Back to Journals » OncoTargets and Therapy » Volume 13
HANR Enhances Autophagy-Associated Sorafenib Resistance Through miR-29b/ATG9A Axis in Hepatocellular Carcinoma
Authors Shi Y, Yang X, Xue X, Sun D, Cai P, Song Q, Zhang B, Qin L
Received 5 September 2019
Accepted for publication 12 December 2019
Published 9 March 2020 Volume 2020:13 Pages 2127—2137
DOI https://doi.org/10.2147/OTT.S229913
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Prof. Dr. Nicola Silvestris
Yang Shi, 1 Xiaohua Yang, 1 Xiaofeng Xue, 1 Ding Sun, 1 Peng Cai, 2 Qingwei Song, 2 Bin Zhang, 2 Lei Qin 1
1Department of General Surgery, The First Affiliated Hospital of Soochow University, Suzhou, People’s Republic of China; 2Department of General Surgery, The Affiliated Hospital of Xuzhou Medical University, Xuzhou, People’s Republic of China
Correspondence: Lei Qin
No. 899, Pinghai Road, Suzhou 215000, People’s Republic of China
Tel +86 1395110345
Email [email protected]
Background: Hepatocellular carcinoma (HCC) is one of the most common malignancies worldwide and chemoresistance is the main obstacle for effective treatments of HCC. Accumulating studies indicated that long non-coding RNAs (lncRNAs) contribute to the chemoresistance of human carcinoma. However, the functional role of HANR in autophagy-mediated chemoresistance of HCC is unknown.
Methods: The expressions of HANR, miR-29b and ATG9A in tissues and cell lines were detected by real-time quantitative PCR (RT-qPCR). The expression of autophagy-related protein LC3-I and LC3-II was evaluated by Western blotting. The cell viability and apoptosis were examined by CCK-8 and flow cytometry, respectively. Bioinformatics analysis and luciferase activity assay were applied to determine the downstream target gene of HANR or miR-29b. Xenograft experiment was used to detect the effect of HANR on tumor growth.
Results: In the present study, we demonstrated that HANR was notably overexpressed in sorafenib-resistant HepG2 (HepG2/sora) and sorafenib-resistant Huh7 (Huh7/sora) cells, and HANR enhanced sorafenib resistance by facilitating autophagy in HepG2/sora and Huh7/sora cells. Furthermore, we demonstrated that miR‑29b could directly interact with HANR and abolished HANR-induced sorafenib resistance by suppressing autophagy in HepG2/sora and Huh7/sora cells. Moreover, ATG9A was validated as a target of miR-29b and its overexpression obviously reversed the inhibitory effect of miR-29b on sorafenib resistance and autophagy. In addition, HANR could act as a competing endogenous RNA (ceRNA) to upregulate ATG9A expression by sponging miR-29b. Hence, HANR increased autophagy-related sorafenib resistance via inhibiting the miR-29b/ATG9A axis in HepG2/sora and Huh7/sora cells, indicating that it may be a potential target to prevent chemoresistance of HCC.
Conclusion: Our study revealed HANR enhanced sorafenib resistance by acting as an autophagy promoter by regulating miR-29b/ATG9A axis in sorafenib‑resistant HCC cells and might provide potential therapeutic strategies for HCC treatment.
Keywords: hepatocellular carcinoma, HCC, autophagy, HANR, miR-29b, ATG9A
Introduction
Hepatocellular carcinoma (HCC) is the second most life-threatening cancer worldwide and the fifth most frequent malignancy globally.1,2 Currently, chemotherapy is one of the main strategies for advanced-stage HCC, and sorafenib is regarded as the first-line systemic drug.3 However, the clinical treatment efficacy of sorafenib is largely restricted due to the emergence of drug resistance.4 Autophagy, a conserved catabolic process, has been reported to contribute to tumor chemotherapy resistance and promote further tumor cell growth, including HCC. For instance, Zhai et al reported autophagy participates in sorafenib resistance and blockade of autophagy sensitizes HCC to sorafenib.5 Therefore, developing approaches to inhibit autophagy and sensitize HCC cells to sorafenib resistance may improve the therapeutic efficacy of HCC patients.
Long noncoding RNAs (lncRNAs) are a class of non-coding transcripts longer than 200 nucleotides, which is closely associated with the regulation of autophagy in certain types of cancer.6,7 For example, lncRNA XIST abrogation attenuates the chemoresistance of non-small cell lung cancer (NSCLC) cells through inhibition of autophagy.8 LncRNA HULC overexpression enhances the chemoresistance of HCC cells through facilitating autophagy.9 HANR is a newly identified HCC associated lncRNA which promotes the tumorigenesis and enhances chemoresistance in HCC.10 However, the regulation of HANR on autophagy-mediated chemoresistance of HCC is unclear.
Our previous study demonstrated that HANR may act as a competing endogenous RNA (ceRNA) for sponging microRNAs to promote cancer development.11 miR-29b, a member of the miR-29 family, which is thoroughly documented as a tumor suppressor in majority of researches.12–14 In addition, miR-29 was reported to play a critical role in the regulation of autophagy. For instance, Yang et al reported that miR-29b inhibits autophagy in pancreatic cancer cells.15 Moreover, miR-29b was shown to enhance cisplatin sensitivity of ovarian cancer, revealed by reducing cell viability and promoting apoptosis, which led us to hypothesize that HANR may regulate autophagy and chemoresistance of HCC by directly interacting with miR-29b.16
In the present study, we found that HANR was overexpressed in sorafenib-resistant HCC cells, and knockdown of HANR could enhance chemosensitivity via inhibiting autophagy. Moreover, we found that HANR promoted autophagy as ceRNA to increase the ATG9A expression by sponging miR-29b. Taken together, our results demonstrated that HANR enhanced the autophagy-induced chemoresistance of HCC cells through inhibiting miR-29b/ATG9A axis, which might provide a potential therapeutic strategy for the treatment of HCC.
Methods
Patients and Tissues
The study was accepted by the Institutional Research Ethics Committee of the Affiliated Hospital of Xuzhou Medical University and written informed consent was obtained from all patients with HCC. In this study, HCC samples and adjacent normal tissues collected from 30 patients who suffered from the sorafenib-based chemotherapy after surgical treatment. The patients were divided into sorafenib-sensitive or -resistant group according to the response evaluation criteria. All samples were snap frozen in liquid nitrogen immediately and stored at −80°C until required.
