")
Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Hemagglutinin-based polyanhydride nanovaccines against H5N1 influenza elicit protective virus neutralizing titers and cell-mediated immunity
Authors Ross K, Loyd H, Wu W, Huntimer L, Ahmed S, Sambol A, Broderick S, Flickinger Z, Rajan K, Bronich T, Mallapragada S, Wannemuehler M , Carpenter S, Narasimhan B
Received 5 August 2014
Accepted for publication 4 October 2014
Published 30 December 2014 Volume 2015:10(1) Pages 229—243
DOI https://doi.org/10.2147/IJN.S72264
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Kathleen A Ross,1 Hyelee Loyd,2 Wuwei Wu,2 Lucas Huntimer,3 Shaheen Ahmed,4 Anthony Sambol,5 Scott Broderick,6 Zachary Flickinger,2 Krishna Rajan,6 Tatiana Bronich,4 Surya Mallapragada,1 Michael J Wannemuehler,3 Susan Carpenter,2 Balaji Narasimhan1
1Chemical and Biological Engineering, Iowa State University, Ames, IA, USA; 2Animal Science, Iowa State University, Ames, IA, USA; 3Veterinary Microbiology and Preventive Medicine, Iowa State University, Ames, IA, USA; 4Pharmaceutical Sciences, University of Nebraska Medical Center, Omaha, NE, USA; 5Pathology and Microbiology, University of Nebraska Medical Center, Omaha, NE, USA; 6Materials Science and Engineering, Iowa State University, Ames, IA, USA
Abstract: H5N1 avian influenza is a significant global concern with the potential to become the next pandemic threat. Recombinant subunit vaccines are an attractive alternative for pandemic vaccines compared to traditional vaccine technologies. In particular, polyanhydride nanoparticles encapsulating subunit proteins have been shown to enhance humoral and cell-mediated immunity and provide protection upon lethal challenge. In this work, a recombinant H5 hemagglutinin trimer (H53) was produced and encapsulated into polyanhydride nanoparticles. The studies performed indicated that the recombinant H53 antigen was a robust immunogen. Immunizing mice with H53 encapsulated into polyanhydride nanoparticles induced high neutralizing antibody titers and enhanced CD4+ T cell recall responses in mice. Finally, the H53-based polyanhydride nanovaccine induced protective immunity against a low-pathogenic H5N1 viral challenge. Informatics analyses indicated that mice receiving the nanovaccine formulations and subsequently challenged with virus were similar to naïve mice that were not challenged. The current studies provide a basis to further exploit the advantages of polyanhydride nanovaccines in pandemic scenarios.
Keywords: polymer, nanoparticle, vaccine, subunit, neutralizing antibody
Introduction
Influenza virus is a major cause of serious respiratory illness and has been responsible for significant morbidity and mortality in humans worldwide. The major strains of influenza A virus circulating in human populations are H3N2 and H1N1, which are associated with seasonal influenza viral infections. In addition, the avian strains H5N1 and H7N9 have been found to infect humans;1 however, they are not currently capable of sustained human-to-human transmission. Should these highly pathogenic avian viruses develop such capability, the disease could spread rapidly, resulting in a global influenza pandemic. Vaccination represents a critical control measure against yearly seasonal influenza viruses and is an essential component of pandemic preparedness plans.2 Nearly all of the current influenza vaccine technologies are based on the use of embryonated eggs and require a relatively long production cycle, resulting in limited manufacturing capacity.2 The response to the 2009 H1N1 pandemic clearly demonstrated the limitations of vaccine production methods with respect to rapid deployment, which led to vaccine shortages. Consequently, there is great interest in developing new technologies for rapid, large-scale production of safe and efficacious influenza vaccines.
Recombinant hemagglutinin (rHA)-based vaccines produced in mammalian or insect cell culture systems are attractive alternatives to egg-based vaccine technologies.3 Production and purification of rHA protein can effectively reduce the vaccine production time and the use of cell culture systems may glycosylate proteins, which is important for the development of neutralizing antibodies against receptor-binding site epitopes.4 Recent studies demonstrated that rHA engineered to form a stable trimeric configuration elicited a protective immune response in vaccinated animals.5–10 Neutralizing antibody levels and protection from disease were enhanced in mice vaccinated with soluble rHA trimers as compared to animals vaccinated with rHA monomers,5,8 indicating the importance of immunogens that mimic those expressed by the infectious agent. Along these lines, the composition and/or number of N-linked glycans on rHA trimers have been shown to modify the level of the protective antibody response.10,11 All of these studies indicate that rHA trimers could be an important component of a subunit vaccine strategy against influenza.
Hemagglutinin (HA)-specific serum antibody titers correlate with protection from disease; however, recombinant proteins are often weak immunogens and require multiple, high-dose immunizations to achieve protection.6,7,12 Polyanhydride nanoparticles have been shown to be a versatile vaccine adjuvant/delivery platform capable of enhancing the immune response to recombinant proteins.13–15 Polyanhydrides are biodegradable materials that provide sustained release kinetics of encapsulated antigens, resulting in long-lived, high-avidity antibody titers, even with suboptimal doses of antigen.13–15 In addition to amplifying humoral immunity, polyanhydride nanoparticle–based vaccines (ie, nanovaccines) have been shown to be immunomodulatory16 and are capable of promoting cell-mediated immunity. Previous work with polyanhydride nanovaccines indicated expansion of antigen-specific CD8+ T cells following nanovaccine immunization, resulting in memory T cell populations that responded to antigen-expressing tumor challenge (CD8+ T cells).17 This broad repertoire of immune responses induced by polyanhydride nanovaccines may prove beneficial for influenza vaccine efficacy because robust cell-mediated responses are often associated with broader protective immunity and directed at conserved epitopes.18,19
Previously, soluble H5 HA trimers (sH53) from H5N1 influenza virus A/Whooper Swan/Mongolia/244/05 were produced using a baculovirus insect cell expression system and shown to maintain their oligomeric structure and antigenicity upon release from polyanhydride nanoparticles.20 In this study, the immunogenicity and virus-neutralizing antibody titer in response to nanovaccine immunization were observed for approximately 2 months, and concluded with an analysis of memory T cell responses. Finally, the efficacy of the nanovaccine formulations was examined using a low-pathogenic, live-viral challenge. The data demonstrate that polyanhydride nanovaccines containing immunogenic HA trimer represent a potentially viable platform for pandemic influenza vaccines.
