infection. However, the risk of HCC persists after HCV treatment among cirrhotic patients. The population of HCV-cured cirrhotic patients is expected to increase. These patients remain at risk for liver disease progression due to non-viral risk factors including obesity, alcohol, diabetes, and other autoimmune diseases. Fatty liver is a particular threat to the long-term healing of cirrhotic patients after HCV cure. These patients need to remain under surveillance for liver complications, including HCC. Since cirrhosis regression is a slow process, the long-term benefits of viral cure among cirrhotic patients are uncertain. This review provided molecular basis on how hepatic pathological adaption to viral insults and the immunological mechanisms associated with development of cirrhosis and HCC occurs during chronic HCV infection. The fibrosis regression and restoration of hepatic injury are important for reduction of HCC risk. The development of novel biomarkers to evaluate the extent of immune restoration and reduction of cellular injuries is needed for early prediction of persistent cirrhosis and HCC. The current DAA-induced viral elimination does not provide immune protection for reinfection among intravenous drug users. The prevention of HCV transmission and reinfection through vaccination is an important aspect of post-cure management and a key to the global elimination of HCV infection.
Abbreviations
AFP, alpha-fetoprotein; ALT, alanine aminotransferase; APC, antigen-presenting cells; AST, aspartate aminotransferase; BMI, body mass index; CMA, chaperone-mediated autophagy; CSC, cancer stem cell; DAA, direct-acting antiviral; DAMP, damage-associated molecular pattern; DMT, DNA methyltransferase; EGFR, epidermal growth factor receptor; ER, endoplasmic reticulum; ERK, extracellular signal-regulated kinase; FGF, fibroblast growth factor; GJIC, gap junctional intracellular communication; GSK3β, glycogen synthase kinase 3β; HAT, histone acetyl transferase; HBV, hepatitis B virus; HCC, hepatocellular carcinoma; HCV, hepatitis C virus; HDAC, histone deacetylase; HIF-1α, hypoxia-inducible factor 1 alpha; HNF4α, hepatocyte nuclear factor 4 alpha; IFN, interferon; IL, interleukin; ISR, integrated stress response; MDM2, mouse double minute 2; MEK, mitogen-activated kinase; miR, microRNA; mTOR, mammalian target of rapamycin; NAFLD, non-alcoholic fatty liver disease; ncRNA, non-coding RNA; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; NRF2, nuclear factor erythroid 2-related factor 2; PAMP, pathogen-associated molecular pattern; PDGF, platelet-derived growth factor; PI3K, phosphoinositide 3-kinase; PRC, polycomb repressive protein complex; RTK, receptor tyrosine kinase; Rb, retinoblastoma protein; RIG-1, retinoic acid-inducible gene-1 receptor; RBV, ribavirin; RNS, reactive nitrogen species; ROS, reactive oxygen species; SOX, sex determining region Y-box; SVR, sustained virological response; TERT, telomerase reverse transcriptase; TLR, toll-like receptor; TGF, transforming growth factor; VEGF, vascular endothelial growth factor.
Acknowledgments
This work was supported by Veterans Affairs Merit Review Grant: 1I01BX004516-01A1, 1P20GM121288 and Louisiana Clinical and Translational Science (LACaTS) Center Grant: U54 GM104940. We acknowledge our students; Venu Reddy and Adriana Lopez for editing this review. We also acknowledge our collaborators who have supplied reagents and supported our research in the hepatitis research laboratory at Tulane University Health Sciences Center.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Sayiner M, Golabi P, Younossi ZM. Disease burden of hepatocellular carcinoma: a global perspective. Dig Dis Sci. 2019;64(4):910–917. doi:10.1007/s10620-019-05537-2
2. Naugler WE, Sakurai T, Kim S, et al. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science. 2007;317(5834):121–124. doi:10.1126/science.1140485
3. Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136(5):E359–E386. doi:10.1002/ijc.29210
4. Ryerson AB, Eheman CR, Altekruse SF, et al. Annual report to the nation on the status of cancer, 1975–2012, featuring the increasing incidence of liver cancer. Cancer. 2016;122(9):1312–1337. doi:10.1002/cncr.v122.9
5. Fattovich G, Stroffolini T, Zagni I, Donato F. Hepatocellular carcinoma in cirrhosis: incidence and risk factors. Gastroenterology. 2004;127(5 Suppl 1):S35–S50. doi:10.1053/j.gastro.2004.09.014
6. El-Serag HB. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology. 2012;142(6):1264. doi:10.1053/j.gastro.2011.12.061
7. Beste LA, Leipertz SL, Green PK, Dominitz JA, Ross D, Ioannou GN. Trends in burden of cirrhosis and hepatocellular carcinoma by underlying liver disease in US veterans, 2001–2013. Gastroenterology. 2015;149(6):1471. doi:10.1053/j.gastro.2015.07.056
8. D’Ambrosio R, Della Corte C, Colombo M. Hepatocellular carcinoma in patients with a sustained response to anti-hepatitis C therapy. Int J Mol Sci. 2015;16(8):19698–19712. doi:10.3390/ijms160819698
9. Strader DB, Wright T, Thomas DL, Seeff LB, American Association for the Study of Liver D. Diagnosis, management, and treatment of hepatitis C. Hepatology. 2004;39(4):1147–1171. doi:10.1002/hep.20119
10. Roche B, Coilly A, Duclos-Vallee JC, Samuel D. The impact of treatment of hepatitis C with DAAs on the occurrence of HCC. Liver Int. 2018;38(Suppl 1):139–145. doi:10.1111/liv.13659
11. Gower E, Estes C, Blach S, Razavi-Shearer K, Razavi H. Global epidemiology and genotype distribution of the hepatitis C virus infection. J Hepatol. 2014;61(1 Suppl):S45–S57. doi:10.1016/j.jhep.2014.07.027
12. El-Serag HB. Hepatocellular carcinoma. N Engl J Med. 2011;365(12):1118–1127. doi:10.1056/NEJMra1001683
13. Bruix J, Sherman M, American Association for the Study of Liver D. Management of hepatocellular carcinoma: an update. Hepatology. 2011;53(3):1020–1022. doi:10.1002/hep.24199
14. Bralet MP, Regimbeau JM, Pineau P, et al. Hepatocellular carcinoma occurring in nonfibrotic liver: epidemiologic and histopathologic analysis of 80 French cases. Hepatology. 2000;32(2):200–204. doi:10.1053/jhep.2000.9033
15. Bullock RE, Zaitoun AM, Aithal GP, Ryder SD, Beckingham IJ, Lobo DN. Association of non-alcoholic steatohepatitis without significant fibrosis with hepatocellular carcinoma. J Hepatol. 2004;41(4):685–686. doi:10.1016/j.jhep.2004.05.008
16. Guzman G, Brunt EM, Petrovic LM, Chejfec G, Layden TJ, Cotler SJ. Does nonalcoholic fatty liver disease predispose patients to hepatocellular carcinoma in the absence of cirrhosis? Arch Pathol Lab Med. 2008;132(11):1761–1766.
17. Paradis V, Zalinski S, Chelbi E, et al. Hepatocellular carcinomas in patients with metabolic syndrome often develop without significant liver fibrosis: a pathological analysis. Hepatology. 2009;49(3):851–859. doi:10.1002/hep.22734
18. Desai A, Sandhu S, Lai JP, Sandhu DS. Hepatocellular carcinoma in non-cirrhotic liver: a comprehensive review. World J Hepatol. 2019;11(1):1–18. doi:10.4254/wjh.v11.i1.1
19. Lok AS, Everhart JE, Wright EC, et al. Maintenance peginterferon therapy and other factors associated with hepatocellular carcinoma in patients with advanced hepatitis C. Gastroenterology. 2011;140(3):840–U230. doi:10.1053/j.gastro.2010.11.050
20. Gawrieh S, Dakhoul L, Miller E, et al. Characteristics, aetiologies and trends of hepatocellular carcinoma in patients without cirrhosis: a United States multicentre study. Aliment Pharmacol Ther. 2019;50(7):809–821. doi:10.1111/apt.v50.7
21. Hoofnagle JH. A step forward in therapy for hepatitis C. New Engl J Med. 2009;360(18):1899–1901. doi:10.1056/NEJMe0901869
22. Fried MW, Shiffman ML, Reddy KR, et al. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. New Engl J Med. 2002;347(13):975–982. doi:10.1056/NEJMoa020047
23. Lange CM, Zeuzem S. IL28B single nucleotide polymorphisms in the treatment of hepatitis C. J Hepatol. 2011;55(3):692–701. doi:10.1016/j.jhep.2011.03.006
24. Scheel TK, Rice CM. Understanding the hepatitis C virus life cycle paves the way for highly effective therapies. Nat Med. 2013;19(7):837–849. doi:10.1038/nm.3248
25. Pawlotsky JM, Feld JJ, Zeuzem S, Hoofnagle JH. From non-A, non-B hepatitis to hepatitis C virus cure. J Hepatol. 2015;62(1 Suppl):S87–S99. doi:10.1016/j.jhep.2015.02.006
26. Lombardi A, Mondelli MU, Hepatitis ESGV, Hepatitis C. Is eradication possible? Liver Int. 2019;39(3):416–426. doi:10.1111/liv.14011
27. van der Meer AJ, Veldt BJ, Feld JJ, et al. Association between sustained virological response and all-cause mortality among patients with chronic hepatitis C and advanced hepatic fibrosis. J Am Med Assoc. 2012;308(24):2584–2593. doi:10.1001/jama.2012.144878
28. Thomas DL. Global control of hepatitis C: where challenge meets opportunity. Nat Med. 2013;19(7):850–858. doi:10.1038/nm.3184
29. Flemming JA, Kim WR, Brosgart CL, Terrault NA. Reduction in liver transplant wait-listing in the era of direct-acting antiviral therapy. Hepatology. 2017;65(3):804–812. doi:10.1002/hep.v65.3
30. Lee YA, Friedman SL. Reversal, maintenance or progression: what happens to the liver after a virologic cure of hepatitis C? Antivir Res. 2014;107:23–30. doi:10.1016/j.antiviral.2014.03.012
31. Dang H, Yeo YH, Yasuda S, et al. Cure with interferon free DAA is associated with increased survival in patients with HCV related HCC from both East and West. Hepatology. 2019. doi:10.1002/hep.30988
32. Salmon D, Mondelli MU, Maticic M, Arends JE. The benefits of hepatitis C virus cure: every rose has thorns. J Viral Hepat. 2018;25(4):320–328. doi:10.1111/jvh.2018.25.issue-4
33. Mangia A, Piazzolla V, Giannelli A, et al. SVR12 rates higher than 99% after sofosbuvir/velpatasvir combination in HCV infected patients with F0-F1 fibrosis stage: a real world experience. PLoS One. 2019;14(5):e0215783.
34. Young K, Liu B, Bhuket T, Gish RG, Wong RJ. Improved liver transplant waitlist mortality and lower risk of disease progression among chronic hepatitis C patients awaiting liver transplantation after the introduction of direct-acting antiviral therapies in the United States. J Viral Hepatitis. 2019;26(3):350–361. doi:10.1111/jvh.2019.26.issue-3
35. Lledo GM, Carrasco I, Benitez-Gutierrez LM, et al. Regression of liver fibrosis after curing chronic hepatitis C with oral antivirals in patients with and without HIV coinfection. Aids. 2018;32(16):2347–2352. doi:10.1097/QAD.0000000000001966
36. Bonacci M, Lens S, Marino Z, et al. Long-term outcomes of patients with HCV-associated cryoglobulinemic vasculitis after virologic cure. Gastroenterology. 2018;155(2):311. doi:10.1053/j.gastro.2018.04.024
37. Mandorfer M, Kozbial K, Schwabl P, et al. Sustained virologic response to interferon-free therapies ameliorates HCV-induced portal hypertension. J Hepatol. 2016;65(4):692–699. doi:10.1016/j.jhep.2016.05.027
38. Nahon P, Bourcier V, Layese R, et al. Eradication of hepatitis C virus infection in patients with cirrhosis reduces risk of liver and non-liver complications. Gastroenterology. 2017;152(1):142. doi:10.1053/j.gastro.2016.09.009
39. Park H, Wang W, Henry L, Nelson DR. Impact of all-oral direct-acting antivirals on clinical and economic outcomes in patients with chronic hepatitis C in the United States. Hepatology. 2019;69(3):1032–1045. doi:10.1002/hep.v69.3
40. Pons M, Rodriguez-Tajes S, Esteban JI, et al. Non-invasive prediction of liver-related events in patients with HCV-associated compensated advanced chronic liver disease after oral antivirals. J Hepatol. 2019;72:472–480.
