")
Back to Journals » Journal of Blood Medicine » Volume 13
Hemophilic Arthropathy: Barriers to Early Diagnosis and Management
Authors Cuesta-Barriuso R , Donoso-Úbeda E , Meroño-Gallut J , Ucero-Lozano R , Pérez-Llanes R
Received 6 May 2022
Accepted for publication 29 September 2022
Published 17 October 2022 Volume 2022:13 Pages 589—601
DOI https://doi.org/10.2147/JBM.S343924
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Martin H Bluth
Rubén Cuesta-Barriuso,1,2 Elena Donoso-Úbeda,3 Javier Meroño-Gallut,4 Roberto Ucero-Lozano,5 Raúl Pérez-Llanes3
1Department of Surgery and Medical-Surgical Specialties, University of Oviedo, Oviedo, Spain; 2Royal Victoria Eugenia Foundation, Madrid, Spain; 3Department of Physiotherapy, Catholic University San Antonio-UCAM, Murcia, Spain; 4Physiotherapy Service, Tu Bienestar 360°, Murcia, Spain; 5Department of Physiotherapy, European University of Madrid, Madrid, Spain
Correspondence: Rubén Cuesta-Barriuso, Department of Surgery and Medical-Surgical Specialties, University of Oviedo, Campus de El Cristo s/n, Faculty of Medicine, Oviedo, 33006, Spain, Tel +34 985 103 386, Email [email protected]
Abstract: Hemophilia is a congenital coagulopathy characterized by a deficiency of one of the clotting factors. It is characterized by the development of hematomas and hemarthrosis, either spontaneously or after minor trauma. The recurrence of hemarthroses leads to progressive and degenerative joint damage from childhood (hemophilic arthropathy). This arthropathy is characterized by disabling physical effects that limit the functionality and quality of life of these patients. Medical progress achieved over the last decade in the drug treatment of hemophilia has improved the medium and long-term prospects of patients with more effective and long-lasting drugs. The universal use of safer, more effective and prolonged prophylactic treatments may promote the prevention of bleeding, and also therefore, of the development of hemarthrosis and joint damage. A number of imaging instruments have been developed for the assessment of hemarthrosis and hemophilic arthropathy, using ultrasound, magnetic resonance imaging and simple radiology. Different physical examination scores and questionnaires allow the assessment of joint health, self-perceived activity and functionality of patients with hemophilia. The approach to these patients should be interdisciplinary. Assessment of the processes that affect pain in these patients and the development of pain education models should be implemented. Expert advice and information to patients with hemophilia should be based on individual functional prevention diagnoses, advice on available therapies and sports practice, as well as health recommendations.
Keywords: hemophilia, joint disease, pain, diagnosis, management
Hemophilia. Overview and Clinical Manifestations
Hemophilia is a genetic bleeding disorder caused by a missing or defective clotting factor.1 Hemophilia is usually inherited via an affected X chromosome with a mutation of the FVIII or FIX gene. However, both genes are prone to new mutations and about 30% of all cases result from spontaneous genetic variants. Prospective studies have shown that more than 50% of newly diagnosed individuals with severe hemophilia have no family history of the disease2 Depending on the missing clotting factor, it can be classified hemophilia A (factor VIII) or hemophilia B (factor IX). Both types of hemophilia can be classified according to the plasma level of existing clotting factor: mild (5–40%), moderate (1–5%) or severe (<1%).3 The severity of bleeding in patients with hemophilia is proportional to the amount of residual factor activity in the blood. In patients with severe hemophilia, bleeding may occur spontaneously or with minor trauma. In patients with moderate or mild hemophilia, bleeding episodes occur after minor surgical interventions, requiring immediate treatment to prevent complications.4
Hemophilia is considered a rare disease. The estimated prevalence for hemophilia A is 24.6:100,000 live births, and 5.0:100,000 live births for hemophilia B.5
Uncontrolled bleeding in critical organs can lead to death in patients with hemophilia. However, the main clinical manifestation is the development of hemarthrosis These intra-articular bleeds are more common in knees, ankles and elbows.1 Similarly, the risk of hemarthrosis increases in cases of severe hemophilia with minimal trauma.6
Repeated bleeding in the same joint eventually leads to synovial hypertrophy, hemosiderin deposits, cartilage destruction and changes in the structure of the subchondral bone. Such hemosiderin deposits can stimulate further synovial hypertrophy and swelling.7 Several in vitro and in vivo animal studies have shown how blood has a direct effect on cartilage due to the combination of mononuclear cells and red blood cells present in the blood.8 The development of hemarthrosis induces chondrocyte apoptosis, which alters the cartilage matrix renewal. Hypervascularization occurs on the synovial tissue, initially synoviocyte hypertrophy, increasing the risk of new bleeding events and inflammatory changes.9 These inflammatory changes contribute to cartilage damage through the production of tissue-destroying enzymes and cytokines10 leading to joint destruction. A recent study by Zheng et al11 has shown how iron is involved in cartilage injury through regulation of FGF23 and SOX9 expression in chondrocytes in patients with hemophilia who have developed hemarthrosis. A high level of COL-18N has been noted in patients with hemophilia A12, which could be used as a potential marker to monitor the development of arthropathy, making it possible to adapt the best treatment to prevent further joint damage. The recurrence of hemarthrosis leads to the development of a degenerative and irreversible process known as hemophilic arthropathy.1,13 As a result, hemophilic arthropathy is accompanied by pain, loss of joint range and muscle atrophy, among other alterations.1 Figure 1 shows the joint damage process induced by the development of hemarthrosis.
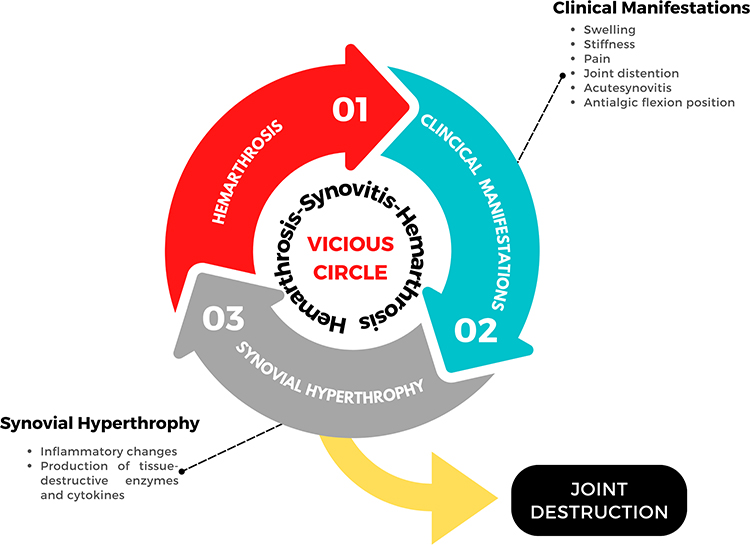 |
Figure 1 The hemarthrosis-synovitis-hemarthrosis vicious circle in hemophilia. |
The gold standard in the prevention of bleeding and hemophilic arthropathy is the intravenous prophylactic administration of the missing clotting factor.1 However, most patients with hemophilia worldwide can only access on-demand pharmacological treatment once bleeding has happened, with the consequent risk of joint damage.3 The main complication related to the treatment of hemophilia is the development of antibodies to clotting factor concentrates. These inhibitors react against the administered substitution factor, increasing the risk of serious bleeding and accelerating the degenerative damage caused by the recurrence of hemarthrosis.14
Prophylactic regimens are customized depending on the hemorrhagic phenotype, the pharmacokinetics of FVIII/FIX or the physical activity performed by the patient15 However, the short half-life of these factor VIII/FIX concentrates implies the need for frequent infusions. In addition, the limitations offered by the minimum blood levels of FVIII/FIX do not ensure complete protection against the development of hemarthrosis in the event of moderate-intensity trauma.16
In the last ten years, new FVIII/FIX concentrates have been designed to prolong their half-life: extended half-life treatment (EHL).17 This extended half-life can reduce the frequency of intravenous infusions by 30–35%,18 maintaining a higher minimum level of FVIII (3%) than with conventional products.19 Similarly, in patients with hemophilia B, a considerable increase in the terminal half-life has been described in pediatric subjects (median: 36.9 h) and over 13 years of age (median: 49.9 h).20
Search Strategy
Clinical trials were searched in various electronic databases: PubMed/MEDLINE, Web of Science, Embase, and the Cochrane Central Register of Controlled Trials (CENTRAL). For the drafting of the review, articles were extracted using the following keywords and search strategies: (hemophilia OR haemophilia), (joint disease OR haemophilic arthropathy), (pain OR pain management), and (diagnosis OR evaluation OR approach). Boolean operators (AND, OR, NOT) were used in the search.