Materials and methods
Plasmids and antibody
Plasmid pHW500 (Genbank DQ659326), obtained from Dr Bruce Janke of Iowa State University, contains the full-length HA gene from HPAI H5N1 influenza virus A/Whooper Swan/Mongolia/244/05 (H5N1). The pHW500 HA gene was modified by replacement of the cognate polybasic cleavage site with that from a low-pathogenic H6N1 avian influenza virus.21 The FDA-VN plasmid, obtained from Dr Carol Weiss of the US Food and Drug Administration, contains the full-length HA from A/Vietnam/1203/2004 (H5N1) (Genbank EF541403) in pCMV/R.22 The pWS-HA was constructed by replacing the H5 Vietnam HA gene in FDA-VN with the HA gene from pHW500. The low-pathogenic cleavage site in pWS-HA was replaced with the polybasic cleavage site from A/Whooper Swan/Mongolia/3/05 (H5N1) (Genbank AB233320.1). The equine infectious anemia lentivirus vector plasmids pEV53B and pSIN6.1ClucW were obtained from John Olsen.23 Hyperimmune swine sera containing high titers of H5 neutralizing antibody was obtained from Dr Bruce Janke.21 H5-specific polyclonal rabbit antisera was obtained from Dr Carol Weiss.22
Cloning and expression of soluble H5 trimer (sH53)
The HA ectodomain (nucleotides 1–1,723) was amplified from pHW500 and modified at the 3′ end by addition of linker sequences, a GCN4pII trimerization domain,8 and His-tag sequences (Figure 1A). The modified gene was cloned into pFastBac I, transformed into DH10Bac cells, and recombinant baculoviruses were generated using Bac-to-Bac Baculovirus Expression System (Thermo Fisher Scientific, Waltham, MA, USA). Sf9 cells (Thermo Fisher Scientific) were infected with recombinant baculovirus and supernatants collected 96 hours after infection were clarified by centrifugation, dialyzed against 10 mM Tris buffer (10 mM Tris, 50 mM NaCl, pH 8.0), and incubated with Ni-NTA beads (Thomas Scientific, Swedesboro, NJ, USA) overnight at 4°C. The beads were washed with 10 mM Tris buffer containing 10 mM imidazole and recombinant sH53 was eluted in 10 mM Tris buffer containing 250 mM imidazole. The eluted proteins were dialyzed against 10 mM Tris buffer to remove imidazole, concentrated, and stored at 4°C for up to 1 month or at −80°C for long-term storage.
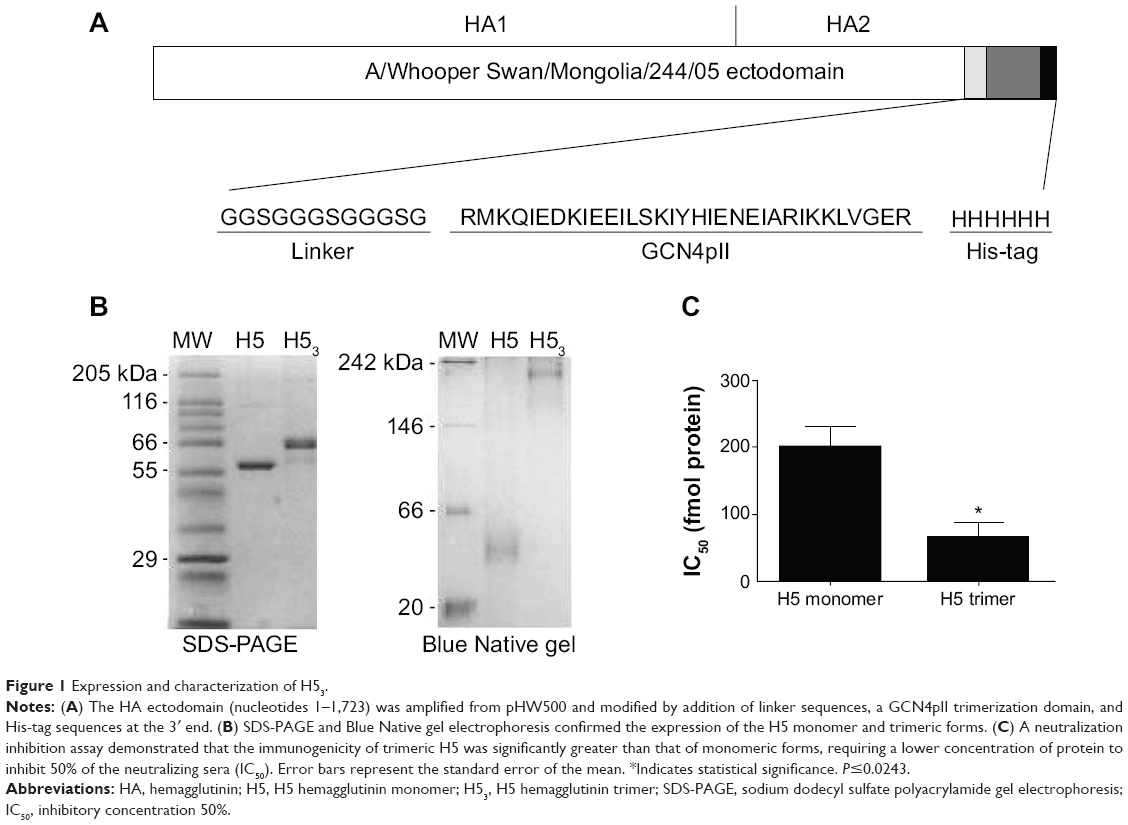 | Figure 1 Expression and characterization of H53. |
HA-pseudotyped reporter virus
HA-pseudotyped viruses were produced in HEK293T cells as described previously.22,24 Briefly, HEK293T cells were cotransfected with 5 μg of pEV-53B encoding the lentiviral core proteins of equine infectious anemia virus, 5.5 μg of the luciferase reporter plasmid plgSIN6.1Luc, and 1 μg of pFDA-VN or pWS-HA plasmid DNA expressing HA from A/Vietnam/1203/2004 (clade 1) or A/Whooper Swan/Mongolia/244/05 (clade 2.2), respectively. At 18 hours post-transfection, cells were incubated with fresh medium containing 7 mU/mL of Vibrio cholerae Type II neuraminidase (NA) (Sigma-Aldrich Co., St Louis, MO, USA) and 10 mM NaB to induce the release of H5-pseudovirions from the surface of the producer cells. Supernatants were collected 48 hours post-transfection, clarified by centrifugation, and stored at −80°C.
Reporter virus pseudotyped with HA from A/Vietnam/1203/2004 (H5-VN-Luc) or A/Whooper Swan/Mongolia/244/05 (H5-WS-Luc) were titered on HEK293T cells. Cells were seeded in 96-well plates at 2×104 cells/well in Dulbecco’s modified Eagle medium supplemented with antibiotics and heat-inactivated fetal bovine serum. The following day, cells were inoculated in triplicate with 10-fold serial dilutions of pseudovirus stock in the presence of 8 μg/mL polybrene. At 48 hours post-transduction, cells were lysed and assayed for luciferase activity using the ONE-Glo Luciferase assay system (Promega Corporation, Fitchburg, WI, USA). Luciferase activity was quantified using a Centro XS3 LB960 illuminometer (Berthold Technologies, Bad Wildbad, Germany) and results reported as relative light units (RLU)/mL supernatant.
Electrophoresis
For protein analyses in denaturing conditions, 1 μg of purified sH53 protein was boiled for 5 minutes in sodium dodecyl sulfate (SDS) loading buffer (50 mM Tris, 1% β-mercaptoethanol, 2% SDS, 0.005% bromophenol blue, and 10% glycerol) and electrophoresed in 10% SDS-PAGE (polyacrylamide gel electrophoresis). For analyses in non-denaturing gels, 3 μg of purified sH53 protein was mixed with Blue Native loading buffer (2 mM EDTA, 20 mM NaCl, 20 mM Bis-Tris, 10% glycerol, and 0.08% Coomassie Blue G-250) and separated on 10% Blue Native PAGE gel containing Bis-Tris, glycerol, and acrylamide in Bis-Tris buffer in the outer chamber and Tricine, Bis-Tris with Coomassie Blue G250 in the inner chamber. Following electrophoresis, gels were stained with Coomassie Blue and imaged with a GelDoc XR+ imaging system (Bio-Rad Laboratories Inc., Hercules, CA, USA).