41. Ioannou GN, Beste LA, Green PK, et al. Increased risk for hepatocellular carcinoma persists up to 10 years after HCV eradication in patients with baseline cirrhosis or high FIB-4 scores. Gastroenterology. 2019;157(5):1264–1278 e1264. doi:10.1053/j.gastro.2019.07.033
42. Nakano M, Koga H, Ide T, et al. Predictors of hepatocellular carcinoma recurrence associated with the use of direct-acting antiviral agent therapy for hepatitis C virus after curative treatment: a prospective multicenter cohort study. Cancer Med-Us. 2019;8(5):2646–2653. doi:10.1002/cam4.2061
43. Pinero F, Mendizabal M, Ridruejo E, et al. Treatment with direct-acting antivirals for HCV decreases but does not eliminate the risk of hepatocellular carcinoma. Liver Int. 2019;39(6):1033–1043. doi:10.1111/liv.2019.39.issue-6
44. Singh S, Nautiyal A, Loke YK. Oral direct-acting antivirals and the incidence or recurrence of hepatocellular carcinoma: a systematic review and meta-analysis. Frontline Gastroente. 2018;9(4):262–270. doi:10.1136/flgastro-2018-101017
45. Nahon P, Layese R, Bourcier V, et al. Incidence of hepatocellular carcinoma after direct antiviral therapy for HCV in patients with cirrhosis included in surveillance programs. Gastroenterology. 2018;155(5):1436. doi:10.1053/j.gastro.2018.07.015
46. Rein DB, Wittenborn JS, Weinbaum CM, Sabin M, Smith BD, Lesesne SB. Forecasting the morbidity and mortality associated with prevalent cases of pre-cirrhotic chronic hepatitis C in the United States. Digest Liver Dis. 2011;43(1):66–72. doi:10.1016/j.dld.2010.05.006
47. Nishiguchi S, Kuroki T, Nakatani S, et al. Randomised trial of effects of interferon-alpha on incidence of hepatocellular carcinoma in chronic active hepatitis C with cirrhosis. Lancet. 1995;346(8982):1051–1055. doi:10.1016/S0140-6736(95)91739-X
48. Singal AK, Singh A, Jaganmohan S, et al. Antiviral therapy reduces risk of hepatocellular carcinoma in patients with hepatitis C virus-related cirrhosis. Clin Gastroenterol Hepatol. 2010;8(2):192–199. doi:10.1016/j.cgh.2009.10.026
49. Lok AS, Seeff LB, Morgan TR, et al. Incidence of hepatocellular carcinoma and associated risk factors in hepatitis C-related advanced liver disease. Gastroenterology. 2009;136(1):138–148. doi:10.1053/j.gastro.2008.09.014
50. El-Serag HB, Kanwal F, Richardson P, Kramer J. Risk of hepatocellular carcinoma after sustained virological response in Veterans with hepatitis C virus infection. Hepatology. 2016;64(1):130–137. doi:10.1002/hep.v64.1
51. Singal AK, Freeman DH, Anand BS. Meta-analysis: interferon improves outcomes following ablation or resection of hepatocellular carcinoma. Aliment Pharmacol Ther. 2010;32(7):851–858. doi:10.1111/j.1365-2036.2010.04414.x
52. Bang CS, Song IH. Impact of antiviral therapy on hepatocellular carcinoma and mortality in patients with chronic hepatitis C: systematic review and meta-analysis. BMC Gastroenterol. 2017;17(1):46. doi:10.1186/s12876-017-0606-9
53. Manthravadi S, Paleti S, Pandya P. Impact of sustained viral response postcurative therapy of hepatitis C-related hepatocellular carcinoma: a systematic review and meta-analysis. Int J Cancer. 2017;140(5):1042–1049. doi:10.1002/ijc.30521
54. Bruno S, Di Marco V, Iavarone M, et al. Survival of patients with HCV cirrhosis and sustained virologic response is similar to the general population. J Hepatol. 2016;64(6):1217–1223. doi:10.1016/j.jhep.2016.01.034
55. Morgan RL, Baack B, Smith BD, Yartel A, Pitasi M, Falck-Ytter Y. Eradication of hepatitis C virus infection and the development of hepatocellular carcinoma a meta-analysis of observational studies. Ann Intern Med. 2013;158(5):329. doi:10.7326/0003-4819-158-5-201303050-00005
56. Rutledge SM, Zheng H, Li DK, Chung RT. No evidence for higher rates of hepatocellular carcinoma after direct-acting antiviral treatment: a meta-analysis. Hepatoma Res. 2019;5:31.
57. Waziry R, Hajarizadeh B, Grebely J, et al. Hepatocellular carcinoma risk following direct-acting antiviral HCV therapy: a systematic review, meta-analyses, and meta-regression. J Hepatol. 2017;67(6):1204–1212. doi:10.1016/j.jhep.2017.07.025
58. Conti F, Buonfiglioli F, Scuteri A, et al. Early occurrence and recurrence of hepatocellular carcinoma in HCV-related cirrhosis treated with direct-acting antivirals. J Hepatol. 2016;65(4):727–733. doi:10.1016/j.jhep.2016.06.015
59. Ravi S, Axley P, Jones D, et al. Unusually high rates of hepatocellular carcinoma after treatment with direct-acting antiviral therapy for hepatitis C related cirrhosis. Gastroenterology. 2017;152(4):911–912. doi:10.1053/j.gastro.2016.12.021
60. Calvaruso V, Cabibbo G, Cacciola I, et al. Incidence of hepatocellular carcinoma in patients with HCV-associated cirrhosis treated with direct-acting antiviral agents. Gastroenterology. 2018;155(2):411–421 e414. doi:10.1053/j.gastro.2018.04.008
61. Cheung MCM, Walker AJ, Hudson BE, et al. Outcomes after successful direct-acting antiviral therapy for patients with chronic hepatitis C and decompensated cirrhosis. J Hepatol. 2016;65(4):741–747. doi:10.1016/j.jhep.2016.06.019
62. Llovet JM, Villanueva A. Liver cancer: effect of HCV clearance with direct-acting antiviral agents on HCC. Nat Rev Gastroenterol Hepatol. 2016;13(10):561–562. doi:10.1038/nrgastro.2016.140
63. Ioannou GN, Green PK, Berry K. HCV eradication induced by direct-acting antiviral agents reduces the risk of hepatocellular carcinoma. J Hepatol. 2017. doi:10.1016/j.jhep.2017.08.030
64. Kanwal F, Kramer J, Asch SM, Chayanupatkul M, Cao Y, El-Serag HB. Risk of hepatocellular cancer in HCV patients treated with direct-acting antiviral agents. Gastroenterology. 2017;153(4):996–1005 e1001. doi:10.1053/j.gastro.2017.06.012
65. Nahon P, Layese R, Bourcier V, et al. Incidence of hepatocellular carcinoma after direct antiviral therapy for HCV in patients with cirrhosis included in surveillance programs. Gastroenterology. 2018;155(5):1436–1450 e1436. doi:10.1053/j.gastro.2018.07.015
66. Singer AW, Reddy KR, Telep LE, et al. Direct-acting antiviral treatment for hepatitis C virus infection and risk of incident liver cancer: a retrospective cohort study. Aliment Pharmacol Ther. 2018;47(9):1278–1287. doi:10.1111/apt.2018.47.issue-9
67. Li DK, Ren Y, Fierer DS, et al. The short-term incidence of hepatocellular carcinoma is not increased after hepatitis C treatment with direct-acting antivirals: an ERCHIVES study. Hepatology. 2018;67(6):2244–2253. doi:10.1002/hep.29707
68. Kozbial K, Moser S, Schwarzer R, et al. Unexpected high incidence of hepatocellular carcinoma in cirrhotic patients with sustained virologic response following interferon-free direct-acting antiviral treatment. J Hepatol. 2016;65(4):856–858. doi:10.1016/j.jhep.2016.06.009
69. Nakao Y, Hashimoto S, Abiru S, et al. Rapidly growing, moderately differentiated HCC: a clinicopathological characteristic of HCC occurrence after IFN-free DAA therapy? J Hepatol. 2018;68(4):854–855. doi:10.1016/j.jhep.2017.11.011
70. Renzulli M, Buonfiglioli F, Conti F, et al. Imaging features of microvascular invasion in hepatocellular carcinoma developed after direct-acting antiviral therapy in HCV-related cirrhosis. Eur Radiol. 2018;28(2):506–513. doi:10.1007/s00330-017-5033-3
71. Reig M, Marino Z, Perello C, et al. Unexpected high rate of early tumor recurrence in patients with HCV-related HCC undergoing interferon-free therapy. J Hepatol. 2016;65(4):719–726. doi:10.1016/j.jhep.2016.04.008
72. Yang JD, Aqel BA, Pungpapong S, Gores GJ, Roberts LR, Leise MD. Direct acting antiviral therapy and tumor recurrence after liver transplantation for hepatitis C-associated hepatocellular carcinoma. J Hepatol. 2016;65(4):859–860. doi:10.1016/j.jhep.2016.06.023
73. Cabibbo G, Petta S, Calvaruso V, et al. Is early recurrence of hepatocellular carcinoma in HCV cirrhotic patients affected by treatment with direct-acting antivirals? A prospective multicentre study. Aliment Pharmacol Ther. 2017;46(7):688–695. doi:10.1111/apt.14256
74. [email protected] AcsgohcEa. Lack of evidence of an effect of direct-acting antivirals on the recurrence of hepatocellular carcinoma: data from three ANRS cohorts. J Hepatol. 2016;65(4):734–740. doi:10.1016/j.jhep.2016.05.045
75. Nagata H, Nakagawa M, Asahina Y, et al. Effect of interferon-based and -free therapy on early occurrence and recurrence of hepatocellular carcinoma in chronic hepatitis C. J Hepatol. 2017;67(5):933–939. doi:10.1016/j.jhep.2017.05.028
76. Huang AC, Mehta N, Dodge JL, Yao FY, Terrault NA. Direct-acting antivirals do not increase the risk of hepatocellular carcinoma recurrence after local-regional therapy or liver transplant waitlist dropout. Hepatology. 2018;68(2):449–461. doi:10.1002/hep.29855
77. Prenner SB, VanWagner LB, Flamm SL, Salem R, Lewandowski RJ, Kulik L. Hepatocellular carcinoma decreases the chance of successful hepatitis C virus therapy with direct-acting antivirals. J Hepatol. 2017;66(6):1173–1181. doi:10.1016/j.jhep.2017.01.020
78. Beste LA, Green PK, Berry K, Kogut MJ, Allison SK, Ioannou GN. Effectiveness of hepatitis C antiviral treatment in a USA cohort of veteran patients with hepatocellular carcinoma. J Hepatol. 2017;67(1):32–39. doi:10.1016/j.jhep.2017.02.027
79. Radhakrishnan K, Di Bisceglie AM, Reddy KR, et al. Impact of hepatocellular carcinoma (HCC) and tumor treatment on sustained virologic response (SVR) rates with direct-acting antiviral (DAA) therapy for hepatitis C: HCV-TARGET results. Hepatology. 2017;66:755a–756a.
80. Nault JC, Colombo M. Hepatocellular carcinoma and direct acting antiviral treatments: controversy after the revolution. J Hepatol. 2016;65(4):663–665. doi:10.1016/j.jhep.2016.07.004
81. Huang P, Liu M, Zang F, et al. The development of hepatocellular carcinoma in HCV-infected patients treated with DAA: a comprehensive analysis. Carcinogenesis. 2018;39(12):1497–1505. doi:10.1093/carcin/bgy099
82. Singal AG, Lim JK, Kanwal F. AGA clinical practice update on interaction between oral direct-acting antivirals for chronic hepatitis C infection and hepatocellular carcinoma: expert review. Gastroenterology. 2019;156(8):2149–2157. doi:10.1053/j.gastro.2019.02.046
83. Hoofnagle JH. Course and outcome of hepatitis C. Hepatology. 2002;36(5 Suppl 1):S21–S29. doi:10.1053/jhep.2002.36227
84. Kaito M, Watanabe S, Tsukiyama-Kohara K, et al. Hepatitis C virus particle detected by immunoelectron microscopic study. J Gen Virol. 1994;75(Pt 7):1755–1760. doi:10.1099/0022-1317-75-7-1755
85. Gastaminza P, Dryden KA, Boyd B, et al. Ultrastructural and biophysical characterization of hepatitis C virus particles produced in cell culture. J Virol. 2010;84(21):10999–11009. doi:10.1128/JVI.00526-10
86. Dubuisson J, Cosset FL. Virology and cell biology of the hepatitis C virus life cycle: an update. J Hepatol. 2014;61(1 Suppl):S3–S13. doi:10.1016/j.jhep.2014.06.031
87. Belouzard S, Cocquerel L, Dubuisson J. Hepatitis C virus entry into the hepatocyte. Cent Eur J Biol. 2011;6(6):933–945.