Limitations in the Physical and Functional Approach and Evaluation in Patients with Hemophilia
The clinical symptoms of acute haemarthrosis are characterized by a rapid swelling where patients usually notice initial symptoms of stiffness, tingling and pain.21 Their physical examination reveals typical signs of swelling, hotness, redness of the surrounding skin, joint strain, muscle contracture and limitation of mobility.22 As a result of the hemorrhagic process, the joint becomes positioned in an antalgic flexion position, compensating for intra-articular pressure to reduce pain. Acute hemarthrosis do not usually cause secondary changes in the synovial cells, so limitations do not usually persist and the joint returns to its normal state.23
Considering the hemarthrosis recurrence pattern in patients with hemophilia, hemarthrosis tends to become chronic, generating chronic synovitis, inflammatory arthritis and progressive hemophilic arthropathy.24 This arthropathy is the cause of the progressive development of chronic pain, limitations in the range of motion and functional alterations causing disability in patients with hemophilia.25 Due to the serious sequelae of hemarthrosis in patients with hemophilia, early diagnosis and treatment is essential to minimize the evolution of arthropathy, with the functional and physical limitations that this process entails. Hence the importance of an early diagnosis and treatment in order to minimize the degenerative evolution to hemophilic arthropathy with the functional and physical limitations that such a process would entail.26
The World Federation of Hemophilia has been recommending for years the assessment of the degree of joint injury using radiological scoring systems and orthopedic exploration.27 The use of diagnostic imaging techniques makes it possible to classify the joint degenerative process into different clinical and pathological stages.28 The aim is to detect joint changes, determine their severity and evaluate the follow-up of therapeutic effects. The most commonly used techniques are ultrasound, simple radiology and magnetic resonance imaging (MRI).
Ultrasound is an inexpensive, easily available and excellent technique for determining the presence of soft tissue inflammation. In the same way, it allows detecting intra-articular fluid and evaluating the cartilage, being a highly useful tool for clinical follow-up and adapting treatments for bleeding and synovitis. Musculoskeletal ultrasound has shown high sensitivity in detecting low concentrations of intra-articular blood and in discriminating between bloody and non-bloody fluids. Thus, ultrasound is an ideal tool for the rapid detection of joint hemorrhage.29 Martinoli et al30 designed a simplified scoring system (HEAD-US) based on an additive scale to define the joint condition and offer a tool to assess the progression of the disease and monitor the outcome of treatment in follow-up studies. It has three markers for the main joints affected (knees, elbows and ankles): synovitis (score 0–2), cartilage (score 0–4) and subchondral bone (score 0–2), with a maximum score of 8 points per joint; the higher the score the more evolved the degenerative joint process. The literature has shown the importance of ultrasound in the early detection of arthropathic changes and its contribution to the clinical approach in patients with hemophilia.31 An association between the Joint tissue Activity and Damage Exam protocol and clinical and functional parameters has been observed,32 supporting the clinical value of musculoskeletal ultrasound as a complementary tool for the diagnosis and treatment of hemophilic arthropathy.
Simple radiology remains one of the most widely used methods for joint evaluation in hemophilic arthropathy, despite its limitations.33 Its main disadvantage is that in the initial stages it underestimates the degree of joint pathology and alterations in soft parts such as synovitis due to its low sensitivity for detecting problems in the synovial membrane and cartilage. Simple radiology should not be the method of choice for early assessment of the disease or to determine the evolution of a short-term treatment.34 Its main utility lies in the evaluation of adult patients with hemophilia who present with advanced joint pathology.35 The scoring system recommended by the WFH is the Pettersson system. It assesses 8 parameters with a maximum score of 13 points; each radiological change is assigned a score according to its severity.36 The higher the score, the poorer the joint condition.
MRI is the gold standard instrument for the study of joint changes in patients with hemophilia.37 It has been shown to be the most useful tool in the analysis of soft tissues and osteochondral alterations, present in the initial stages of hemophilic arthropathy. However, its high costs, its lower accessibility, the time required for its evaluation and its difficult applicability in children, limit its use and periodic follow-ups to determine the evolution of acute and chronic processes.26 The most widely accepted methods proposed for assessing joint damage with magnetic resonance imaging include the Denver scale38 and the European scale.39
The Denver group created a 5-phase joint evaluation scale assessing the presence of effusion or hemarthrosis, synovial hypertrophy with hemosiderin deposits, presence of cysts, bone erosions and cartilage destruction with joint impingement. The score is determined on the basis of the most significant observable deterioration with a range of 0 to 10 points. For its part, the European classification collects information on similar criteria with a more detailed reading procedure. The evaluation criteria are the existence or not of subchondral cysts, erosion, chondral lesion, joint effusion or hemarthrosis, synovial hypertrophy and hemosiderin deposition. This additive scale has a range of 0 to 28 points where the higher the score, the greater the joint destruction.
In relation to the diagnosis of the degree of joint injury by orthopedic examination, the World Federation of Hemophilia considers the use of different tools.
The Hemophilia Joint Health Score (HJHS) version 2.1, evaluates the joint condition of knees, ankles and elbows, in patients with hemophilia. This scale was designed to assess joint deterioration in pediatric patients with hemophilia,40 although a study has recently been published that shows its validity in the adult population.41 This measuring instrument measures 8 items: Swelling, Duration of swelling, Muscle Atrophy and Strength, Crepitus, Mobility and Joint Pain. The scoring range of this additive scale is 0–20 points in each joint.