Polymer and nanoparticle synthesis
Diacids based on 1,8-bis(p-carboxyphenoxy)-3,6-dioxaoctane (CPTEG) and 1,6-bis(p-carboxyphenoxy)hexane (CPH) were synthesized as previously described25,26 using 1,6-dibromohexane, 1-methyl-2-pyrrolidinone, hydroxybenzoic acid, N,N-dimethylacetamide, and tri-ethylene glycol (Sigma-Aldrich Co.); acetic acid, acetone, acetonitrile, dimethyl formamide, potassium carbonate, sulfuric acid, and toluene (Thermo Fisher Scientific, Waltham, MA, USA); 4-p-fluorobenzonitrile (Apollo Scientific, Bredbury, UK). Following diacid synthesis, 20:80 CPTEG:CPH copolymer was synthesized by melt polycondensation.26 The final composition of the polymer (23:77 CPTEG:CPH), molecular weight (6,055 Da), and purity were determined with 1H nuclear magnetic resonance spectroscopy (VXR 300 MHz; Varian, Palo Alto, CA, USA). H53-loaded nanoparticles were synthesized using a water-oil-oil double emulsion process.27 First, 4.5 mg of sH53 was concentrated into 100 μL of nanopure water. The solution of sH53 was then added to 45 mg of 20:80 CPTEG:CPH dissolved in 2.25 mL of methylene chloride and homogenized for 90 seconds. The H53-loaded nanoparticles were precipitated by pouring the solution into 562.5 mL of chilled pentane and collected via vacuum filtration. Blank nanoparticles and 1% polyinosinic-polycytidylic acid (poly I:C) (InvivoGen, San Diego, CA, USA)-loaded nanoparticles were synthesized similarly without water. Scanning electron microscopy (Quanta 250; FEI, Hillsboro, OR, USA) was used to characterize the size and morphology of the nanoparticles, which were found to be consistent with previous work.13 The encapsulation efficiency of protein was determined by degrading 10 mg of nanoparticles in 250 μL of 40 mM sodium hydroxide and quantifying the total protein released using a microBCA protein kit (Pierce, Rockford, IL, USA).13
Mice
Female BALB/c mice were purchased from Harlan Laboratories (Indianapolis, IN, USA). All mice were housed under specific pathogen-free conditions where all bedding, caging, water, and feed were sterilized prior to use. Animal procedures were conducted with the approval of the Iowa State University and University of Nebraska Medical Center Institutional Animal Care and Use Committees.
Immunizations
For each formulation, mice were administered a total of 10 μg H53. As outlined in Table 1, a portion of the 10 μg H53 dose was encapsulated into 50 μg polyanhydride nanoparticles, and delivered with or without 250 μg nanoparticles encapsulating poly I:C. Blank (ie, no encapsulated protein or poly I:C) nanoparticles were used to keep the total amount of nanoparticles (300 μg) constant among the different vaccine regimens. In addition, control formulations composed of 10 μg of soluble H53 (sH53) alone, 10 μg of sH53 delivered with blank nanoparticles, or 10 μg of sH53 in conjunction with 10 μg of monophosphoryl lipid A (MPLA) from Salmonella enterica (Sigma-Aldrich Co.) were administered. All formulations were suspended in 250 μL (subcutaneous [SC] immunization) or 50 μL (intranasal [IN] immunization) of sterile saline. Formulations containing nanoparticles were sonicated for 30 seconds to ensure dispersion of particle aggregates before immunization. SC immunizations were administered at the nape of the neck; IN immunizations were carried out using droplet admission via pipettor after administration of ketamine/xylazine chemical anesthetic. For “prime/boost/boost” regimens, booster immunizations were prepared and administered the same way as primary immunizations at days 21 and 42. Serum samples were obtained at the time points indicated via saphenous vein bleeding.
 | Table 1 H53 vaccine formulations |
Neutralization assay
Neutralizing antibody assays were carried out using HA-pseudotyped reporter virus as described previously.22,24 HEK293T cells were seeded in a 96-well plate at 2×104 cells/well and grown for 24 hours. Sera samples were serially diluted three-fold in culture medium containing 8 μg/mL polybrene (Sigma-Aldrich Co.), and mixed with an equal volume of diluted pseudovirus stock containing 5×104 RLU/mL. After incubation at 37°C for 1 hour, virus and serum mixtures were added to the cells. Infectivity was evaluated 48 hours after transduction using the One-glo Luciferase assay system (Promega Corporation). The percentage neutralization was calculated as (1− [virus + sera RLU/virus only RLU]) ×100. The percentage neutralization for each sera dilution was plotted and neutralizing titers were reported as infectious dose 50 (ID50), calculated as the reciprocal of the serum dilution that neutralized 50% of the virus.
A neutralization inhibition assay was used to evaluate the immunogenic properties of sH53 protein. Ten-fold serial dilutions of sH5 trimer or monomer were incubated at 37°C for 30 minutes with a 1:5,000 dilution of pig H5-antiserum (Dr Bruce Janke, Iowa State University). Following pre-incubation with H5 protein, 50,000 RLU of H5-WS-Luc was added and the samples incubated for an additional 1 hour at 37°C and inoculated onto HEK293T cells in 96-well plates. At 48 hours after transduction, the levels of luciferase in the transfected cells were evaluated using the One-Glo luciferase assay system (Promega Corporation) and percentage neutralization was calculated as described above. The percentage neutralization at each protein concentration was plotted and the half maximal inhibitory concentration (IC50) was calculated as the concentration of soluble H5 protein that inhibited 50% of the neutralizing activity of the diluted pig sera.
Flow cytometry for T cell memory populations
Brachial and axillary lymph nodes were harvested 63 days postimmunization and homogenized into single cell suspensions in complete tissue culture medium. Single cell populations were labeled with 2.5 μM 5-(and-6)-carboxyfluorescein diacetate, succinimidyl ester (5[6]-CFDA, SE; CFSE; Thermo Fisher Scientific, Waltham, MA, USA). Cells (2.5×105) were incubated in 96-well U-bottom plates with 0.5 μg of sH53 antigen for 96 hours. Cells were aspirated and cell surface marker expression and proliferation were evaluated using flow cytometry (BD FACSCanto; BD Biosciences, San Jose, CA, USA). Cell suspensions were blocked for nonspecific antibody binding using 0.1 mg/mL Rat IgG (Sigma-Aldrich Co.) and 10 μg/mL mouse anti-CD16/32 (eBioscience, San Diego, CA, USA). Fluorescently conjugated antibodies specific for murine CD4 and CD8 (eBioscience) were diluted in FACS buffer and were used to quantify specific cell populations.
