Back to Journals » Journal of Blood Medicine » Volume 6
Hematopoietic stem cell transplantation in sickle cell disease: patient selection and special considerations
Received 1 April 2015
Accepted for publication 11 May 2015
Published 10 July 2015 Volume 2015:6 Pages 229—238
DOI https://doi.org/10.2147/JBM.S60515
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Martin Bluth
Monica Bhatia,1 Sujit Sheth2
1Division of Pediatric Hematology/Oncology/Stem Cell Transplantation, Columbia University Medical Center, 2Division of Pediatric Hematology and Oncology, Weill Cornell Medical College, New York, NY, USA
Abstract: Hematopoietic stem cell transplantation remains the only curative treatment currently in use for patients with sickle cell disease (SCD). The first successful hematopoietic stem cell transplantation was performed in 1984. To date, approximately 1,200 transplants have been reported. Given the high prevalence of this disorder in Africa, and its emergence in the developed world through immigration, this number is relatively small. There are many reasons for this; primary among them are the availability of a donor, the risks associated with this complex procedure, and the cost and availability of resources in the developing world. Of these, it is fair to say that the risks associated with the procedure have steadily decreased to the point where, if currently performed in a center with experience using a matched sibling donor, overall survival is close to 100% and event-free survival is over 90%. While there is little controversy around offering hematopoietic stem cell transplantation to symptomatic SCD patients with a matched sibling donor, there is much debate surrounding the use of this modality in “less severe” patients. An overview of the current state of our understanding of the pathology and treatment of SCD is important to show that our current strategy is not having the desired impact on survival of homozygous SCD patients, and should be changed to significantly impact the small proportion of these patients who have matched siblings and could be cured, especially those without overt clinical manifestations. Both patient families and providers must be made to understand the progressive nature of SCD, and should be encouraged to screen full siblings of patients with homozygous SCD for their potential to be donors. Matched siblings should be referred to an experienced transplant center for evaluation and counseling. In this review, we will discuss the rationale for these opinions and make recommendations for patient selection.
Keywords: sickle cell disease, morbidity, stem cell transplantation, patient selection, matched sibling donor
Introduction
Sickle cell disease (SCD) is a devastating condition which affects virtually every tissue in the body, causing significant morbidity and resulting in a markedly reduced life expectancy. Recent longitudinal studies have shown that the median life expectancy for individuals in the US with homozygous SCD is less than 40 years, approximately half that of the general population.1
Currently, we have limited options in the treatment of SCD, including transfusion therapy, hydroxyurea (HU), and the only definitive curative modality–hematopoietic stem cell transplantation (HSCT). HSCT is a complex and expensive treatment, with some mortality and short- and long-term morbidity, but recent advances have greatly improved outcomes.
Recommendations
- At this time, all patients with homozygous SCD and matched sibling donors (MSDs) should be evaluated for HSCT regardless of symptomatology. The donor stem cell source is usually umbilical cord blood (UCB) or bone marrow, but may include peripheral blood.
- Families of patients with SCD should be thoroughly educated on the severity and of SCD, the likelihood of reduced life expectancy, and the pros and cons of HSCT, so that they may be able to make a truly informed decision.
- HSCT using alternate donor sources should be considered experimental, and such transplants must be done in controlled settings such as research studies with regulatory/Data and Safety Monitoring Board oversight.
- Families of children with SCD should be encouraged to have UCB collected and stored (in the public domain, utilizing resources available at no cost for these patient families) for possible future use as a source for stem cells for HSCT.
- The experience of HSCT in those with hemoglobin SC disease or Hb S-β-thalassemia is limited, and as such, should be reserved only for those with MSDs and severe disease manifestations such as outlined in the initial trials with homozygous SCD.
This review will examine the rationale for these recommendations, and describe recent advances, which have made HSCT a more viable and acceptable treatment.
Rationale for HSCT in SCD
Initially considered a treatment modality for patients with severe SCD, HSCT has been more widely employed as a curative therapy as experience and expertise has grown. However, there remains some controversy about which patients should undergo the procedure. In the context of treating SCD, the decision to not treat patients with mild disease with HSCT has been mainly based on the potential for toxicity and long-term complications. The debate has focused on the balance between disease severity and the risks of the procedure itself. Both of these will be discussed in this review, so as to provide a basis for a more rational approach to patient selection.
Severity of SCD
Homozygous SCD has wide phenotypic variability in its clinical manifestations with a spectrum that ranges from the relatively asymptomatic to severe clinical disease with multiple organs affected early in the course. However, there is no debate that the disease is invariably progressive, causing irreversible organ damage despite the absence of overt symptoms, and resulting in a markedly reduced life expectancy. While most hematologists agree that patients with clinically severe SCD should be evaluated for HSCT, others have attempted to define “less severe” disease as being the absence of clinical manifestations.2 After an extensive review, and citing literature, Kassim and DeBaun3 concluded that “the perception of asymptomatic sickle cell disease is a misnomer”. There is extensive published data on the myriad complications of SCD, its progressive course and the far from optimal impact of improved supportive care on life expectancy. While several groups have attempted to define markers which predict severity,4,5 this has not been consistently validated.
Our understanding of the natural history of SCD has been greatly advanced by organized prospective cooperative studies starting in infancy.6 We know that renal dysfunction and neuropsychiatric abnormalities begin as early as infancy, with progressive involvement of other organs such as the brain (overt and silent infarcts), spleen (sequestration and autosplenectomy), lungs (acute chest syndrome and pulmonary hypertension), and other tissues like bone (avascular necrosis) and eyes (sickle retinopathy). In fact, no organ system is spared in this disease. These effects on the vascular system are particularly devastating when they occur in the brain, the lungs, and the eyes.
