Back to Journals » Clinical and Experimental Gastroenterology » Volume 19
Helicobacter pylori and Cancer: What’s the Link?
Authors Merola E ![]() , Pes GM
, Pes GM ![]() , Graham DY
, Graham DY ![]() , Dore MP
, Dore MP
Received 29 August 2025
Accepted for publication 23 December 2025
Published 7 January 2026 Volume 2026:19 495588
DOI https://doi.org/10.2147/CEG.S495588
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Vipul Yagnik
Elettra Merola,1 Giovanni Mario Pes,1 David Yates Graham,2,3 Maria Pina Dore1,2
1Department of Medicine, Surgery and Pharmacy, University of Sassari, Sassari, Italy; 2Baylor College of Medicine, One Baylor Plaza, Houston, TX, USA; 3Department of Medicine, Michael E. DeBakey Veterans Affairs Medical Center and Baylor College of Medicine, Houston, TX, USA
Correspondence: David Yates Graham, Department of Medicine, Michael E. DeBakey Veterans Affairs Medical Center and Baylor College of Medicine, 2002 Holcombe Blvd (111D), Houston, TX, 77030, USA, Email [email protected]
Abstract: Helicobacter pylori (H. pylori) is a human bacterial pathogen that causes one of the most common chronic bacterial infections worldwide. The microorganism has been classified by the International Agency for Research on Cancer as a Group I carcinogen. While the etiological link to gastric cancer is well established, the precise molecular and cellular mechanisms driving this transformation are highly complex and incompletely understood. Fundamentally, the infection results from the chronic presence of acute on chronic gastric mucosal inflammation. H. pylori pathogenicity is increased by bacterial virulence factors including the cytotoxin-associated gene A (CagA) and Vacuolating cytotoxin A (VacA) which may interfere with the host’s cell communication and create a pro-tumorigenic microenvironment. Host microRNAs (miRNAs) may amplify these effects by modulating immune responses, enhancing oncogenic signalling. Despite the proven benefits of H. pylori eradication in reducing cancer risk, especially in high-incidence regions, rising antibiotic resistance and host-related variables impede its global implementation. Recent advances in genomics and multi-omics profiling potentially offer new opportunities for targeted prevention. Moreover, emerging evidence suggests H. pylori may also negatively influence immunotherapy outcomes, underscoring its broader relevance in cancer treatment planning. By synthesizing molecular insights, epidemiological trends, and clinical data, this narrative review examines the multifaceted pathways through which H. pylori contributes to gastric carcinogenesis, integrating current knowledge on microbial virulence, host signalling disruption, immune modulation, and epigenetic remodelling.
Keywords: Helicobacter pylori, gastric cancer, pathogenesis, CagA, VacA, virulence
Introduction
Gastric cancer remains a significant global health concern. According to the latest estimates from the International Agency for Research on Cancer (IARC), gastric cancer is the fifth most diagnosed malignancy and the fourth leading cause of cancer-related mortality worldwide.1,2 Although its overall incidence has declined in recent decades, particularly in high-income countries, substantial geographic disparities persist. Eastern Asia, notably Mongolia, Japan, and South Korea, continues to bear a disproportionately high burden of gastric cancer, reflecting a combination of regional exposures and host-specific susceptibilities.1–3
Helicobacter pylori (H. pylori), a spiral-shaped Gram-negative bacterium, is the principal etiologic agent for non-cardia gastric cancer. First isolated by Warren and Marshall in 1982, in 1994 H. pylori was classified as a Group I carcinogen by the IARC due to its strong association with gastric adenocarcinoma.2,4 The bacterium infects roughly half of the global population, where it induces chronic gastritis, which may progress through a well-established histopathological sequence, known as Correa’s cascade, that proceeds from inflammation to atrophy, intestinal metaplasia, dysplasia, and ultimately gastric adenocarcinoma.2,4,5
Despite the high global prevalence of H. pylori infections, only a fraction of individuals, depending on the geographic area, develop gastric cancer. Multiple factors are involved including bacterial virulence determinants, most notably the cytotoxin-associated gene A (CagA), host genetic polymorphisms, immune responses, dietary influences, and environmental stressors.5–8 For example, CagA-positive strains are more frequently associated with malignancy, possibly due to their ability to subvert intracellular signalling pathways, such as SHP2, ERK, and β-catenin, thereby promoting epithelial proliferation and inflammation (Figure 1).3,7
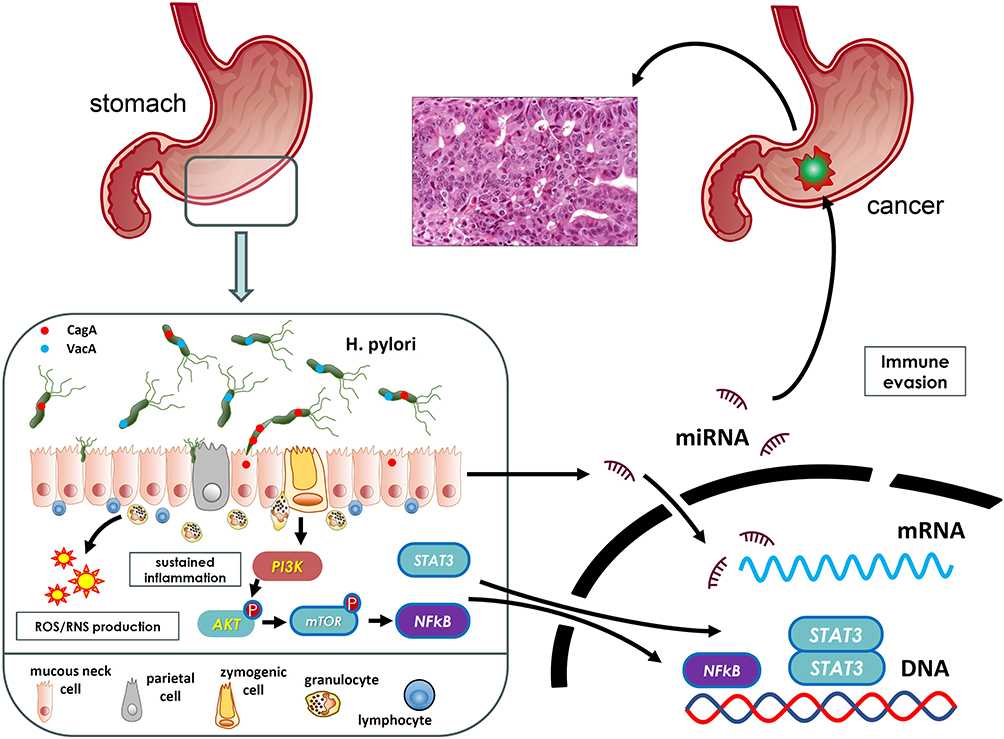 |
Figure 1 Chronic Helicobacter pylori infection initiates a cascade of molecular and cellular events that promote gastric cancer. Key virulence factors, such as CagA and VacA, are introduced into gastric epithelial cells, triggering the dysregulation of several molecular pathways. These include altered expression of microRNAs, activation of NF-κB and STAT3 signalling, sustained inflammation, oncogenic signalling, and immune evasion. Together, these processes reprogram epithelial physiology and facilitate the multistep progression toward malignant transformation. |
Recent evidence suggests that the oncogenic role of H. pylori may extend beyond the infection itself, as chronic colonization also perturbs the gastric microbiota, leading to dysbiosis and the enrichment of other potentially pathogenic species, including Fusobacterium and Peptostreptococcus. This shift may also drive inflammation, genotoxicity, and mucosal barrier dysfunction, even after H. pylori has been eradicated, a concept known as the “hit-and-run” hypothesis.7,8