Low-pathogenic viral challenge and clinical evaluation
The efficacy of H53-based nanoparticle vaccines were evaluated in mice that were challenged at 63 days postimmunization with the low pathogenicity influenza virus A/H5N1 VNH5N1-PR8CDC-RG, obtained from the Centers for Disease Control and Prevention (Atlanta, GA, USA). This is a PR-8-derived reassortant virus that contains the HA and NA genes of H5N1 A/Vietnam/1203/04, a clade 1 virus. Mice were anesthetized with 20 mg/mL of xylazine and 100 mg/mL of ketamine (1:4 ratio) and inoculated intranasally with 2.7×103 tissue culture infectious doses 50% (TCID50) virus in 30 μL phosphate-buffered saline. A control group consisting of naïve (ie, no vaccine), no challenge (ie, no infection) mice were housed similarly throughout the duration of the study. Three days after challenge, half of the mice were euthanized with 600 μL of 20 mg/mL xylazine and 100 mg/mL ketamine delivered via intraperitoneal injection. Bronchoalveolar lavage (BAL) fluid was collected as described previously28 and lung tissue was collected for inflammatory cytokine and viral load quantitation (described below). The remaining mice were monitored for weight loss for 2 weeks postchallenge before being removed from study.
Virus load quantitation
Following the procedure of Alsharifi et al lung tissue was preserved in 3 mL of RNAlater Stabilizing Reagent (Qiagen NV, Venlo, the Netherlands).29 Tissue was held submerged in the RNAlater for 3 days at 4°C. Tissues were then removed from the RNAlater, weighed, and cut into 30 mg pieces and individually frozen at −80°C in 1.5 mL microcentrifuge tubes. For the extraction process (total RNA), individual 30 mg tissue pieces were homogenized in 200 μL buffer RLT (RNeasy Mini Kit, Qiagen NV) using a disposable pellet pestle (Thermo Fisher Scientific) in conjunction with a cordless motor (Thermo Fisher Scientific). An additional 400 μL of buffer RLT was added to each tube after homogenization was completed. Tissue was extracted into 60 μL final volume in sterile, RNase-free H2O (Qiagen NV) and frozen at −80°C until polymerase chain reaction (PCR) was performed. Samples containing the extracted RNA were thawed, mixed well, and the total RNA concentration was determined, in duplicate measurements, using the Nanodrop method for RNA content. Total RNA concentration for each sample was adjusted to 40 μg/μL, and 5 μL was used as the template for the PCR reaction. PCR was performed on an Applied Biosystems 7500 Fast Real-Time PCR System, on the standard mode, using AgPath-ID One-Step RT-PCR Reagents (Thermo Fisher Scientific, Waltham, MA, USA) in conjunction with the Fast-Track Diagnostics FTD-21-96/12 Kit (Junglinster, Luxembourg), which contains bome mosaic virus (BMV) internal PCR extraction control, positive control, and primer/probes for universal influenza A antigen. For the standard curve, normal, non-influenza–challenged mice lungs (naïve controls) were homogenized using the procedure outlined above. RNA from stock influenza A H5 virus was extracted using the Qiagen QiAmp Viral RNA Kit Mini Kit (Qiagen NV). Extracted RNA was quantified using the Nanodrop procedure. For the standard curve, ten-fold dilutions of the H5-extracted viral RNA were mixed with extracted RNA from the normal mouse lungs that had been standardized to 40 ng/μL. Standard curves were obtained with each set of PCR reactions.
Cytokine analysis of BAL fluid
BAL fluid samples collected 3 days post-viral challenge were analyzed for the inflammatory cytokines IL-6, IP-10, MIG, G-CSF, IFN-g, MCP-1, KC, and MIP-2 using a MILLIPLEX® MAP assay kit (EMD Millipore, Billerica, MA, USA). The assay was performed according to manufacturer instructions. Briefly, 25 μL of BAL fluid, 25 μL assay buffer, and 25 μL MILLIPLEX MAP beads were added to each well of a 96-well plate. After shaking overnight at 4°C, the plate was washed and incubated with 25 μL/well detection antibody for 1 hour at room temperature. Following, 25 μL/well of streptavidin phycoerythrin was added for an additional 30 minutes. The plate was washed once more before measuring fluorescence intensity on a Bio-Plex 200 system (Bio-Rad Laboratories Inc.).
Principal component analysis
Previously, principal component analysis (PCA) was used to identify pathogen-mimicking capabilities of polyanhydride nanoparticles in comparison to bacteria.30 In this study, PCA was used to mathematically capture the similarities among the different vaccine formulations tested and their immune responses. A linear data dimensionality reduction method31–33 was used to condense eight-dimensional data (neutralizing antibody titer, viral load, and six cytokine concentrations) into two dimensions (ie, principal components [PC]). In this work, PC1 was a linear combination of viral load and cytokine concentration(s), while PC2 primarily captured neutralizing antibody titer. At least 90% of the variance was captured between these two PCs. Similarities among data were mathematically represented as the proximity between data points (ie, data points close together are more similar than data points farther apart).
Statistics
Statistical significance among formulations (P≤0.05) was determined by one-way analysis of variance (ANOVA) followed by Tukey’s post-test using Graph Pad Prism (v6.01; Graph Pad Software, Inc., La Jolla, CA, USA).
Results
Characterization of soluble H5 trimer protein (sH53)
A recombinant baculovirus containing the ectodomain of HA from A/Whooper Swan/Mongolia/244/05 (Figure 1A) was expressed in Sf9 insect cells and the secreted soluble H5 trimer (sH53) protein was purified from culture supernatant by affinity chromatography using nickel agarose beads. Analyses of the purified protein by electrophoresis in SDS-PAGE and Blue Native gels indicated the predominant form of sH53 protein was trimeric (Figure 1B). A neutralization inhibition assay was used to determine whether the sH53 retained neutralizing epitopes of native virus (Figure 1C). In this assay, convalescent pig sera containing high titers of H5-neutralizing antibody was incubated with serial dilutions of sH53 or sH5 monomer and tested for neutralizing activity against H5-WS-Luc pseudotyped reporter virus. The HA presented in its native trimeric state has been found to be more immunogenic than the monomeric form.5,8 Similarly, we observed that while both the monomeric and trimeric forms of sH5 inhibited neutralizing antibody at fmol concentrations, the inhibitory activity of the sH53 trimer was about 2.5-fold higher than that of the monomer.
Immunogenicity of soluble recombinant H5 HA trimer
BALB/c mice (6–8 weeks) were immunized either subcutaneously or intranasally with 10 μg of soluble protein at 0, 21, and 42 days and sera collected at 21, 42, and 63 days postimmunization were tested for neutralizing antibody against the homologous H5-WS-Luc pseudovirus. No neutralizing antibody was detected at day 21 following primary immunization (Figure 2A). By day 42, the majority of mice in both immunization groups had detectable neutralizing antibody titers (ie, >50) that were greater than protective titers reported in the literature.7,8 However, the response was highly variable among individual mice, with ID50 titers ranging from undetectable to greater than 1,000. Neutralization titers significantly increased following a second boost (day 63), with mean neutralization antibody titers of greater than 10,000. In addition, there was less variability in the neutralizing antibody titers among individual mice, especially those immunized subcutaneously.