The Gilbert screening protocol42 is a tool recommended by the World Federation of Hemophilia for the measurement of joint deterioration. This protocol consists of 7 items linked to anatomical destruction, biomechanical alterations and deformity. Each item has a value of 0, 1 or 2 points, so that the final value is presented in a range between 0 and 12 points, in knee and ankle, and from 0 to 10 in elbow, a higher score indicating greater joint deterioration. Among its limitations we find that it may not be appropriate in the assessment of young children and in mild arthropathies as it is not very sensitive to slight joint changes.43
The Hemophilia Activities List (HAL) is a questionnaire used for the measurement of participation and performance in daily activities of patients with hemophilia.44,45 With this instrument, the impact of the disease on self-perceived functional abilities in patients with hemophilia is evaluated, through 42 items grouped into seven domains: Lying down, sitting, kneeling and standing; Functions of the legs; Functions of the arms; Use of transportation; Self-care; Household tasks; and Leisure activities and sports. This scale has a score range from 0 to 100 points, where a low score indicates a greater impact of hemophilia on self-perceived functional abilities.
The Functional Independence Score in Hemophilia (FISH)46 is a questionnaire developed to measure the degree of disability in patients with hemophilia with three categories: Self-care (assesses the ability to perform basic activities of daily living; Transfers (assesses the ability to sit and stand); and Mobility (walking and step climbing). Eight activities (Eating; Dressing; Dressing; Transferring from chair; Squatting; Walking; Climbing stairs; and Running) are measured and scored from 1 to 4 with a maximum score of 32 points. The purpose of this instrument is to measure what the person with hemophilia can actually do, and not what he or she should be able to do or might be able to do if the circumstances were different. It is also used to evaluate the change or evolution of these patients over time or after a therapeutic intervention.
For its part, the Haemophilia & Exercise Project Test Questionnaire (HEP-Test-Q) is a measuring instrument that evaluates subjective physical performance.47 This Likert-like scale has five domains: Physical condition (comparing current physical activity to a year earlier), Mobility, Strength and coordination, Resistance and Body perception.
Figure 2 shows the main characteristics of the radiological and physical measuring instruments used in patients with hemophilia.
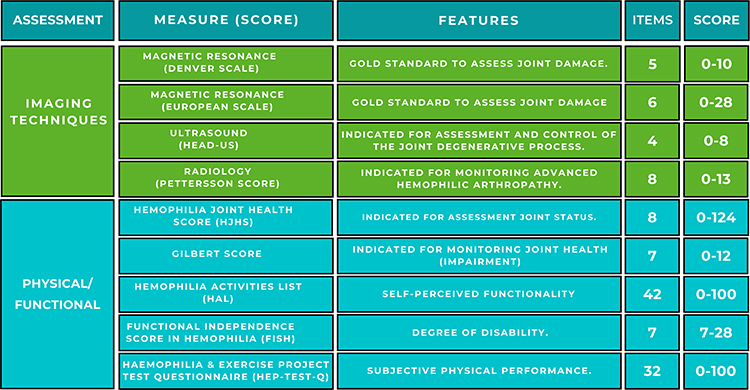 |
Figure 2 Measuring instruments used in the radiological and physical assessment of patients with hemophilia. |
Evaluation and Approach of Pain in Patients with Hemophilia
The International Association for the Study of Pain (IASP) defines pain as an unpleasant sensory and emotional experience associated with or similar to that associated with actual or potential tissue damage.48 Pain is recognized as a personal experience, influenced by biological, psychological and social factors. In the same way, it identifies nociception and pain, and addresses the learning of pain through life experience.
With this concept in mind, we must consider how to assess pain in a patient with hemophilia and how to treat it. Acute and chronic pain in patients with hemophilia has been evaluated with self-reporting tools.49 One-dimensional (analog or numerical) scales have been used to quantify the intensity of pain, and some non-specific multidimensional scales such as the McGill Pain Questionnaire or the Brief Pain Inventory.50
Specific (Hemophilia-Specific Quality of Life Index [Haem-a-Qol])51 and generic scales (36 and 12-Item Short Form Health Survey [SF-36 and SF-12]52 have been used to assess the quality of life of these patients. Other scales used are those that evaluate determining psychosocial factors in the experience of pain, such as catastrophism (Pain catastrophizing scale), kinesiophobia or fear of movement (Tampa Scale of Kinesiophobia), or anxiety (State-Trait Anxiety inventory questionnaire [STAI].53
The Multidimensional Hemophilia Pain Questionnaire [MHPQ]50 is a specific scale for patients with hemophilia, developed from a biopsychosocial perspective. This scale evaluates different pain dimensions (location, duration, frequency, trigger factors, intensity, interference in the patient’s life, management strategies used by patients and by professionals, and satisfaction). Although this scale is very comprehensive, it does not assess other psychosocial aspects such as kinesiophobia or catastrophism.
The use of specific scales improves sensitivity with respect to the study population. However, they prevent comparing the situation of these patients with other populations with similar pathologies. This makes it difficult to find normative values in a population without pathology with which to compare results.
The use of these scales and questionnaires offer valuable information regarding the patient’s painful experience and should be part of the assessment process. However, they do not report aspects such as the functioning of the nervous system or the processing of information whose output is the patient’s pain. Neither do these scales assess the descending inhibitory mechanisms of the patient. It would further be important to establish if there is nociceptive information that is being perceived and relevant to the brain and its bodily representation.54
The literature discloses multiple studies focused on the evaluation and treatment of pain in patients with hemophilia.55 By contrast, no evaluation models are proposed that assess the processes that influence pain. A number of clinical practice guidelines propose a treatment based on analgesics56 understanding pain explained by its intensity, and not as an experience influenced by brain inputs and outputs. Although Auerswald et al57 pointed out the absence of a multidisciplinary approach by pain management specialists, much progress is still to be made. Health professionals treating patients with hemophilia should receive education in pain and this could facilitate a transition from the obsolete Cartesian stimulus-response approach to a biopsychosocial model, a more popular approach to pain today.
Acute pain in hemophilia is associated with the development of joint bleeding, although imaging techniques have shown how it can develop in the absence of hemarthrosis.58 Acute pain linked to hemarthrosis and, therefore, clearly having a nociceptive mechanism, is treated conservatively according to the RICE protocol (Rest, Ice, Compression, Elevation). This approach does not differ from that used in most acute injuries that involve bleeding or swelling. However, doubts have been raised on the use of ice due to its interference in the clotting process.58 Along with the administration of clotting factor replacement, the administration of analgesic drugs (paracetamol, acetaminophen, COX-2 inhibitors) and, where indicated, arthrocentesis is recommended to reduce bleeding as soon as possible.55
The pharmacological approach to chronic pain includes the use of antidepressants, antiepileptic drugs, NSAIDS, opioids and paracetamol.55 Ultimately, with the progression of the joint degenerative process, orthopedic surgery and the use of orthoses are widely supported therapeutic proposals. The above is useful when there are movement-related nociceptive mechanisms involved in the patient’s pain.