From a public health standpoint, the preventive strategy of H. pylori eradication to prevent gastric cancer has gained global momentum. Randomized controlled trials and long-term cohort studies conducted in high-risk regions such as Taiwan, China, Japan, and Korea have shown that timely eradication can significantly reduce both primary and metachronous gastric cancer incidence.2 Notably, the large-scale MITS trial in Linqu County, China, which enrolled over 180,000 participants, demonstrated that community-wide H. pylori eradication lowered gastric cancer incidence and mortality, particularly when treatment occurred at younger ages.2 A population H. pylori eradication for a high-risk Taiwanese population aged 30 years or older dwelling on Matsu Islands (2004–2018) was effective in reducing gastric cancer incidence 53% (95% CI 30% to 69%, p<0.001).9 Meta-analyses further support these findings, confirming both risk reduction and cost-effectiveness in endemic areas.2,10
Taken together, H. pylori acts as both a microbial initiator and an ecological disruptor in the development of gastric carcinogenesis.
By synthesizing molecular insights, epidemiological trends, and clinical data, this narrative review examines the multifaceted pathways through which H. pylori contributes to gastric carcinogenesis, integrating current knowledge on microbial virulence, host signalling disruption, and epigenetic remodelling. Differently from most of previous reviews, our overview will also analyse current controversies regarding the clinical impact of H. pylori infection, focusing on different response to eradication therapy, as well as to immunotherapy in cancer.
Epidemiology of H. pylori Infection
H. pylori infection remains one of the most widespread chronic pathogenic bacterial infections globally, affecting over 50% of the world’s population. However, the prevalence varies considerably by region, reflecting differences in sanitation, socioeconomic status, public health infrastructure, and generational patterns of exposure.1 The highest prevalence is observed in low- and middle-income countries (LMICs), particularly in Africa, South America, and parts of Asia, where early childhood transmission is common due to overcrowding and inadequate hygiene practices.1,4 In contrast, high-income regions such as Western Europe, North America, and Oceania have experienced a marked decline in prevalence over recent decades, driven by improvements in sanitation, public health interventions, and possibly widespread antibiotic use.1,4
Historically, H. pylori infection has been nearly ubiquitous in many populations. Currently, for example, Eastern Asia, which includes Mongolia, Japan, and South Korea, continues to carry a disproportionate burden in terms of both H. pylori prevalence and associated gastric cancer risk, and remains a top priority for targeted screening and eradication efforts.1
Beyond geography and age, socioeconomic status is a consistent risk factor for H. pylori infections. Limited access to clean water, household overcrowding, and lower parental education levels are all strongly associated with higher infection rates across both developing and developed settings. These findings highlight the necessity for context-specific interventions that address not only medical treatment but also the broader social determinants of health.1,4
Recent meta-analyses reinforce these epidemiological trends. A 2023 pooled analysis of 224 population-based studies estimated that adult global prevalence has declined from approximately 58% in the 1980s to 43% during 2011–2022, reflecting an average annual reduction of 0.5 percentage points.11 The steepest recent declines have been recorded in the WHO African Region and the Americas, although Africa still exhibits the highest absolute prevalence, exceeding 60% in many surveys.11,12
Age-stratified data underscore the importance of early-life transmission. A 2022 systematic review of pediatric cohorts reported global prevalences of 26% in children aged ≤6 years, 34% in those aged 7–12 years, and 42% among adolescents aged 13–18 years. Notably, prevalence in children from LMICs was nearly double that of their peers in high-income countries.13 Given that H. pylori infection, once acquired, generally persists for decades, current pediatric infection rates are strong predictors of future adult disease burden. Although direct incidence data are limited, the observed cohort decline in many regions suggests that new adult infections are now rare (<1% per year) in high-income countries but remain relatively common in young children within high-prevalence LMICs.
Encouragingly, several recent studies from Africa and Asia indicate emerging declines in pediatric prevalence, likely reflecting the combined effects of improved water, sanitation, and hygiene measures along with broader access to eradication therapy.11–13
Importantly, socioeconomic disparities in H. pylori infection persist even within high-income nations. Among 1.1 million US veterans tested between 1999 and 2018, overall seroprevalence was 25.8%, but rose to approximately 40% in non-Hispanic Black individuals and 37% in Hispanic individuals, nearly double the prevalence observed in non-Hispanic Whites.14 These disparities mirror established risk factors such as overcrowded living conditions, shared sleeping spaces, unfiltered water use, low parental education, persistence, and immigration from high-risk countries.
Collectively, these findings highlight the complexity of H. pylori epidemiology and underscore the need for regionally tailored strategies to identify, treat, and prevent infection.
