Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 16
GSK-3β Inhibitors Attenuate the PM2.5-Induced Inflammatory Response in Bronchial Epithelial Cells
Authors Zou W ![]() , Ye D, Liu S, Hu J, Zhu T, He F, Ran P
, Ye D, Liu S, Hu J, Zhu T, He F, Ran P
Received 6 July 2021
Accepted for publication 5 October 2021
Published 14 October 2021 Volume 2021:16 Pages 2845—2856
DOI https://doi.org/10.2147/COPD.S327887
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Min Zhang
Weifeng Zou,1 Dong Ye,2 Sha Liu,3 Jinxing Hu,1 Tao Zhu,1 Fang He,4 Pixin Ran5
1State Key Laboratory of Respiratory Disease, Guangzhou Chest Hospital, Guangzhou, Guangdong, People’s Republic of China; 2State Key Laboratory of Respiratory Disease, Guangzhou Medical University, Guangzhou, Guangdong, People’s Republic of China; 3The Second Hospital, University of South China, Hengyang, Hunan, People’s Republic of China; 4School of Basic Medical Sciences, Guangzhou Medical University, Guangzhou, Guangdong, People’s Republic of China; 5State Key Laboratory of Respiratory Diseases, National Clinical Research Center for Respiratory Diseases, Guangzhou Institute of Respiratory Health, The First Affiliated Hospital of Guangzhou Medical University, Guangzhou, Guangdong, People’s Republic of China
Correspondence: Pixin Ran
State Key Laboratory of Respiratory Diseases, National Clinical Research Center for Respiratory Diseases, Guangzhou Institute of Respiratory Health, The First Affiliated Hospital of Guangzhou Medical University, 151 Yanjiang Road, Yuexiu District, Guangzhou, Guangdong, 510120, People’s Republic of China
Tel +86 13922765811
Email [email protected]
Background and Purpose: PM2.5-associated airway inflammation has recently been recognized as pivotal to the development of COPD. Aberrant glycogen synthase kinase (GSK)-3β signaling is linked to the inflammatory response. Therefore, we investigated the effects of GSK-3β inhibitors on the PM2.5-induced inflammatory response in bronchial epithelial cells.
Methods: The production of phosphorylated GSK-3β (p-GSK-3β) was analyzed by immunohistochemistry with PM2.5-induced mice. HBECs were treated with various inhibitors targeting GSK-3β or JNK before PM2.5 stimulation. The production of GSK-3β signaling was analyzed by Western blotting. Inflammatory cytokine production was detected by qRT–PCR and ELISA.
Results: PM2.5 exposure caused lung inflammation, upregulated serum concentrations of HMGB1 and IL-6, decreased IL-10 expression, and significantly attenuated p-GSK-3β production in mice. HBECs exposed to PM2.5 showed significantly reduced p-GSK-3β production, an increased ratio of p-JNK/JNK, increased NF-κB activation and IκB degradation, and upregulated the inflammatory cytokines HMGB1 and IL-6. Intervention with GSK-3β inhibitors TDZD-8 and SB216763 significantly suppressed PM2.5-induced outcomes. Moreover, the JNK inhibitor SP600125 also reduced the level of NF-κB phosphorylation induced by PM2.5. The differences in the levels of inflammation-related cytokines in the TDZD-8 groups were greater than those in the SB216763 groups.
Conclusion: Inhibition of GSK-3β weakens the PM2.5-induced inflammatory response by regulating the JNK/NF-κB signaling pathway in bronchial epithelial cells.
Keywords: PM2.5, COPD, GSK-3β, inflammatory response, HBECs
Introduction
With the increasing speed of urbanization and industrialization, environmental pollution caused by PM2.5 has challenged public health and resulted in an enormous economic burden. Many epidemiological studies have shown that PM2.5 exposure is correlated with an enhanced risk of chronic obstructive pulmonary disease (COPD),1,2 which is characterized by progressive fixed airflow because of airway inflammation.3 The rates of morbidity and mortality of COPD have gradually increased over the past decade. Therefore, there is an urgent need to study the cellular mechanisms of PM2.5-induced COPD.