In terms of the physiotherapeutic approach, various therapeutic alternatives have been proposed. Passive and analgesic interventions, such as the use of laser, massage, heat or cold, have shown limited evidence in the approach to chronic pain.59,60 Approaches aimed at causing structural changes or addressing nociceptive or tissue mechanisms such as manual therapy or myofascial release have shown good results in the short and medium term. Similarly, interventions focused on tackling central mechanisms through educational programs, and the approach to the patient’s descending inhibitory mechanisms through aerobic exercise, and strength or proprioception have demonstrated relevant results.60
However, different clinical guidelines and publications stress on the pharmacological approach to pain in patients with hemophilia, using analgesics and anti-inflammatory drugs.55 In the same way, physiotherapy is outlined as a therapeutic tool focusing only on passive techniques, centered on the patient as a passive target. This approach limits the active role of the patient as an active participant necessary to achieve changes in the process and improve his or her painful experience. It should not be forgotten that psychosocial factors have an impact on the painful experience of hemophilia patients.53
If chronic pain is understood as a disease in itself, and as something dysfunctional that will affect people’s lives,61 progress must be made in the treatment of patients with hemophilia. Future research should consider the treatment of chronic pain in patients with hemophilia accounting for the mechanisms involved in the patient’s pain. A summary of the approach to pain in patients with hemophilia is shown in Figure 3.
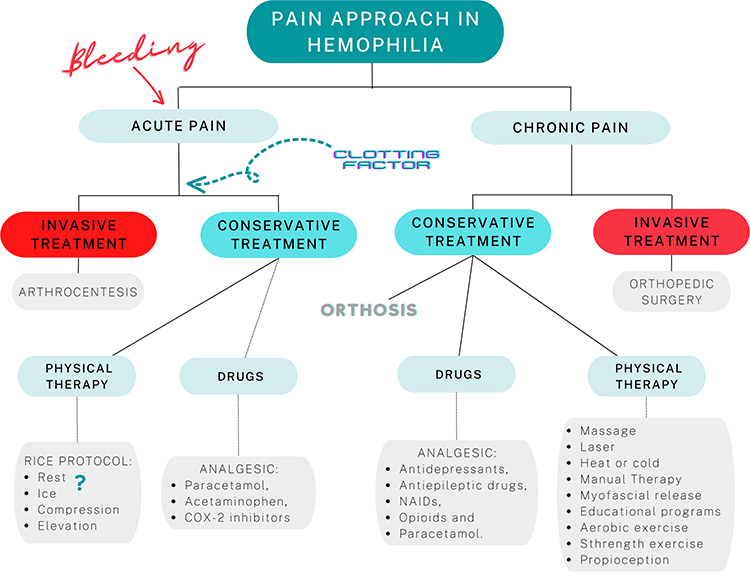 |
Figure 3 Multidisciplinary approach to pain in patients with hemophilia. |
Quality of Life and Psychosocial Factors in Patients with Hemophilia
The World Health Organization62 defines quality of life as “an individual’s perception of their position in life in the context of the culture and value systems in which they live and in relation to their goals, expectations, standards and concerns”. In the same way as pharmacological treatment is needed, objective health measures are required in aspects such as the joint condition and the quality of life of hemophilia patients. In recent decades, the assessment of quality of life has increasingly focused on the inclusion of items and domains that assess the doctor-patient relationship, in order to quantify the impact of the disease and its treatment. Over time, generic and specific hemophilia questionnaires have been designed and validated, which measure the impact of the disease and treatment from the patient’s perspective.63
The psychosocial predictors of patients with hemophilia have a greater influence on the quality of life assessment than the clinical predictors. However, psychosocial treatment is deficient and poorly implemented, even in countries where pharmacological treatment is novel.64
The severity phenotype of the pathology affects the quality of life of patients with hemophilia A and B, showing a poorer quality of life in patients with severe hemophilia, compared to those with mild or moderate phenotypes.65 Patients with hemophilia who report pain have limitations in their mobility and autonomy, with increased anxiety, poorer quality of life and frustration due to restrictions in the performance of activities of daily living.66 However, therapeutic advances in the approach to patients with hemophilia and the generalization of prophylactic treatment have allowed to improve life expectancy and health-related quality of life in these patients.67
The development of hemophilic arthropathy and joint pain are predictors of a deteriorated perception of physical health68 limiting the type of work and activities in these patients, especially when the lower limbs are involved, causing a high prevalence of mental health disorders.69 The clinical situation and psychosocial variables affect the perceived quality of life of patients with hemophilia. The absence of a relationship between the patients’ joint condition and their perceived quality of life prompts the need to investigate the effectiveness of interventions aimed at improving quality of life.70
The literature that addresses other psychosocial factors such as catastrophism or kinesiophobia in patients with hemophilia is limited, thus limiting the knowledge of these factors and their possible approach. Kinesiophobia, catastrophism and anxiety are determining factors in the painful experience in patients with chronic pain and their approach may be key for the treatment of patients with hemophilia.53 Therefore, addressing the psychosocial factors that affect pain should be a priority for the multidisciplinary team, to establish the association between intensity of pain and functionality.
New Perspectives in the Approach and Management of Patients with Hemophilia
The development of extended-life factors and strategies to replace the function of procoagulant factors through drugs administered subcutaneously, have meant a paradigm shift in the treatment of hemophilia. In the near future, gene therapy, which has shown a long-lasting effect for increasing the levels of the missing factor, poses a favorable horizon towards the reduction and even elimination of hemorrhagic symptoms.71 Although gene therapy with adeno-associated vectors (AAV) has shown some extent of efficacy in patients with hemophilia A, it is somewhat limited due to AAV-induced cellular stress, immunogenicity and lower durability of gene expression. Recently, Kaminski et al72 linked altered liver-targeted gene transfer to maladaptive structural changes of the hepatic endothelium associated with FVIII deficiency in mice. Despite the limitations of the predictive value of preclinical immunogenicity testing, progress has been made in developing the principles of safe and effective genetic drug design in animal models.73 In June 2022, the European Medicines Agency has recommended granting a conditional marketing authorization in the European Union for roxaparvovec valoctocogene gene therapy for the treatment of severe hemophilia A in adults who do not have factor VIII inhibitors or antibodies against adeno-associated virus serotype 5 (AAV5). Several clinical trials are currently investigating the safety and efficacy of gene therapy in hemophilia A (NCT05454774) and hemophilia B (NCT05203679). These advances can change the natural history of the disease and, consequently, the clinical manifestations of hemophilia. This promising future in terms of the significant reduction of muscle and joint bleeding will result in the improvement of the perceived quality of life in these patients.
These changes in the medical approach mean that the future management of hemophilic arthropathy will likewise change radically. In many cases arthropathy development will be minor, if at all, being similar to that present in the general population. Therefore, the therapeutic possibilities are expected to be greater and more effective in pediatric and young patients in whom a deterioration of the locomotor system has not yet been established, and who will benefit from these new therapies.1
Faced with this paradigm shift, future clinical strategies should be oriented in two directions: a) adult patients who are over the 30 years old and who have hemophilic arthropathy and, b) younger patients, beneficiaries of the new treatments and who have not developed arthropathy or to a minor extent.
In the case of patients with an already established arthropathy, we must refer to the existing scientific literature to follow the guidelines for safe and effective therapeutic programs. These should be aimed at controlling the symptomatology related to pain, joint mobility, decreased functionality and quality of life. The effectiveness of laser combined with exercise and hydrotherapy and dry exercise has been proven, in relation to the management of joint pain. Similarly, physical exercise can promote a reduction in pain perception, improving range of motion and muscle strength in patients with hemophilia.74,75 Other physiotherapy approaches, aimed at tackling tissue damage through myofascial therapy,76,77 joint traction78 and home exercises.75 They have also shown their safety and efficacy in decreasing the frequency of hemarthrosis, improving range of motion and perceived joint pain.