Molecular Pathways in H. pylori-Related Carcinogenesis
H. pylori has coexisted with humans for at least 60,000 years, tracing back to early human migration patterns. This prolonged host-pathogen interaction has led to substantial co-evolution, resulting in the development of genetically diverse H. pylori strains and shaping the evolution of the human immune system, particularly innate immunity.10,15 As the first line of defense, innate immunity mounts rapid, non-specific responses that bridge to the more targeted mechanisms of adaptive immunity.16
H. pylori expresses a range of conserved molecular structures, including lipopolysaccharides, flagellin A, fucose, and nucleic acids such as DNA and single-stranded RNA. These microbial patterns are detected by host innate immune receptors, triggering a cascade of inflammatory responses characterized by vasodilation, increased vascular permeability, and the recruitment of immune cells to contain and eliminate the pathogen.3
Despite this effective surveillance system, H. pylori has evolved sophisticated mechanisms to evade immune detection, allowing persistent colonization and chronic inflammation,16 conditions that markedly increase the risk of malignant transformation. Compounding this challenge is the global rise in antibiotic resistance, particularly to macrolides. In several regions, resistance rates have reached or exceeded critical thresholds, compromising eradication efforts and amplifying the public health burden of H. pylori-associated diseases, including gastric cancer.3
The molecular landscape driving the progression from chronic gastritis to gastric cancer is complex and remains incompletely understood. Multiple signaling pathways are dysregulated throughout this process. H. pylori plays the central role in orchestrating this disruption, possibly through its virulence factors or more likely indirectly via host-derived inflammatory mediators. This multifaceted interference reprograms epithelial cell function and promotes oncogenic transformation, with extensive pathway crosstalk accelerating the onset of gastric tumorigenesis (Figure 1).
Virulence Factors
H. pylori exhibits remarkable genomic diversity, characterized by pronounced geographic variation and genetic plasticity that facilitate its long-term colonization of the stomach and its pathogenic potential. Key virulence factors are summarized in Table 1. Among the principal determinants encoded by this heterogeneous genome are the cag pathogenicity island (cagPAI), the vacuolating cytotoxin A (VacA), and additional components that enable survival and adhesion. Together, these elements help orchestrate mucosal injury and may also directly promote gastric carcinogenesis.17
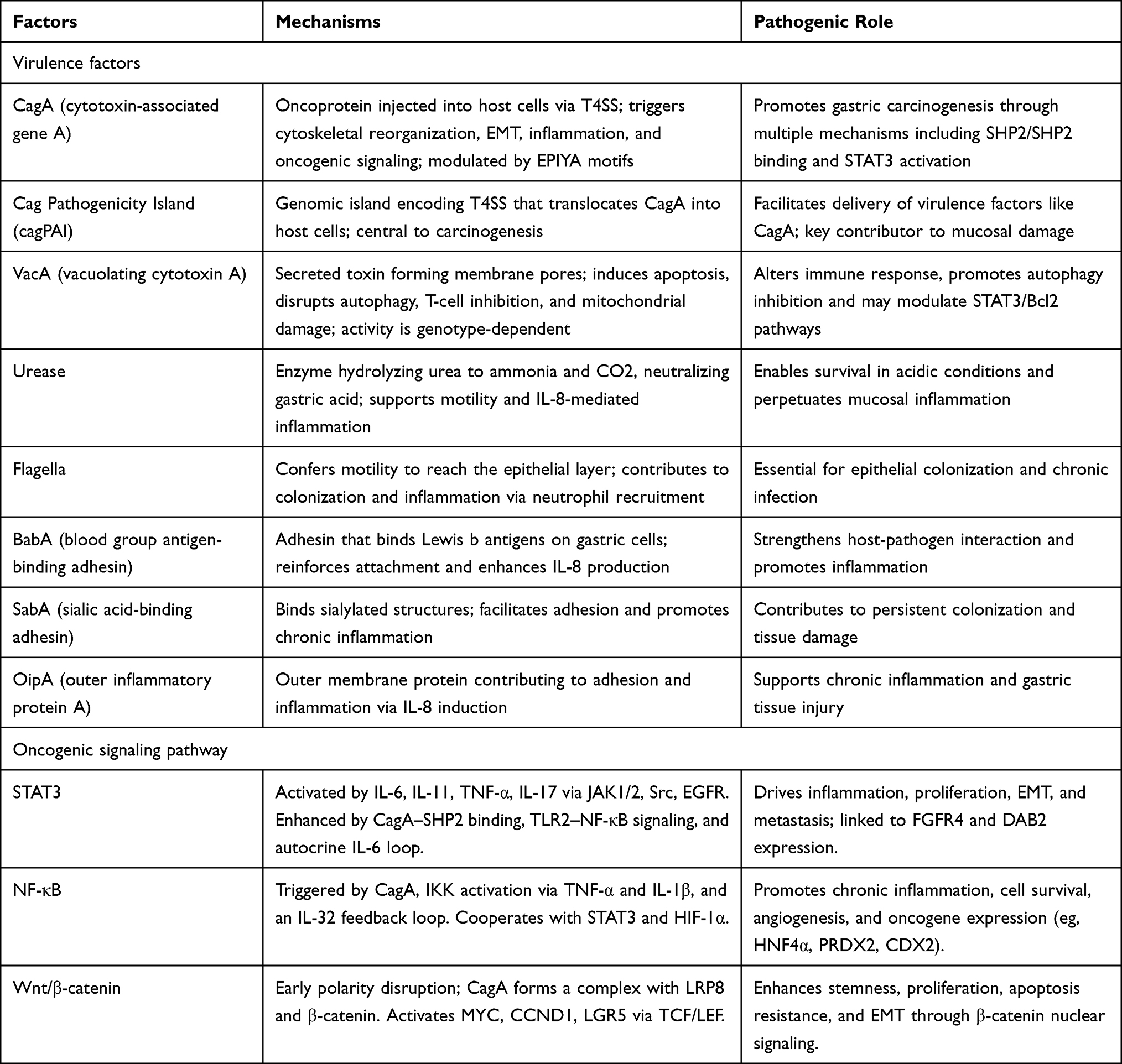 |
Table 1 Helicobacter pylori Virulence Factors and Oncogenic Signalling Involved in Related Carcinogenesis |
The cagPAI is a ~40-kb chromosomal segment comprising 31 genes, widely conserved across H. pylori lineages. It encodes a type IV secretion system (T4SS) responsible for translocating the oncoprotein CagA into gastric epithelial cells. Once delivered, CagA (120–140 K) is phosphorylated by host tyrosine kinases at conserved EPIYA motifs. Phosphorylated CagA engages SH2-domain-containing protein tyrosine phosphatase-2 (SHP2), activating downstream signaling cascades that remodel the actin cytoskeleton, destabilize tight junctions, suppress apoptosis, and drive epithelial-to-mesenchymal transition (EMT). Concurrently, CagA triggers pro-inflammatory signaling, recruits immune cells, and reshapes the tumor microenvironment, underscoring its pivotal role in gastric cancer development.18,19 Virulence is modulated by the number and type of EPIYA motifs; four variants are defined (A, B, C, D). Western strains commonly carry EPIYA-C, while East Asian strains harbor EPIYA-D, which exhibits higher SHP2 affinity and stronger oncogenic potential. CagA also interacts with SHIP2, enhancing intracellular delivery and amplifying oxidative and hypoxic stress. Additionally, strains with multiple EPIYA-C repeats upregulate key oncogenes, offering alternate routes to malignant transformation.4
VacA is a secreted pore-forming cytotoxin expressed by all H. pylori strains, although some produce inactive or less active forms of the toxin. The N-terminal p33 domain forms anion-selective channels in host membranes, while the C-terminal p55 domain mediates epithelial binding and induces apoptosis.4 VacA activity is genotype-specific, with four known hypervariable regions and distinct allelic combinations correlating with clinical phenotypes. Beyond epithelial vacuolization, VacA impairs T cell activation and proliferation, disrupts mitochondrial function and autophagic flux, induces cytochrome c release, activates the intrinsic apoptotic cascade and interferes with cell polarity. It also promotes IL-8 overproduction and disrupts intracellular amino acid pools, further contributing to autophagy inhibition.4 Functional synergy between VacA and CagA enhances bacterial adaptation under iron-limited gastric conditions and may exacerbate carcinogenesis, in part by modulating autophagic responses. Moreover, VacA has been shown to suppress signal transducer and activator of transcription 3 (STAT3), Bcl-2, and Bcl-XL expression in a dose-dependent manner, implicating it in inflammatory pathway remodeling.4,20