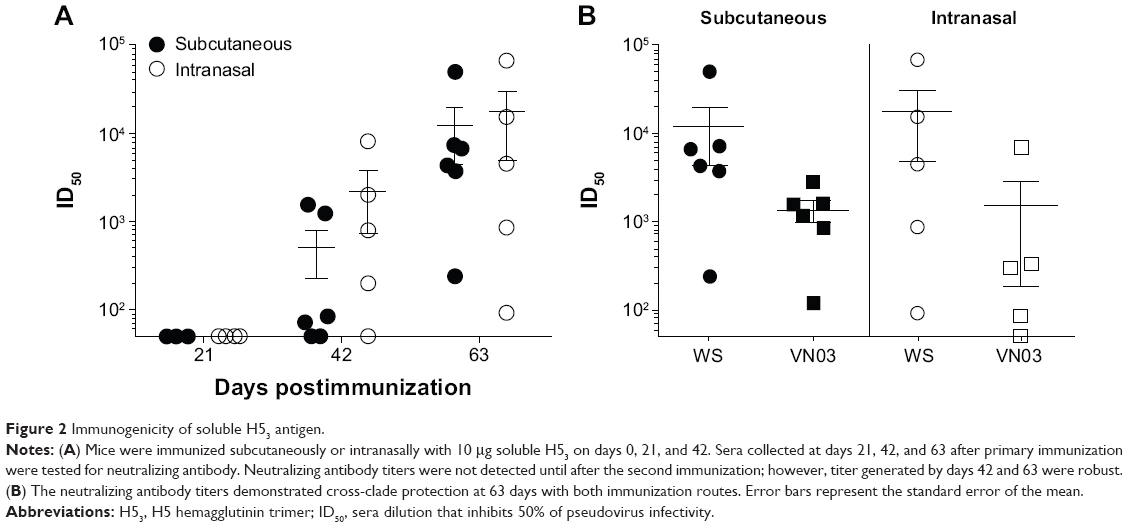 | Figure 2 Immunogenicity of soluble H53 antigen. |
As highly pathogenic H5N1 has continued to spread in avian and mammalian hosts, different lineages have emerged that are now classified into distinct, but phylogenetically related clades.1 To examine the cross-clade breadth of the neutralizing antibody response elicited with A/Whooper Swan/Mongolia/244/05-based sH53 (clade 2.2), day 63 sera was tested for neutralizing activity against H5-VN-Luc, a luciferase reporter virus pseudotyped with HA from the A/Vietnam/1203/2004 HA, an H5 clade 1 strain. Although the cross-clade neutralization titers were 2–10-fold lower than titers to homologous virus, serum samples from all mice were able to neutralize Vietnam/1203 pseudotyped virus, with mean neutralizing antibody titers of 1,000 (Figure 2B). Together, these data indicate that nonadjuvanted sH53 antigen is able to elicit high titers of H5 cross-clade neutralizing antibody when administered using a prime/boost/boost regimen. Consistent with previous reports,6,7,12 multiple immunizations of sH53 recombinant protein were required to generate consistently high neutralizing antibody titers.
Neutralizing antibody response in mice vaccinated with H53-based nanovaccines
Our previous studies showed that polyanhydride nanovaccines enhanced the immune response to recombinant proteins.13–15 Therefore, we examined the immune response of sH53 delivered with and/or encapsulated into polyanhydride nanoparticles (Table 1). Mice were immunized subcutaneously with either a single-dose regimen or a prime/boost/boost regimen of three immunizations 21 days apart. Serum was collected at 42 and 63 days postimmunization and tested for neutralizing antibody titers against H5-WS-Luc. As we observed above, sH53 alone elicited low neutralizing titers in the absence of booster immunizations, with most mice having ID50 titers of less than 100 through 63 days postimmunization (Figure 3). Higher titers were observed using a single-dose immunization regimen with sH53 in the presence of adjuvant, with MPLA eliciting higher neutralizing antibody titers than any of the nanovaccine formulations or sH53 alone. In the absence of booster immunizations, the neutralizing antibody titers in sera from mice immunized with nanovaccine formulations was lower than that from the MPLA-treated mice at 42 days, with the exception of the formulation including poly I:C; however, there were a greater number of single-dose, nanoparticle-vaccinated mice with detectable neutralizing antibodies than in mice vaccinated once with sH53 alone. Administration of one or two booster vaccinations resulted in enhanced neutralizing antibody titers in all the formulations. The highest and most consistent titers were observed after receiving three immunizations (prime/boost/boost regimen, day 63), with little difference observed among the different formulations (Figure 3).
 | Figure 3 The neutralizing antibody responses to H53-polyanhydride nanovaccines were examined 42 and 63 days post-primary immunization. |
Cell-mediated immune response
Cell-mediated immune responses are often associated with broader protective immunity and can play an important role in protection against antigenically diverse strains of influenza.18,19 Therefore, we examined the proliferative T cell populations on day 63 (ie, 21 days after the last immunization) in mice receiving the prime/boost/boost regimen (Figure 4). Draining lymph nodes were removed and homogenized to single cell suspensions and labeled with CFSE to observe proliferating cell populations. After 96 hours of ex vivo stimulation with sH53, CD4+ T cells were quantified via flow cytometry. A significant expansion of CD4+ T cells was observed in all groups of vaccinated mice as compared to mice receiving saline alone. In addition, the inclusion of poly I:C within the nanovaccine formulation resulted in significantly higher numbers of CD4+ T cells as compared to mice immunized with sH53 alone or sH53 adjuvanted with MPLA. These data suggest that inclusion of nanoparticles encapsulating poly I:C to induce an appropriate innate immune response may enhance antigen-specific adaptive immune responses.
 | Figure 4 Enhanced CD4+ T cell memory with poly I:C polyanhydride nanovaccine. |
H53 vaccination protects against live-viral challenge
To examine the efficacy of H53-loaded nanoparticles in protection from clinical disease, mice were vaccinated with various nanovaccine formulations (Table 1) using a prime/boost/boost regimen and challenged at day 63 with the low-pathogenic VNH5N1-PR8/CDC-RG, a reverse genetics-derived influenza virus containing the HA and NA genes of A/Vietnam/1203/04 (H5N1) virus in the genetic background of the high-growth master strain A/Puerto Rico/8/34 (H1N1). Pre-challenge antibody response to the various vaccine regimens was evaluated in serum collected 1 week before challenge. All vaccinated groups had high levels of total anti-H53 IgG, with no significant difference among the groups. Similar to our earlier studies, all vaccinated mice had detectable neutralizing antibody titers of greater than 100 to the clade 2.2 H5-WS-Luc pseudotyped reporter virus, with mean titers among the group ranging between 3,000–4,000 (Figure S1).
The body weight of each mouse was observed for 14 days postchallenge. All of the vaccine formulations protected mice from challenge with body weight increasing postchallenge similar to naïve mice that were not challenged (Figure 5A). In contrast, mice receiving saline immunizations began to lose a significant proportion of their body weight approximately 5 days postchallenge. Saline-administered mice continued to lose approximately 20% of their total weight by 8 days postchallenge before recovering. Based on clinical response, as measured by weight loss, all vaccine regimens induced a protective immune response and there was no significant difference among the various vaccine regimens.