As noted for the evaluation and management of pain, the patient’s psychosocial and individual aspects should give him or her an active role in the process of changing the painful experience.53 This consideration, together with suitable education in pain based on neuroscience, offers a new therapeutic perspective in the management of chronic pain. In this way, the approach to hemophilic arthropathy would follow the assumptions already noted for other osteoarthritis processes.79 Similarly, improvements have been identified in psychosocial variables related to pain catastrophizing and kinesiophobia.80 García-Dasí et al81 observed an improvement in patients’ pain after a cognitive behavioral therapy approach, although further studies are needed to provide more information on this type of approach.
In the younger population groups, linked to the new drug treatments, a preventive approach associated with suitable education and counseling for the patient and their family environment is proposed. Promoting healthy habits and physical activity in children with hemophilia is essential at each stage of development and growth. In the postnatal period, adequate psychomotor stimulation should be used to favor the child’s motor and social skills. In preschool and childhood it is recommended to encourage play and exploration of activities that stimulate and promote movement. An interest in physical or sports activity is generated in school children, at which time the relevant decisions must be analyzed and made in relation to the type of activity to be carried out. In this section it is essential that the interdisciplinary team assesses to what extent the corresponding prophylactic measures favor the hematological coverage of these patients.
These approaches are of great importance. Patients with hemophilia show an increased risk of low bone mass during childhood, due to reduced physical activity and other factors such as increased urinary calcium loss and impaired bone metabolism. These metabolic alterations predispose subjects to osteoporosis later on in life.82 This phenomenon, together with lower vitamin D levels compared to the general population,83 is a risk factor in young people with hemophilia and may contribute to increased skeletal fragility.
A low level of physical activity is a risk factor for the reduction of lumbar and coxofemoral bone mass in people with hemophilia,84 and can be prevented through physical activity from childhood.85 In this population it is essential to educate in order to avoid sedentary lifestyle and obesity, which act as negative stimuli for the locomotor system. In many cases, this inactivity can be reinforced by the family environment through overprotective behaviors and hypervigilance. Physical activity is effective for improving the musculoskeletal clinical symptoms of hemophilia, with a low incidence of related adverse events. Similarly, physical activity can improve endogenous clotting factors after high-intensity activities that may reduce the risk of bleeding in patients with mild and moderate hemophilia,60 as well as prothrombin time in adults with hemophilia,86 with greater changes in those individuals presenting elevated prothrombin time during exercise.
Expert advice and information to patients with hemophilia should be based on individual functional prevention diagnoses, advice on available therapies and sports practice, and health recommendations. In this way, the safety and effectiveness of the activity and physical fitness of these patients can be improved, without increasing the bleeding rate and maintaining joint function.87 It is recommended that such advice, the prescription of therapeutic exercise and the recommendation on sports activities, be guided by a physiotherapist of reference within the framework of the interdisciplinary group. However, it is essential that this measure be taken in consensus with the patient and his or her family. For this to be successful, it is essential to consider the specific features of the activity, offering all the available information to guarantee safety, and promote the prevention of exercise-related bleeding. This advice can be based on that guidelines provided by Bérubé et al88 who pointed out that behaviors, attitudes and subjective rules present in the educational and formative processes of children largely explain the intention to engage in physical activity in the future. Figure 4 shows a summary of the new perspectives in the approach and management of patients with hemophilia.
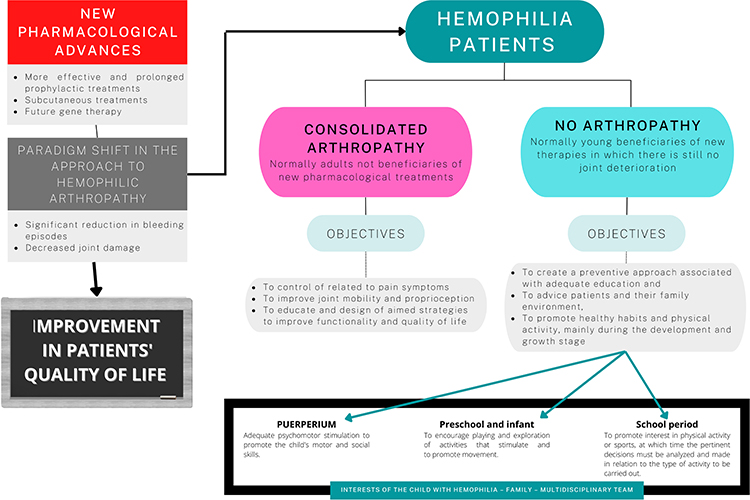 |
Figure 4 New perspectives in the approach and management of patients with hemophilia. |
Conclusions
Progress made in the pharmacological treatment of patients with hemophilia has significantly improved the life prospects of these patients. The interdisciplinary approach should focus on the total prevention of hemarthrosis and of the development of hemophilic arthropathy. In children and adolescents, the chances of success in the prevention of joint damage is favored by recent medical progress. For patients with already established hemophilic arthropathy, it is necessary to change the therapeutic model, with the patients actively participating in their treatment. The adoption of an active role by patients can facilitate a better control of the disability associated with hemophilic arthropathy and their quality of life.
Data Sharing Statement
All data are available within the manuscript and may further be available upon reasonable request to the corresponding author.
Consent for Publication
All Authors approved the present submission.
Author Contributions
All authors made substantial contributions to the conception and design, acquisition of data, or analysis and interpretation of data; took part in the drafting of the article or revising it critically for important intellectual content; agreed to submit the paper to this journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Funding
This research received no external funding.
Disclosure
Dr. Rubén Cuesta-Barriuso received consultancy honoraria and speaker bureau from Takeda, Sobi and Roche; Dr. Elena Donoso-Úbeda declares no conflicts of interest to disclose; Dr. Javier Meroño-Gallut declares no conflicts of interest to disclose; Mr. Roberto Ucero-Lozano declares no conflicts of interest to disclose; Dr. Raúl Pérez-Llanes declares no conflicts of interest to disclose.
References
1. Gualtierotti R, Solimeno LP, Peyvandi F. Hemophilic arthropathy: current knowledge and future perspectives. J Thromb Haemost. 2021;19(9):2112–2121. doi:10.1111/jth.15444
2. Calvez T, Chambost H, Claeyssens-Donadel S, et al. Recombinant factor VIII products and inhibitor development in previously untreated boys with severe hemophilia A. Blood. 2014;124:23. doi:10.1182/blood-2014-07-586347
3. Hotea I, Brinza M, Blag C, et al. Current therapeutic approaches in the management of hemophilia-a consensus view by the Romanian Society of Hematology. Ann Transl Med. 2021;9(13):1091. doi:10.21037/atm-21-747
4. Clausen N, Petrini P, Claeyssens-Donadel S, Gouw SC, Liesner R; PedNet and Research of Determinants of Inhibitor development (RODIN) Study Group. Similar bleeding phenotype in young children with haemophilia A or B: a cohort study. Haemophilia. 2014;20(6):747–755. doi:10.1111/hae.12470
5. Iorio A, Stonebraker JS, Chambost H, et al. Establishing the prevalence and prevalence at birth of hemophilia in males. Ann Intern Med. 2019;171(8):540–546. doi:10.7326/M19-1208
6. Lobet S, Timmer M, Königs C, et al. The role of physiotherapy in the new treatment landscape for haemophilia. J Clin Med. 2021;10(13):2822. doi:10.3390/jcm10132822
7. Prasetyo M, Mongan AE, Chozie NA, Prihartono J, Setiawan SI. Hemosiderin deposition evaluation in hemophilic ankle joints: association between US finding and gradient-recalled echo MR imaging sequence. Insights Imaging. 2021;12(1):107. doi:10.1186/s13244-021-01050-1
8. Roosendaal G, Vianen ME, van den Berg HM, Lafeber FP, Bijlsma JW. Cartilage damage as a result of hemarthrosis in a human in vitro model. J Rheumatol. 1997;24(7):1350–1354.