It is clear that not all individuals with SCD have all of these complications, but besides a higher fetal Hb level, there is little evidence to support any other markers of severity, making it difficult to plan treatment prospectively.
Some children may experience episodic vaso-occlusive pain crises, life-threatening complications such as ischemic stroke, splenic sequestration, and acute chest syndrome, and others may just have chronic anemia, impaired growth, but none of the other more severe complications. However, it is clear that chronic organ damage begins in childhood, with the majority of children with homozygous SCD being functionally asplenic by 5 years of age.7
Effects on the brain are particularly devastating. The Cooperative Study of Sickle Cell Disease reported in 2001 that 10% of children with homozygous SCD had overt strokes.8 Additionally, over twice the number of children with overt strokes had silent infarcts; these children manifest significantly lower neurocognitive functioning compared to those with normal magnetic resonance imaging (MRI).9 A more recent study corroborates this, confirming that 27% of children have silent infarcts by age 6, and the proportion rises to 37% by age 14, associated with a loss of global intellectual function.10 The landmark Stroke Prevention (STOP) trial, published in 1998, demonstrated the utility of a new tool, transcranial Doppler (TCD) ultrasonography for predicting stroke risk, and showed that regular transfusion therapy could prevent most children with elevated TCD flow velocities from having overt strokes.11 No predictor of silent infarcts has yet been identified, and children would need to have MRI studies periodically (often with sedation, further increasing risk) to detect these lesions. Further, most recent data suggest that while regular transfusion therapy may prevent a first or recurrent overt stroke, this therapy is not able to prevent progression of silent infarcts in most children.12 While approximately half of these children did not have progression, no clear markers of risk for progression were identified. The neurocognitive effects have been extensively studied in children and adults. A large study of 255 children with homozygous SCD found that children with silent infarcts had lower full scale, verbal and performance intelligence quotients (IQs) than those with normal MRI, and very importantly, that these progressively declined with age.9 A large adult study in 19–55-year-old patients with SCD, stratified by age, and with appropriate age-matched controls, also showed significantly lower full scale, performance, and verbal IQ.13
SCD is progressive in all individuals, albeit at different rates. Adults continue to have pain crises, may have acute chest syndrome, develop pulmonary hypertension and have hemorrhagic strokes. Most live with chronic organ dysfunction, and many are disabled by severe pain, chronic bone disease, leg ulcers, and retinopathy. All of these account for the markedly reduced life expectancy in patients with homozygous SCD.
Life expectancy and quality of life
In 1994, the Cooperative Study of Sickle Cell Disease (CSSCD) described the progress made in treating SCD with rapidly improving life expectancy in the US, and predicted that the median survival would be well over 40 years by the year 2000.6 As a result of improvements in care, including penicillin prophylaxis, vaccination against Streptococcus pneumoniae, better recognition and treatment of complications like acute chest syndrome and stroke, and better education of families and better access to expert medical care, almost 94% of children with SCD are reaching adulthood14 with the mortality rate in children with homozygous SCD being 0.52 per 100 patient-years. Despite these dramatic improvements, by 2006, the mean age at death was still only 39 years for all comers,1 with only 35% of individuals reaching the age of 45.
Figure 1 follows the age at death in cohorts between 1979 and 2006. One can see from this figure that the mortality curve has shifted to the right, with improved life expectancy, essentially eliminating the peak in early childhood. However, the shape of the curve has not changed. The rapid rise in mortality, which previously occurred in the first part of the second decade, now occurs in the third decade. Several studies have shown that mortality shows a sharp increase in early adulthood,1,14,15 and no clear causal factors are evident. The lack of significant impact on mortality in adults may reflect a stall or deceleration in our progress caring for these patients, particularly in preventing or treating complications related to progression of the underlying pathophysiologic basis of SCD – red cell sickling and vascular damage.
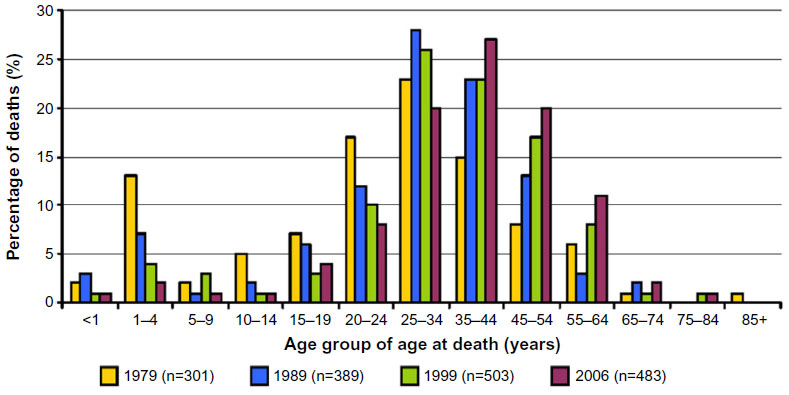 | Figure 1 Change in age at death in individuals with sickle cell disease between 1979 and 2006. |
“Adults and children with sickle cell disease have significantly impaired HRQL [health related quality of life] that is comparable to or worse than other chronic diseases […] even in their baseline state of health”.