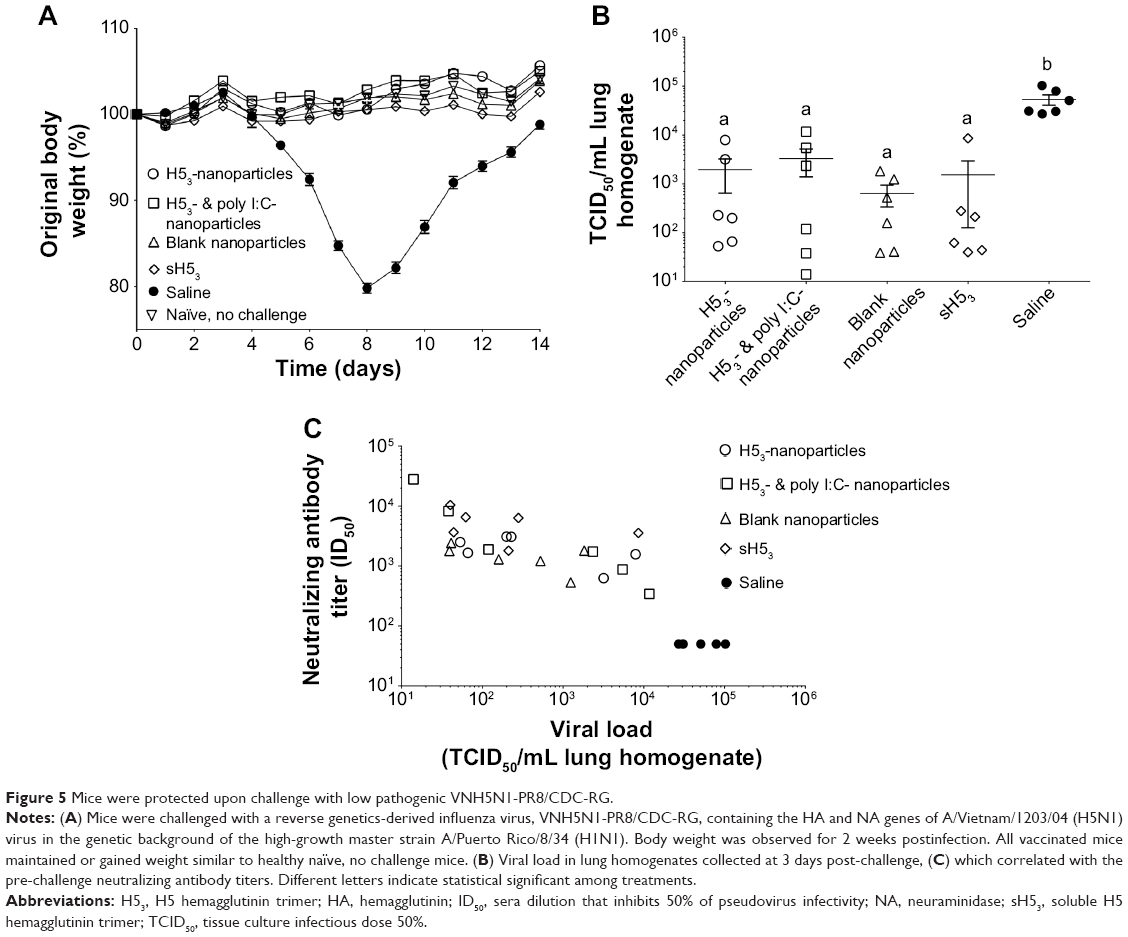 | Figure 5 Mice were protected upon challenge with low pathogenic VNH5N1-PR8/CDC-RG. |
At 3 days postchallenge, half of the mice in each group were sacrificed and lung homogenates were assayed for virus load using quantitative reverse transcription PCR as described previously.34 All vaccinated mice showed significant reduction in virus load upon challenge as compared to the virus load in the saline-administered mice (Figure 5B). A wide mouse-to-mouse variability in virus load was observed for all vaccine regimens, with no significant differences among the groups in mean virus loads. However, a correlation between pre-challenge (day 56 post-primary immunization) neutralizing antibody titer and virus load (day 3 postchallenge) was observed (Figure 5C).
Finally, a multiplexed assay was used to quantitate the concentrations of inflammatory cytokines (IL-6, IP-10, G-CSF, IFN-γ, MCP-1, KC, and MIP-2) present in BAL fluid. Vaccinated mice had low levels of inflammatory cytokines 3 days postchallenge similar to the naïve, no challenge controls (Figure S2). In contrast, mice receiving saline had significantly greater concentrations of most inflammatory cytokines postchallenge with the exception of IFN-γ (data not shown).
Clinical signs postchallenge in mice vaccinated with H53-nanoparticles are similar to healthy naïve, noninfected mice
PCA was utilized to capture the complex relationships between the neutralizing antibody titer, viral load, and six inflammatory cytokine measurements discussed above. Following infection, saline-administered mice (ie, immunologically naïve) had the most unique behavior because this was the only treatment group with any significant PC1 values (Figure 6A). The PC1 dimension captured the largest amount of independent data and was a linear combination of viral load and cytokine concentration. In contrast, the PC2 dimension was primarily captured by neutralizing antibody titer. While all of the H53 vaccine formulations demonstrated similar PC1 values, mice receiving the ‘H53- & poly I:C-nanoparticle’ formulation or ‘sH53’ showed the most significant PC2 values, or variability in neutralizing antibody among animals. Therefore, saline-administered mice have the behavior most unlike the remaining formulations, and neutralizing antibody titer is the most unique of the descriptors used in the analysis.
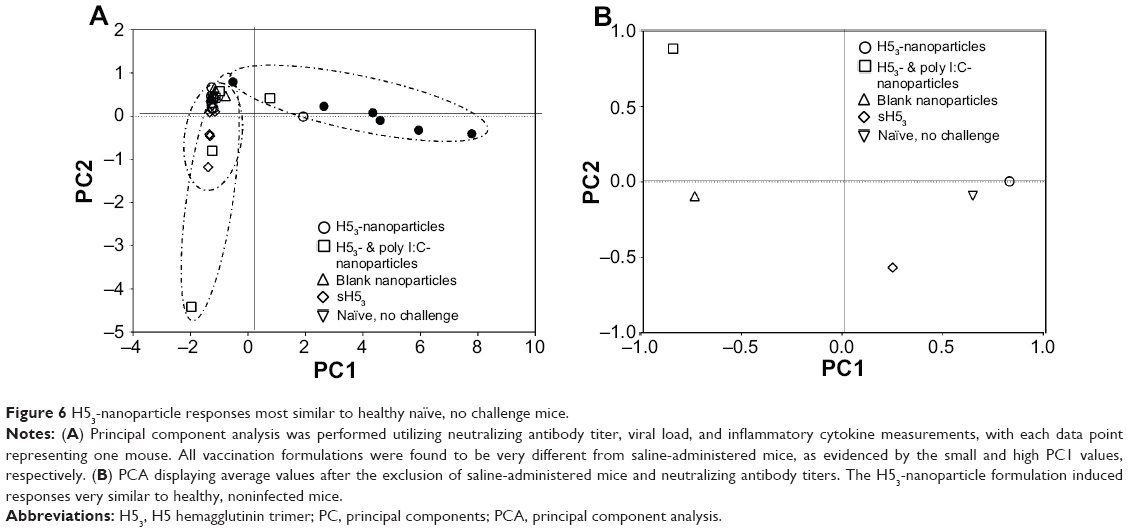 | Figure 6 H53-nanoparticle responses most similar to healthy naïve, no challenge mice. |
In addition to the primary correlations uncovered by correlations to saline-administered mice and neutralizing antibody titers, more subtle relationships in the data remained hidden. Thus, the analysis was repeated after excluding the primary correlations (ie, saline-administered mice and neutralizing antibody titer) and is shown in Figure 6B, where only the average value of each treatment is displayed for clarity. The ‘H53-nanoparticle’ formulation induced a response postchallenge similar to the healthy naïve, nonchallenged (ie, control) mice, as seen by the close proximity of the average data points (Figure 6B). Mice receiving soluble H53 alone (sH53) also resulted in responses similar to the control mice, but less so than the ‘H53-nanoparticle’ formulation. This analysis demonstrated that all the vaccine formulations resulted in a significant improvement in clinical responses when compared to the saline-treated, infected mice, while the ‘H53-nanoparticle’ formulation induced responses most similar to healthy control mice.