88. Blanchard E, Belouzard S, Goueslain L, et al. Hepatitis C virus entry depends on clathrin-mediated endocytosis. J Virol. 2006;80(14):6964–6972. doi:10.1128/JVI.00024-06
89. Meertens L, Bertaux C, Dragic T. Hepatitis C virus entry requires a critical postinternalization step and delivery to early endosomes via clathrin-coated vesicles. J Virol. 2006;80(23):11571–11578. doi:10.1128/JVI.01717-06
90. Paul D, Madan V, Bartenschlager R. Hepatitis C virus RNA replication and assembly: living on the fat of the land. Cell Host Microbe. 2014;16(5):569–579. doi:10.1016/j.chom.2014.10.008
91. Bartenschlager R, Penin F, Lohmann V, Andre P. Assembly of infectious hepatitis C virus particles. Trends Microbiol. 2011;19(2):95–103. doi:10.1016/j.tim.2010.11.005
92. Huang H, Sun F, Owen DM, et al. Hepatitis C virus production by human hepatocytes dependent on assembly and secretion of very low-density lipoproteins. Proc Natl Acad Sci USA. 2007;104(14):5848–5853. doi:10.1073/pnas.0700760104
93. Berger C, Romero-Brey I, Radujkovic D, et al. Daclatasvir-like inhibitors of NS5A block early biogenesis of hepatitis C virus-induced membranous replication factories, independent of RNA replication. Gastroenterology. 2014;147(5):1094–1105 e1025. doi:10.1053/j.gastro.2014.07.019
94. Alazard-Dany N, Denolly S, Boson B, Cosset FL. Overview of HCV life cycle with a special focus on current and possible future antiviral targets. Viruses. 2019;11(1). doi:10.3390/v11010030
95. Vescovo T, Refolo G, Vitagliano G, Fimia GM, Piacentini M. Molecular mechanisms of hepatitis C virus-induced hepatocellular carcinoma. Clin Microbiol Infect. 2016;22(10):853–861. doi:10.1016/j.cmi.2016.07.019
96. Mitchell JK, Lemon SM, McGivern DR. How do persistent infections with hepatitis C virus cause liver cancer? Curr Opin Virol. 2015;14:101–108. doi:10.1016/j.coviro.2015.09.003
97. Wang M, Kaufman RJ. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature. 2016;529(7586):326–335. doi:10.1038/nature17041
98. Green DR, Galluzzi L, Kroemer G. Metabolic control of cell death. Science. 2014;345(6203):1466.
99. Hofmann S, Krajewski M, Scherer C, et al. Complex lipid metabolic remodeling is required for efficient hepatitis C virus replication. Bba-Mol Cell Biol L. 2018;1863(9):1041–1056. doi:10.1016/j.bbalip.2018.06.002
100. Kubes P, Mehal WZ. Sterile Inflammation in the Liver. Gastroenterology. 2012;143(5):1158–1172. doi:10.1053/j.gastro.2012.09.008
101. Luedde T, Schwabe RF. NF-kappaB in the liver–linking injury, fibrosis and hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. 2011;8(2):108–118. doi:10.1038/nrgastro.2010.213
102. Donato MF, Arosio E, Monti V, et al. Proliferating cell nuclear antigen assessed by a computer-assisted image analysis system in patients with chronic viral hepatitis and cirrhosis. Dig Liver Dis. 2002;34(3):197–203. doi:10.1016/S1590-8658(02)80193-1
103. Freeman A, Hamid S, Morris L, et al. Improved detection of hepatocyte proliferation using antibody to the pre-replication complex: an association with hepatic fibrosis and viral replication in chronic hepatitis C virus infection. J Viral Hepat. 2003;10(5):345–350. doi:10.1046/j.1365-2893.2003.00454.x
104. Falkowski O, An HJ, Ianus IA, et al. Regeneration of hepatocyte ‘buds’ in cirrhosis from intrabiliary stem cells. J Hepatol. 2003;39(3):357–364. doi:10.1016/S0168-8278(03)00309-X
105. Sen B, Rastogi A, Nath R, et al. Senescent hepatocytes in decompensated liver show reduced UPR(MT) and its key player, CLPP, attenuates senescence in vitro. Cell Mol Gastroenterol Hepatol. 2019;8(1):73–94. doi:10.1016/j.jcmgh.2019.03.001
106. Dash S, Aydin Y, Wu T. Integrated stress response in hepatitis C promotes Nrf2-related chaperone-mediated autophagy: a novel mechanism for host-microbe survival and HCC development in liver cirrhosis. Semin Cell Dev Biol. Epub 2019 Aug 8. doi:10.1016/j.semcdb.2019.07.015
107. Tian Y, Garcia G, Bian Q, et al. Mitochondrial stress induces chromatin reorganization to promote longevity and UPR(mt). Cell. 2016;165(5):1197–1208. doi:10.1016/j.cell.2016.04.011
108. Schito L, Rey S. Cell-autonomous metabolic reprogramming in hypoxia. Trends Cell Biol. 2018;28(2):128–142. doi:10.1016/j.tcb.2017.10.006
109. Matilainen O, Quiros PM, Auwerx J. Mitochondria and epigenetics - crosstalk in homeostasis and stress. Trends Cell Biol. 2017;27(6):453–463. doi:10.1016/j.tcb.2017.02.004
110. Chandra PK, Gunduz F, Hazari S, et al. Impaired expression of type I and type II interferon receptors in HCV-associated chronic liver disease and liver cirrhosis. PLoS One. 2014;9(9):e108616. doi:10.1371/journal.pone.0108616
111. Aydin Y, Stephens CM, Chava S, et al. Chaperone-mediated autophagy promotes beclin1 degradation in persistently infected hepatitis C virus cell culture. Am J Pathol. 2018;188(10):2339–2355. doi:10.1016/j.ajpath.2018.06.022
112. Bao L, Chandra PK, Moroz K, et al. Impaired autophagy response in human hepatocellular carcinoma. Exp Mol Pathol. 2014;96(2):149–154. doi:10.1016/j.yexmp.2013.12.002
113. Chava S, Lee C, Aydin Y, et al. Chaperone-mediated autophagy compensates for impaired macroautophagy in the cirrhotic liver to promote hepatocellular carcinoma. Oncotarget. 2017;8(25):40019–40036. doi:10.18632/oncotarget.v8i25
114. Inami Y, Waguri S, Sakamoto A, et al. Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J Cell Biol. 2011;193(2):275–284. doi:10.1083/jcb.201102031
115. Bartolini D, Dallaglio K, Torquato P, Piroddi M, Galli F. Nrf2-p62 autophagy pathway and its response to oxidative stress in hepatocellular carcinoma. Transl Res. 2018;193:54–71. doi:10.1016/j.trsl.2017.11.007
116. Ally A, Balasundaram M, Carlsen R, Cancer Genome Atlas Research Network. Electronic address wbe, Cancer Genome Atlas Research N. Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell. 2017;169(7):1327–1341 e1323. doi:10.1016/j.cell.2017.05.046
117. Kanda T, Goto T, Hirotsu Y, Moriyama M, Omata M. Molecular mechanisms driving progression of liver cirrhosis towards hepatocellular carcinoma in chronic hepatitis B and C infections: a review. Int J Mol Sci. 2019;20(6):1358. doi:10.3390/ijms20061358
118. Zucman-Rossi J, Villanueva A, Nault JC, Llovet JM. Genetic landscape and biomarkers of hepatocellular carcinoma. Gastroenterology. 2015;149(5):1226. doi:10.1053/j.gastro.2015.05.061
119. Tornesello ML, Buonaguro L, Izzo F, Buonaguro FM. Molecular alterations in hepatocellular carcinoma associated with hepatitis B and hepatitis C infections. Oncotarget. 2016;7(18):25087–25102. doi:10.18632/oncotarget.v7i18
120. Thorgeirsson SS, Grisham JW. Molecular pathogenesis of human hepatocellular carcinoma. Nat Genet. 2002;31(4):339–346. doi:10.1038/ng0802-339
121. Guichard C, Amaddeo G, Imbeaud S, et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat Genet. 2012;44(6):694–U120. doi:10.1038/ng.2256
122. Chochi Y, Kawauchi S, Nakao M, et al. A copy number gain of the 6p arm is linked with advanced hepatocellular carcinoma: an array-based comparative genomic hybridization study. J Pathol. 2009;217(5):677–684. doi:10.1002/path.2491
123. Midorikawa Y, Yamamoto S, Tsuji S, et al. Allelic imbalances and homozygous deletion on 8p23.2 for stepwise progression of hepatocarcinogenesis. Hepatology. 2009;49(2):513–522. doi:10.1002/hep.22698
124. Totoki Y, Tatsuno K, Covington KR, et al. Trans-ancestry mutational landscape of hepatocellular carcinoma genomes. Nat Genet. 2014;46(12):1267–1273. doi:10.1038/ng.3126
125. Schulze K, Imbeaud S, Letouze E, et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat Genet. 2015;47(5):505–U106.
126. Nault JC, Ningarhari M, Rebouissou S, Zucman-Rossi J. The role of telomeres and telomerase in cirrhosis and liver cancer. Nat Rev Gastro Hepat. 2019;16(9):544–558. doi:10.1038/s41575-019-0165-3
127. Donaires FS, Scatena NF, Alves-Paiva RM, et al. Telomere biology and telomerase mutations in cirrhotic patients with hepatocellular carcinoma. PLoS One. 2017;12(8):e0183287. doi:10.1371/journal.pone.0183287
128. Pezzuto F, Buonaguro L, Buonaguro FM, Tornesello ML. Frequency and geographic distribution of TERT promoter mutations in primary hepatocellular carcinoma. Infect Agents Cancer. 2017;12. doi:10.1186/s13027-017-0138-5
129. Oh BK, Kim H, Park YN, et al. High telomerase activity and long telomeres in advanced hepatocellular carcinomas with poor prognosis. Lab Invest. 2008;88(2):144–152. doi:10.1038/labinvest.3700710
130. Zhu ZW, Tran H, Mathahs MM, Moninger TO, Schmidt WN. HCV Induces telomerase reverse transcriptase, increases its catalytic activity, and promotes caspase degradation in infected human hepatocytes. PLoS One. 2017;12(1):e0166853.