This was the conclusion of a meta-analysis of 39 (five prospective) surveys of health-related quality of life measures in SCD analyzed, using a generic instrument.16 SCD patients had poorer physical, social, and school functioning, and to make matters worse, the decline was progressive as the child grew older, consistent with the neurocognitive changes reviewed previously. When compared with their healthy peers, adolescents and young adults functioned at 58%, 70%, and 48% physical, social, and school function, respectively.17 In this study, just 7% of respondents believed that an affected child’s life may be shortened by more than 10 years, two-thirds of parents believed that the disease would get better, 83% believed that it would not prevent their child from achieving his/her goals, and 86% actually believed that it would not shorten one’s life. These data indicate a significant lack of awareness or understanding on the part of parents of children with SCD, and possibly a lack of appropriate education about the disease and its course by their child’s health care providers, this being emphatically echoed in a National Institutes of Health (NIH) consensus statement.18 This may have a tremendous impact on the decision-making ability of parents as well as providers, and may impede them accessing more definitive therapies such as HU or HSCT.
Current approach to definitive treatment in SCD and the need for expansion of the curative treatment option
Our ability to predict the severity of SCD in individual patients is limited. We are able to predict the development of overt stroke in children through the use of TCD, but not silent infarcts. We know that a higher level of fetal Hb predicts less severe vaso-occlusive crises and fewer episodes of acute chest syndrome. Similarly, we are unable to predict the onset or progression of pulmonary hypertension, hemorrhagic strokes, and chronic organ dysfunction in adults. As a result, the mainstay of the management of SCD remains supportive care. Infection prophylaxis, management of pain crises, and specific treatment of acute complications with intermittent transfusions as clinically indicated are all supportive.
Regular red cell transfusion therapy may alleviate most symptoms of SCD, and may alter the natural progression of the disease. However, it does not prevent the development or progression of silent infarcts in all patients,10,12 and other complications such as avascular necrosis and moyamoya disease are known to progress while on transfusions.19 Moreover, in children who receive transfusions as prophylaxis for stroke (abnormal TCD) the SWiTCH study showed that risk reverts back if transfusions are discontinued.20 Other long-term complications of regular transfusions such as alloimmunization and iron overload have their own morbidity and make this regimen far from ideal.
HU has been in use in SCD since the early 1990s, with several trials showing good efficacy in both children and adults,21–24 but is still not universal. In the BABY HUG trial, a multi center randomized placebo-controlled trial with 193 children (aged 9–18 months), found that the HU group had significantly fewer episodes of vasoocclusive crisis (VOC) and acute chest syndrome (ACS) and required fewer transfusions than the placebo control group.25 However, despite the 2-decade long experience with HU use, the demonstration of clear benefit and the lack of significant toxicity, a 2008 Consensus Statement issued by the NIH in the US concluded that HU was underutilized.18
HSCT is the only curative treatment currently available. Gene therapy shows promise as a future curative therapeutic option, and early human trials are underway, but long-term efficacy remains to be seen.
In summary, current conventional non-HSCT treatments are not effective in reducing morbidity and mortality (supportive care alone), have significant barriers and side effects (transfusion therapy), or we have inadequate knowledge of long-term effects on organ function (HU). Based on this discussion, a safe and effective treatment with curative intent would be the most optimal to prevent progression of SCD from infancy though childhood and adolescence to adulthood, thus reducing morbidity and improving life expectancy to rates that we have come to accept as indicators of progress in the developed world.
HSCT
The first clinical trials for children with SCD undergoing HSCT were based on data from transplants performed for those with β-thalassemia major. For children with β-thalassemia major, the clinical course is consistent and often predictable, making donor availability the only deciding factor before HSCT. In contrast, the unpredictable course of SCD and the lack of effective measures to identify severe disease phenotypes makes deciding who should receive HSCT, and when, a perpetual quandary.
According to the Center for International Blood and Marrow Transplant Research and the European Society for Blood and Marrow Transplantation, approximately 1,200 patients with SCD have undergone HSCT (Center for International Blood and Marrow Transplant Research, European Society for Blood and Marrow Transplantation personal communication, 2014) since the first report of the curative effect of this procedure in a patient with SCD and acute leukemia in 1984.26 Significant reductions in infection rates, graft failure, graft versus host disease (GVHD), and long-term organ toxicity, through advances in supportive care, modified conditioning regimens and better prophylaxis for rejection and GVHD have accounted for these results. An in-depth review of progress of HSCT in SCD was published in 2011.27
Early experience is best reviewed through the experience reported in three series from the US,28 Belgium,29 and France.30 Most of the almost 200 patients (aged 0.9–23 years) had an HLA-MSD and underwent myeloablative conditioning followed by HSCT. All three studies reported similar results, with overall survival (OS) rates of 92%–94% and an event-free survival (EFS) of 82%–86%, with an approximately 7% transplant-related mortality, mostly due to infection. Within the Belgian series, a cohort of asymptomatic children in the Belgian series (14/50), had notably better results compared to those with symptomatic disease, with an OS of 100% vs 88% and EFS 93% vs 76%, suggesting even in the 1990s that asymptomatic patients with MSDs may have greater benefit from early transplant.29 A review of recently published studies (Table 1) demonstrates continued improvement in outcomes, particularly in MSD transplants, but also in all HSCT for SCD. Among myeloablative conditioning regimens, a 15%–20% rate of GVHD has been observed with some deaths directly attributable to GVHD.27 Despite the intensity of these regimens, some patients continue to demonstrate a mixture of both donor and recipient hematopoietic cells, termed mixed donor chimerism.28 Another key finding from the French group was that the addition of anti-thymocyte globulin to the conditioning regimen greatly decreased the rejection rate from 22.6% to 3% with an improvement in EFS to 95%.31
 | Table 1 Review of reported results of related hematopoietic stem cell transplantation (HSCT) in sickle cell disease |
Earlier concern for inadequate production of HbA in recipients with mixed chimerism was also dispelled, supporting the rationale for reduced intensity conditioning regimens using conventional agents at lower doses.28–30 The mixed donor chimerism states seen with these regimens are able to produce sufficient donor type Hb reversing the SCD phenotype as well as minimize the risk of GVHD.32 Non-myeloablative regimens may result in less acute and long-term organ toxicity and also expand the option of HSCT to older patients with SCD who may have some compromised organ function and may not be able to tolerate full myeloablation.27,33 Targeted immunoablation, allowing engraftment to proceed through a donor alloimmune response has also been used successfully, primarily in patients with MSDs.34–36
Based on these data, outcomes for MSD HSCT have improved significantly, and in our opinion enough to warrant wider use in homozygous SCD patients. However, it is estimated that only 14%–20% of individuals with SCD have unaffected MSDs. To expand the donor pool so that all eligible patients with SCD could be offered this curative therapy, alternative donor sources have been explored.