Discussion
The design of effective vaccines for the prevention of influenza is a challenging task. Despite best efforts, the 2013–2014 US seasonal influenza vaccine was shown to be ~60% effective in patients.35 In the case of pandemic influenza, such as H5N1, the efficacy of vaccines can be even further reduced. Currently, the approved H5N1 influenza vaccine consists of two large doses (90 μg) administered 28 days apart, but is only 45% effective.36 In this work, we examined an alternative to conventional influenza vaccines through the use of polyanhydride nanoparticles. By encapsulating H5 antigen into polyanhydride nanoparticles, the foundation has been laid for the design of pandemic influenza vaccines with improved efficacy by demonstrating reduced variability in the neutralizing antibody titer, enhanced T cell responses, and greater stability.20
As mentioned above, the recent seasonal influenza vaccine is approximately 60% effective; however, this estimate has a 95% confidence interval of 5%–68%,35 which is a substantial range that indicates the large (and unacceptable) variability of the host responses to the vaccine. Variability of vaccine effectiveness can be influenced by a large number of factors, including age, obesity, nutrition, or other disease(s) present in the host.37–39 While the vaccine formulations used in this work performed similarly, one of the strengths of the polyanhydride nanovaccine is illustrated in its ability to reduced immune response variability. Immunizations with H53-nanoparticles were shown to induce more consistent neutralizing antibody titers with less variability compared to other formulations (Figures 3 and S1). In addition, PCA, which incorporated the neutralizing antibody titer, viral load, and inflammatory cytokine data postchallenge, indicated that H53-nanoparticle vaccination led to consistent immune responses (Figure 6A). Similarly, H53-nanoparticle immunized mice did not exhibit clinical signs of disease postchallenge and were the most similar to healthy (naïve, noninfected) mice (Figure 6B).
Aside from variability in immune responses, many vaccines struggle to stimulate T cell immunity. Although humoral immunity is often sufficient to provide protection against homologous strains of influenza, cell-mediated immunity is necessary when confronting heterologous variants.18 CD8+ T cells secrete antiviral cytokines and often have a broader epitope spectrum than that of the humoral response.18,19 While not fully understood, CD4+ T cells play a role in protection against heterologous influenza infection by producing cytokines to restrict viral replication and even providing some lytic activity as well.18,19,40 Vaccines that can stimulate both humoral and cell-mediated immunity would likely have an advantage in cross-clade protection. The H53 antigen used in this work demonstrated the induction of cross-reactive neutralizing antibodies when delivered alone (Figure 2). However, when delivered in the context of polyanhydride nanoparticles, CD4+ T cell proliferation was enhanced upon ex vivo stimulation with antigen (Figure 4). The nanovaccine formulation containing poly I:C induced the greatest proliferation of CD4+ T cells. It is known that poly I:C signaling through TLR3 induces the production of several Th1 and Th2 cytokines, as well as type I interferons,41 which may explain the T cell expansion observed in Figure 4.
Finally, we believe that the ability to encapsulate influenza antigens into polyanhydride nanoparticles will provide additional benefits, including dose sparing and enhanced shelf-life characteristics.15,42 While many of the nanoparticle formulations used in this study performed similarly to soluble antigen delivered alone, it is likely that the 10 μg dose of the highly immunogenic H53 antigen masked the advantages provided by the polyanhydride nanoparticles. Previously, it has been demonstrated that encapsulation of a suboptimal dose of antigen into polyanhydride particles induced similar antibody titers to administration of 64-fold higher soluble antigen alone.15 The ability to reduce antigen per dose (and therefore increase vaccine availability) and optimize single-dose formulations will be paramount in providing rapid protection upon a pandemic outbreak of H5N1. Finally, polyanhydride nanovaccines exhibit release of immunogenic protein even when stored at room temperature for 1 year.42 This ability for a vaccine to be stored at room temperature would be a major benefit in terms of maintaining stockpiles of pandemic vaccines.
Conclusion
The studies presented herein demonstrate the strong immunogenic properties of the recombinant H53 antigen developed for these studies as well as the immune responses to H53-loaded polyanhydride nanovaccines. All formulations were able to induce detectable mean neutralizing antibody titers 42 days postvaccination. The formulation including poly I:C also conferred the additional advantages of enhanced antibody titers at earlier time points and induction of proliferative T cell responses. All vaccine formulations were able to induce protection against a low-pathogenic, live-viral challenge. Of interest, PCA demonstrated that after viral challenge, the mice immunized with the H53-nanoparticle formulation were as healthy as naïve, nonchallenged mice and this formulation induced less variable immune responses. The current studies provide a foundation to further exploit the advantages of polyanhydride nanoparticle-based subunit vaccines against influenza.
Acknowledgments
The authors wish to acknowledge financial support from the US Army Medical Research and Materiel Command (grant numbers W81XWH-09-1-0386 and W81XWH-10-1-0806).
Disclosure
The authors report no conflicts of interest in this work.