131. Zhu Z, Wilson AT, Gopalakrishna K, Brown KE, Luxon BA, Schmidt WN. Hepatitis C virus core protein enhances Telomerase activity in Huh7 cells. J Med Virol. 2010;82(2):239–248. doi:10.1002/jmv.v82:2
132. Sherr CJ, McCormick F. The RB and p53 pathways in cancer. Cancer Cell. 2002;2(2):103–112. doi:10.1016/S1535-6108(02)00102-2
133. Mitchell JK, Midkiff BR, Israelow B, et al. Hepatitis C virus indirectly disrupts DNA damage-induced p53 responses by activating protein kinase R. mBio. 2017;8(2). doi:10.1128/mBio.00121-17
134. Munakata T, Liang Y, Kim S, et al. Hepatitis C virus induces E6AP-dependent degradation of the retinoblastoma protein. PLoS Pathog. 2007;3(9):1335–1347. doi:10.1371/journal.ppat.0030139
135. Munakata T, Nakamura M, Liang Y, Li K, Lemon SM. Down-regulation of the retinoblastoma tumor suppressor by the hepatitis C virus NS5B RNA-dependent RNA polymerase. Proc Natl Acad Sci U S A. 2005;102(50):18159–18164. doi:10.1073/pnas.0505605102
136. Aydin Y, Chatterjee A, Chandra PK, et al. Interferon-alpha-induced hepatitis C virus clearance restores p53 tumor suppressor more than direct-acting antivirals. Hepatol Commun. 2017;1(3):256–269. doi:10.1002/hep4.1025
137. Aydin Y, Chedid M, Chava S, et al. Activation of PERK-Nrf2 oncogenic signaling promotes Mdm2-mediated Rb degradation in persistently infected HCV culture. Sci Rep. 2017;7(1):9223. doi:10.1038/s41598-017-10087-6
138. Wang W, Pan Q, Fuhler GM, Smits R, Peppelenbosch MP. Action and function of Wnt/beta-catenin signaling in the progression from chronic hepatitis C to hepatocellular carcinoma. J Gastroenterol. 2017;52(4):419–431. doi:10.1007/s00535-016-1299-5
139. Liu J, Ding X, Tang J, et al. Enhancement of canonical Wnt/beta-catenin signaling activity by HCV core protein promotes cell growth of hepatocellular carcinoma cells. PLoS One. 2011;6(11):e27496. doi:10.1371/journal.pone.0027496
140. Park CY, Choi SH, Kang SM, et al. Nonstructural 5A protein activates beta-catenin signaling cascades: implication of hepatitis C virus-induced liver pathogenesis. J Hepatol. 2009;51(5):853–864. doi:10.1016/j.jhep.2009.06.026
141. Street A, Macdonald A, Crowder K, Harris M. The Hepatitis C virus NS5A protein activates a phosphoinositide 3-kinase-dependent survival signaling cascade. J Biol Chem. 2004;279(13):12232–12241. doi:10.1074/jbc.M312245200
142. Higgs MR, Lerat H, Pawlotsky JM. Hepatitis C virus-induced activation of beta-catenin promotes c-Myc expression and a cascade of pro-carcinogenetic events. Oncogene. 2013;32(39):4683–4693. doi:10.1038/onc.2012.484
143. Komposch K, Sibilia M. EGFR signaling in liver diseases. Int J Mol Sci. 2015;17(1). doi:10.3390/ijms17010030
144. Sigismund S, Avanzato D, Lanzetti L. Emerging functions of the EGFR in cancer. Mol Oncol. 2018;12(1):3–20. doi:10.1002/mol2.2018.12.issue-1
145. Bergeron JJ, Di Guglielmo GM, Dahan S, Dominguez M, Posner BI. Spatial and temporal regulation of receptor tyrosine kinase activation and intracellular signal transduction. Annu Rev Biochem. 2016;85:573–597. doi:10.1146/annurev-biochem-060815-014659
146. Kira S, Nakanishi T, Suemori S, Kitamoto M, Watanabe Y, Kajiyama G. Expression of transforming growth factor alpha and epidermal growth factor receptor in human hepatocellular carcinoma. Liver. 1997;17(4):177–182. doi:10.1111/j.1600-0676.1997.tb00803.x
147. Ito Y, Takeda T, Sakon M, et al. Expression and clinical significance of erb-B receptor family in hepatocellular carcinoma. Br J Cancer. 2001;84(10):1377–1383. doi:10.1054/bjoc.2000.1580
148. Daveau M, Scotte M, Francois A, et al. Hepatocyte growth factor, transforming growth factor alpha, and their receptors as combined markers of prognosis in hepatocellular carcinoma. Mol Carcinog. 2003;36(3):130–141. doi:10.1002/mc.10103
149. Buckley AF, Burgart LJ, Sahai V, Kakar S. Epidermal growth factor receptor expression and gene copy number in conventional hepatocellular carcinoma. Am J Clin Pathol. 2008;129(2):245–251. doi:10.1309/WF10QAAED3PP93BH
150. Divella R, Daniele A, Gadaleta C, et al. Circulating transforming growth factor-beta and epidermal growth factor receptor as related to virus infection in liver carcinogenesis. Anticancer Res. 2012;32(1):141–145.
151. Diao J, Pantua H, Ngu H, et al. Hepatitis C virus induces epidermal growth factor receptor activation via CD81 binding for viral internalization and entry. J Virol. 2012;86(20):10935–10949. doi:10.1128/JVI.00750-12
152. Igloi Z, Kazlauskas A, Saksela K, Macdonald A, Mankouri J, Harris M. Hepatitis C virus NS5A protein blocks epidermal growth factor receptor degradation via a proline motif- dependent interaction. J Gen Virol. 2015;96(8):2133–2144. doi:10.1099/vir.0.000145
153. Hu TH, Li CX. Convergence between Wnt-beta-catenin and EGFR signaling in cancer. Mol Cancer. 2010;9(1):236. doi:10.1186/1476-4598-9-236
154. Beenken A, Mohammadi M. The FGF family: biology, pathophysiology and therapy. Nat Rev Drug Discov. 2009;8(3):235–253.
155. Gauglhofer C, Sagmeister S, Schrottmaier W, et al. Up-regulation of the fibroblast growth factor 8 subfamily in human hepatocellular carcinoma for cell survival and neoangiogenesis. Hepatology. 2011;53(3):854–864. doi:10.1002/hep.24099
156. Cotler SJ, Hay N, Xie H, et al. Immunohistochemical expression of components of the Akt-mTORC1 pathway is associated with hepatocellular carcinoma in patients with chronic liver disease. Digest Dis Sci. 2008;53(3):844–849. doi:10.1007/s10620-007-9934-x
157. Villanueva A, Chiang DY, Newell P, et al. Pivotal role of mTOR signaling in hepatocellular carcinoma. Gastroenterology. 2008;135(6):1972–1983. doi:10.1053/j.gastro.2008.08.008
158. Sahin F, Kannangai R, Adegbola O, Wang J, Su G, Torbenson M. mTOR and P70 S6 kinase expression in primary liver neoplasms. Clin Cancer Res. 2004;10(24):8421–8425. doi:10.1158/1078-0432.CCR-04-0941
159. Zhou LD, Huang Y, Li JD, Wang ZM. The mTOR pathway is associated with the poor prognosis of human hepatocellular carcinoma. Med Oncol. 2010;27(2):255–261. doi:10.1007/s12032-009-9201-4
160. Wouters BG, Koritzinsky M. Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat Rev Cancer. 2008;8(11):851–864. doi:10.1038/nrc2501
161. Sengupta S, Peterson TR, Sabatini DM. Regulation of the mTOR complex 1 pathway by nutrients, growth factors, and stress. Mol Cell. 2010;40(2):310–322. doi:10.1016/j.molcel.2010.09.026
162. He YP, Nakao HH, Tan SL, et al. Subversion of cell signaling pathways by hepatitis C virus nonstructural 5A protein via interaction with Grb2 and P85 phosphatidylinositol 3-kinase. J Virol. 2002;76(18):9207–9217. doi:10.1128/JVI.76.18.9207-9217.2002
163. Wang JD, Tong WY, Zhang XN, et al. Hepatitis C virus non-structural protein NS5A interacts with FKBP38 and inhibits apoptosis in Huh7 hepatoma cells. FEBS Lett. 2006;580(18):4392–4400. doi:10.1016/j.febslet.2006.07.002
164. Paternostro C, David E, Novo E, Parola M. Hypoxia, angiogenesis and liver fibrogenesis in the progression of chronic liver diseases. World J Gastroenterol. 2010;16(3):281–288. doi:10.3748/wjg.v16.i3.281
165. Di Bonzo LV, Novo E, Cannito S, et al. Angiogenesis and liver fibrogenesis. Histol Histopathol. 2009;24(10):1323–1341. doi:10.14670/HH-24.1323
166. Semela D, Dufour JF. Angiogenesis and hepatocellular carcinoma. J Hepatol. 2004;41(5):864–880. doi:10.1016/j.jhep.2004.09.006
167. Nishida N, Yano H, Nishida T, Kamura T, Kojiro M. Angiogenesis in cancer. Vasc Health Risk Manag. 2006;2(3):213–219. doi:10.2147/vhrm.2006.2.issue-3
168. Whittaker S, Marais R, Zhu AX. The role of signaling pathways in the development and treatment of hepatocellular carcinoma. Oncogene. 2010;29(36):4989–5005. doi:10.1038/onc.2010.236
169. Mas VR, Maluf DG, Archer KJ, Yanek KC, Fisher RA. Angiogenesis soluble factors as hepatocellular carcinoma noninvasive markers for monitoring hepatitis C virus cirrhotic patients awaiting liver transplantation. Transplantation. 2007;84(10):1262–1271. doi:10.1097/01.tp.0000287596.91520.1a
170. Hassan M, Selimovic D, El-Khattouti A, et al. Hepatitis C virus-mediated angiogenesis: molecular mechanisms and therapeutic strategies. World J Gastroenterol. 2014;20(42):15467–15475. doi:10.3748/wjg.v20.i42.15467
171. Hassan M, Selimovic D, Ghozlan H, Abdel-Kader O. Hepatitis C virus core protein triggers hepatic angiogenesis by a mechanism including multiple pathways. Hepatology. 2009;49(5):1469–1482. doi:10.1002/hep.22849
172. Allis CD, Jenuwein T. The molecular hallmarks of epigenetic control. Nat Rev Genet. 2016;17(8):487–500. doi:10.1038/nrg.2016.59
173. Nehme Z, Pasquereau S, Herbein G. Control of viral infections by epigenetic-targeted therapy. Clin Epigenetics. 2019;11(1):55. doi:10.1186/s13148-019-0654-9
174. Lieberman PM. Epigenetics and genetics of viral latency. Cell Host Microbe. 2016;19(5):619–628. doi:10.1016/j.chom.2016.04.008
175. Boiani M, Scholer HR. Regulatory networks in embryo-derived pluripotent stem cells. Nat Rev Mol Cell Biol. 2005;6(11):872–884. doi:10.1038/nrm1744
176. Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis. 2010;31(1):27–36. doi:10.1093/carcin/bgp220
177. Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150(1):12–27. doi:10.1016/j.cell.2012.06.013
178. Aloia L, Di Stefano B, Di Croce L. Polycomb complexes in stem cells and embryonic development. Development. 2013;140(12):2525–2534. doi:10.1242/dev.091553
179. Schuettengruber B, Martinez AM, Iovino N, Cavalli G. Trithorax group proteins: switching genes on and keeping them active. Nat Rev Mol Cell Bio. 2011;12(12):799–814. doi:10.1038/nrm3230
180. van Vlerken LE, Hurt EM, Hollingsworth RE. The role of epigenetic regulation in stem cell and cancer biology. J Mol Med. 2012;90(7):791–801. doi:10.1007/s00109-012-0917-9
181. Tysnes BB. Tumor-initiating and -propagating cells: cells that we would like to identify and control. Neoplasia. 2010;12(7):506–515. doi:10.1593/neo.10290
182. Takebe N, Ivy SP. Controversies in cancer stem cells: targeting embryonic signaling pathways. Clin Cancer Res. 2010;16(12):3106–3112. doi:10.1158/1078-0432.CCR-09-2934
183. Jones PA, Issa JP, Baylin S. Targeting the cancer epigenome for therapy. Nat Rev Genet. 2016;17(10):630–641. doi:10.1038/nrg.2016.93
184. Ali N, Allam H, May R, et al. Hepatitis C virus-induced cancer stem cell-like signatures in cell culture and murine tumor xenografts. J Virol. 2011;85(23):12292–12303. doi:10.1128/JVI.05920-11
185. Machida K, Tsukamoto H, Mkrtchyan H, et al. Toll-like receptor 4 mediates synergism between alcohol and HCV in hepatic oncogenesis involving stem cell marker Nanog. P Natl Acad Sci USA. 2009;106(5):1548–1553. doi:10.1073/pnas.0807390106
186. Feng QH, Stern JE, Hawes SE, Lu H, Jiang MJ, Kiviat NB. DNA methylation changes in normal liver tissues and hepatocellular carcinoma with different viral infection. Exp Mol Pathol. 2010;88(2):287–292. doi:10.1016/j.yexmp.2010.01.002
187. Feng QH, Das T, Huang J, Feng Z, Gretch D. Epigenetic mechanisms in hepatitis C virus-associated hepatocellular carcinoma: a potential new link between stem cells, virology and cancer. Am Med J. 2013;4(1):21–35.