Published studies have indicated that abnormal glycogen synthase kinase-3 (GSK-3) activity contributes to the pathological processes of COPD.4 GSK3 is a serine-threonine protein kinase that is constitutively active in cells and has two isoforms in mammals: GSK-3a and GSK-3β. Phosphorylation of an N-terminal serine residue (Ser9) in GSK-3β suppresses GSK-3 activity and attenuates its activity to change cell function.5 GSK-3β is a contributing factor in regulating the activity of numerous factors, including c-Jun,6 β-catenin,7 NF-κB and STAT.8 Recently, GSK-3β has emerged as a key regulator of the inflammatory response,9 and it may represent an important downstream effector of oxidant-mediated signaling during COPD inflammation.10 Moreover, an increasing number of studies have shown that PM2.5 exposure increases airway inflammatory cytokine production in COPD. Such discoveries are the basis for the hypothesis that GSK-3β may play an important role in regulating the airway inflammation caused by PM2.5 in COPD.
Several previous studies showed that GSK-3β inhibition alleviated the inflammatory response induced by a wide range of extracellular stimuli.11 However, it is currently uncertain whether GSK-3β inhibitors could suppress the airway inflammation of COPD caused by PM2.5. As the first line of defense against inhaled harmful particles, HBECs play a key role in the development of COPD.12 Here, different GSK-3β inhibitors, the small-molecule ATP competitive inhibitor SB216763 and the non-ATP competitive inhibitor TDZD-8, were employed to intervene in the GSK-3β pathway in HBECs.
Materials and Methods
Cell Culture and Treatment
The 16HBE human bronchial epithelial cell line was obtained from Guangzhou Medical University (Guangzhou, China), an agent of ATCC. The 16HBECs were treated with DMEM (Sigma–Aldrich, Louis, USA) supplemented with 10% FBS and 1% penicillin (Sigma–Aldrich, Louis, USA), and then, the cells were pretreated with TDZD-8, SB216763 or SP600125 (Sigma–Aldrich, Louis, USA) for 30 mins and treated with different concentrations of PM2.5 (5–20 μg/mL).
Cell Counting Kit-8 (CCK8) Cell Viability Assay
HBECs were treated with PM2.5 (5, 10, 20, or 40 μg/mL) for 24 hours. HBEC viability was measured by CCK-8 assay (Dojindo, Kumamoto, Japan) according to the manufacturer’s instructions, and the optical density (OD) was measured at 490 nm.
Collection and Extraction of PM2.5
The details of the PM2.5 collection and analysis methods were obtained as previously described.13 Briefly, the collection of traffic ambient PM2.5 was prepared with aerodynamic impactors, and the extracts were obtained with DMSO. The physicochemical characterizations were analyzed through gravimetric analysis. The mean concentrations of polynuclear aromatic hydrocarbons (PAHs) and n-alkanes in PM2.5 were 108.453 μg/g and 18,670.883 μg/g, respectively, with a final PAH recovery of 44.62%; the mean concentrations of these DMSO extracts were 48.392 μg/g and 164.675 μg/g, respectively.
Animals and Sample Lung Tissues
Twenty female C57BL mice were randomly fed in the Laboratory Animal Center of Guangzhou Medical University. The experiment was approved by the Institutional Animal Care and Use Committee of Guangzhou Medical University and performed according to the regulations designated by the Chinese Association for Laboratory Animal Science Policy. The mice were randomly allocated to the PBS and PM2.5 concentration groups (100 μg/20 μL) twice per week for 4 weeks by tracheal drip. Mice were sacrificed by intraperitoneal injection of sodium pentobarbital (100 mg/kg), and then paraffin-embedded lung tissues were stained for hematoxylin and eosin (H&E) and immunohistochemistry using standard techniques.
Histology and Immunohistochemistry
These sections of lung tissue were stained by H&E for morphometric detection. The sections were stained with p-GSK-3β antibodies (1:100, Santa Cruz, USA) and then with peroxidase (HRP)-conjugated anti-mouse IgG according to the manufacturer’s protocols. Expression was evaluated by confocal microscopy at 200× magnification. We quantified the OD of p-GSK-3β-positive cells using a semiquantitative scoring system.
Quantitative Reverse Transcription Polymerase Chain Reaction
Total RNA from HBECs was extracted using the RNeasy plus mini kit (Qiagen) according to the manufacturer’s instructions. GAPDH served as an internal standard. The primer sequences were as follows: GAPDH, 5′-ATCACTGCCACCCAGAAG-3′ and 5′-TCCACGACGGACACATTG-3′; IL-6, 5′-GCCTTCGGTCCAGTTGCC-3′ and 5′-GCGCAGAATGAGATGAGTTGTCATG-3′; HMGB1, 5ʹ-AATACGAAAAGGATATTGCT-3ʹ and 5ʹ-GCGCTAGAACCAACTTAT-3.