UCB
In recent years, UCB, which contains a rich source of hematopoietic stem and progenitor cells, has been used successfully as an alternative allogeneic donor source to treat a variety of pediatric genetic, hematologic, immunologic, and oncologic disorders.
Related donor cord blood has been used with equal success compared to related donor marrow for HSCT in patients with SCD, with one series of eleven patients reporting 100% OS and 90% EFS, with low rates of both acute and chronic GVHD.37 In light of the success seen with related cord blood transplants, there has been a growing push toward banking cord blood in those families with children affected by SCD. Viacord®, in conjunction with the Children’s Hospital Oakland Research Institute, has developed The Sibling Connection Program, which includes cord blood and cord tissue stem collection, processing, and 5 years storage at no cost to expectant parents who have a child with an underlying condition that can be cured by HSCT.
The use of unrelated cord blood units for HSCT has been investigated for patients with SCD with disappointing results to date. While the greater tolerance of HLA disparity and lower overall incidence of GVHD make cord blood an attractive alternative for patients without sibling donors, studies have shown high rates of primary graft failure despite several different non-myeloablative conditioning regimens.38 In a recent study of eight patients, using a myeloablative regimen with unrelated cord blood grafts, the OS was 62.5% and the EFS 50%, making these results unacceptable.39 Although a slightly higher rate of donor engraftment was seen with this regimen, there was a longer delay in immune reconstitution and higher transplant-related mortality was observed. Additional strategies to enhance engraftment, such as using double cord blood units or ex vivo expansion of cord blood stem cells, are under investigation.
Alternative stem cell sources such as matched unrelated donors (MUDs) are currently being studied with several multicenter trials being conducted to assess the feasibility and safety of MUD transplants for symptomatic patients with SCD who lack an MSD. Although this approach may afford some patients without an MSD the possibility of HCT, the requirement of an 8/8 HLA-MUD is also limiting. The National Marrow Donor Program and Center for International Blood and Marrow Transplant Research have used HLA data from their donor and cord blood registries to predict the likelihood of locating an MUD within the worldwide registries. The likelihood of finding a donor varies considerably and is based on ethnic and racial groups. The probability of finding an 8/8 HLA-MUD is best among Caucasians of European decent (75%) and the worst among blacks of South and Central American decent (16%), with patients of Middle Eastern or North African descent and African-Americans and patients of African descent having probabilities of 46%, 19%, and 18%, respectively. This is disappointing, because the highest prevalence of SCD remains in Africa, and the low likelihood of finding an unrelated donor in patients of African descent will severely limit the availability of this curative treatment to most of these patients.40
Haploidentical donor transplantation has shown some promise and the success of this modality would expand the possibility of HSCT to nearly all patients. In a recent series of 19 SCD patients with SCD undergoing HSCT, 14 had haploidentical donors.41 Although graft failure (primary and secondary) was seen in six of these 14 patients, all were alive with a median follow-up of 711 days. Six of the eight patients who engrafted are completely off immunosuppression with no evidence of GVHD. In another study using haploidentical HSCT in SCD, disease-free survival was only 38% with an OS of 75%.42 While these early reports do show some promise, much progress remains to be made to improve outcomes before this approach can be more broadly used.
Preimplantation genetic diagnosis with HLA matching
Many patients and families remain interested in pursuing the currently accepted option of MSD HSCT despite the absence of an MSD. For such patients, an alternative strategy using assisted reproductive technology has been developed in the past decade. Such technology would identify an unaffected, HLA-matched embryo with the goal of using this future child’s cord blood after birth for HSCT in the affected sibling. Pre-implantation genetic diagnosis (PGD) combined with HLA matching was first reported for a family with a child diagnosed with Fanconi Anemia in 2001.43 Recent data suggest a success rate of 70% when selecting for an HLA-matched embryo; however the rate of success falls to 57% when the goal is to select for embryos unaffected by a genetic mutation.44 The largest barrier to procuring a matched sibling cord blood unit via PGD is cost. Often it takes several cycles to achieve a successful pregnancy with an unaffected, HLA-matched sibling, and at a cost of US$15,000–$20,000/cycle, a family may spend as much as $60,000 (currently not reimbursed by insurance) with no assurance that they will be successful.45 However, PGD does offer another option for families who do not have an MSD and are concerned about the higher risks of unrelated donor transplantation.