188. Takebe N, Harris PJ, Warren RQ, Ivy SP. Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog pathways. Nat Rev Clin Oncol. 2011;8(2):97–106. doi:10.1038/nrclinonc.2010.196
189. Iwai A, Takegami T, Shiozaki T, Miyazaki T. Hepatitis C virus NS3 protein can activate the notch-signaling pathway through binding to a transcription factor, SRCAP. PLoS One. 2011;6(6):e20718. doi:10.1371/journal.pone.0020718
190. Eissenberg JC, Wong M, Chrivia JC. Human SRCAP and Drosophila melanogaster DOM are homologs that function in the notch signaling pathway. Mol Cell Biol. 2005;25(15):6559–6569. doi:10.1128/MCB.25.15.6559-6569.2005
191. Kageyama R, Ohtsuka T, Kobayashi T. The Hes gene family: repressors and oscillators that orchestrate embryogenesis. Development. 2007;134(7):1243–1251. doi:10.1242/dev.000786
192. Briscoe J, Therond PP. The mechanisms of Hedgehog signalling and its roles in development and disease. Nat Rev Mol Cell Biol. 2013;14(7):416–429. doi:10.1038/nrm3598
193. Machado MV, Diehl AM. Hedgehog signalling in liver pathophysiology. J Hepatol. 2018;68(3):550–562. doi:10.1016/j.jhep.2017.10.017
194. Choi SS, Bradrick S, Qiang G, et al. Up-regulation of hedgehog pathway is associated with cellular permissiveness for hepatitis C virus replication. Hepatology. 2011;54(5):1580–1590. doi:10.1002/hep.24576
195. Wienken M, Dickmanns A, Nemajerova A, et al. MDM2 associates with polycomb repressor complex 2 and enhances stemness-promoting chromatin modifications independent of p53. Mol Cell. 2016;61(1):68–83. doi:10.1016/j.molcel.2015.12.008
196. Stefanska B, Huang J, Bhattacharyya B, et al. Definition of the landscape of promoter DNA hypomethylation in liver cancer. Cancer Res. 2011;71(17):5891–5903. doi:10.1158/0008-5472.CAN-10-3823
197. Nishida N, Kudo M, Nagasaka T, Ikai I, Goel A. Characteristic patterns of altered DNA methylation predict emergence of human hepatocellular carcinoma. Hepatology. 2012;56(3):994–1003. doi:10.1002/hep.25706
198. Archer KJ, Mas VR, Maluf DG, Fisher RA. High-throughput assessment of CpG site methylation for distinguishing between HCV-cirrhosis and HCV-associated hepatocellular carcinoma. Mol Genet Genomics. 2010;283(4):341–349. doi:10.1007/s00438-010-0522-y
199. Shen J, Wang S, Zhang YJ, et al. Genome-wide DNA methylation profiles in hepatocellular carcinoma. Hepatology. 2012;55(6):1799–1808. doi:10.1002/hep.25569
200. Hung SY, Lin HH, Yeh KT, Chang JG. Histone-modifying genes as biomarkers in hepatocellular carcinoma. Int J Clin Exp Patho. 2014;7(5):2496–2507.
201. Lachenmayer A, Toffanin S, Cabellos L, et al. Combination therapy for hepatocellular carcinoma: additive preclinical efficacy of the HDAC inhibitor panobinostat with sorafenib. J Hepatol. 2012;56(6):1343–1350. doi:10.1016/j.jhep.2012.01.009
202. Bandiera S, Pfeffer S, Baumert TF, Zeisel MB. miR-122–a key factor and therapeutic target in liver disease. J Hepatol. 2015;62(2):448–457. doi:10.1016/j.jhep.2014.10.004
203. Plissonnier ML, Herzog K, Levrero M, Zeisel MB. Non-coding RNAs and hepatitis C virus-induced hepatocellular carcinoma. Viruses. 2018;10(11). doi:10.3390/v10110591
204. Wu M, Lin Z, Li X, et al. HULC cooperates with MALAT1 to aggravate liver cancer stem cells growth through telomere repeat-binding factor 2. Sci Rep. 2016;6:36045. doi:10.1038/srep36045
205. Cheng D, Deng J, Zhang B, et al. LncRNA HOTAIR epigenetically suppresses miR-122 expression in hepatocellular carcinoma via DNA methylation. EBioMedicine. 2018;36:159–170. doi:10.1016/j.ebiom.2018.08.055
206. Aydin Y, Kurt R, Song K, et al. Hepatic stress response in HCV infection promotes STAT3-mediated inhibition of HNF4A-miR-122 feedback loop in liver fibrosis and cancer progression. Cancers (Basel). 2019;11(10):1407. doi:10.3390/cancers11101407
207. Gao B, Jeong WI, Tian Z. Liver: an organ with predominant innate immunity. Hepatology. 2008;47(2):729–736. doi:10.1002/hep.22034
208. Crispe IN. The Liver as a Lymphoid Organ. Annu Rev Immunol. 2009;27:147–163. doi:10.1146/annurev.immunol.021908.132629
209. Norris S, Collins C, Doherty DG, et al. Resident human hepatic lymphocytes are phenotypically different from circulating lymphocytes. J Hepatol. 1998;28(1):84–90. doi:10.1016/S0168-8278(98)80206-7
210. Shuai ZW, Leung MWY, He XS, et al. Adaptive immunity in the liver. Cell Mol Immunol. 2016;13(3):354–368. doi:10.1038/cmi.2016.4
211. Chan YK, Gack MU. Viral evasion of intracellular DNA and RNA sensing. Nat Rev Microbiol. 2016;14(6):360–373. doi:10.1038/nrmicro.2016.45
212. Martinez-Esparza M, Tristan-Manzano M, Ruiz-Alcaraz AJ, Garcia-Penarrubia P. Inflammatory status in human hepatic cirrhosis. World J Gastroenterol. 2015;21(41):11522–11541. doi:10.3748/wjg.v21.i41.11522
213. Jouan L, Melancon P, Rodrigue-Gervais IG, et al. Distinct antiviral signaling pathways in primary human hepatocytes and their differential disruption by HCV NS3 protease. J Hepatol. 2010;52(2):167–175. doi:10.1016/j.jhep.2009.11.011
214. Park SH, Rehermann B. Immune responses to HCV and other hepatitis viruses. Immunity. 2014;40(1):13–24. doi:10.1016/j.immuni.2013.12.010
215. Boltjes A, Movita D, Boonstra A, Woltman AM. The role of Kupffer cells in hepatitis B and hepatitis C virus infections. J Hepatol. 2014;61(3):660–671. doi:10.1016/j.jhep.2014.04.026
216. Miller K, McArdle S, Gale MJ, et al. Effects of the hepatitis C virus core protein on innate cellular defense pathways. J Interferon Cytokine Res. 2004;24(7):391–402. doi:10.1089/1079990041535647
217. Hosomura N, Kono H, Tsuchiya M, et al. HCV-related proteins activate Kupffer cells isolated from human liver tissues. Dig Dis Sci. 2011;56(4):1057–1064. doi:10.1007/s10620-010-1395-y
218. Chang S, Dolganiuc A, Szabo G. Toll-like receptors 1 and 6 are involved in TLR2-mediated macrophage activation by hepatitis C virus core and NS3 proteins. J Leukoc Biol. 2007;82(3):479–487. doi:10.1189/jlb.0207128
219. Zhu H, Liu C. Interleukin-1 inhibits hepatitis C virus subgenomic RNA replication by activation of extracellular regulated kinase pathway. J Virol. 2003;77(9):5493–5498. doi:10.1128/JVI.77.9.5493-5498.2003
220. Zhu H, Shang X, Terada N, Liu C. STAT3 induces anti-hepatitis C viral activity in liver cells. Biochem Biophys Res Commun. 2004;324(2):518–528. doi:10.1016/j.bbrc.2004.09.081
221. Khakoo SI, Soni PN, Savage K, et al. Lymphocyte and macrophage phenotypes in chronic hepatitis C infection. Correlation with disease activity. Am J Pathol. 1997;150(3):963–970.
222. McGuinness PH, Painter D, Davies S, McCaughan GW. Increases in intrahepatic CD68 positive cells, MAC387 positive cells, and proinflammatory cytokines (particularly interleukin 18) in chronic hepatitis C infection. Gut. 2000;46(2):260–269. doi:10.1136/gut.46.2.260
223. Negash AA, Ramos HJ, Crochet N, et al. IL-1beta production through the NLRP3 inflammasome by hepatic macrophages links hepatitis C virus infection with liver inflammation and disease. PLoS Pathog. 2013;9(4):e1003330. doi:10.1371/journal.ppat.1003330
224. Tu Z, Pierce RH, Kurtis J, Kuroki Y, Crispe IN, Orloff MS. Hepatitis C virus core protein subverts the antiviral activities of human Kupffer cells. Gastroenterology. 2010;138(1):305–314. doi:10.1053/j.gastro.2009.09.009
225. Abe T, Kaname Y, Hamamoto I, et al. Hepatitis C virus nonstructural protein 5A modulates the toll-like receptor-MyD88-dependent signaling pathway in macrophage cell lines. J Virol. 2007;81(17):8953–8966. doi:10.1128/JVI.00649-07
226. Chung H, Watanabe T, Kudo M, Chiba T. Hepatitis C virus core protein induces homotolerance and cross-tolerance to Toll-like receptor ligands by activation of Toll-like receptor 2. J Infect Dis. 2010;202(6):853–861. doi:10.1086/653018
227. Szabo G, Dolganiuc A. Hepatitis C and innate immunity: recent advances. Clin Liver Dis. 2008;12(3):675–692. doi:10.1016/j.cld.2008.03.003
228. Chung H, Watanabe T, Kudo M, Chiba T. Correlation between hyporesponsiveness to Toll-like receptor ligands and liver dysfunction in patients with chronic hepatitis C virus infection. J Viral Hepatitis. 2011;18(10):E561–E567. doi:10.1111/jvh.2011.18.issue-10
229. Lin WY, Kim SS, Yeung E, et al. Hepatitis C virus core protein blocks interferon signaling by interaction with the STAT1 SH2 domain. J Virol. 2006;80(18):9226–9235. doi:10.1128/JVI.00459-06
230. Meylan E, Curran J, Hofmann K, et al. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature. 2005;437(7062):1167–1172. doi:10.1038/nature04193
231. Miyazaki M, Kanto T, Inoue M, et al. Impaired cytokine response in myeloid dendritic cells in chronic hepatitis C virus infection regardless of enhanced expression of toll-like receptors and retinoic acid inducible gene-I. J Med Virol. 2008;80(6):980–988. doi:10.1002/(ISSN)1096-9071
232. Doherty DG. Immunity, tolerance and autoimmunity in the liver: a comprehensive review. J Autoimmun. 2016;66:60–75. doi:10.1016/j.jaut.2015.08.020
233. Gao B. Basic liver immunology. Cell Mol Immunol. 2016;13(3):265–266. doi:10.1038/cmi.2016.09
234. Amadei B, Urbani S, Cazaly A, et al. Activation of natural killer cells during acute infection with hepatitis C virus. Gastroenterology. 2010;138(4):1536–1545. doi:10.1053/j.gastro.2010.01.006
235. Khakoo SI, Thio CL, Martin MP, et al. HLA and NK cell inhibitory receptor genes in resolving hepatitis C virus infection. Science. 2004;305(5685):872–874. doi:10.1126/science.1097670
236. Ahlenstiel G, Martin MP, Gao X, Carrington M, Rehermann B. Distinct KIR/HLA compound genotypes affect the kinetics of human antiviral natural killer cell responses. J Clin Invest. 2008;118(3):1017–1026. doi:10.1172/JCI32400