Cell Culture
The human HCC cell lines (HepG2 and Huh7) and 293T cells were obtained from ATCC. Cells were maintained in DMEM with 10% fetal bovine serum (FBS) in humidified 5% CO2 at 37°C. HepG2 and Huh7 cell lines were incubated with slowly increasing concentrations of sorafenib (0.5–10 µg/mL) for more than 6 months to obtain sorafenib-resistant HepG2 (HepG2/sora) cells and sorafenib-resistant Huh7 (Huh7/sora) cells.
Cell Transfection
The shRNA specific to HANR (shHANR) and its negative control (shNC), miR-29b mimics and its negative control (miR-NC), miR-29b inhibitor and its negative control (inh-miR-NC), were synthesized by GenePharma (Shanghai, China). For overexpression of HANR and ATG9A, the HANR and ATG9A cDNA were cloned into pCDNA3.1 vector by GenePharma, respectively. Transfection was conducted with Lipofectamine 2000 following the manufacturer’s instructions.
Western Blot
Briefly, the cultured cells were lysed by RIPA lysate to extract total protein. Equal quantities of protein separated by SDS-PAGE were electrophoretically transferred to nitrocellulose membranes. After blocking with 5% bovine serum albumin for 1 h at room temperature, the proteins were incubated with primary antibodies against ATG9A (ab108338, EPR2450(2), 1:2000 dilution; Abcam), LC3B (ab51520, 1:3000 dilution; Abcam, containing LC3‑I and LC3‑II), or β-actin (ab8227, 1:2000 dilution; Abcam) at 4°C overnight. After being washed, membranes were incubated with horseradish peroxidase‑conjugated goat anti-rabbit (ab6721, 1:5000 dilution; Abcam) for 2 h at room temperature and the proteins were detected by an enhanced chemiluminescence detection system (ECL).
RT-qPCR
Total RNA of cultured cells was extracted using TRIzol reagent (Invitrogen, CA, USA) and cDNA was then synthesized using the reverse transcription kit (Takara, Tokyo, Japan). Then the mRNA expression was measured by SYBR Green PCR Kit (Takara Bio Inc, Japan). 2−ΔΔCt method was used for gene expression quantification. The following primers were used in RT-qPCR experiments:
HANR-F: 5′-AAGTACCAGGCAGTGACAGC-3′; HANR-R: 5′-TTCTCCACGTTCTTCTCGGC-3′; miR-29b F: 5′-UAGCACCATCTGAAATCGGTTA-3′; miR-29b R: 5′-ACCGTGCTCGACTTTCCGG-3′; ATG9A-F 5′-CCCCAGTACTGCCACCTTTA-3′; ATG9A-R 5′-ACAGCCTGACCTGCTCATCT-3′; GAPDH-F: 5′-GAGTCAACGGATTTGGTCGT-3′; GAPDH-R: 5′-TTGATTTTGGAGGGATCTCG-3′; U6-F: 5′-CTCGCTTCGGCAGCACA-3′; U6-R: 5′-AACGCTTCACGAATTTGCGT-3′.
Cell Proliferation Assay
Cell proliferation was performed by CCK-8 assay to evaluate the drug resistance. After transfection, cells were seeded into 96-well plates and incubated in CCK-8 solution for 4 h. The optical density value was measured at 450 nm.
Dual Luciferase Reporter Assay
Mutant HANR and ATG9A were generated using site-directed mutagenesis method. MiR-29b mimics, miR-29b inhibitor or NC (negative control) mimics were co-transfected with pmirGlo-NC, pmirGlo-HANR-mut or pmirGlo-HANR-wt into 293T cells. MiR-29b mimics, miR-29b inhibitor or NC were co-transfected with pmirGlo-NC, pmirGlo-ATG9A-3ʹUTR-wt or pmirGlo-ATG9A-3ʹUTR-mut into 293T cells. The relative luciferase activities were evaluated by dual-luciferase reporter assay kit (Promega, USA).
Xenograft Experiment
8 male BABL/c nude mice (6 weeks old) were maintained under specific pathogen-free conditions and randomly divided into 4 groups, and were housed individually in microisolator ventilated cages (temperature, 26–28°C; 40–60% humidity and ventilation 10–15 times/h) with free access to water and food. BABL/c nude mice were injected subcutaneously with HepG2 cells stably transfected with HANR overexpression plasmid or vector control and treated with sorafenib. On the 30th day, the mice were sacrificed by cervical dislocation after deep anesthesia with 2% isoflurane (Baxter Healthcare Corporation) to obtain the tumors. The tumors were photographed and tumor weights were measured.
Statistical Analysis
Data were presented as the mean ± S.D. from at least 3 independent repeats. Statistical analysis was performed using SPSS 13.0 software. Comparison between groups was done by using double-sided Student’s test. P<0.05 was considered statistically significant.
Results
HANR Is Upregulated in Sorafenib‑Resistant HCC Tissues and Cells
We first detected the expression of HANR in HCC patient tissues. The data of RT-qPCR assay revealed that the expression of HANR was significantly upregulated in HCC tissues compared with that in normal tissues and sorafenib-resistant HCC tissues showed higher expression of HANR than HCC-sensitive tissues (Figure 1A). Subsequently, sorafenib‑resistant HCC cell lines (HepG2/sora and Huh7/sora) were established to explore whether HANR was associated with sorafenib resistance in HCC. As shown in Figure 1B, the IC50 of sorafenib was markedly increased in HepG2/sora and Huh7/sora cells compared with that in the corresponding parental cell lines (HepG2 and Huh7, respectively), indicating that sorafenib-resistant HCC cell lines were successfully established. It was further demonstrated that HANR expression was markedly increased in HepG2/sora and Huh7/sora compared with the parental cell lines, respectively (Figure 1C). To explore the effect of HANR on sorafenib resistance of HCC cells, HepG2 and Huh7 cells were transfected with HANR overexpression plasmid. The transfection efficacy was confirmed by RT-qPCR (Figure 1D). Subsequently, HCC cells were exposed with sorafenib for 24 h. CCK-8 assay showed that sorafenib led to obvious reduction of cell viability, but was reversed by overexpression of HANR in HepG2 and Huh7 cells (Figure 1E and F), indicating that HANR markedly enhanced the resistance of HCC cells to sorafenib. To further investigate the effect of HANR on sorafenib resistance in vivo, xenograft tumor experiment was conducted. The results showed that tumor weight was obviously decreased after sorafenib treatment compared with those in control group. However, addition of HANR weakened the suppressive effect of sorafenib on tumor growth (Figure 1G).