9. Roosendaal G, Mauser-Bunschoten EP, De Kleijn P, et al. Synovium in haemophilic arthropathy. Haemophilia. 1998;4(4):502–505. doi:10.1046/j.1365-2516.1998.440502.x
10. Roosendaal G, Vianen ME, Wenting MJ, et al. Iron deposits and catabolic properties of synovial tissue from patients with haemophilia. J Bone Joint Surg Br. 1998;80(3):540–545. doi:10.1302/0301-620X.80B3.0800540
11. Zheng L, Han Z, Luo D, et al. FGF23 and SOX9 expression in hemophilic cartilage: in vitro studies of the effects of iron. Haemophilia. 2022. doi:10.1111/hae.14623
12. Tantawy AAG, Elsherif NHK, Mostafa S, Safwat NA, El Seteha KAES. Endothelial specific isoform of type XVIII collagen (COL-18N): a marker of vascular integrity in haemophilic arthropathy. Haemophilia. 2022. doi:10.1111/hae.14593
13. Wojdasiewicz P, Poniatowski ŁA, Nauman P, et al. Cytokines in the pathogenesis of hemophilic arthropathy. Cytokine Growth Factor Rev. 2018;39:71–91. doi:10.1016/j.cytogfr.2017.11.003
14. Ljung R, Auerswald G, Benson G, et al. Inhibitors in haemophilia A and B: management of bleeds, inhibitor eradication and strategies for difficult-to-treat patients. Eur J Haematol. 2019;102(2):111–122. doi:10.1111/ejh.13193
15. Sørensen B, Auerswald G, Benson G, et al. Rationale for individualizing haemophilia care. Blood Coagul Fibrinolysis. 2015;26(8):849–857. doi:10.1097/MBC.0000000000000225
16. Castaman G, Linari S. Prophylactic versus on-demand treatments for hemophilia: advantages and drawbacks. Expert Rev Hematol. 2018;11(7):567–576. doi:10.1080/17474086.2018.1486704
17. Ragni MV. New and emerging agents for the treatment of hemophilia: focus on extended half-life recombinant clotting proteins. Drugs. 2015;75(14):1587–1600. doi:10.1007/s40265-015-0451-5
18. Berntorp E, Andersson NG. Prophylaxis for hemophilia in the era of extended half-life factor VIII/factor IX products. Semin Thromb Hemost. 2016;42(5):518–525. doi:10.1055/s-0036-1571315
19. Mahlangu J, Powell JS, Ragni MV, et al. Phase 3 study of recombinant factor VIII Fc fusion protein in severe hemophilia A. Blood. 2014;123(3):317–325. doi:10.1182/blood-2013-10-529974
20. Tardy B, Lambert T, Chamouni P, et al. Revised terminal half-life of nonacog alfa as derived from extended sampling data: a real-world study involving 64 haemophilia B patients on nonacog alfa regular prophylaxis. Haemophilia. 2022;28(4):542–547. doi:10.1111/hae.14560
21. Forsyth AL, Rivard GÉ, Valentino LA, et al. Consequences of intra-articular bleeding in haemophilia: science to clinical practice and beyond. Haemophilia. 2012;18(Suppl 4):112–119. doi:10.1111/j.1365-2516.2012.02835.x
22. Arnold WD, Hilgartner MW. Hemophilic arthropathy. Current concepts of pathogenesis and management. J Bone Joint Surg Am. 1977;59(3):287–305.
23. Stein H, Duthie RB. The pathogenesis of chronic haemophilic arthropathy. J Bone Joint Surg Br. 1981;63B(4):601–609. doi:10.1302/0301-620X.63B4.7298694
24. Rodríguez-Merchán EC. Pathogenesis, early diagnosis, and prophylaxis for chronic hemophilic synovitis. Clin Orthop Relat Res. 1997;343:6–11.
25. Pacheco-Serrano A, Lucena-Anton D, Moral-Muñoz J. Rehabilitación física en pacientes con artropatía hemofílica: revisión sistemática y metaanálisis sobre dolor. Rev Colomb Reumatol. 2021;28(2):124–133.
26. Stephensen D, Taylor S, Bladen M, Drechsler WI. Relationship between physical function and biomechanical gait patterns in boys with haemophilia. Haemophilia. 2016;22(6):e512–e518. doi:10.1111/hae.13118
27. Srivastava A, Santagostino E, Dougall A, et al. WFH guidelines for the management of hemophilia. Haemophilia. 2020;26(S6):1–158. doi:10.1111/hae.14046
28. Goddard NJ, Mann H. Diagnosis of haemophilic synovitis. Haemophilia. 2007;13(Suppl 3):14–19. doi:10.1111/j.1365-2516.2007.01535.x
29. Nguyen S, Lu X, Ma Y, Du J, Chang EY, von Drygalski A. Musculoskeletal ultrasound for intra-articular bleed detection: a highly sensitive imaging modality compared with conventional magnetic resonance imaging. J Thromb Haemost. 2018;16(3):490–499. doi:10.1111/jth.13930
30. Martinoli C, Della Casa Alberighi O, Di Minno G, et al. Development and definition of a simplified scanning procedure and scoring method for Haemophilia Early Arthropathy Detection with Ultrasound (HEAD-US). Thromb Haemost. 2013;109(6):1170–1179. doi:10.1160/TH12-11-0874
31. Draghi F. The value of ultrasonography in detecting early arthropathic changes and contribution to the clinical approach in patients of hemophilia the value of ultrasonography in patients of hemophilia. J Clin Ultrasound. 2022;50(3):433–434. doi:10.1002/jcu.23165
32. Mesleh Shayeb A, Barnes RFW, Hanacek C, et al. Quantitative measurements of haemophilic joint tissues by point-of-care musculoskeletal ultrasound: associations with clinical and functional joint outcome parameters. Haemophilia. 2021;27(5):866–875. doi:10.1111/hae.14368
33. Cifrián C, Aparisi F. Evaluación de la artropatía hemofílica: nuevos criterios radiológicos. Haematológica. 2005;90(Suppl 4):32–39.