Beyond CagA and VacA, H. pylori relies on a broad array of auxiliary virulence factors that promote persistent colonization and mucosal damage. Adaptation to the gastric lumen’s acidic environment (pH ≈ 1–2) is mediated by urease and flagella. Urease catalyzes the hydrolysis of urea into ammonia and carbon dioxide, locally buffering gastric acidity to create a niche suitable for bacterial survival.21 The resulting ammonia also supports flagellar motility by reducing proton flux stress, and both urease and ammonia stimulate Interleukin-8 (IL-8) release, intensifying neutrophilic inflammation. Flagella enable motility through the mucus layer toward the epithelial surface, an essential step for stable colonization.22
Another possibly critical group of virulence factors includes outer membrane adhesins such as blood group antigen-binding adhesin (BabA), sialic acid-binding adhesin (SabA), and outer inflammatory protein (OipA), which bind to Lewis antigens or sialylated structures on host epithelial cells. These interactions strengthen bacterial attachment and further enhance IL-8 secretion, promoting chronic inflammation.23 Together, this integrated network of acid-resistance systems, motility apparatus, and outer membrane adhesins helps explain how H. pylori withstands the hostile gastric environment, sustains immune activation, and contributes to the pathophysiological cascade leading to gastric carcinogenesis.
Oncogenic Signalling
Oncogenic signaling pathways implicated in H. pylori-related carcinogenesis are summarized in Table 1. Among these, STAT3 signaling emerges as a central mediator of H. pylori-associated gastric tumorigenesis. Activation begins when pro-inflammatory cytokines such as IL-6, IL-11, Tumor necrosis factor- α (TNF-α), or IL-17 engage their respective receptors, triggering downstream kinases including JAK1/2, Src family kinases, and epidermal Growth Factor Receptor (EGFR). These kinases phosphorylate STAT3, which then dimerizes, translocates into the nucleus, and modulates transcriptional programs governing inflammation, proliferation, survival, epithelial-to-mesenchymal transition (EMT), and metastatic potential.24 H. pylori reinforces this signaling through several mechanisms. First, an autocrine IL-6 loop is established, whereby H. pylori infection upregulates epithelial IL-6 expression, leading to sustained extracellular stimulation of the JAK–STAT3 axis.25 Second, CagA translocation into host cells enables its binding and activation of SHP2, producing persistent STAT3 phosphorylation and a broad transcriptional response that favors oncogenesis.4 Third, H. pylori engages Toll-like receptor 2 (TLR2), enhancing NF-κB–mediated inflammation, which synergistically amplifies STAT3 signaling.26 Fourth, STAT3 interacts with other oncogenic modules, upregulating DAB2 via the SRC-YAP1 pathway and promoting Fibroblast Growth Factor Receptor 4 (FGFR4) expression, further contributing to malignant transformation. Interestingly, certain H. pylori strains can also suppress elements of the JAK–STAT pathway, possibly as a strategy to evade immune detection. Together, these findings underscore persistent STAT3 activation as a key molecular link between H. pylori infection and gastric cancer and highlight the therapeutic potential of targeting this pathway.
The NF-κB pathway, although classically associated with inflammation, also plays a direct oncogenic role in H. pylori-mediated gastric tumorigenesis. CagA enhances NF-κB activity by interacting with host cellular components that facilitate its nuclear translocation and transcriptional activation. H. pylori infection also stimulates the IKK kinase complex via elevated levels of TNF-α, IL-1β, and other pro-inflammatory cytokines, leading to phosphorylation of IκB and the release of active NF-κB dimers. Furthermore, an autoregulatory loop involving IL-32 perpetuates NF-κB signaling, creating a chronic inflammatory environment. Activated NF-κB drives the expression of numerous oncogenic effectors, including transcription factors, antioxidants, and differentiation regulators, which together enhance cell cycle progression, inhibit apoptosis, and promote angiogenesis.27 Additionally, NF-κB signaling converges with other pathways, such as STAT3 and HIF-1α, reinforcing a pro-tumorigenic microenvironment.28,29 Given NF-κB’s dual role in inflammation and cancer, elucidating H. pylori-specific triggers and downstream targets may uncover novel therapeutic strategies for gastric cancer prevention.
Another critical oncogenic mechanism linked to H. pylori infection is the persistent activation of the Wnt/β-catenin pathway. Early during infection, H. pylori disrupts epithelial cell polarity by interfering with a Wnt-regulated polarity program, enhancing stem cell proliferation and enteroendocrine differentiation. CagA plays a pivotal role in forming a complex with β-catenin, which stabilizes β-catenin and promotes its nuclear translocation.30 In the nucleus, β-catenin partners with transcription factors to activate genes such as MYC, CCND1, and LGR5, thereby accelerating the cell cycle, preventing apoptosis, and fostering stem cell-like traits.31 In addition, β-catenin activates EMT programs by inducing transcription factors that drive motility, invasion, and metastatic behavior.32 Together, these interconnected signaling events emphasize the central role of Wnt/β-catenin dysregulation in H. pylori-driven gastric carcinogenesis.