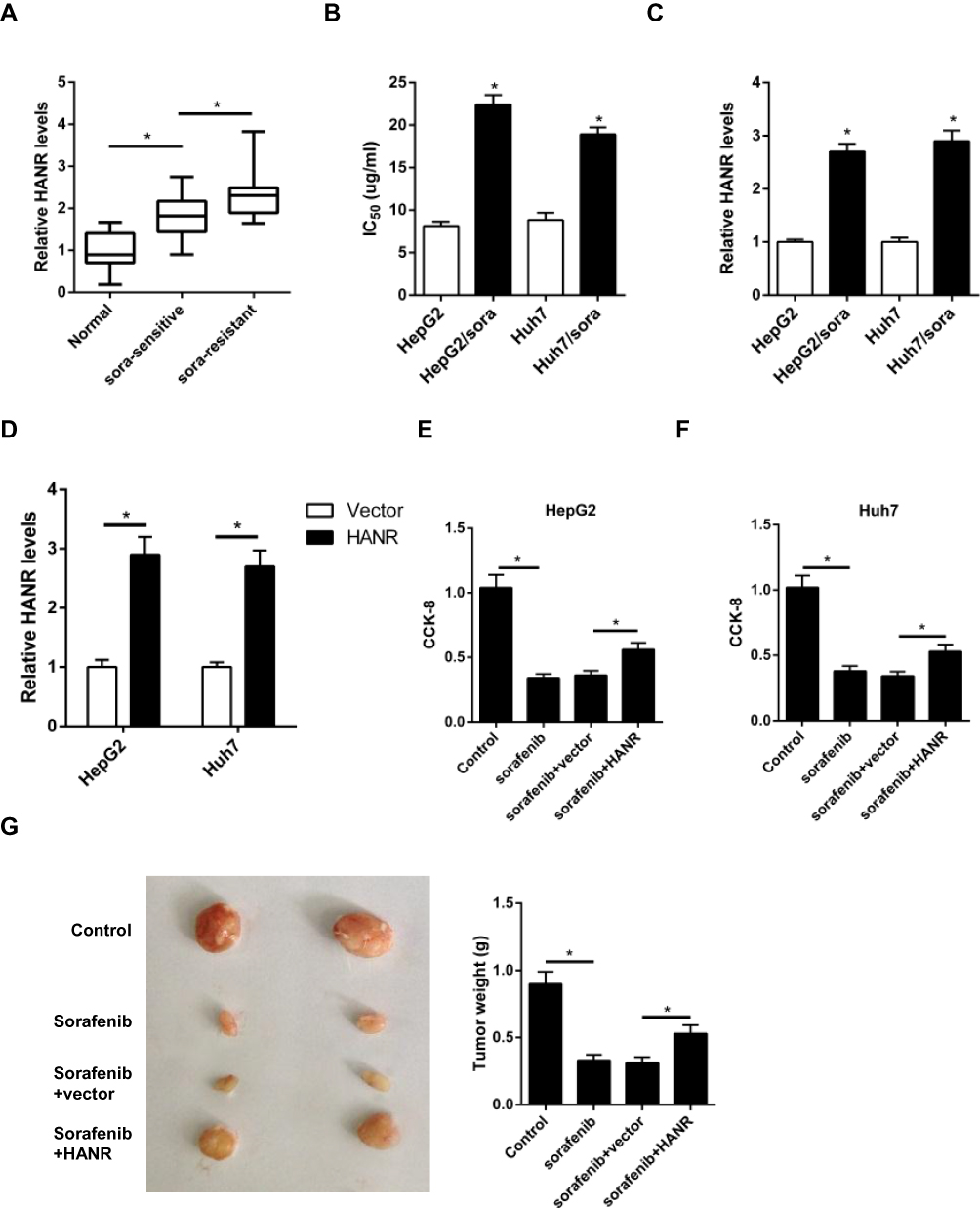 |
Figure 1 HANR expression is enhanced in HCC-sensitive or -resistant tissues and cells. (A) RT-qPCR shows the expression of HANR in HCC samples and para-tumor normal tissues. (B) CCK‑8 assay shows IC50 value of HepG2, HepG2/sora, Huh7 and Huh7/sora cells. (C) RT-qPCR shows the relative expression levels of HANR in HepG2, HepG2/sora, Huh7 and Huh7/sora cells. (D) RT-qPCR shows the relative expression levels of HANR in HepG2 and Huh7 cells transfected with vector or HANR overexpression plasmid. (E, F) CCK-8 assay shows the effect of HANR overexpression on cell viability in HepG2 and Huh7 cells with HANR transfection and sorafenib treatment. (G) Xenograft assay shows tumor growth and tumor weight was detected at the end point. The data were presented as mean ± SD (*P < 0.05). |
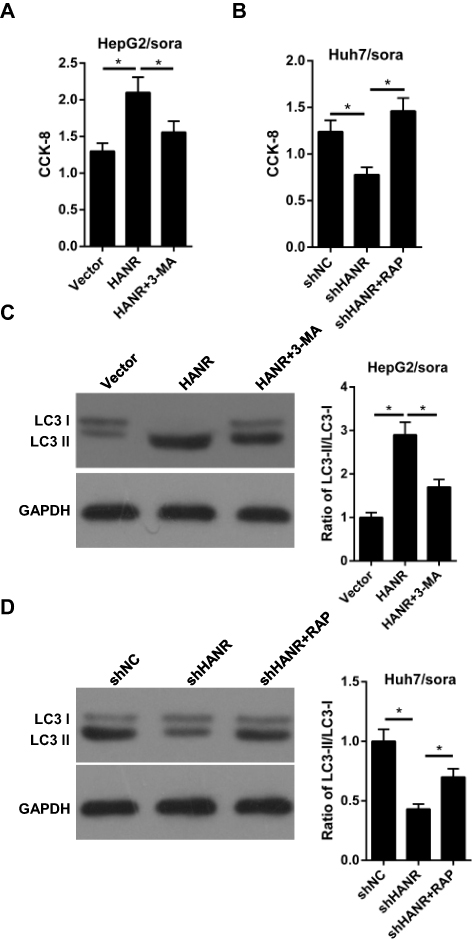 |
Figure 2 Inhibition of autophagy reduces HANR-regulated sorafenib resistance of HCC cells. (A) CCK-8 assay shows the cell viability of HepG2/sora cells transfected with vector, HANR and HANR+3-MA. (B) CCK-8 assay shows the cell viability of Huh7/sora cells transfected with shNC, shHANR and shHANR+RAP. (C) Western blotting shows the LC3-II/LC3-I protein ratio in HepG2/sora cells transfected with vector, HANR and HANR+3-MA. (D) Western blotting shows the LC3-II/LC3-I protein ratio in Huh7/sora cells transfected with shNC, shHANR and shHANR+RAP. The data were presented as mean ± SD (*P < 0.05). |
Inhibition of Autophagy Partially Reduces HANR-Regulated Sorafenib Resistance of HCC Cells
To further verify the role of autophagy in HANR-related sorafenib resistance, we used the autophagy pathway inhibitor 3-methyladenine (3-MA) and the autophagy inducer rapamycin (RAP), which could inhibit and promote autophagy, respectively. CCK-8 assay illuminated that 3-MA effectively reduced HANR-mediated sorafenib resistance in HepG2/sora cells (Figure 2A). By contrast, the resistance in Huh7/sora was notably enhanced in shHANR-transfected Huh7/sora cells treated with RAP (Figure 2B). Moreover, Western blot analysis showed that 3-MA markedly abolished HANR-induced autophagy in HepG2/sora cells (Figure 2C). By contrast, RAP alleviated the inhibitory effect of shHANR on autophagy in Huh7/sora cells (Figure 2D). In summary, the results demonstrated that autophagy promoted HANR-related sorafenib resistance of HCC cells.