237. Ahlenstiel G, Titerence RH, Koh C, et al. Natural killer cells are polarized toward cytotoxicity in chronic hepatitis c in an interferon-alfa-dependent manner. Gastroenterology. 2010;138(1):325–335. doi:10.1053/j.gastro.2009.08.066
238. De Maria A, Fogli M, Mazza S, et al. Increased natural cytotoxicity receptor expression and relevant IL-10 production in NK cells from chronically infected viremic HCV patients. Eur J Immunol. 2007;37(2):445–455. doi:10.1002/(ISSN)1521-4141
239. Oliviero B, Varchetta S, Paudice E, et al. Natural killer cell functional dichotomy in chronic hepatitis B and chronic hepatitis C virus infections. Gastroenterology. 2009;137(3):
240. Ahlenstiel G, Edlich B, Hogdal LJ, et al. Early changes in natural killer cell function indicate virologic response to interferon therapy for hepatitis C. Gastroenterology. 2011;141(4):
241. Miyagi T, Gil MP, Wang X, Louten J, Chu WM, Biron CA. High basal STAT4 balanced by STAT1 induction to control type 1 interferon effects in natural killer cells. J Exp Med. 2007;204(10):2383–2396. doi:10.1084/jem.20070401
242. Serti E, Chepa-Lotrea X, Kim YJ, et al. Successful interferon-free therapy of chronic hepatitis C virus infection normalizes natural killer cell function. Gastroenterology. 2015;149(1):190–200 e192. doi:10.1053/j.gastro.2015.03.004
243. Strunz B, Hengst J, Deterding K, et al. Chronic hepatitis C virus infection irreversibly impacts human natural killer cell repertoire diversity. Nat Commun. 2018;9(1):2275. doi:10.1038/s41467-018-04685-9
244. Doherty DG, O’Farrelly C. Innate and adaptive lymphoid cells in the human liver. Immunol Rev. 2000;174:5–20. doi:10.1034/j.1600-0528.2002.017416.x
245. Ahmad A, Alvarez F. Role of NK and NKT cells in the immunopathogenesis of HCV-induced hepatitis. J Leukoc Biol. 2004;76(4):743–759. doi:10.1189/jlb.0304197
246. Swain MG. Hepatic NKT cells: friend or foe? Clin Sci (Lond). 2008;114(7):457–466. doi:10.1042/CS20070328
247. Sprent J, Surh CD. T cell memory. Annu Rev Immunol. 2002;20:551–579. doi:10.1146/annurev.immunol.20.100101.151926
248. Park SH, Veerapu NS, Shin EC, et al. Subinfectious hepatitis C virus exposures suppress T cell responses against subsequent acute infection. Nat Med. 2013;19(12):1638–1642. doi:10.1038/nm.3408
249. Grakoui A, Shoukry NH, Woollard DJ, et al. HCV persistence and immune evasion in the absence of memory T cell help. Science. 2003;302(5645):659–662. doi:10.1126/science.1088774
250. Rehermann B, Thimme R. Insights from antiviral therapy into immune responses to hepatitis B and C virus infection. Gastroenterology. 2019;156(2):369–383. doi:10.1053/j.gastro.2018.08.061
251. Kasprowicz V, Kang YH, Lucas M, et al. Hepatitis C Virus (HCV) sequence variation induces an HCV-specific T-cell phenotype analogous to spontaneous resolution. J Virol. 2010;84(3):1656–1663. doi:10.1128/JVI.01499-09
252. Sekyere SO, Suneetha PV, Kraft ARM, et al. A heterogeneous hierarchy of co-regulatory receptors regulates exhaustion of HCV-specific CD8 T cells in patients with chronic hepatitis C. J Hepatol. 2015;62(1):31–40. doi:10.1016/j.jhep.2014.08.008
253. Blackburn SD, Shin H, Haining WN, et al. Coregulation of CD8(+) T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat Immunol. 2009;10(1):29–37. doi:10.1038/ni.1679
254. Wedemeyer H, He XS, Nascimbeni M, et al. Impaired effector function of hepatitis C virus-specific CD8(+) T cells in chronic hepatitis C virus infection. Journal of Immunology. 2002;169(6):3447–3458. doi:10.4049/jimmunol.169.6.3447
255. Schurich A, Pallett LJ, Jajbhay D, et al. Distinct metabolic requirements of exhausted and functional virus-specific CD8 T cells in the same host. Cell Rep. 2016;16(5):1243–1252. doi:10.1016/j.celrep.2016.06.078
256. Kared H, Fabre T, Bedard N, Bruneau J, Shoukry NH. Galectin-9 and IL-21 mediate cross-regulation between Th17 and treg cells during acute hepatitis C. PLoS Pathog. 2013;9(6). doi:10.1371/journal.ppat.1003422
257. Urbani S, Amadei B, Fisicaro P, et al. Outcome of acute hepatitis C is related to virus-specific CD4 function and maturation of antiviral memory CD8 responses. Hepatology. 2006;44(1):126–139. doi:10.1002/(ISSN)1527-3350
258. Kasprowicz V, Wiesch JSZ, Kuntzen T, et al. High level of PD-1 expression on hepatitis C virus (HCV)-specific CD8(+) and CD4(+) T cells during acute HCV infection, irrespective of clinical outcome. J Virol. 2008;82(6):3154–3160. doi:10.1128/JVI.02474-07
259. Radziewicz H, Ibegbu CC, Fernandez ML, et al. Liver-infiltrating lymphocytes in chronic human hepatitis C virus infection display an exhausted phenotype with high levels of PD-1 and low levels of CD127 expression. J Virol. 2007;81(6):2545–2553. doi:10.1128/JVI.02021-06
260. Penna A, Pilli M, Zerbini A, et al. Dysfunction and functional restoration of HCV-specific CD8 responses in chronic hepatitis C virus infection. Hepatology. 2007;45(3):588–601. doi:10.1002/(ISSN)1527-3350
261. Golden-Mason L, Palmer B, Klarquist J, Mengshol JA, Castelblanco N, Rosen HR. Upregulation of PD-1 expression on circulating and intrahepatic hepatitis C virus-specific CD8(+) T cells associated with reversible immune dysfunction. J Virol. 2007;81(17):9249–9258. doi:10.1128/JVI.00409-07
262. Tacke RS, Lee HC, Goh C, et al. Myeloid suppressor cells induced by hepatitis C virus suppress T-cell responses through the production of reactive oxygen species. Hepatology. 2012;55(2):343–353. doi:10.1002/hep.24700
263. Sreenarasimhaiah J, Jaramillo A, Crippin J, Lisker-Melman M, Chapman WC, Mohanakumar T. Lack of optimal T-cell reactivity against the hepatitis C virus is associated with the development of fibrosis/cirrhosis during chronic hepatitis. Hum Immunol. 2003;64(2):224–230. doi:10.1016/S0198-8859(02)00781-4
264. Osburn WO, Snider AE, Wells BL, et al. Clearance of hepatitis C infection is associated with the early appearance of broad neutralizing antibody responses. Hepatology. 2014;59(6):2140–2151. doi:10.1002/hep.v59.6
265. Pestka JM, Zeisel MB, Blaser E, et al. Rapid induction of virus-neutralizing antibodies and viral clearance in a single-source outbreak of hepatitis C. Proc Natl Acad Sci U S A. 2007;104(14):6025–6030. doi:10.1073/pnas.0607026104
266. Abdel-Hakeem MS, Shoukry NH. Protective immunity against hepatitis C: many shades of gray. Front Immunol. 2014;5:274.
267. Mankowski MC, Kinchen VJ, Wasilewski LN, et al. Synergistic anti-HCV broadly neutralizing human monoclonal antibodies with independent mechanisms. Proc Natl Acad Sci U S A. 2018;115(1):E82–E91. doi:10.1073/pnas.1718441115
268. Mohanty A, Salameh S, Butt AA. Impact of direct acting antiviral agent therapy upon extrahepatic manifestations of hepatitis C virus infection. Curr Hiv-Aids Rep. 2019;16(5):389–394. doi:10.1007/s11904-019-00466-1
269. Eming SA, Wynn TA, Martin P. Inflammation and metabolism in tissue repair and regeneration. Science. 2017;356(6342):1026–1030. doi:10.1126/science.aam7928
270. Koyama Y, Brenner DA. Liver inflammation and fibrosis. J Clin Invest. 2017;127(1):55–64. doi:10.1172/JCI88881
271. Anstee QM, Reeves HL, Kotsiliti E, Govaere O, Heikenwalder M. From NASH to HCC: current concepts and future challenges. Nat Rev Gastroenterol Hepatol. 2019;16(7):411–428.
272. Grivennikov SI, Greten FR, Karin M. Immunity, Inflammation, and Cancer. Cell. 2010;140(6):883–899. doi:10.1016/j.cell.2010.01.025
273. Nakagawa H, Maeda S, Yoshida H, et al. Serum IL-6 levels and the risk for hepatocarcinogenesis in chronic hepatitis C patients: an analysis based on gender differences. Int J Cancer. 2009;125(10):2264–2269. doi:10.1002/ijc.v125:10
274. Aroucha DCBL, Do Carmo RF, Moura P, et al. High tumor necrosis factor-alpha/interleukin-10 ratio is associated with hepatocellular carcinoma in patients with chronic hepatitis C. Cytokine. 2013;62(3):421–425. doi:10.1016/j.cyto.2013.03.024
275. Ramzan M, Sturm N, Decaens T, et al. Liver-infiltrating CD8(+) lymphocytes as prognostic factor for tumour recurrence in hepatitis C virus-related hepatocellular carcinoma. Liver Int. 2016;36(3):434–444. doi:10.1111/liv.12927
276. Piconese S, Timperi E, Pacella I, et al. Human OX40 tunes the function of regulatory T cells in tumor and nontumor areas of hepatitis C virus-infected liver tissue. Hepatology. 2014;60(5):1494–1507. doi:10.1002/hep.v60.5
277. Collins JM, Raphael KL, Terry C, et al. Hepatitis B virus reactivation during successful treatment of hepatitis C virus with sofosbuvir and simeprevir. Clin Infect Dis. 2015;61(8):1304–1306. doi:10.1093/cid/civ474
278. Perello C, Fernandez-Carrillo C, Londono MC, et al. Reactivation of herpesvirus in patients with hepatitis C treated with direct-acting antiviral agents. Clin Gastroenterol H. 2016;14(11):1662. doi:10.1016/j.cgh.2016.05.016
279. Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med. 2003;348(17):1625–1638. doi:10.1056/NEJMoa021423
280. Dyson J, Jaques B, Chattopadyhay D, et al. Hepatocellular cancer: the impact of obesity, type 2 diabetes and a multidisciplinary team. J Hepatol. 2014;60(1):110–117. doi:10.1016/j.jhep.2013.08.011
281. Huang TS, Lin CL, Lu MJ, et al. Diabetes, hepatocellular carcinoma, and mortality in hepatitis C-infected patients: a population-based cohort study. J Gastroenterol Hepatol. 2017;32(7):1355–1362. doi:10.1111/jgh.13670
282. Raff EJ, Kakati D, Bloomer JR, Shoreibah M, Rasheed K, Singal AK. Diabetes mellitus predicts occurrence of cirrhosis and hepatocellular cancer in alcoholic liver and non-alcoholic fatty liver diseases. J Clin Transl Hepatol. 2015;3(1):9–16.