Chronic Inflammation and DNA Damage
Oxidative stress likely constitutes the central mechanism in H. pylori-driven gastric tumorigenesis. The accumulation of reactive oxygen and nitrogen species (ROS/RNS) overwhelms the gastric mucosa’s antioxidant defenses and disrupts epithelial metabolic homeostasis, thereby promoting malignant transformation.4 This oxidative imbalance arises through both direct and indirect mechanisms.33 Directly, bacterial metabolism generates superoxide and hydrogen peroxide, while H. pylori virulence factors, such as CagA and VacA, exacerbate intracellular ROS production. Indirectly, chronic infection elicits a prolonged inflammatory response, leading to the recruitment of neutrophils and macrophages, which are potent sources of ROS/RNS.
Recent evidence suggests that in the context of impaired DNA repair mechanisms, H. pylori infection induces even more profound oxidative stress and genotoxicity, including double-strand breaks, point mutations, chromosomal rearrangements, ribosomal degradation, digestion damage, and microsatellite instability, ultimately culminating in malignant transformation.34
H. pylori also enhances the expression of host pro-oxidant enzymes such as inducible nitric oxide synthase (iNOS) or enzymes dependent on Nicotinamide Adenine Dinucleotide Phosphate oxidase (NADPH), further intensifying oxidative injury. Simultaneously, the bacterium impairs host DNA repair systems, particularly nucleotide excision repair, and depletes key antioxidants, including vitamins C and E, from the gastric epithelium. Host cells already compromised in DNA repair are particularly vulnerable to oxidative stress upon H. pylori colonization, further increasing their susceptibility to cancerogenesis.35,36
Inflammation is likely the fundamental key contributor to H. pylori-associated carcinogenesis. Chemokine-mediated immune cell recruitment, mainly of neutrophils and macrophages, results in abundant ROS/RNS production, driving DNA damage, mutational burden, and genomic instability. In parallel, tumor-associated macrophages secrete matrix metalloproteinases, which degrade extracellular matrix components and facilitate the invasion of cancer cells.4
Apoptotic dysregulation represents another hallmark of H. pylori-associated gastric cancer. H. pylori modulates epithelial cell apoptosis to promote tumor cell survival and progression. Although H. pylori can induce apoptosis during acute infection phases, especially in vitro, it also suppresses apoptosis during chronic colonization, thereby allowing the survival of genetically altered cells.37 Several mechanisms underlie this dual effect. Urease, beyond its role in acid neutralization, binds class II MHC molecules on epithelial cells, initiating immune-mediated apoptosis.38,39 Furthermore, H. pylori upregulates anti-apoptotic proteins, such as Bcl-2 and Bcl-xL, while suppressing pro-apoptotic members, like Bax and Bad, thereby favoring cellular survival. The activation of NF-κB by H. pylori increases the transcription of anti-apoptotic genes, including c-IAP2 and survivin, while simultaneously suppressing p53 expression and function, thereby disabling a critical DNA damage response pathway. This ability to both induce and inhibit apoptosis reflects H. pylori’s adaptability and stage-specific control over host cell fate. By preventing the elimination of damaged or mutated cells, H. pylori facilitates the clonal expansion and progression of tumors.4,29
Disruption of normal cell cycle regulation may also contribute to uncontrolled proliferation in the gastric epithelium of H. pylori-infected individuals. H. pylori induces the overexpression of cyclins such as cyclin D1 and cyclin E, as well as cyclin-dependent kinases (CDKs), which collectively accelerate cell cycle progression.40 Concurrently, it downregulates tumor suppressor proteins and CDK inhibitors, including p21 and p27, skewing the balance toward hyperproliferation. The resulting cell cycle dysregulation not only drives tumor growth but also compromises genome integrity by impairing DNA damage checkpoints, permitting the accumulation of oncogenic mutations, and further enhancing cancer risk.4
Emerging Mechanisms in H. pylori-Related Carcinogenesis and Response to Therapy
Investigations continue to reveal novel mechanisms by which H. pylori may contribute to gastric cancer. Epigenetic modifications, including DNA hypermethylation and histone alterations, play a pivotal role in silencing tumor suppressor genes such as p14ARF and FOXD3.41 Moreover, hypermethylation of connexin genes (Cx32, Cx43) disrupts gap junctional communication and facilitates tumor progression. At the same time, aberrant demethylation of GNB4 enhances oncogenesis via the Hippo-YAP1 signalling pathway.42
H. pylori also influences the composition of the gut microbiota, thereby modulating host immune responses and metabolic pathways. In addition, the tumor microenvironment is shaped by interactions with immune cells, extracellular vesicles, microRNAs (miRNAs), and long non-coding RNAs (lncRNAs), contributing to a milieu conducive to malignant transformation.4
H. pylori infection triggers a dual immunological effect, driving both inflammation and immune tolerance.16 Features such as increased tumor mutational burden and microsatellite instability, coupled with CD103+ CD8+ T cell infiltration, may explain better immunotherapy outcomes in H. pylori-positive gastric cancer. Additionally, H. pylori alters the gut microbiota composition, reducing the number of beneficial short-chain fatty acid producers and enriching pro-carcinogenic taxa, which further influences systemic immune responses.4
H. pylori also induces PD-L1 expression in gastric epithelial cells by activating the NF-κB, JAK/STAT, and PI3K/Akt pathways, enabling immune evasion. Concurrently, the bacterium suppresses dendritic cell activity and cytokine production, thereby undermining innate immunity and diminishing the effectiveness of immunotherapies.43,44 Clinical data suggest that H. pylori-positive advanced gastric cancer patients may experience reduced responses and shorter progression-free survival when treated with PD-1 inhibitors.45 While H. pylori fosters an inflamed microenvironment in gastric cancer, its effects may hinder immunotherapy in other cancers such as esophageal and colorectal carcinomas.46 Two key studies, by Jia et al and Chen et al, shed light on this complex relationship.46,47 Jia et al46 analysed data from over 10,000 cancer patients, some of whom received anti-PD-1/PD-L1 therapies. In gastric cancer, H. pylori-positive individuals exhibited improved progression-free survival and a trend toward enhanced overall survival. These patients showed increased PD-L1 expression and greater infiltration of non-exhausted CD8+ T cells. Transcriptomic analyses suggested that H. pylori-positive tumors possess immunologically active profiles, although in mismatch repair-deficient colorectal cancers, H. pylori infection correlated with poorer responses, illustrating cancer type-specific immune modulation.46
Chen et al47 expanded these findings using multiplex immunohistochemistry to characterize tumor-infiltrating immune cells. Their results identified immune cell signatures, such as CD4+FoxP3−PD-L1+, CD8+PD-1-LAG3-, and CD68+STING+, that predicted treatment response and survival. The spatial distribution of CD8+PD-1+LAG3− cells was particularly prognostic, underscoring the clinical utility of comprehensive immune profiling.