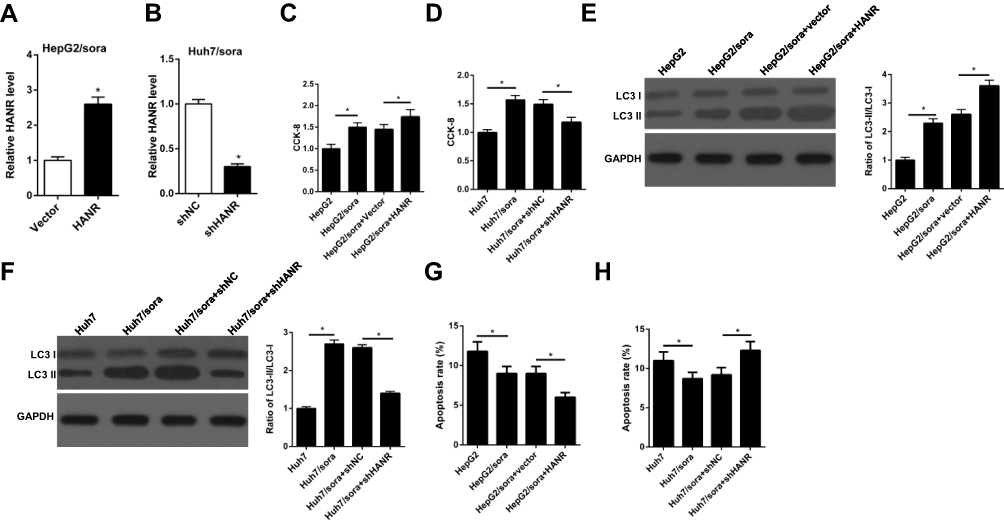 |
Figure 3 HANR overexpression enhances sorafenib resistance of HCC cells by promoting autophagy. (A) RT-qPCR shows the relative expression levels of HANR in HepG2/sora cells transfected with vector or HANR overexpression plasmid. (B) RT-qPCR shows the relative expression levels of HANR in Huh7/sora cells transfected with shNC or shHANR. (C, D) CCK-8 assay shows the cell viability of HepG2/sora and Huh7/sora cells in different transfected groups. (E, F) Western blotting shows the LC3-II/LC3-I protein ratio in HepG2/sora and Huh7/sora cells in different transfected groups. (G, H) Flow cytometry shows the cell apoptosis rate of HepG2/sora and Huh7/sora cells in different transfected groups. The data were presented as mean ± SD (*P < 0.05). |
HANR Inhibits Sorafenib Susceptibility of HCC Cells by Promoting Autophagy
To investigate the role of HANR in sorafenib-resistant HCC cells, sorafenib‑resistant HepG2 cells transfected with HANR overexpression plasmid (Huh7/sora + HANR) and Huh7 cells transfected with shHANR (Huh7/sora +shHANR) were established. RT-qPCR analysis showed that HANR expression was effectively upregulated in HANR overexpressed HepG2/sora cells, but downregulated in HANR-silenced Huh7/sora cells (Figure 3A and B). Furthermore, CCK-8 assay demonstrated that HepG2/sora and Huh7/sora cells enhanced the resistance to sorafenib compared with their respective parental cells (Figure 3C and D). Moreover, HANR overexpression obviously increased the resistance of HepG2/sora cells to sorafenib (Figure 3C). Conversely, HANR-depleted Huh7/sora significantly reduced sorafenib resistance of Huh7/sora cells (Figure 3D). To explore the effect of HANR on autophagy, the expression of autophagy-related protein LC3-I and LC3-II was evaluated by Western blotting. As shown in Figure 3E and F, the ratio of LC3-II/LC3-I was increased in HepG2/sora cells and Huh7/sora cells compared with HepG2 and Huh7 cells, respectively. Moreover, HANR overexpression increased the ratio in HepG2/sora cells, while HANR knockdown reduced the ratio in Huh7/sora (Figure 3E and F). Flow cytometry showed that the cell apoptosis was significantly reduced in sorafenib-resistant HCC cell lines (Figure 3G and H). Moreover, HANR overexpression inhibited cell apoptosis in HepG2/sora, while HANR knockdown facilitated cell apoptosis in Huh7/sora cells (Figure 3G and H). In summary, the above data demonstrated that HANR enhanced sorafenib resistance of HCC by inducing autophagy.
HANR Inhibits miR-29b Expression
Increasing evidence shows that lncRNAs could competitively bind to microRNAs through complementary base pairing to block their expression. By performing bioinformatics analysis on starBase (http://starbase.sysu.edu.cn), we found that miR-29b was a candidate gene of HANR (Figure 4A). To confirm the binding between HANR and miR-29b, we performed the luciferase reporter analysis. As shown in Figure 4B, miR-29b mimics obviously weakened the luciferase activity of HANR reporter vector, while had no influence on mutant HANR. Moreover, we found that miR-29b significantly reduced the HANR expression, while miR-29b inhibitor increased HANR expression (Figure 4C). These data indicated that HANR could directly target miR-29b to inhibit its expression.