34. Gupta S, Garg K, Singh J. Assessment of musculoskeletal function and its correlation with radiological joint score in children with hemophilia A. Indian J Pediatr. 2015;82(12):1101–1106. doi:10.1007/s12098-015-1759-6
35. Berntorp E, Gomperts E, Hoots K, Wong WY. The next generation of hemophilia treatment specialists. Semin Thromb Hemost. 2006;32(Suppl 2):39–42. doi:10.1055/s-2006-946914
36. Pettersson H, Ahlberg A, Nilsson IM. A radiologic classification of hemophilic arthropathy. Clin Orthop Relat Res. 1980;149:153–159.
37. Doria AS, Lundin B, Miller S, et al. Reliability and construct validity of the compatible MRI scoring system for evaluation of elbows in haemophilic children. Haemophilia. 2008;14(2):303–314. doi:10.1111/j.1365-2516.2007.01602.x
38. Nuss R, Kilcoyne RF, Geraghty S, et al. MRI findings in haemophilic joints treated with radiosynoviorthesis with development of an MRI scale of joint damage. Haemophilia. 2000;6(3):162–169. doi:10.1046/j.1365-2516.2000.00383.x
39. Lundin B, Pettersson H, Ljung R. A new magnetic resonance imaging scoring method for assessment of haemophilic arthropathy. Haemophilia. 2004;10(4):383–389. doi:10.1111/j.1365-2516.2004.00902.x
40. Feldman BM, Funk SM, Bergstrom BM, et al. Validation of a new pediatric joint scoring system from the International Hemophilia Prophylaxis Study Group: validity of the hemophilia joint health score. Arthritis Care Res. 2011;63(2):223–230. doi:10.1002/acr.20353
41. St-Louis J, Abad A, Funk S, et al. The hemophilia joint health score version 2.1 validation in adult patients study: a multicenter international study. Res Pract Thromb Haemost. 2022;6(2):e12690. doi:10.1002/rth2.12690
42. Gilbert MS. Prophylaxis: musculoskeletal evaluation. Semin Hematol. 1993;30(3 Suppl 2):3–6.
43. World Federation of Haemophilia. WFH Physical Examination Score (aka Gilbert score). eLearning Platform. Available from: https://elearning.wfh.org/resource/wfh-physical-examination-score-aka-gilbert-score/.
44. van Genderen FR, van Meeteren NLU, van der Bom JG, et al. Functional consequences of haemophilia in adults: the development of the Haemophilia Activities List. Haemophilia. 2004;10(5):565–571. doi:10.1111/j.1365-2516.2004.01016.x
45. van Genderen FR, Westers P, Heijnen L, et al. Measuring patients’ perceptions on their functional abilities: validation of the Haemophilia Activities List. Haemophilia. 2006;12(1):36–46. doi:10.1111/j.1365-2516.2006.01186.x
46. Poonnoose PM, Manigandan C, Thomas R, et al. Functional Independence Score in Haemophilia: a new performance-based instrument to measure disability. Haemophilia. 2005;11(6):598–602. doi:10.1111/j.1365-2516.2005.01142.x
47. von Mackensen S, Czepa D, Herbsleb M, Hilberg T. Development and validation of a new questionnaire for the assessment of subjective physical performance in adult patients with haemophilia–the HEP-Test-Q. Haemophilia. 2010;16(1):170–178. doi:10.1111/j.1365-2516.2009.02112.x
48. Raja SN, Carr DB, Cohen M, et al. The revised international association for the study of pain definition of pain: concepts, challenges, and compromises. Pain. 2020;161(9):1976–1982. doi:10.1097/j.pain.0000000000001939
49. Stromer W, Pabinger I, Ay C, et al. Pain management in hemophilia: expert recommendations. Wien Klin Wochenschr. 2021;133(19–20):1042–1056. doi:10.1007/s00508-020-01798-4
50. Paredes AC, Costa P, Almeida A, Pinto PR. A new measure to assess pain in people with haemophilia: the Multidimensional Haemophilia Pain Questionnaire (MHPQ). PLoS One. 2018;13(11):e0207939. doi:10.1371/journal.pone.0207939
51. Ferreira AA, Leite ICG, Bustamante-Teixeira MT, et al. Health-related quality of life in hemophilia: results of the Hemophilia-Specific Quality of Life Index (Haem-a-Qol) at a Brazilian blood center. Rev Bras Hematol Hemoter. 2013;35(5):314–318. doi:10.5581/1516-8484.20130108
52. Shah RM, Banahan BF, Holmes ER, et al. An evaluation of the psychometric properties of the sf-12v2 health survey among adults with hemophilia. Health Qual Life Outcomes. 2018;16(1):229. doi:10.1186/s12955-018-1059-8
53. Ucero-Lozano R, López-Pina JA, Ortiz-Pérez A, Cuesta-Barriuso R. The relationship between chronic pain and psychosocial aspects in patients with haemophilic arthropathy. A cross-sectional study. Haemophilia. 2022;28(1):176–182. doi:10.1111/hae.14469
54. Sündermann O, Rydberg K, Linder L, Linton SJ. “When I feel the worst pain, I look like shit” - body image concerns in persistent pain. Scand J Pain. 2018;18(3):379–388. doi:10.1515/sjpain-2017-0163
55. Rodriguez-Merchan EC, De la Corte-Rodriguez H. Pain management in people with hemophilia in childhood and young adulthood. Expert Rev Hematol. 2021;14(6):525–535. doi:10.1080/17474086.2021.1935852
56. Álvarez Román M, Bernardo Gutierrez Á, Berrueco Moreno R, et al. Guías españolas para el manejo del paciente con hemofilia[Spanish guidelines for the management of patients with hemophilia]; 2022.