283. Li X, Xu H, Gao Y, Pan M, Wang L, Gao P. Diabetes mellitus increases the risk of hepatocellular carcinoma in treatment-naive chronic hepatitis C patients in China. Medicine (Baltimore). 2017;96(13):e6508. doi:10.1097/MD.0000000000006508
284. Rao H, Wu E, Fu S, et al. The higher prevalence of truncal obesity and diabetes in American than Chinese patients with chronic hepatitis C might contribute to more rapid progression to advanced liver disease. Aliment Pharmacol Ther. 2017;46(8):731–740. doi:10.1111/apt.14273
285. Donato F, Tagger A, Gelatti U, et al. Alcohol and hepatocellular carcinoma: the effect of lifetime intake and hepatitis virus infections in men and women. Am J Epidemiol. 2002;155(4):323–331. doi:10.1093/aje/155.4.323
286. Hagstrom H. Alcohol consumption in concomitant liver disease: how much is too much? Curr Hepatol Rep. 2017;16(2):152–157. doi:10.1007/s11901-017-0343-0
287. Singal AK, Anand BS. Mechanisms of synergy between alcohol and hepatitis C virus. J Clin Gastroenterol. 2007;41(8):761–772. doi:10.1097/MCG.0b013e3180381584
288. Kruse RL, Kramer JR, Tyson GL, et al. Clinical outcomes of hepatitis B virus coinfection in a united states cohort of hepatitis C virus-infected patients. Hepatology. 2014;60(6):1871–1878. doi:10.1002/hep.v60.6
289. Kramer JR, Kowalkowski MA, Duan ZG, Chiao EY. The effect of HIV viral control on the incidence of hepatocellular carcinoma in veterans with hepatitis C and HIV coinfection. Jaids-J Acq Imm Def. 2015;68(4):456–462. doi:10.1097/QAI.0000000000000494
290. Ioannou GN, Bryson CL, Weiss NS, Scott JD, Boyko EJ. The prevalence of cirrhosis and hepatocellular carcinoma in patients with human immunodeficiency virus infection: are the data found in this sample applicable to other settings? Reply. Hepatology. 2013;57(6):2545. doi:10.1002/hep.26171
291. Gjaerde LI, Shepherd L, Jablonowska E, et al. Trends in incidences and risk factors for hepatocellular carcinoma and other liver events in HIV and hepatitis C virus-coinfected individuals from 2001 to 2014: a multicohort study. Clin Infect Dis. 2016;63(6):821–829. doi:10.1093/cid/ciw380
292. McBurney MI, Van Soest PJ, Jeraci JL. Colonic carcinogenesis: the microbial feast or famine mechanism. Nutr Cancer. 1987;10(1–2):23–28. doi:10.1080/01635588709513937
293. Dalmasso G, Cougnoux A, Delmas J, Darfeuille-Michaud A, Bonnet R. The bacterial genotoxin colibactin promotes colon tumor growth by modifying the tumor microenvironment. Gut Microbes. 2014;5(5):675–680. doi:10.4161/19490976.2014.969989
294. Kipanyula MJ, Seke Etet PF, Vecchio L, Farahna M, Nukenine EN, Nwabo Kamdje AH. Signaling pathways bridging microbial-triggered inflammation and cancer. Cell Signal. 2013;25(2):403–416. doi:10.1016/j.cellsig.2012.10.014
295. Kofahi HM, Taylor NG, Hirasawa K, Grant MD, Russell RS. Hepatitis C virus infection of cultured human hepatoma cells causes apoptosis and pyroptosis in both infected and bystander cells. Sci Rep. 2016;6:37433. doi:10.1038/srep37433
296. Vinken M, Vanhaecke T, Papeleu P, Snykers S, Henkens T, Rogiers V. Connexins and their channels in cell growth and cell death. Cell Signal. 2006;18(5):592–600. doi:10.1016/j.cellsig.2005.08.012
297. Nicholson B, Dermietzel R, Teplow D, Traub O, Willecke K, Revel JP. Two homologous protein components of hepatic gap junctions. Nature. 1987;329(6141):732–734. doi:10.1038/329732a0
298. Vinken M, Henkens T, De Rop E, Fraczek J, Vanhaecke T, Rogiers V. Biology and pathobiology of gap junctional channels in hepatocytes. Hepatology. 2008;47(3):1077–1088. doi:10.1002/hep.22049
299. Krutovskikh V, Mazzoleni G, Mironov N, et al. Altered homologous and heterologous gap-junctional intercellular communication in primary human liver tumors associated with aberrant protein localization but not gene mutation of connexin 32. Int J Cancer. 1994;56(1):87–94. doi:10.1002/ijc.2910560116
300. Omori Y, Krutovskikh V, Mironov N, Tsuda H, Yamasaki H. Cx32 gene mutation in a chemically induced rat liver tumour. Carcinogenesis. 1996;17(9):2077–2080. doi:10.1093/carcin/17.9.2077
301. Kawasaki Y, Omori Y, Li Q, et al. Cytoplasmic accumulation of connexin32 expands cancer stem cell population in human HuH7 hepatoma cells by enhancing its self-renewal. Int J Cancer. 2011;128(1):51–62. doi:10.1002/ijc.v128:1
302. Sinyuk M, Mulkearns-Hubert EE, Reizes O, Lathia J. Cancer connectors: connexins, gap junctions, and communication. Front Oncol. 2018;8:646. doi:10.3389/fonc.2018.00646
303. Prise KM, O’Sullivan JM. Radiation-induced bystander signalling in cancer therapy. Nat Rev Cancer. 2009;9(5):351–360. doi:10.1038/nrc2603
304. Verma N, Tiku AB. Significance and nature of bystander responses induced by various agents. Mutat Res. 2017;773:104–121. doi:10.1016/j.mrrev.2017.05.003
305. Eghbali B, Kessler JA, Reid LM, Roy C, Spray DC. Involvement of gap junctions in tumorigenesis: transfection of tumor cells with connexin 32 cDNA retards growth in vivo. Proc Natl Acad Sci U S A. 1991;88(23):10701–10705. doi:10.1073/pnas.88.23.10701
306. Evert M, Ott T, Temme A, Willecke K, Dombrowski F. Morphology and morphometric investigation of hepatocellular preneoplastic lesions and neoplasms in connexin32-deficient mice. Carcinogenesis. 2002;23(5):697–703. doi:10.1093/carcin/23.5.697
307. Naus CC, Laird DW. Implications and challenges of connexin connections to cancer. Nat Rev Cancer. 2010;10(6):435–441. doi:10.1038/nrc2841
308. Aasen T, Mesnil M, Naus CC, Lampe PD, Laird DW. Gap junctions and cancer: communicating for 50 years. Nat Rev Cancer. 2017;17(1):74. doi:10.1038/nrc.2016.142
309. Ioannou GN, Green PK, Beste LA, Mun EJ, Kerr KF, Berry K. Development of models estimating the risk of hepatocellular carcinoma after antiviral treatment for hepatitis C. J Hepatol. 2018;69(5):1088–1098. doi:10.1016/j.jhep.2018.07.024
310. D’Amico G, Garcia-Tsao G, Pagliaro L. Natural history and prognostic indicators of survival in cirrhosis: a systematic review of 118 studies. J Hepatol. 2006;44(1):217–231. doi:10.1016/j.jhep.2005.10.013
311. Akhtar E, Manne V, Saab S. Cirrhosis regression in hepatitis C patients with sustained virological response after antiviral therapy: a meta-analysis. Liver Int. 2015;35(1):30–36. doi:10.1111/liv.12576
312. Arif A, Levine RA, Sanderson SO, et al. Regression of fibrosis in chronic hepatitis C after therapy with interferon and ribavirin. Digest Dis Sci. 2003;48(7):1425–1430. doi:10.1023/A:1024196201684
313. Poynard T, McHutchison J, Manns M, et al. Impact of pegylated interferon alfa-2b and ribavirin on liver fibrosis in patients with chronic hepatitis C. Gastroenterology. 2002;122(5):1303–1313. doi:10.1053/gast.2002.33023
314. Shiratori Y, Imazeki F, Moriyama M, et al. Histologic improvement of fibrosis in patients with hepatitis C who have sustained response to interferon therapy. Ann Intern Med. 2000;132(7):517–524. doi:10.7326/0003-4819-132-7-200004040-00002
315. Grgurevic I, Bozin T, Madir A. Hepatitis C is now curable, but what happens with cirrhosis and portal hypertension afterwards? Clin Exp Hepatol. 2017;3(4):181–186. doi:10.5114/ceh.2017.71491
316. Rockey DC. Fibrosis reversal after hepatitis C virus elimination. Curr Opin Gastroen. 2019;35(3):137–144. doi:10.1097/MOG.0000000000000524
317. D’Ambrosio R, Aghemo A, Rumi MG, et al. A morphometric and immunohistochemical study to assess the benefit of a sustained virological response in hepatitis C virus patients with cirrhosis. Hepatology. 2012;56(2):532–543. doi:10.1002/hep.25606
318. D’Ambrosio R, Aghemo A, Fraquelli M, et al. The diagnostic accuracy of Fibroscan (R) for cirrhosis is influenced by liver morphometry in HCV patients with a sustained virological response. J Hepatol. 2013;59(2):251–256. doi:10.1016/j.jhep.2013.03.013
319. Bachofner JA, Valli PV, Kroger A, et al. Direct antiviral agent treatment of chronic hepatitis C results in rapid regression of transient elastography and fibrosis markers fibrosis-4 score and aspartate aminotransferase-platelet ratio index. Liver Int. 2017;37(3):369–376. doi:10.1111/liv.2017.37.issue-3
320. Sporea I, Lupusoru R, Mare R, et al. Dynamics of liver stiffness values by means of transient elastography in patients with HCV liver cirrhosis undergoing interferon free treatment. J Gastrointest Liver. 2017;26(2):145–150.
321. Pockros P, Crissien-Martinez AM, Frenette C, et al. Degree of liver fibrosis regression predicted by transient elastography after cure of chronic hepatitis C with direct acting antivirals is overestimated but confirmed by liver biopsy. J Hepatol. 2017;66(1):S108. doi:10.1016/S0168-8278(17)30475-0
322. Afdhal N, Everson GT, Calleja JL, et al. Effect of viral suppression on hepatic venous pressure gradient in hepatitis C with cirrhosis and portal hypertension. J Viral Hepatitis. 2017;24(10):823–831. doi:10.1111/jvh.2017.24.issue-10
323. Fehily SR, Papaluca T, Thompson AJ. Long-term impact of direct-acting antiviral agent therapy in HCV cirrhosis: critical review. Semin Liver Dis. 2019;39(3):341–353. doi:10.1055/s-0039-1685538
324. Tacke F, Boeker KHW, Klinker H, et al. Baseline risk factors determine lack of biochemical response after SVR in chronic hepatitis C patients treated with DAAs. Liver Int. 2019;40:539–548.