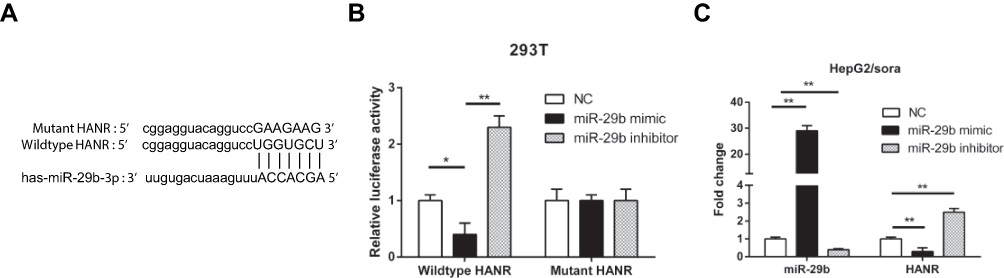 |
Figure 4 HANR inhibits miR-29b expression by direct interaction. (A) Bioinformatic prediction of binding site of miR-29b by HANR. (B) Luciferase reporter assay shows miR-29b bind to wildtype HANR, not mutant HANR in 293T cells. (C) RT-qPCR shows the expression levels of miR-29b and HANR in HepG2/sora cells transfected with NC, miR-29b mimics or miR-29b inhibitor. The data were presented as mean ± SD (*P < 0.05, **p < 0.01). |
miR-29b Attenuates HANR-Induced Sorafenib Resistance by Inhibition of Autophagy in Sorafenib-Resistant HCC Cells
Since HANR inhibits miR-29b expression by direct interaction, we performed rescue experiments to investigate whether miR-29b was involved in HANR-induced sorafenib resistance. First, we introduced vector, HANR, HANR+miR-NC or HANR+miR-29b into HepG2/sora cells, while Huh7/sora cells were transfected with shNC, shHANR, shHANR+inh-miR-NC or shHANR+inh-miR-29b. The data indicated that miR-29b mimics attenuated sorafenib resistance of HANR-transfected HepG2/sora cells (Figure 5A). Conversely, miR-29b inhibitor partially restored sorafenib resistance of shHANR-transfected Huh7/sora cells (Figure 5B). Subsequently, Western blot analysis showed that miR-29b mimics markedly abolished HANR-induced autophagy in HepG2/sora cells, which was proved by the decreased ratio of LC3‑II/LC3‑I (Figure 5C). By contrast, miR‑29b inhibitor alleviated the inhibitory effect of shHANR on autophagy in Huh7/sora cells (Figure 5D). Therefore, these results revealed that miR‑29b weakened HANR-induced sorafenib resistance by suppressing autophagy in sorafenib-resistant HCC cells.
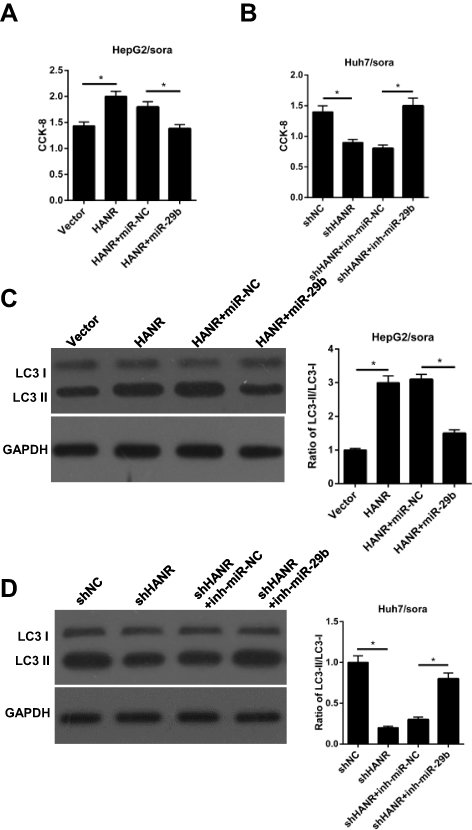 |
Figure 5 Restoration of miR-29b abrogates HANR-induced sorafenib resistance by inhibiting autophagy in sorafenib-resistant HCC cells. (A) CCK-8 assay shows the cell viability of HepG2/sora cells transfected with vector, HANR, HANR+miR-NC, or HANR+miR-29b. (B) CCK-8 assay shows the cell viability of Huh7/sora cells transfected with shNC, shHANR, shHANR+inh-miR-NC, or shHANR+inh-miR-29b. (C, D) Western blotting shows the LC3-II/LC3-I protein ratio in HepG2/sora and Huh7/sora cells in different transfected groups. The data were presented as mean ± SD (*P < 0.05). |
miR-29b Enhances Sorafenib Sensitivity by Directly Interacting with ATG9A in Sorafenib-Resistant HCC Cells
Through using TargetScan (http://www.targetscan.org), ATG9A was predicted as a downstream target of miR-29b (Figure 6A). Luciferase reporter assay indicated that miR-29b mimics significantly reduced the luciferase activity of the wildtype 3ʹ-UTR of ATG9A, but had no effect on the mutant 3ʹ-UTR of ATG9A, which confirmed a direct interaction between miR-29b and ATG9A (Figure 6B). To determine the effect of ATG9A on sorafenib sensitivity, HepG2 and Huh7 cells were transfected with ATG9A overexpression plasmid. The transfection efficacy was validated by qRT-PCR assay (Figure 6C). CCK-8 assay showed that sorafenib significantly reduced cell viability, but was reversed by overexpression of ATG9A in HepG2 and Huh7 cells (Figure 6D and E). To explore whether the miR-29b-induced chemosensitivity was regulated by ATG9A, HepG2/sora cells were transfected with miR-NC, miR-29b mimics, miR-29b+pcDNA or miR-29b mimics+ATG9A, and Huh7/sora cells were transfected with inh-miR-NC, miR-29b inhibitor, inh-miR-29b+siNC or inh-miR-29b+siATG9A. RT-qPCR analysis indicated that miR-29b mimics inhibited ATG9A expression in HepG2/sora (Figure 6F), while miR-29b inhibitor upregulated ATG9A expression in Huh7/sora cells (Figure 6G). Moreover, ATG9A overexpression reversed the inhibitory effect of miR-29b on ATG9A expression in HepG2/sora (Figure 6F). By contrast, ATG9A depletion abolished inh-miR-29b-induced ATG9A upregulation in Huh7/sora cells (Figure 6G). In addition, miR-29b mimics attenuated sorafenib resistance and decreased the ratio of LC3-II/LC3-I in HepG2/sora cells, indicating that miR-29b enhanced chemosensitivity and inhibited autophagy, while these effects were abrogated by ATG9A overexpression (Figure 6H and J). Conversely, the opposite effect was occurred in Huh7/sora cells co-transfected with inh-miR-29b and siATG9A (Figure 6I and K). In conclusion, our results demonstrated that ATG9A promoted autophagy in sorafenib-resistant cell lines and ATG9A restoration or silence abrogated the effect of miR-29b mimics or miR-29b inhibitor on the sorafenib-resistant cell lines.