57. Auerswald G, Dolan G, Duffy A, et al. Pain and pain management in haemophilia. Blood Coagul Fibrinolysis. 2016;27(8):845–854. doi:10.1097/MBC.0000000000000571
58. Gualtierotti R, Tafuri F, Arcudi S, et al. Current and emerging approaches for pain management in hemophilic arthropathy. Pain Ther. 2022;11(1):1–15. doi:10.1007/s40122-021-00345-x
59. El-Shamy SM, Abdelaal AAM. Efficacy of pulsed high-intensity laser therapy on pain, functional capacity, and gait in children with haemophilic arthropathy. Disabil Rehabil. 2018;40(4):462–468. doi:10.1080/09638288.2016.1261416
60. Groen WG, den Uijl IEM, van der Net J, Grobbee DE, de Groot PG, Fischer K. Protected by nature? Effects of strenuous physical exercise on FVIII activity in moderate and mild haemophilia A patients: a pilot study. Haemophilia. 2013;19(4):519–523. doi:10.1111/hae.12111
61. Treede RD, Rief W, Barke A, et al. Chronic pain as a symptom or a disease: the IASP classification of chronic pain for the International Classification of Diseases (ICD-11). Pain. 2019;160(1):19–27. doi:10.1097/j.pain.0000000000001384
62. The WHOQOL Group. The Development of the World Health Organization Quality of Life Assessment Instrument (the WHOQOL). Orley J, Kuyken W, editors. Quality of Life Assessment: International Perspectives. Springer Berlin Heidelberg; 1994:41–57. doi:10.1007/978-3-642-79123-9_4
63. Aledort L, Bullinger M, von Mackensen S, et al. Why should we care about quality of life in persons with haemophilia? Haemophilia. 2012;18(3):e154–157. doi:10.1111/j.1365-2516.2012.02771.x
64. Cuesta-Barriuso R, Torres-Ortuño A, Nieto-Munuera J, López-Pina JA. Quality of life, perception of disease and coping strategies in patients with hemophilia in Spain and El Salvador: a comparative study. Patient Prefer Adherence. 2021;15:1817–1825. doi:10.2147/PPA.S326434
65. Miners AH, Sabin CA, Tolley KH, Jenkinson C, Kind P, Lee CA. Assessing health-related quality-of-life in individuals with haemophilia. Haemophilia. 1999;5(6):378–385. doi:10.1046/j.1365-2516.1999.00347.x
66. Brodin E, Sunnerhagen KS, Baghaei F, Törnbom M. Persons with haemophilia in Sweden- experiences and strategies in everyday life. a single centre study. PLoS One. 2015;10(10):e0139690. doi:10.1371/journal.pone.0139690
67. Kempton CL, Makris M, Holme PA. Management of comorbidities in haemophilia. Haemophilia. 2021;27(Suppl 3):37–45. doi:10.1111/hae.14013
68. Carroll L, Benson G, Lambert J, Benmedjahed K, Zak M, Lee XY. Real-world utilities and health-related quality-of-life data in hemophilia patients in France and the United Kingdom. Patient Prefer Adherence. 2019;13:941–957. doi:10.2147/PPA.S202773
69. Buckner TW, Batt K, Quon D, et al. Assessments of pain, functional impairment, anxiety, and depression in US adults with hemophilia across patient-reported outcome instruments in the Pain, Functional Impairment, and Quality of Life (P-FiQ) study. Eur J Haematol. 2018;100(Suppl 1):5–13. doi:10.1111/ejh.13027
70. Ucero-Lozano R, López-Pina JA, Ortiz-Pérez A, Cuesta-Barriuso R. Quality of life and its predictors among adult patients with haemophilic arthropathy. An observational study. BMC Musculoskelet Disord. 2021;22(1):448. doi:10.1186/s12891-021-04319-0
71. Páramo JA. Tratamiento de la hemofilia: de la terapia sustitutiva a la terapia génica[Hemophilia treatment: from replacement therapy to gene therapy]. Medicina Clínica. 2021;157(12):583–587. doi:10.1016/j.medcli.2021.04.031
72. Kaminski TW, Ju EM, Gudapati S, et al. Defenestrated endothelium delays liver-directed gene transfer in hemophilia A mice. Blood Adv. 2022;6(12):3729–3734. doi:10.1182/bloodadvances.2021006388
73. Lundgren TS, Denning G, Stowell SR, Spencer HT, Doering CB. Pharmacokinetic analysis identifies a factor VIII immunogenicity threshold after AAV gene therapy in hemophilia A mice. Blood Adv. 2022;6(8):2628–2645. doi:10.1182/bloodadvances.2021006359
74. Calatayud J, Pérez-Alenda S, Carrasco JJ, et al. Safety and effectiveness of progressive moderate-to-vigorous intensity elastic resistance training on physical function and pain in people with hemophilia. Phys Ther. 2020;100(9):1632–1644. doi:10.1093/ptj/pzaa106
75. Cuesta-Barriuso R, Torres-Ortuño A, Nieto-Munuera J, López-Pina JA. Effectiveness of an educational physiotherapy and therapeutic exercise program in adult patients with hemophilia: a randomized controlled trial. Arch Phys Med Rehabil. 2017;98(5):841–848. doi:10.1016/j.apmr.2016.10.014
76. Cuesta-Barriuso R, Pérez-Llanes R, Donoso-Úbeda E, López-Pina JA, Meroño-Gallut J. Effects of myofascial release on frequency of joint bleedings, joint status, and joint pain in patients with hemophilic elbow arthropathy: a randomized, single-blind clinical trial. Medicine. 2021;100(20):e26025. doi:10.1097/MD.0000000000026025
77. Donoso-Úbeda E, Meroño-Gallut J, López-Pina JA, Cuesta-Barriuso R. Effect of manual therapy in patients with hemophilia and ankle arthropathy: a randomized clinical trial. Clin Rehabil. 2020;34(1):111–119. doi:10.1177/0269215519879212
78. Cuesta‐Barriuso R, Gómez‐Conesa A, López‐Pina JA. The effectiveness of manual therapy in addition to passive stretching exercises in the treatment of patients with haemophilic knee arthropathy: a randomized, single‐blind clinical trial. Haemophilia. 2021;27(1). doi:10.1111/hae.14181
79. Fernandes L, Storheim K, Sandvik L, Nordsletten L, Risberg MA. Efficacy of patient education and supervised exercise vs patient education alone in patients with Hip osteoarthritis: a single blind randomized clinical trial. Osteoarthritis Cartilage. 2010;18(10):1237–1243. doi:10.1016/j.joca.2010.05.015
80. Lluch E, Dueñas L, Falla D, et al. Preoperative pain neuroscience education combined with knee joint mobilization for knee osteoarthritis: a randomized controlled trial. Clin J Pain. 2018;34(1). doi:10.1097/AJP.0000000000000511
81. García-Dasí M, Pérez-Alenda S, Carrasco JJ, et al. Effects of a non-pharmacological approach for chronic pain management in patients with haemophilia: efficacy of cognitive-behavioural therapy associated with physiotherapy. Haemophilia. 2021;27(3):e357–e367. doi:10.1111/hae.14284
82. Ranta S, Viljakainen H, Mäkipernaa A, Mäkitie O. Hypercalciuria in children with haemophilia suggests primary skeletal pathology. Br J Haematol. 2011;153(3):364–371. doi:10.1111/j.1365-2141.2011.08639.x
83. Ashritha A, Delhi Kumar CG, Sahoo J, Nalini P. Evaluation of bone mineral density in children with hemophilia: an observational case-control study. J Pediatr Hematol Oncol. 2019;41(7):511–514. doi:10.1097/MPH.0000000000001554
84. Tlacuilo-Parra A, Morales-Zambrano R, Tostado-Rabago N, Esparza-Flores MA, Lopez-Guido B, Orozco-Alcala J. Inactivity is a risk factor for low bone mineral density among haemophilic children. Br J Haematol. 2008;140(5):562–567. doi:10.1111/j.1365-2141.2007.06972.x
85. Khawaji M, Astermark J, Akesson K, Berntorp E. Physical activity for prevention of osteoporosis in patients with severe haemophilia on long-term prophylaxis. Haemophilia. 2010;16(3):495–501. doi:10.1111/j.1365-2516.2009.02186.x
86. Beltrame LGN, Abreu L, Almeida J, Boullosa DA. The acute effect of moderate intensity aquatic exercise on coagulation factors in haemophiliacs. Clin Physiol Funct Imaging. 2015;35(3):191–196. doi:10.1111/cpf.12145
87. Sondermann J, Herbsleb M, Stanek FD, Gabriel H, Kentouche K. Health promotion for young patients with haemophilia. Hamostaseologie. 2017;37(2):107–116. doi:10.5482/HAMO-15-02-0007
88. Bérubé S, Cloutier-Bergeron A, Amesse C, Sultan S. Understanding adherence to treatment and physical activity in children with hemophilia: the role of psychosocial factors. Pediatr Hematol Oncol. 2017;34(1):1–9. doi:10.1080/08880018.2016.1260669
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.