References
Subbarao K, Klimov A, Katz J, et al. Characterization of an avian influenza A (H5N1) virus isolated from a child with fatal respiratory illness. Science. 1998;279(5349):393–396. | ||
Nichol KL, Treanor JJ. Vaccines for seasonal and pandemic influenza. J Infect Dis. 2006;194 Suppl 2:S111–S118. | ||
Palese P, Shaw ML. Orthomyxoviridae: the viruses and their replication. In: Knipe DM, Howley PM, editors. Fields Virology. Philadelphia, PA: Lippincott-Raven Publishers; 2007:1647–1689. | ||
Osterhaus A, Fouchier R, Rimmelzwaan G. Towards universal influenza vaccines? Philos Trans R Soc Lond B Biol Sci. 2011;366(1579):2766–2773. | ||
Cox MM, Patriarca PA, Treanor J. FluBlok, a recombinant hemagglutinin influenza vaccine. Influenza Other Respir Viruses. 2008;2(6):211–219. | ||
Wei CJ, Xu L, Kong WP, et al. Comparative efficacy of neutralizing antibodies elicited by recombinant hemagglutinin proteins from avian H5N1 influenza virus. J Virol. 2008;82(13):6200–6208. | ||
Cornelissen LA, de Vries RP, de Boer-Luijtze EA, Rigter A, Rottier PJ, de Haan CA. A single immunization with soluble recombinant trimeric hemagglutinin protects chickens against highly pathogenic avian influenza virus H5N1. PLoS One. 2010;5(5):e10645. | ||
Weldon WC, Wang BZ, Martin MP, Koutsonanos DG, Skountzou I, Compans RW. Enhanced immunogenicity of stabilized trimeric soluble influenza hemagglutinin. PLoS One. 2010;5(9). pii: e12466. | ||
Loeffen WL, de Vries RP, Stockhofe N, et al. Vaccination with a soluble recombinant hemagglutinin trimer protects pigs against a challenge with pandemic (H1N1) 2009 influenza virus. Vaccine. 2011;29(8):1545–1550. | ||
de Vries RP, Smit CH, de Bruin E, et al. Glycan-dependent immunogenicity of recombinant soluble trimeric hemagglutinin. J Virol. 2012;86(21):11735–11744. | ||
Wang CC, Chen JR, Tseng YC, et al. Glycans on influenza hemagglutinin affect receptor binding and immune response. Proc Natl Acad Sci U S A. 2009;106(43):18137–18142. | ||
Prabakaran M, Madhan S, Prabhu N, Qiang J, Kwang J. Gastrointestinal delivery of baculovirus displaying influenza virus hemagglutinin protects mice against heterologous H5N1 infection. J Virol. 2010;84(7):3201–3209. | ||
Ulery BD, Kumar D, Ramer-Tait AE, Metzger DW, Wannemuehler MJ, Narasimhan B. Design of a protective single-dose intranasal nanoparticle-based vaccine platform for respiratory infectious diseases. PLoS One. 2011;6(3):e17642. | ||
Kipper MJ, Shen E, Determan AS, Narasimhan B. Design of an injectable system based on bioerodible polyanhydride microspheres for sustained drug delivery. Biomaterials. 2002;23(22):4405–4412. | ||
Huntimer L, Wilson Welder JH, Ross K, et al. Single immunization with a suboptimal antigen dose encapsulated into polyanhydride microparticles promotes high titer and avid antibody responses. J Biomed Mater Res B Appl Biomater. 2013;101(1):91–98. | ||
Kipper MJ, Wilson JH, Wannemuehler MJ, Narasimhan B. Single dose vaccine based on biodegradable polyanhydride microspheres can modulate immune response mechanism. J Biomed Mater Res A. 2006;76(4):798–810. | ||
Huntimer LM, Ross KA, Darling RJ, et al. Polyanhydride nanovaccine platform enhances antigen-specific cytotoxic T cell responses. Technology. 2014;2(2):171–175. | ||
Woodland DL, Hogan RJ, Zhong W. Cellular immunity and memory to respiratory virus infections. Immunol Res. 2001;24(1):53–67. | ||
Thomas PG, Keating R, Hulse-Post DJ, Doherty PC. Cell-mediated protection in influenza infection. Emerg Infect Dis. 2006;12(1):48–54. | ||
Ross KA, Loyd H, Wu W, et al. Structural and antigenic stability of H5N1 hemagglutinin trimer upon release from polyanhydride nanoparticles. J Biomed Mater Res A. 2014;102(11):4161–4168. | ||
Buehler J, Lager K, Vincent A, Miller C, Thacker E, Janke B. Issues encountered in development of enzyme-linked immunosorbent assay for use in detecting influenza A virus subtype H5N1 exposure in swine. J Vet Diagn Invest. 2014;26(2):277–281. | ||
Wang W, Xie H, Ye Z, Vassell R, Weiss CD. Characterization of lentiviral pseudotypes with influenza H5N1 hemagglutinin and their performance in neutralization asssays. J Virol Methods. 2010;165(2):305–310. | ||
Olsen JC. Gene transfer vectors derived from equine infectious anemia virus. Gene Ther. 1998;5(11):1481–1487. | ||
Tallmadge RL, Brindly MA, Salmans J, Mealey RH, Maury W, Carpenter S. Development and characterization of an equine infectious anemia virus Env-pseudotyped reporter virus. Clin Vaccine Immunol. 2008;15(7):138–140. | ||
Conix A. Poly[1,3-bis(p-carboxyphenoxy)-propane anhydride]. Macromol Synth. 1966;2:95–98. | ||
Torres MP, Vogel BM, Narasimhan B, Mallapragada SK. Synthesis and characterization of novel polyanhydrides with tailored erosion mechanisms. J Biomed Mater Res A. 2006;76(1):102–110. | ||
Determan AS, Graham JR, Pfeiffer KA, Narasimhan B. The role of microsphere fabrication methods on the stability and release kinetics of ovalbumin encapsulated in polyanhydride microspheres. J Microencapsul. 2006;23(8):832–843. | ||
Chavez-Santoscoy AV, Huntimer LM, Ramer-Tait AE, Wannemuehler M, Narasimhan B. Harvesting murine alveolar macrophages and evaluating cellular activation induced by polyanhydride nanoparticles. J Vis Exp. 2012;(64):e3883. | ||
Alsharifi M, Furuya Y, Bowden TR, et al. Intranasal flu vaccine protective against seasonal and H5N1 avian influenza infections. PLoS One. 2009;4(4):e5336. | ||
Ulery BD, Petersen LK, Phanse Y, et al. Rational design of pathogen-mimicking amphiphilic materials as nanoadjuvants. Sci Rep. 2011;1:198. | ||
Jolliffee IT. Principal Component Analysis. Secaucus, NJ: Springer; 2002. | ||
Korenius T, Laurikkala J, Juhola M. On principal component analysis, cosine, and Euclidean measures in information retrieval. Inf Sci. 2007;177(22):4893–4905. | ||
Broderick SR, Aourag H, Rajan K. Data mining of Ti–Al semi-empirical parameters for developing reduced order models. Physica B. 2011;406(11):2055–2060. | ||
McDonald RS, Sambol AR, Heimbuch BK, Brown TL, Hinrichs SH, Wander JD. Proportional mouse model for aerosol infection by influenza. J Appl Micro. 2012;113(4):767–778. | ||
Centers for Disease Control and Prevention (CDC) [webpage on the Internet]. Vaccine effectiveness-how well does the flu vaccine work? Available from: http://www.cdc.gov/flu/about/qa/vaccineeffect.htm. Accessed June 12, 2014. | ||
US Food and Drug Administration. H5N1 influenza virus vaccine, manufactured by Sanofi Pasteur, Inc. questions and answers. Available from http://www.fda.gov/BiologicsBloodVaccines/Vaccines/QuestionsaboutVaccines/ucm080753.htm. Accessed May 14, 2014. | ||
McElhaney JE. Influenza vaccine responses in older adults. Ageing Res Rev. 2011;10(3):379–388. | ||
Haynes L, Swain SL. Why aging T cells fail: Implications for vaccination. Immunity. 2006;24(6):663–666. | ||
Sheridan PA, Paich HA, Handy J, et al. Obesity is associated with impaired immune response to influenza vaccination in humans. Int J Obes (Lond). 2012;36(8):1072–1077. | ||
McKinstry KK, Strutt TM, Swain SL. Hallmarks of CD4 T cell immunity against influenza. J Intern Med. 2011;269(5):507–518. | ||
Ichinohe T, Kawaguchi A, Tamura S, et al. Intranasal immunization with H5N1 vaccine plus Poly I:Poly C12U, a Toll-like receptor agonist, protects mice against homologous and heterologous virus challenge. Microbes Infect. 2007;9(11):1333–1340. | ||
Petersen LK, Phase Y, Ramer-Tait AE, Wannemuehler MJ, Narasimhan B. Amphiphilic polyanhydride nanoparticles stabilize Bacillus anthracis protective antigen. Mol Pharm. 2012;9(4):874–882. |
Supplementary materials
 | Figure S1 Prime/boost/boost immunized mice induced protective neutralizing antibody titers. |
 | Figure S2 Low levels of inflammatory cytokines were present in immunized mice postchallenge. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.