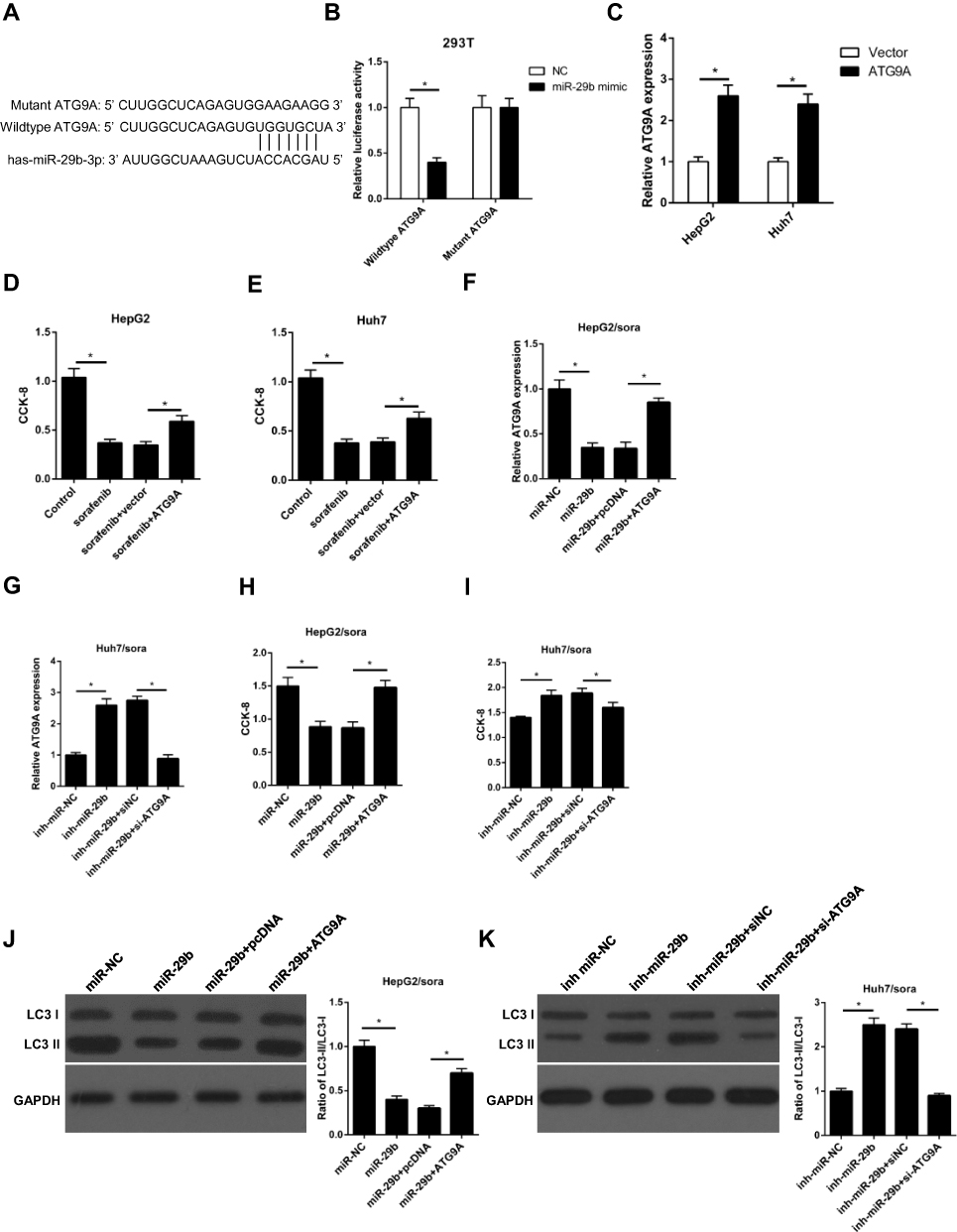 |
Figure 6 miR-29b enhances sorafenib sensitivity by directly interacting with ATG9A in sorafenib-resistant HCC cells. (A) Predicted binding sites between miR‑29b and the ATG9A 3ʹ‑untranslated region by TargetScan online website. (B) Luciferase reporter assay shows miR-29b bind to wildtype ATG9A, not mutant ATG9A in 293T cells. (C) RT-qPCR shows the relative expression levels of ATG9A in HepG2 and Huh7 cells transfected with vector or ATG9A. (D, E) CCK-8 assay shows the effect of ATG9A overexpression on cell viability in HepG2 and Huh7 cells with ATG9A transfection and sorafenib treatment. (F) RT-qPCR shows the relative expression of HepG2/sora cells transfected with miR-NC, miR-29b, miR-29b+pcDNA, or miR-30b+ATG9A. (G) RT-qPCR shows the relative expression of Huh7/sora cells transfected with inh-miR-NC, inh-miR-29b, inh-miR-29b+siNC, or inh-miR-29b+si-ATG9A. (H, I) CCK-8 assay shows the cell viability of HepG2/sora and Huh7/sora cells in different transfected groups. (J, K) Western blotting shows the LC3-II/LC3-I protein ratio in HepG2/sora and Huh7/sora cells in different transfected groups. The data were presented as mean ± SD (*P < 0.05). |
HANR Modulates ATG9A Expression by Sponging miR-29b
We further explore whether HANR could act as a ceRNA to regulate ATG9A expression in HepG2/sora and Huh7/sora cells. RT-qPCR analysis showed that ectopic expression of HANR upregulated the expression of ATG9A in HepG2/sora cells, while this effect was abolished by miR‑29b overexpression (Figure 7A). By contrast, HANR knockdown downregulated the expression of ATG9A and miR-29b inhibitor partially restored the inhibitory effect of shHANR on ATG9A in Huh7/sora cells (Figure 7B). In summary, these data demonstrated that HANR served as ceRNA of miR‑29b to upregulate ATG9A expression in sorafenib‑resistant HCC cell lines.