Long-term effects of HSCT
There has also been debate about the long-term toxicities from HSCT. The rates of most of the other more severe complications, including rejection and GVHD, have been significantly reduced as demonstrated by the high EFS rates as summarized in Table 1. However, fertility has long remained an area of concern for both patients and their families and sometimes deters them from pursuing HSCT. The risk of infertility after HSCT depends on many factors, most notably the inclusion of radiation, in the conditioning regimen, gonadotoxic chemotherapeutic agents, and stage of pubertal development at the time of HSCT.46.47 Long-term follow-up evaluations of SCD patients who underwent HSCT with myeloablative conditioning found that, of the evaluable patients, 9/13 males had normal follicle-stimulating hormone and luteinizing hormone levels, but only 3/13 had normal testosterone levels.47 Among females, 8/14 had primary ovarian failure.47 Improvements in reproductive medicine techniques have offered more individuals the option of fertility preservation. Although there is limited data, fertility preservation has been successful in both males (cryopreservation of sperm or testicular tissue) and females (preservation of embryos, mature oocytes, and ovarian tissue) with SCD prior to HSCT.48 While these options do afford patients with SCD undergoing HSCT the possibility of successful pregnancies, one must be mindful of the fact that these procedures can be quite costly and may not be covered by insurance. One hopes that with the use of non-myeloablative regimens and improvement of techniques to improve fertility preservation, the benefits of HSCT to those with SCD will outweigh the risks.
Patient selection for HSCT in SCD
Currently, patient selection for non-investigational HSCT in SCD is still mostly based on the inclusion criteria from some of the early studies of transplantation.49 This includes patients with homozygous SCD who have had severe or recurrent complications of SCD, and have an HLA-MSD. While initial studies included only patients with debilitating clinical events, such as central nervous system involvement (stroke or neurological event lasting >24 hours, abnormal brain MRI, or increased velocities on TCD) or recurrent ACS and VOC requiring frequent hospitalizations, osteonecrosis, red cell alloimmunization, and lung disease in adults have been added to the generally accepted inclusion criteria. Typically, children tend to be selected over adults as they have less end organ damage, and are therefore at lower risk for transplant-related morbidity.
The rationale for this approach has been related to the risk of morbidity and mortality associated with the procedure itself, which is even higher in the non-MSD setting. While we agree that the latter scenario has not yet progressed adequately to an acceptable morbidity risk, in the MSD setting, as described in the earlier section, tremendous progress has been made, with OS and EFS in a very acceptable range. It is important to keep in mind that these HSCT mortality rates are from series including recipients who already had preexisting morbidity, and other potential risk factors such as alloimmunization. If asymptomatic children who have none of these complications undergo MSD HSCT, the results would be excellent, as has already been demonstrated in the small Belgian cohort.29 There has also been debate about the long-term toxicities from HSCT, especially the risk for infertility and increased risk of future malignancies. Keeping in mind the lower risk of these complications with the use of more non-myeloablative regimens, and the higher overall risk of morbidity (including infertility, and impotence in males suffering priapism) and mortality without HSCT, the balance between these risks may favor HSCT.
Therefore, we would recommend that siblings of all patients with homozygous SCD be HLA-tested to see if they are a match, and if they are, that the patient should be evaluated for HSCT. We recommend this be done in early childhood, and be irrespective of whether the child is symptomatic or not. The discussion above should provide adequate justification for such an approach.
The family should then be thoroughly apprised of the risk of the procedure itself and a balanced view of the risks of living with SCD itself should be provided. Patients and families should be encouraged to consider all treatment options even in the absence of current or chronic symptoms. With the current transplant-related mortality rate in children undergoing HSCT from MSDs being less than the 6.1% overall sickle cell-related mortality by age 18 years,14 one could conclude that it is more likely that a child would succumb to complications related to SCD if he/she does not undergo HSCT than if he/she does.
Families should also be made aware of the Viacord®/Children’s Hospital Oakland Research Institute UCB repository, and encouraged to have UCB collected and stored from future pregnancies, because of the excellent results from HSCT using matched sibling UCB as well.
At present, we do not recommend HSCT for patients without an MSD who do not have “severe” disease with clinical symptomatology.
For those patients with symptoms, consideration for unrelated HSCT must be in the investigational setting only, with stringent oversight and defined inclusion and exclusion criteria.
At present, we do not have enough data on the morbidity and mortality of other forms of SCD, such as Hb S-β-thalassemia or hemoglobin SC disease and cannot make any recommendations for HSCT in such patients.
Discussion
In the authors’ opinion, the severe morbidity and markedly increased mortality of homozygous SCD justifies more aggressive treatments with curative intent. As reviewed above, progress treating this disease has made a significant impact on morality rates in children, with over 90% reaching 18 years of age. However, mortality rises steeply after this time, a result of chronic organ damage and dysfunction, which starts in childhood. At the same time, results of MSD HSCT have improved dramatically, especially when the recipient is young or treated before any significant organ complications having developed. Despite this, hematologists remain reluctant to recommend HSCT, particularly in “less severe” homozygous SCD patients, who have not had many complications. Yet it is well known that the concept of “less severe” is a fallacy. The same hematologists would likely recommend HSCT for β-thalassemia, a non fatal condition where regularly transfused and effectively chelated patients have an excellent chance of near normal life expectancy in the 21st century. SCD is a progressive condition with a high prevalence of ongoing brain injury, organ damage, and a markedly shortened life expectancy. Currently, childhood mortality from homozygous SCD in the US is higher than that from MSD HSCT, and the latter is decreasing. The excellent results of MSD HSCT from single institutions must also be replicated in other institutions, through large multi center trials with consistent protocols. Further, when considering recent MSD HSCT trials with excellent survival, it is important to keep in mind that the follow-up in these trials may not be very long, and the “transplant-related” mortality may increase with time if long-term complications were to occur (such as secondary malignancy from chemotherapy exposure). However, the incidence of these would likely be small.