The miRNAs also play a pivotal role in H. pylori-driven carcinogenesis (Figure 2). These small non-coding RNAs regulate immune responses by modulating inflammatory signalling and immune cell function, as well as targeting bacterial virulence genes such as VacA and BabA. They influence cytokine and chemokine expression and have demonstrated diagnostic and prognostic utility across H. pylori-related pathologies.48
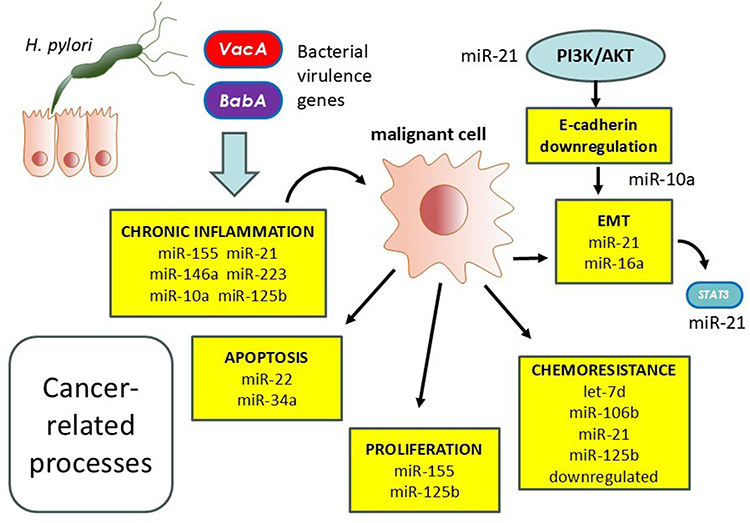 |
Figure 2 Upon Helicobacter pylori infection, altered microRNA (miRNA) expression occurs within the gastric mucosa, affecting both immune responses and gastric epithelial cell behaviour. On the one hand, dysregulated miRNAs compromise host immune defences and facilitate immune evasion. On the other, they enhance epithelial cell proliferation and survival. These effects converge to promote carcinogenic mechanisms, ultimately contributing to gastric cancer development. |
While H. pylori infection stimulates the release of proinflammatory cytokines, maintaining chronic inflammation and contributing to mucosal injury, specific miRNAs, including miR-155, miR-146a, miR-21, miR-223, miR-10a, and miR-125b, modulate this inflammatory milieu.49 Aberrant miRNA expression promotes cell proliferation, suppresses apoptosis, disrupts cell cycle control, and enhances metastatic potential through mechanisms like E-cadherin downregulation and PI3K/AKT pathway activation.48 Moreover, miRNAs contribute to chemoresistance. Upregulation of miR-21 and downregulation of miR-34a and miR-125b have been linked to reduced chemotherapy sensitivity and impaired DNA repair.50,51 Circulating miRNAs, such as miR-21, let-7d, and miR-106b, are being explored as non-invasive biomarkers for early gastric cancer detection and prognostication.48
Targeting miRNAs holds therapeutic promise. Experimental models demonstrate that modulating miRNAs, including miR-22, miR-143-3p, and miR-146a, can suppress tumor growth, promote apoptosis, and reverse drug resistance.48,52
Together, these findings emphasize the diverse and dynamic roles of miRNAs in H. pylori-associated gastric cancer. Their diagnostic, prognostic, and therapeutic potential offers promising avenues for advancing precision oncology in infection-driven malignancies.
Conclusions and Future Directions
Inflammation is likely the main key contributor to H. pylori-associated carcinogenesis, with mechanisms encompassing dysregulation of the cell cycle, manipulation of apoptotic pathways, disruption of autophagy, induction of oxidative stress, and epigenetic reprogramming.
H. pylori also participates in extensive crosstalk with host oncogenic signaling pathways, creating a complex and multifactorial network that underlies gastric tumorigenesis.
These findings collectively underscore the importance of H. pylori screening in cancer prevention and highlight the need for precision-based interventions that account for individual risk profiles, regional patterns of antimicrobial resistance, and the cost-effectiveness of screening within specific populations.
Prioritized research areas in this field should include: (1) elucidation of host-pathogen molecular interactions that drive carcinogenesis; (2) characterization of H. pylori genotypes and their influence on disease progression and therapeutic response; (3) investigation of H. pylori-induced immune modulation in the context of cancer immunotherapy; (4) identification of therapeutic targets within disrupted host signaling pathways; and (5) development of effective preventive strategies, including vaccination programs and optimized eradication protocols.
Funding
Dr. Graham is supported in part by the Office of Research and Development Medical Research Service Department of Veterans Affairs, Public Health Service grant DK56338, which funds the Texas Medical Center Digestive Diseases Center and by the Cancer Prevention and Research Institute of Texas (RP220127).
Disclosure
Dr. Graham is an unpaid consultant for RedHill Biopharma and has previously received research support for the culture of Helicobacter pylori. He has also been a consultant with Janssen Research & Development regarding potential gastrointestinal effects of drugs under development and has collaborated on research projects with American Molecular Laboratories regarding molecular diagnostics for H. pylori. The authors report no other conflicts of interest in this work.