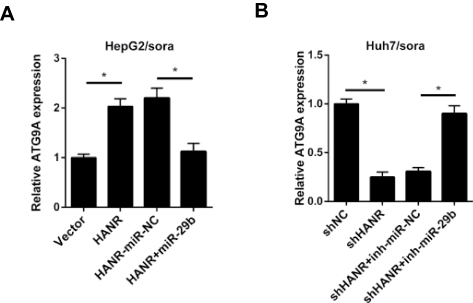 |
Figure 7 HANR modulates ATG9A expression by sponging miR-29b. (A) RT-qPCR shows the relative expression of ATG9A in HepG2/sora cells transfected with pcDNA, HANR, HANR-miR-NC, or HANR+miR-29b. (B) RT-qPCR shows the relative expression of ATG9A in Huh7/sora cells transfected with shNC, shHANR, shHANR+inh-miR-NC, or shHANR+inh-miR-29b.The data were presented as mean ± SD (*P < 0.05). |
Discussion
Currently, chemotherapy is considered as the primary therapeutic strategy for advanced HCC. Although the advancements have been made in the development of novel chemotherapeutic drugs, the therapeutic efficacy is still unsatisfactory due to the emergence of drug resistance. Therefore, understanding novel molecular mechanisms underlying HCC chemoresistance is very imperative.
LncRNA is an important regulatory molecular in various cancers, including HCC.17–19 Increasing evidence indicated the expression level of lncRNA is closely correlated with treatment efficacy.20–22 Moreover, some studies also demonstrated that lncRNA is deeply involved in autophagy-associated chemoresistance.9,23 In our study, we found that HANR was significantly overexpressed in sorafenib-resistant HCC cells. Furthermore, we demonstrated that HANR overexpression enhanced sorafenib resistance of HepG2/sora cells by promoting autophagy. By contrast, HANR knockdown reduced the resistance of Hub7/sora cells to sorafenib by inhibiting autophagy. Therefore, we identified the suppressive role of HANR in sorafenib-resistant HCC cells.
Numerous studies reported that lncRNAs could exert an oncogenic role in human cancers by working as ceRNAs. For example, lncRNA TDRG1 promotes cervical cancer progression by targeting miR-326.24 LncRNA MIR31HG acts as a ceRNA of miR-193b to promote the progression of pancreatic ductal adenocarcinoma.25 MiR-29b was reported to exert anti-tumor activity in several tumor types.26–28 Moreover, downregulation of miR-29b induces anticancer drug resistance in ovarian cancer.16 The abovementioned findings indicated that miR-29b may regulate sorafenib resistance and autophagy. In this study, we revealed that miR‑29b could directly interact with HANR by bioinformatics predictions and luciferase reporter assay, and its overexpression abrogated HANR-induced sorafenib resistance by inhibiting autophagy in sorafenib-resistant HCC cells. These data indicated that miR-29b attenuates HANR-induced sorafenib resistance by inhibiting autophagy in sorafenib-resistant HCC cells.
Autophagy has been reported to act either oncogenic or antitumor in cancer development. Endo et al reported that autophagy can protect cancer cells against cell death through catabolic pathway,29 while Pan et al indicated autophagy promotes cancer cell apoptosis via degradation of fundamental cellular organelles.30 Even though autophagy plays a controversial role in cancer progression, autophagy inhibition may be an effective therapeutic strategy for chemoresistance in HCC cells.31,32 Therefore, modulating the expression of autophagy-related (ATG) genes may be a potential method for decreasing drug chemoresistance in HCC. There are more than 36 ATG genes are primarily involved in the autophagy process.33 Among which, we identified ATG9A is a downstream target gene of miR-29b. miR-29b attenuated chemoresistance and inhibited autophagy, while these effects were abrogated by ATG9A overexpression in HepG2/sora cells (Figure 4E and F). By contrast, miR-29b inhibitor enhanced chemoresistance and promoted autophagy, while ATG9A silencing reversed the effect of miR-29b inhibitor on chemoresistance and autophagy in Huh7/sora cells. Moreover, we demonstrated that HANR upregulated ATG9A expression by sponging miR-29b. These findings indicated that HANR increased autophagy-related sorafenib resistance via inhibiting the miR-29b/ATG9A axis in sorafenib-resistant HCC cells.
In conclusion, our study revealed HANR enhanced sorafenib resistance by acting as an autophagy promoter through sponging miR-29b to upregulate ATG9A expression in sorafenib‑resistant HCC cells. Our study provided a new sight on autophagy-induced chemoresistance in sorafenib-resistant HCC cells and potential therapeutic strategies for HCC treatment.
Author Contributions
This study was conceived and designed by LQ and YS. The experiments were carried out by XY, XX and PC. The manuscript was prepared by DS, QS and BZ. All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors declare that they have no competing interests to disclose.
References
1. El-Serag HB. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology. 2012;142:1264–1273 e1261. doi:10.1053/j.gastro.2011.12.061
2. Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in globocan 2012. Int J Cancer. 2015;136(5):E359–E386. doi:10.1002/ijc.29210
3. Forner A, Reig M, Bruix J. Hepatocellular carcinoma. Lancet. 2018;391:1301–1314. doi:10.1016/S0140-6736(18)30010-2
4. Avila M, Berasain C. Making sorafenib irresistible: in vivo screening for mechanisms of therapy resistance in hepatocellular carcinoma hits on Mapk14. Hepatology. 2015;61(5):1755–1757. doi:10.1002/hep.27739
5. Zhai B, Hu F, Jiang X, et al. Inhibition of Akt reverses the acquired resistance to sorafenib by switching protective autophagy to autophagic cell death in hepatocellular carcinoma. Mol Cancer Ther. 2014;13:1589–1598. doi:10.1158/1535-7163.MCT-13-1043
6. Bartonicek N, Maag JL, Dinger ME. Long noncoding RNAs in cancer: mechanisms of action and technological advancements. Mol Cancer. 2016;15:43. doi:10.1186/s12943-016-0530-6
7. Yin Q, Feng W, Shen X, Ju S. Regulatory effects of lncRNA and miRNAs on autophagy in malignant tumorigenesis. Biosci Rep. 2018;38. doi:10.1042/BSR20180516
8. Sun W, Zu Y, Fu X, Deng Y. Knockdown of lncRNA-Xist enhances the chemosensitivity of Nsclc cells via suppression of autophagy. Oncol Rep. 2017;38:3347–3354. doi:10.3892/or.2017.6056
9. Xiong H, Ni Z, He J, et al. lncRNA Hulc triggers autophagy via stabilizing Sirt1 and attenuates the chemosensitivity of Hcc cells. Oncogene. 2017;36:3528–3540. doi:10.1038/onc.2016.521
10. Xiao J, Lv Y, Jin F, et al. lncRNA HANR promotes tumorigenesis and increase of chemoresistance in hepatocellular carcinoma. Cell Physiol Biochem. 2017;43(5):1926–1938. doi:10.1159/000484116
11. Shi Y, Yang X, Xue X, et al. HANR promotes hepatocellular carcinoma progression via miR-214/Ezh2/Tgf-Beta axis. Biochem Biophys Res Commun. 2018;506:189–193. doi:10.1016/j.bbrc.2018.10.038
12. Chen L, Zhang S, Wu J, et al. Circrna_100290 plays a role in oral cancer by functioning as a sponge of the miR-29 family. Oncogene. 2017;36:4551–4561. doi:10.1038/onc.2017.89
13. Wang CY, Ren JB, Liu M, Yu L. Targeting miR-29 induces apoptosis of osteosarcoma Mg-63 cells via regulation of Tgf-Beta1/Puma signal. Eur Rev Med Pharmacol Sci. 2016;20:3552–3560.