Based on current guidelines, holdovers from the 1990s (Walters et al),49 HSCT in SCD is only for those with “severe” disease, often after they have had an overt stroke or multiple episodes of ACS or VOC. HSCT at this point is not only more complicated because of organ damage, but silent infarcts and other neurological complications may already have occurred. Recently an expert international panel recommended that symptomatic children with SCD undergo early (pre school age) HSCT if they had MSDs, but no mention was made of the “asymptomatic” or “less severe patient” with SCD.50 One barrier to the greater use of HSCT remains the lack of knowledge about the natural history of SCD, as has been shown by multiple surveys. Therefore, the authors feel strongly about recommendations 1 and 2. HSCT should be discussed early in the course of the relationship between hematologist and patient/family. If a patient with homozygous SCD has an MSD and is eligible for HSCT, this must be offered, and the sooner the evaluation begins the better, so that if a transplant is planned, it could be done before significant organ damage has occurred. If the patient fully understands the pros and cons, and does not consent, we suggest there remain an ongoing dialogue about this modality as further progress is made in the field of HSCT. With the sharp increase in interest in this curative therapy, parental and patient acceptance of the risks of the procedure has also increased, with 45% of parents likely accepting this procedure for their child, and 35% of adolescents accepting of it for themselves, if recommended by their hematologist (Roth et al).17 Further, early analysis of health-related quality of life in children following HSCT showed a significant improvement within just 1 year of HSCT.51
Admittedly, HSCT is associated with some risk and by no means are the authors minimizing the complications that can accompany the procedure. Certainly the potential for infertility, life-threatening infections or chronic GVHD would justify pause for many patients and their families.52 However, with the advent of non-myeloablative conditioning regimens and improved supportive care therapies, these risks have dramatically decreased such that outcomes with non-myeloablative regimens are similar to, if not better than, those seen with myeloablative regimens. It is also important to be mindful of the complications of SCD itself such as the higher overall risk of morbidity (including infertility, and impotence in males suffering priapism) and mortality without HSCT. Additionally, as has been shown in the earlier studies, all stem cell sources (bone marrow, cord blood, and peripheral blood) have been used with similar outcomes and relatively few complications in both the adult and pediatric setting.
As previously discussed, donor availability still remains one of the major limiting factors in offering this modality to more individuals. As SCD is a disease which primarily affects those of African or Hispanic descent who are not well represented in the donor registries, more and more people should be encouraged to join registries to make this treatment a more viable option for all afflicted with this condition. The repository for UCB mentioned above should be expressly contacted when the mother of a child with SCD is having another child. Cord blood is stored free of charge for 5 years by this registry and will be made available if compatible for an affected sibling for HSCT.
While the use of alternate donor transplants in SCD cannot be considered standard of care, there is certainly enough data to advocate for further clinical investigative trials, such that this treatment modality may one day be considered first line therapy as opposed to retrieval therapy.
While most of the information regarding HSCT and SCD has focused on those with homozygous SCD, it would be remiss not to mention those with hemoglobin SC disease or HbS-β-thalassemia. Historically, these patients have less severe disease; however, HSCT should be considered in those with an MSD who display the more severe manifestations such as those outlined by the Multicenter Collaborative Trial.49
Finally, from the conclusions of the NIH Consensus Panel,18 and the survey of parents and children with SCD,17 it is clear that hematologists who care for patients with SCD could be better informed about treatment options, their pros, cons, and results, and should pass this information on to the patient families so that they are up to date about the options for treatment, including HU and HSCT, and in the future, gene therapy.
Summary and conclusion
It is time that we change our eligibility criteria for HSCT in SCD. We have provided justification for expanding the role of this curative treatment option for all patients with homozygous SCD who have an MSD, irrespective of whether they have “less severe” disease or have had one or more of the devastating complications of SCD. In our opinion, to say that MSD HSCT is still an experimental therapy, or that the risks associated with it are unacceptable and that it cannot be recommended for asymptomatic patients is no longer valid. The overall risk of mortality with HSCT in this specific setting is lower than the risk of mortality before age 18 without HSCT and should be a convincing argument for expanding its use. However, alternative donor HSCT and other modifications of the standard protocol must still be considered experimental and should be performed in an investigational setting. At this time, we do not have adequate data on mortality and complications of other forms of SCD, and cannot make specific recommendations for those patients.
Disclosure
The authors report no conflicts of interest in this work.