References
1. Inoue M. Epidemiology of gastric cancer-changing trends and global disparities. Cancers. 2024;16(17):2948. doi:10.3390/cancers16172948
2. Liu Z, Xu H, You W, Pan K, Li W. eradication for primary prevention of gastric cancer: progresses and challenges. J Natl Cancer Cent. 2024;4(4):299–11. doi:10.1016/j.jncc.2024.06.006
3. Zhang Y, Yan Z, Jiao Y, Feng Y, Zhang S, Yang A. Innate immunity in Helicobacter pylori infection and gastric oncogenesis. Helicobacter. 2025;30(2):e70015. doi:10.1111/hel.70015
4. Duan Y, Xu Y, Dou Y, Xu D. Helicobacter pylori and gastric cancer: mechanisms and new perspectives. J Hematol Oncol. 2025;18(1):10. doi:10.1186/s13045-024-01654-2
5. Shah SAR, Mumtaz M, Sharif S, Mustafa I, Nayila I. Helicobacter pylori and gastric cancer: currentinsights and nanoparticle-based interventions. RSC Adv. 2025;15(7):5558–5570. doi:10.1039/d4ra07886a
6. Raza Y, Mubarak M, Memon MY, Alsulaimi MS. Update on molecular pathogenesis of. World J Gastrointest Pathophysiol. 2025;16(2):107052. doi:10.4291/wjgp.v16.i2.107052
7. Ono A, Tanaka S, Sawada N, et al. Helicobacter pylori eradication and gastric cancer prevention in a pooled analysis of large-scale cohort studies in Japan. Sci Rep. 2025;15(1):21307. doi:10.1038/s41598-025-00713-z
8. Zeng R, Gou H, Lau HCH, Yu J. Stomach microbiota in gastric cancer development and clinical implications. Gut. 2024;73(12):2062–2073. doi:10.1136/gutjnl-2024-332815
9. Chiang TH, Chang WJ, Chen SL, et al. Mass eradication of. Gut. 2021;70(2):243–250. doi:10.1136/gutjnl-2020-322200
10. Zhang X, Arnold IC, Müller A. Mechanisms of persistence, innate immune activation and immunomodulation by the gastric pathogen Helicobacter pylori. Curr Opin Microbiol. 2020;54:1–10. doi:10.1016/j.mib.2020.01.003
11. Li Y, Choi H, Leung K, Jiang F, Graham DY, Leung WK. Global prevalence of Helicobacter pylori infection between 1980 and 2022: a systematic review and meta-analysis. Lancet Gastroenterol Hepatol. 2023;8(6):553–564. doi:10.1016/S2468-1253(23)00070-5
12. Wondmagegn YM, Girmay G, Amare GA, et al. Prevalence of intestinal parasites and Helicobacter pylori co-infection in people with gastrointestinal symptoms in Africa: a systematic review and meta-analysis. BMC Infect Dis. 2025;25(1):20. doi:10.1186/s12879-024-10432-x
13. Yuan C, Adeloye D, Luk TT, et al. The global prevalence of and factors associated with Helicobacter pylori infection in children: a systematic review and meta-analysis. Lancet Child Adolesc Health. 2022;6(3):185–194. doi:10.1016/S2352-4642(21)00400-4
14. Shah SC, Halvorson AE, Lee D, et al. Helicobacter pylori burden in the United States according to individual demographics and geography: a nationwide analysis of the veterans healthcare system. Clin Gastroenterol Hepatol. 2024;22(1):42–50.e26. doi:10.1016/j.cgh.2023.05.016
15. Moodley Y, Linz B, Bond RP, et al. Age of the association between Helicobacter pylori and man. PLoS Pathog. 2012;8(5):e1002693. doi:10.1371/journal.ppat.1002693
16. Dore MP, Pes GM. Trained immunity and trained tolerance: the case of Helicobacter pylori infection. Int J Mol Sci. 2024;25(11):5856. doi:10.3390/ijms25115856
17. Thorell K, Muñoz-Ramírez ZY, Wang D, et al. The Helicobacter pylori genome project: insights into H. pylori population structure from analysis of a worldwide collection of complete genomes. Nat Commun. 2023;14(1):8184. doi:10.1038/s41467-023-43562-y
18. Wu X, Zhao Y, Zhang H, et al. Mechanism of regulation of the Helicobacter pylori Cagβ ATPase by CagZ. Nat Commun. 2023;14(1):479. doi:10.1038/s41467-023-36218-4
19. Wang H, Zhao M, Shi F, Zheng S, Xiong L, Zheng L. A review of signal pathway induced by virulent protein CagA of Helicobacter pylori. Front Cell Infect Microbiol. 2023;13:1062803. doi:10.3389/fcimb.2023.1062803
20. Abdullah M, Greenfield LK, Bronte-Tinkew D, Capurro MI, Rizzuti D, Jones NL. VacA promotes CagA accumulation in gastric epithelial cells during Helicobacter pylori infection. Sci Rep. 2019;9(1):38. doi:10.1038/s41598-018-37095-4
21. Ansari S, Yamaoka Y. Virulence factors exploiting gastric colonization and its pathogenicity. Toxins. 2019;11(11):677. doi:10.3390/toxins11110677
22. Gu H. Role of Flagella in the Pathogenesis of Helicobacter pylori. Curr Microbiol. 2017;74(7):863–869. doi:10.1007/s00284-017-1256-4
23. Sallas ML, Dos Santos MP, Orcini WA, et al. Status (on/off) of oipA gene: their associations with gastritis and gastric cancer and geographic origins. Arch Microbiol. 2019;201(1):93–97. doi:10.1007/s00203-018-1580-5
24. Heinrich PC, Behrmann I, Haan S, Hermanns HM, Müller-Newen G, Schaper F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem J. 2003;374(Pt 1):1–20. doi:10.1042/BJ20030407
25. Yang J, Chatterjee-Kishore M, Staugaitis SM, et al. Novel roles of unphosphorylated STAT3 in oncogenesis and transcriptional regulation. Cancer Res. 2005;65(3):939–947. doi:10.1158/0008-5472.939.65.3
26. Tye H, Kennedy CL, Najdovska M, et al. STAT3-driven upregulation of TLR2 promotes gastric tumorigenesis independent of tumor inflammation. Cancer Cell. 2012;22(4):466–478. doi:10.1016/j.ccr.2012.08.010
27. Wang S, Chen Z, Zhu S, et al. PRDX2 protects against oxidative stress induced by H. pylori and promotes resistance to cisplatin in gastric cancer. Redox Biol. 2020;28:101319. doi:10.1016/j.redox.2019.101319