14. Shin J, Shim HG, Hwang T, et al. Restoration of miR-29b exerts anti-cancer effects on glioblastoma. Cancer Cell Int. 2017;17:104. doi:10.1186/s12935-017-0476-9
15. Yang S, Wang X, Contino G, et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011;25:717–729. doi:10.1101/gad.2016111
16. Sugio A, Iwasaki M, Habata S, et al. Bag3 upregulates Mcl-1 through downregulation of miR-29b to induce anticancer drug resistance in ovarian cancer. Gynecol Oncol. 2014;134:615–623. doi:10.1016/j.ygyno.2014.06.024
17. Huo X, Han S, Wu G, et al. Dysregulated long noncoding RNAs (LncRNAs) in hepatocellular carcinoma: implications for tumorigenesis, disease progression, and liver cancer stem cells. Mol Cancer. 2017;16:165. doi:10.1186/s12943-017-0734-4
18. Momen-Heravi F, Bala S. Emerging role of non-coding Rna in oral cancer. Cell Signal. 2018;42:134–143. doi:10.1016/j.cellsig.2017.10.009
19. Li Z, Dou P, Liu T, He S. Application of long noncoding RNAs in osteosarcoma: biomarkers and therapeutic targets. Cell Physiol Biochem. 2017;42:1407–1419. doi:10.1159/000479205
20. Qu L, Ding J, Chen C, et al. Exosome-transmitted Lncarsr promotes sunitinib resistance in renal cancer by acting as a competing endogenous RNA. Cancer Cell. 2016;29:653–668. doi:10.1016/j.ccell.2016.03.004
21. Fan Y, Shen B, Tan M, et al. Long non-coding RNA Uca1 increases chemoresistance of bladder cancer cells by regulating Wnt signaling. FEBS J. 2014;281(7):1750–1758. doi:10.1111/febs.12737
22. Ozes AR, Miller DF, Ozes ON, et al. Nf-Kappab-hotair axis links DNA damage response, chemoresistance and cellular senescence in ovarian cancer. Oncogene. 2016;35:5350–5361. doi:10.1038/onc.2016.75
23. Chen YM, Liu Y, Wei HY, Lv KZ, Fu PF. Large intergenic non-coding RNA-Ror reverses gemcitabine-induced autophagy and apoptosis in breast cancer cells. Oncotarget. 2016;7:59604–59617. doi:10.18632/oncotarget.10730
24. Jiang H, Liang M, Jiang Y, et al. The lncRNA Tdrg1 promotes cell proliferation, migration and invasion by targeting miR-326 to regulate Mapk1 expression in cervical cancer. Cancer Cell Int. 2019;19:152. doi:10.1186/s12935-019-0872-4
25. Yang H, Liu P, Zhang J, et al. Long noncoding Rna miR31hg exhibits oncogenic property in pancreatic ductal adenocarcinoma and is negatively regulated by miR-193b. Oncogene. 2016;35:3647–3657. doi:10.1038/onc.2015.430
26. Wang T, Hou J, Jian S, et al. miR-29b negatively regulates Mmp2 to impact gastric cancer development by suppress gastric cancer cell migration and tumor growth. J Cancer. 2018;9:3776–3786. doi:10.7150/jca.26263
27. Ding D, Li C, Zhao T, Li D, Yang L, Zhang B. lncRNA H19/miR-29b-3p/Pgrn axis promoted epithelial-mesenchymal transition of colorectal cancer cells by acting on Wnt signaling. Mol Cells. 2018;41:423–435. doi:10.14348/molcells.2018.2258
28. Wang L, Wang Z, Huang L, Wu C, Zhang B. Mir-29b suppresses proliferation and mobility by targeting Sox12 and Dnmt3b in pancreatic cancer. Anticancer Drugs. 2019;30:281–288. doi:10.1097/CAD.0000000000000719
29. Endo S, Nakata K, Ohuchida K, et al. Autophagy is required for activation of pancreatic stellate cells, associated with pancreatic cancer progression and promotes growth of pancreatic tumors in mice. Gastroenterology. 2017;152(6):1492–1506e1424. doi:10.1053/j.gastro.2017.01.010
30. Pan Z, Chen Y, Liu J, et al. Design, synthesis, and biological evaluation of polo-like kinase 1/Eukaryotic elongation factor 2 kinase (Plk1/Eef2k) dual inhibitors for regulating breast cancer cells apoptosis and autophagy. Eur J Med Chem. 2018;144:517–528. doi:10.1016/j.ejmech.2017.12.046
31. Shi YH, Ding ZB, Zhou J, et al. Targeting autophagy enhances sorafenib lethality for hepatocellular carcinoma via er stress-related apoptosis. Autophagy. 2011;7:1159–1172. doi:10.4161/auto.7.10.16818
32. Guo XL, Li D, Hu F, et al. Targeting autophagy potentiates chemotherapy-induced apoptosis and proliferation inhibition in hepatocarcinoma cells. Cancer Lett. 2012;320:171–179. doi:10.1016/j.canlet.2012.03.002
33. Shu CW, Liu PF, Huang CM. High throughput screening for drug discovery of autophagy modulators. Comb Chem High Throughput Screen. 2012;15:721–729. doi:10.2174/138620712803519734
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.