References
Hassell KL. Population estimates of sickle cell disease in the US. Am J Prev Med. 2010;38(4 Suppl):S512–S521. | |
Nickel RS, Hendrickson JE, Haight AE. The ethics of a proposed study of hematopoietic stem cell transplant for children with “less severe” sickle cell disease. Blood. 2014;124(6):861–866. | |
Kassim AA, DeBaun MR. The case for and against initiating either hydroxyurea therapy, blood transfusion therapy or hematopoietic stem cell transplant in asymptomatic children with sickle cell disease. Expert Opin Pharmacother. 2014;15(3):325–336. | |
Miller ST, Sleeper LA, Pegelow CH, et al. Prediction of adverse outcomes in children with sickle cell disease. N Eng J Med. 2000;342(2):83–89. | |
Quinn CT, Lee NJ, Shull EP, Ahmad N, Rogers ZR, Buchanan GR. Prediction of adverse out- comes in children with sickle cell anemia: a study of the Dallas Newborn Cohort. Blood. 2008;111(2):544–548. | |
Platt OS, Brambilla DJ, Rosse WF, et al. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med. 1994;330(23):1639–1644. | |
Brousse V, Buffet P, Rees D. The spleen and sickle cell disease: the sick(led) spleen. Br J Haematol. 2014;166(2):165–176. | |
Miller ST, Macklin EA, Pegelow CH, et al. Silent infarction as a risk factor for overt stroke in children with sickle cell anemia: a report from the Cooperative Study of Sickle Cell Disease. J Pediatr. 2001;139(3):385–390. | |
Wang W, Enos L, Gallagher D, et al. Neuropsychologic performance in school-aged children with sickle cell disease: a report from the Cooperative Study of Sickle Cell Disease. J Pediatr. 2001;139(3):391–397. | |
DeBaun MR, Armstrong FD, McKinstry RC, Ware RE, Vichinsky E, Kirkham FJ. Silent cerebral infarcts: a review on a prevalent and progressive cause of neurologic injury in sickle cell anemia. Blood. 2012; 119(20):4587–4596. | |
Adams RJ, McKie VC, Hsu L, et al. Prevention of a first stroke by transfusions in children with sickle cell anemia and abnormal results on transcranial Doppler ultrasonography. N Engl J Med. 1998;339(1):5–11. | |
DeBaun MR, Gordon M, McKinstry RC, et al. Controlled trial of transfusions for silent cerebral infarcts in sickle cell anemia. N Engl J Med. 2014;371(8):699–710. | |
Vichinsky EP, Neumayr LD, Gold JI, et al. Neuropsychological dysfunction and neuroimaging abnormalities in neurologically intact adults with sickle cell anemia. JAMA. 2010;303:1823–1831. | |
Quinn CT, Rogers ZR, McCavit TL, Buchanan GR. Improved survival of children and adolescents with sickle cell disease. Blood. 2010; 115(17):3447–3452. | |
DeBaun MR, Telfair J. Transition and sickle cell disease. Pediatrics. 2012;130(5):926–935. | |
Panepinto JA, Bonner M. Health-related quality of life in sickle cell disease: past, present, and future. Pediatr Blood Cancer. 2012;59(2):377–385. | |
Roth M, Krystal J, Manwani D, Driscoll C, Ricafort R. Stem cell transplant for children with sickle cell anemia: parent and patient interest. Biol Blood Marrow Transplant. 2012;18(11):1709–1715. | |
Brawley OW, Cornelius LJ, Edwards LR, et al. National Institutes of Health Consensus Development Conference statement: hydroxyurea treatment for sickle cell disease. Ann Intern Med. 2008;148(12):932–938. | |
Bishop S, Matheus MG, Abboud MR, et al. Effect of chronic transfusion therapy on progression of neurovascular pathology in pediatric patients with sickle cell anemia. Blood Cells Mol Dis. 2011;47(2):125–128. | |
Abboud MR, Yim E, Musallam KM, Adams RJ; STOP II Study Investigators. Discontinuing prophylactic transfusions increases the risk of silent brain infarction in children with sickle cell disease: data from STOP II. Blood. 2011;118(4):894–898. | |
Charache S, Terrin ML, Moore RD, et al. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia. N Engl J Med. 1995;332(20):1317–1322. | |
Steinberg MH, Barton F, Castro O, et al. Effect of hydroxyurea on mortality and morbidity in adult sickle cell anemia: risks and benefits up to 9 years of treatment. JAMA. 2003;289(13):1645–1651. | |
Steinberg MH, McCarthy WF, Castro O, et al. The risks and benefits of long-term use of hydroxyurea in sickle cell anemia: A 17.5 year follow-up. Am J Hematol. 2010;85(6):403–408. | |
Voskaridou E, Christoulas D, Bilalis A, et al. The effect of prolonged administration of hydroxyurea on morbidity and mortality in adult patients with sickle cell syndromes: results of a 17-year, single-center trial (LaSHS). Blood. 2010;115(12):2354–2363. | |
Wang WC, Ware RE, Miller ST, et al. Hydroxycarbamide in very young children with sickle-cell anaemia: a multicentre, randomised, controlled trial (BABY HUG). Lancet. 2011;377(9778):1663–1672. | |
Johnson FL, Look AT, Gockerman J, Ruggiero MR, Dalla-Pozza L, Billings FT 3rd. Bone-marrow transplantation in a patient with sickle-cell anemia. N Engl J Med. 1984;311(12):780–783. | |
Hsieh MM, Fitzhugh CD, Tisdale JF. Allogeneic hematopoietic stem cell transplantation for sickle cell disease: the time is now. Blood. 2011;118(5):1197–1207. | |
Walters MC, Patience M, Leisenring W, et al. Stable mixed hematopoietic chimerism after bone marrow transplantation for sickle cell anemia. Biol Blood Marrow Transplant. 2001;7(12):665–673. | |
Vermylen C, Cornu G, Ferster A, et al. Haematopoietic stem cell transplantation for sickle cell anaemia: the first 50 patients transplanted in Belgium. Bone Marrow Transplant. 1998;22(1):1–6. | |