28. Huang S, Bucana CD, Van Arsdall M, Fidler IJ. Stat1 negatively regulates angiogenesis, tumorigenicity and metastasis of tumor cells. Oncogene. 2002;21(16):2504–2512. doi:10.1038/sj.onc.1205341
29. Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006;441(7092):431–436. doi:10.1038/nature04870
30. Takahashi-Kanemitsu A, Lu M, Knight CT, et al. The Helicobacter pylori CagA oncoprotein disrupts Wnt/PCP signaling and promotes hyperproliferation of pyloric gland base cells. Sci Signal. 2023;16(794):eabp9020. doi:10.1126/scisignal.abp9020
31. Song P, Gao Z, Bao Y, et al. Wnt/β-catenin signaling pathway in carcinogenesis and cancer therapy. J Hematol Oncol. 2024;17(1):46. doi:10.1186/s13045-024-01563-4
32. Peng Y, Qin Y, Zhang X, et al. MiRNA-20b/SUFU/Wnt axis accelerates gastric cancer cell proliferation, migration and EMT. Heliyon. 2021;7(4):e06695. doi:10.1016/j.heliyon.2021.e06695
33. Ding SZ, Minohara Y, Fan XJ, et al. Helicobacter pylori infection induces oxidative stress and programmed cell death in human gastric epithelial cells. Infect Immun. 2007;75(8):4030–4039. doi:10.1128/IAI.00172-07
34. Sulo P, Šipková B. DNA diagnostics for reliable and universal identification of Helicobacter pylori. World J Gastroenterol. 2021;27(41):7100–7112. doi:10.3748/wjg.v27.i41.7100
35. Chaturvedi R, Asim M, Romero-Gallo J, et al. Spermine oxidase mediates the gastric cancer risk associated with Helicobacter pylori CagA. Gastroenterology. 2011;141(5):
36. McNamara KM, Sierra JC, Latour YL, et al. Spermine oxidase promotes Helicobacter pylori-mediated gastric carcinogenesis through acrolein production. Oncogene. 2025;44(5):296–306. doi:10.1038/s41388-024-03218-7
37. Palrasu M, Zaika E, Paulrasu K, et al. Helicobacter pylori pathogen inhibits cellular responses to oncogenic stress and apoptosis. PLoS Pathog. 2022;18(6):e1010628. doi:10.1371/journal.ppat.1010628
38. Fan X, Gunasena H, Cheng Z, et al. Helicobacter pylori urease binds to class II MHC on gastric epithelial cells and induces their apoptosis. J Immunol. 2000;165(4):1918–1924. doi:10.4049/jimmunol.165.4.1918
39. Galmiche A, Rassow J, Doye A, et al. The N-terminal 34 kDa fragment of Helicobacter pylori vacuolating cytotoxin targets mitochondria and induces cytochrome c release. EMBO J. 2000;19(23):6361–6370. doi:10.1093/emboj/19.23.6361
40. Li SP, Chen XJ, Sun AH, Zhao JF, Yan J. CagA(+) H. pylori induces Akt1 phosphorylation and inhibits transcription of p21(WAF1/CIP1) and p27(KIP1) via PI3K/Akt1 pathway. Biomed Environ Sci. 2010;23(4):273–278. doi:10.1016/S0895-3988(10)60063-3
41. Xi Y, Zhang XL, Luo QX, et al. regulates stomach diseases by activating cell pathways and DNA methylation of host cells. Front Cell Dev Biol. 2023;11:1187638. doi:10.3389/fcell.2023.1187638
42. Liu D, Liu Y, Zhu W, et al. Helicobacter pylori-induced aberrant demethylation and expression of GNB4 promotes gastric carcinogenesis via the Hippo-YAP1 pathway. BMC Med. 2023;21(1):134. doi:10.1186/s12916-023-02842-6
43. Zhu Y, Zhu F, Ba H, Chen J, Bian X. Helicobacter pylori infection and PD-L1 expression in gastric cancer: a meta-analysis. Eur J Clin Invest. 2023;53(2):e13880. doi:10.1111/eci.13880
44. Oster P, Vaillant L, Riva E, et al. infection has a detrimental impact on the efficacy of cancer immunotherapies. Gut. 2022;71(3):457–466. doi:10.1136/gutjnl-2020-323392
45. Magahis PT, Maron SB, Cowzer D, et al. Impact of. J Immunother Cancer. 2023;11(10). doi:10.1136/jitc-2023-007699
46. Jia K, Chen Y, Xie Y, et al. and immunotherapy for gastrointestinal cancer. Innovation. 2024;5(2):100561. doi:10.1016/j.xinn.2023.100561
47. Chen Y, Jia K, Sun Y, et al. Predicting response to immunotherapy in gastric cancer via multi-dimensional analyses of the tumour immune microenvironment. Nat Commun. 2022;13(1):4851. doi:10.1038/s41467-022-32570-z
48. Mansour RM, El-Sayyad GS, Abulsoud AI, et al. The role of miRNAs in pathogenesis, diagnosis, and therapy of Helicobacter pylori infection, gastric cancer-causing bacteria: special highlights on nanotechnology-based therapy. Microb Pathog. 2025;205:107646. doi:10.1016/j.micpath.2025.107646
49. Xu H, Huang K, Shi M, et al. MicroRNAs in Helicobacter pylori-infected gastric cancer: function and clinical application. Pharmacol Res. 2024;205:107216. doi:10.1016/j.phrs.2024.107216
50. Yang SM, Huang C, Li XF, Yu MZ, He Y, Li J. miR-21 confers cisplatin resistance in gastric cancer cells by regulating PTEN. Toxicology. 2013;306:162–168. doi:10.1016/j.tox.2013.02.014
51. Yang F, Li QJ, Gong ZB, et al. MicroRNA-34a targets Bcl-2 and sensitizes human hepatocellular carcinoma cells to sorafenib treatment. Technol Cancer Res Treat. 2014;13(1):77–86. doi:10.7785/tcrt.2012.500364
52. Li S, Liang X, Ma L, et al. MiR-22 sustains NLRP3 expression and attenuates H. pylori-induced gastric carcinogenesis. Oncogene. 2018;37(7):884–896. doi:10.1038/onc.2017.381
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Helicobacter pylori Infection Combined with OLGA and OLGIM Staging Systems for Risk Assessment of Gastric Cancer: A Retrospective Study in Eastern China
Wu M, Feng S, Qian M, Wang S, Zhang K
Risk Management and Healthcare Policy 2022, 15:2243-2255
Published Date: 30 November 2022
Factors Associated with Malignant Gastric Ulcers: A 10-year Retrospective Cohort Study
Zayad A, Yahia Y, Chandra P, Obeidat I, Safieh M, Alnakhala S, Saffo HA
International Journal of General Medicine 2026, 19:593454
Published Date: 29 May 2026