325. Hamdane N, Juhling F, Crouchet E, et al. HCV-induced epigenetic changes associated with liver cancer risk persist after sustained virologic response. Gastroenterology. 2019;156(8):2313. doi:10.1053/j.gastro.2019.02.038
326. Perez S, Kaspi A, Domovitz T, et al. Hepatitis C virus leaves an epigenetic signature post cure of infection by direct-acting antivirals. PLoS Genet. 2019;15(6):e1008181. doi:10.1371/journal.pgen.1008181
327. Li MM, Tang YQ, Gong YF, et al. Development of an oncogenic dedifferentiation SOX signature with prognostic significance in hepatocellular carcinoma. BMC Cancer. 2019;19(1):851. doi:10.1186/s12885-019-6041-2
328. Santangelo L, Bordoni V, Montaldo C, et al. Hepatitis C virus direct-acting antivirals therapy impacts on extracellular vesicles microRNAs content and on their immunomodulating properties. Liver Int. 2018;38(10):1741–1750. doi:10.1111/liv.13700
329. Koberle V, Waidmann O, Kronenberger B, et al. Serum microRNA-122 kinetics in patients with chronic hepatitis C virus infection during antiviral therapy. J Viral Hepatitis. 2013;20(8):530–535. doi:10.1111/jvh.2013.20.issue-8
330. Villani R, Facciorusso A, Bellanti F, et al. DAAs rapidly reduce inflammation but increase serum VEGF level: a rationale for tumor risk during anti-HCV treatment. PLoS One. 2016;11(12):e0167934. doi:10.1371/journal.pone.0167934
331. Faillaci F, Marzi L, Critelli R, et al. Liver angiopoietin-2 is a key predictor of de novo or recurrent hepatocellular cancer after hepatitis C virus direct-acting antivirals. Hepatology. 2018;68(3):1010–1024. doi:10.1002/hep.29911
332. Meissner EG, Wu D, Osinusi A, et al. Endogenous intrahepatic IFNs and association with IFN-free HCV treatment outcome. J Clin Invest. 2014;124(8):3352–3363. doi:10.1172/JCI75938
333. Alao H, Cam M, Keembiyehetty C, et al. Baseline intrahepatic and peripheral innate immunity are associated with hepatitis C virus clearance during direct-acting antiviral therapy. Hepatology. 2018;68(6):2078–2088. doi:10.1002/hep.29921
334. Sung PS, Lee EB, Park DJ, et al. Interferon-free treatment for hepatitis C virus infection induces normalization of extrahepatic type I interferon signaling. Clin Mol Hepatol. 2018;24(3):302–310. doi:10.3350/cmh.2017.0074
335. Debes JD, van Tilborg M, Groothuismink ZMA, et al. Levels of cytokines in serum associate with development of hepatocellular carcinoma in patients with HCV infection treated with direct-acting antivirals. Gastroenterology. 2018;154(3):515. doi:10.1053/j.gastro.2017.10.035
336. Golden-Mason L, Rosen HR. Natural killer cells: multifaceted players with key roles in hepatitis C immunity. Immunol Rev. 2013;255(1):68–81. doi:10.1111/imr.2013.255.issue-1
337. Golden-Mason L, McMahan RH, Kriss MS, et al. Early and late changes in natural killer cells in response to ledipasvir/sofosbuvir treatment. Hepatol Commun. 2018;2(4):364–375. doi:10.1002/hep4.1166
338. Spaan M, van Oord G, Kreefft K, et al. Immunological analysis during interferon-free therapy for chronic hepatitis C virus infection reveals modulation of the natural killer cell compartment. J Infect Dis. 2016;213(2):216–223. doi:10.1093/infdis/jiv391
339. Stevenson TJ, Barbour Y, McMahon BJ, et al. Observed changes in natural killer and T cell phenotypes with evaluation of immune outcome in a longitudinal cohort following sofosbuvir-based therapy for chronic hepatitis C infection. Open Forum Infect Dis. 2019;6(6):ofz223. doi:10.1093/ofid/ofz223
340. Kamimura H, Yamagiwa S, Tsuchiya A, et al. Reduced NKG2D ligand expression in hepatocellular carcinoma correlates with early recurrence. J Hepatol. 2012;56(2):381–388. doi:10.1016/j.jhep.2011.06.017
341. Chu PS, Nakamoto N, Taniki N, et al. On-treatment decrease of NKG2D correlates to early emergence of clinically evident hepatocellular carcinoma after interferon-free therapy for chronic hepatitis C. PLoS One. 2017;12(6). doi:10.1371/journal.pone.0179096
342. Li JJ, Pan K, Gu MF, et al. Prognostic value of soluble MICA levels in the serum of patients with advanced hepatocellular carcinoma. Chin J Cancer. 2013;32(3):141–148. doi:10.5732/cjc.012.10025
343. Goto K, Kato N. MICA SNPs and the NKG2D system in virus-induced HCC. J Gastroenterol. 2015;50(3):261–272. doi:10.1007/s00535-014-1000-9
344. Sachdeva M, Chawla YK, Arora SK. Dendritic cells: the warriors upfront-turned defunct in chronic hepatitis C infection. World J Hepatol. 2015;7(19):2202–2208. doi:10.4254/wjh.v7.i19.2202
345. Crosignani A, Riva A, Della Bella S. Analysis of peripheral blood dendritic cells as a non-invasive tool in the follow-up of patients with chronic hepatitis C. World J Gastroenterol. 2016;22(4):1393–1404. doi:10.3748/wjg.v22.i4.1393
346. Dou L, Ono Y, Chen YF, Thomson AW, Chen XP. Hepatic dendritic cells, the tolerogenic liver environment, and liver disease. Semin Liver Dis. 2018;38(2):170–180. doi:10.1055/s-0038-1646949
347. Laursen TL, Siggard CB, Kazankov K, et al. Rapid and persistent decline in soluble CD163 with successful direct-acting antiviral therapy and associations with chronic hepatitis C histology. Scand J Gastroenterol. 2018;53(8):986–993. doi:10.1080/00365521.2018.1481996
348. Kostadinova L, Shive CL, Zebrowski E, et al. Soluble markers of immune activation differentially normalize and selectively associate with improvement in AST, ALT, albumin, and transient elastography during IFN-free HCV therapy. Pathog Immun. 2018;3(1):149–163. doi:10.20411/pai.v3i1
349. Bolte FJ, O’Keefe AC, Webb LM, et al. Intra-Hepatic depletion of mucosal-associated invariant T cells in hepatitis C virus-induced liver inflammation. Gastroenterology. 2017;153(5):1392. doi:10.1053/j.gastro.2017.07.043
350. Hengst J, Strunz B, Deterding K, et al. Nonreversible MAIT cell-dysfunction in chronic hepatitis C virus infection despite successful interferon-free therapy. Eur J Immunol. 2016;46(9):2204–2210. doi:10.1002/eji.v46.9
351. Cannizzo ES, Cerrone M, Merlini E, et al. Successful direct-acting antiviral therapy in HIV/HCV co-infected patients fails to restore circulating mucosal-associated invariant T cells. Eur J Immunol. 2019;49(7):1127–1129. doi:10.1002/eji.201948152
352. Walker MR, Leung P, Eltahla AA, et al. Clearance of hepatitis C virus is associated with early and potent but narrowly-directed, Envelope-specific antibodies. Sci Rep. 2019;9(1):13300. doi:10.1038/s41598-019-49454-w
353. Burchill MA, Golden-Mason L, Wind-Rotolo M, Rosen HR. Memory re-differentiation and reduced lymphocyte activation in chronic HCV-infected patients receiving direct-acting antivirals. J Viral Hepatitis. 2015;22(12):983–991. doi:10.1111/jvh.2015.22.issue-12
354. Wieland D, Kemming J, Schuch A, et al. TCF1(+) hepatitis C virus-specific CD8(+) T cells are maintained after cessation of chronic antigen stimulation. Nat Commun. 2017;8. doi:10.1038/ncomms15050
355. Aregay A, Owusu Sekyere S, Deterding K, et al. Elimination of hepatitis C virus has limited impact on the functional and mitochondrial impairment of HCV-specific CD8+ T cell responses. J Hepatol. 2019;71(5):889–899. doi:10.1016/j.jhep.2019.06.025
356. Vranjkovic A, Deonarine F, Kaka S, Angel JB, Cooper CL, Crawley AM. Direct-acting antiviral treatment of HCV infection does not resolve the dysfunction of circulating CD8(+) T-cells in advanced liver disease. Front Immunol. 2019;10:1926. doi:10.3389/fimmu.2019.01926
357. Schulze Zur Wiesch J, Ciuffreda D, Lewis-Ximenez L, et al. Broadly directed virus-specific CD4+ T cell responses are primed during acute hepatitis C infection, but rapidly disappear from human blood with viral persistence. J Exp Med. 2012;209(1):61–75. doi:10.1084/jem.20100388
358. Keoshkerian E, Hunter M, Cameron B, et al. Hepatitis C-specific effector and regulatory CD4 T-cell responses are associated with the outcomes of primary infection. J Viral Hepat. 2016;23(12):985–993. doi:10.1111/jvh.2016.23.issue-12
359. Claassen MA, de Knegt RJ, Janssen HL, Boonstra A. Retention of CD4+ CD25+ FoxP3+ regulatory T cells in the liver after therapy-induced hepatitis C virus eradication in humans. J Virol. 2011;85(11):5323–5330. doi:10.1128/JVI.02551-10
360. Spaan M, Claassen MA, Hou J, Janssen HL, de Knegt RJ, Boonstra A. The intrahepatic T cell compartment does not normalize years after therapy-induced hepatitis C virus eradication. J Infect Dis. 2015;212(3):386–390. doi:10.1093/infdis/jiv059
361. Langhans B, Nischalke HD, Kramer B, et al. Increased peripheral CD4(+) regulatory T cells persist after successful direct-acting antiviral treatment of chronic hepatitis C. J Hepatol. 2017;66(5):888–896. doi:10.1016/j.jhep.2016.12.019
362. Ghosh A, Mondal RK, Romani S, et al. Persistent gamma delta T-cell dysfunction in chronic HCV infection despite direct-acting antiviral therapy induced cure. J Viral Hepatitis. 2019;26(9):1105–1116.
363. Comarmond C, Garrido M, Pol S, et al. Direct-acting antiviral therapy restores immune tolerance to patients with hepatitis C virus-induced cryoglobulinemia vasculitis. Gastroenterology. 2017;152(8):2052. doi:10.1053/j.gastro.2017.02.037
364. Saadoun D, Pol S, Ferfar Y, et al. Efficacy and safety of sofosbuvir plus daclatasvir for treatment of HCV-associated cryoglobulinemia vasculitis. Gastroenterology. 2017;153(1):49. doi:10.1053/j.gastro.2017.03.006
365. Chang LY, Li Y, Kaplan DE. Hepatitis C viraemia reversibly maintains subset of antigen-specific T-bet plus tissue-like memory B cells. J Viral Hepatitis. 2017;24(5):389–396. doi:10.1111/jvh.12659
366. Castera L, Forns X, Alberti A. Non-invasive evaluation of liver fibrosis using transient elastography. J Hepatol. 2008;48(5):835–847. doi:10.1016/j.jhep.2008.02.008
367. Lin ZH, Xin YN, Dong QJ, et al. Performance of the aspartate aminotransferase-to-platelet ratio index for the staging of hepatitis C-related fibrosis: an updated meta-analysis. Hepatology. 2011;53(3):726–736. doi:10.1002/hep.24105
368. Sterling RK, Lissen E, Clumeck N, et al. Development of a simple noninvasive index to predict significant fibrosis in patients with HIV/HCV coinfection. Hepatology. 2006;43(6):1317–1325. doi:10.1002/(ISSN)1527-3350
369. European Association for Study of Liver. EASL-ALEH Clinical Practice Guidelines: non-invasive tests for evaluation of liver disease severity and prognosis. J Hepatol. 2015;63(1):237–264. doi:10.1016/j.jhep.2015.04.006
370. Singal A, Volk ML, Waljee A, et al. Meta-analysis: surveillance with ultrasound for early-stage hepatocellular carcinoma in patients with cirrhosis. Aliment Pharmacol Ther. 2009;30(1):37–47. doi:10.1111/apt.2009.30.issue-1
371. Holmberg SD, Spradling PR, Moorman AC, Denniston MM. Hepatitis C in the United States. New Engl J Med. 2013;368(20):1859–1861. doi:10.1056/NEJMp1302973
372. Cox AL. MEDICINE. Global control of hepatitis C virus. Science. 2015;349(6250):790–791. doi:10.1126/science.aad1302
373. Sarrazin C, Isakov V, Svarovskaia ES, et al. Late relapse versus hepatitis C virus reinfection in patients with sustained virologic response after sofosbuvir-based therapies. Clin Infect Dis. 2017;64(1):44–52. doi:10.1093/cid/ciw676
374. Abdel-Hakeem MS, Bedard N, Murphy D, Bruneau J, Shoukry NH. Signatures of protective memory immune responses during hepatitis C virus reinfection. Gastroenterology. 2014;147(4):870–U261. doi:10.1053/j.gastro.2014.07.005
375. Abdel-Hakeem MS, Boisvert M, Bruneau J, Soudeyns H, Shoukry NH. Selective expansion of high functional avidity memory CD8 T cell clonotypes during hepatitis C virus reinfection and clearance. PLoS Pathog. 2017;13(2):e1006191. doi:10.1371/journal.ppat.1006191
376. Osburn WO, Fisher BE, Dowd KA, et al. Spontaneous control of primary hepatitis C Virus infection and immunity against persistent reinfection. Gastroenterology. 2010;138(1):315–324. doi:10.1053/j.gastro.2009.09.017
377. Choo QL, Kuo G, Ralston R, et al. Vaccination of chimpanzees against infection by the hepatitis-C virus. P Natl Acad Sci USA. 1994;91(4):1294–1298. doi:10.1073/pnas.91.4.1294
378. Meunier JC, Gottwein JM, Houghton M, et al. Vaccine-induced cross-genotype reactive neutralizing antibodies against hepatitis C virus. J Infect Dis. 2011;204(8):1186–1190. doi:10.1093/infdis/jir511
379. Frey SE, Houghton M, Coates S, et al. Safety and immunogenicity of HCV E1E2 vaccine adjuvanted with MF59 administered to healthy adults. Vaccine. 2010;28(38):6367–6373. doi:10.1016/j.vaccine.2010.06.084
380. Folgori A, Capone S, Ruggeri L, et al. A T-cell HCV vaccine eliciting effective immunity against heterologous virus challenge in chimpanzees. Nat Med. 2006;12(2):190–197. doi:10.1038/nm1353
381. Barnes E, Folgori A, Capone S, et al. Novel adenovirus-based vaccines induce broad and sustained T cell responses to HCV in man. Sci Transl Med. 2012;4(115):115ra1–115ra1. doi:10.1126/scitranslmed.3003155
382. Swadling L, Capone S, Antrobus RD, et al. A human vaccine strategy based on chimpanzee adenoviral and MVA vectors that primes, boosts, and sustains functional HCV-specific T cell memory. Sci Transl Med. 2014;6:261. doi:10.1126/scitranslmed.3009185
383. Dash S, Aydin Y, Stephens CM. Viral Polymerases: Structures, Functions and Roles as Antiviral Drug Targets. In: Gupta SP, editor. Chapter 8 - Hepatitis C Virus NS5B RNA-Dependent RNA Polymerase Inhibitor: An Integral Part of HCV Antiviral Therapy. Cambridge, MA: Academic Press. 2019;211–235.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.