Bernaudin F. Resultats et indications actuelles de l’allogreffe de moelle dans la drepanocytose [Results and current indications of bone marrow allograft in sickle cell disease]. Pathol Biol (Paris). 1999;47(1):59–64. French. | |
Bernaudin F, Socie G, Kuentz M, et al. Long-term results of related myeloablative stem-cell transplantation to cure sickle cell disease. Blood. 2007;110(7):2749–2756. | |
Wu CJ, Gladwin M, Tisdale J, et al. Mixed haematopoietic chimerism for sickle cell disease prevents intravascular haemolysis. Br J Haematol. 2007;139(3):504–507. | |
Shenoy S. Hematopoietic stem cell transplantation for sickle cell disease: current practice and emerging trends. Hematology Am Soc Hematol Educ Program. 2011;2011:273–279. | |
Shenoy S, Grossman WJ, DiPersio J, et al. A novel reduced-intensity stem cell transplant regimen for nonmalignant disorders. Bone Marrow Transplant. 2005;35(4):345–352. | |
Krishnamurti L, Kharbanda S, Biernacki MA, et al. Stable long-term donor engraftment following reduced-intensity hematopoietic cell transplantation for sickle cell disease. Biol Blood Marrow Transplant. 2008;14(11):1270–1278. | |
Hsieh MM, Fitzhugh CD, Weitzel RP, et al. Nonmyeloablative HLA-Matched Sibling Allogeneic Hematopoietic Stem Cell Transplantation for Severe Sickle Cell Phenotype. JAMA. 2014;312(1):48–56. | |
Locatelli F, Rocha V, Reed W, et al. Related umbilical cord blood transplantation in patients with thalassemia and sickle cell disease. Blood. 2003;101(6):2137–2143. | |
Kamani NR, Walters MC, Carter S, et al. Unrelated donor cord blood transplantation for children with severe sickle cell disease: results of one cohort from the phase II study from the Blood and Marrow Transplant Clinical Trials Network (BMT CTN). Biol Blood Marrow Transplant. 2012;18(8):1265–1272. | |
Radhakrishnan K, Bhatia M, Geyer MB, et al. Busulfan, Fludarabine and Alemtuzumab Conditioning and Unrelated Cord Blood Transplantation in Children with Sickle Cell Disease. Biol Blood Marrow Transplant. 2013;19(4):676–677. | |
Gragert L, Eapen M, Williams E, et al. HLA match likelihoods for hematopoietic stem-cell grafts in the US registry. N Engl J Med. 2014; 371(4):339–348. | |
Bolanos-Meade J, Fuchs EJ, et al. HLA-haploidentical bone marrow transplantation with posttransplant cyclophosphamide expands the donor pool for patients with sickle cell disease. Blood. 2012;120(22):4285–4291. | |
Dallas MH, Triplett B, Shook DR, et al. Long-term outcome and evaluation of organ function in pediatric patients undergoing haploidentical and matched related hematopoietic cell transplantation for sickle cell disease. Biol Blood and Marrow Transplant. 2013;19(5):820–830. | |
Verlinsky Y, Rechitsky S, Schoolcraft W, Strom C, Kuliev A. Preimplantation diagnosis for Fanconi anemia combined with HLA matching. JAMA. 2001;285(24):3130–3133. | |
Kahraman S, Beyazyurek C, Yesilipek MA, et al. Successful haematopoietic stem cell transplantation in 44 children from healthy siblings conceived after preimplantation HLA matching. Reprod Biomed Online. 2014;29(3):340–351. | |
Morgan ER, Girod J, Rinehart JS. Having a child to save a sibling: reassessing risks and benefits of creating stem cell donors. Pediatr Blood Cancer. 2007;48(3):249–253. | |
Smith-Whitley K. Reproductive issues in sickle cell disease. Blood. 2014;124(24):3538–3543. | |
Walters MC, Hardy K, Edwards S, et al. Pulmonary, gonadal, and central nervous system status after bone marrow transplantation for sickle cell disease. Biol Blood Marrow Transplant. 2010;16(2):263–272. | |
Practice Committee of American Society for Reproductive Medicine. Fertility preservation in patients undergoing gonadotoxic therapy or gonadectomy: a committee opinion. Fertil Steril. 2013;100(5):1214–1223. | |
Walters MC, Patience M, Leisenring W, et al. Bone marrow transplantation for sickle cell disease. N Engl J Med. 1996;335(6):369–376. | |
Angelucci E, Matthes-Martin S, Baronciani D, et al. Hematopoietic stem cell transplantation in thalassemia major and sickle cell disease: indications and management recommendations from an international expert panel. Haematologica. 2014;99(5):811–820. | |
Bhatia M, Kolva E, Cimini L, et al. Health-related quality of life after allogeneic hematopoietic stem cell transplantation for sickle cell disease. Biol Blood Marrow Transplant. 2015;21(4):666–672. | |
Chakrabarti S, Bareford D. A survey on patient perception of reduced-intensity transplantation in adults with sickle cell disease. Bone Marrow Transplant. 2007;39(8):447–451. | |
Iannone R, Casella JF, Fuchs EJ, et al. Results of minimally toxic nonmyeloablative transplantation in patients with sickle cell anemia and beta-thalassemia. Biol Blood Marrow Transplant. 2003;9(8):519–528. | |
Panepinto JA, Walters MC, Carreras J, et al. Matched-related donor transplantation for sickle cell disease: report from the Center for International Blood and Transplant Research. Br J Haematol. 2007;137(5):479–485. | |
Matthes-Martin S, Lawitschka A, Fritsch G, et al. Stem cell transplantation after reduced-intensity conditioning for sickle cell disease. Eur J Haematol. 2013;90(4):308–312. | |
Bhatia M, Jin Z, Baker C, et al. Reduced toxicity, myeloablative conditioning with BU, fludarabine, alemtuzumab and SCT from sibling donors in children with sickle cell disease. Bone Marrow Transplant. 2014;49(7):913–920. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.