Western Blot Analysis
Total proteins were extracted using a protein extraction kit (Invitrogen, Carlsbad, USA), and Western blotting was performed according to the manufacturer’s instructions. Antibody concentrations were applied as follows: p-GSK-3β antibody (1:1000), p-JNK antibody (1:1000), p-NF-κB p65 antibody (1:1000), IκBα antibody (1:1000) and β-actin (1:1000, Santa Cruz, USA). The results are expressed relative to the control (control, set as 1.0).
ELISAs
The concentrations of IL-10, IL-6 and HMGB1 (R&D Systems, Minneapolis, MN, USA) in mouse serum and cell culture medium were detected using ELISA kits following the manufacturer’s protocol. A standard curve was prepared with standard solution, and the absolute concentrations were detected according to the standard curve.
Statistical Analysis
Statistical analysis was performed with SPSS 25.0 statistical software. Group data are expressed as the means ± standard deviations (SDs). For data with a normal distribution, comparisons among multiple groups were performed with ANOVAs. Two-group comparisons were performed by Student’s t-test. p<0.05 was considered to be significant.
Results
PM2.5 Induced Inflammatory Changes and Reduced the Expression of p-GSK-3β in the Lungs of Mice
As depicted in Figure 1A, H&E staining analysis showed that PM2.5 exposure caused the upregulation of inflammatory cells in the lungs of mice. ELISA results revealed that the expression levels of the inflammatory mediators HMGB1 and IL-6 were significantly higher in the PM2.5 group; however, the expression of the anti-inflammatory cytokine IL-10 displayed an opposite trend compared with that of HMGB1 and IL-6 in the serum (Figure 1B). Moreover, immunostaining and Western blot data revealed that phosphorylation of serine-threonine kinase (Ser9) of GSK-3β (p-GSK-3β) expression was markedly lower in the PM2.5 group than in the control group (Figures 1C–E).
 |
Figure 1 PM2.5 induced inflammatory changes and reduced the expression of p-GSK-3β in the lungs of mice. Abbreviations: PM2.5, particulate matter<2.5 μm. Notes: (A) H&E staining showed that PM2.5 exposure caused the upregulation of inflammatory cells. (B) ELISA results showed that PM2.5 exposure increased the expression of HMGB1 and IL-6 and decreased the expression of IL-10. (C) Immunohistochemical staining showed that PM2.5 exposure reduced the expression of p-GSK-3β in lung tissues (original magnification 200×, bar 100 μm). (D) The OD of p-GSK-3β-positive cells were lower in PM2.5-treated mice than in untreated mice. (E) Western blot analysis showed that PM2.5 exposure reduced the expression of p-GSK-3β in lung tissues. #P<0.05, compared to control group, n=5. |
Effect of PM2.5 on the HBEC Viability
HBEC viability was measured using a CCK-8 assay to evaluate the cytotoxicity of PM2.5. After incubation with various concentrations of PM2.5 (0, 5, 10, 20, and 40 μg/mL) for 24 h, the CCK-8 assay results showed that the cell viability did not markedly change after 24 h of PM2.5 (5–20 μg/mL) treatment, but the cell viability decreased after treatment with 40 μg/mL PM2.5 (Figure 2). Concentrations of PM2.5 (5–20 μg/mL) were selected for further experiments.
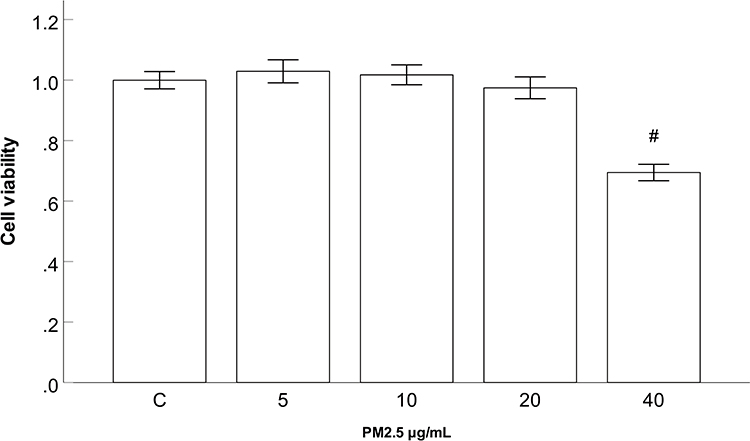 |
Figure 2 Effect of PM2.5 on HBEC viability. Abbreviations: PM2.5, particulate matter<2.5 μm. Notes: CCK-8 assay results showed that cell viability did not markedly change after 24 h of PM2.5 (5–20 μg/mL) treatment, but cell viability decreased after treatment with 40 μg/mL PM2.5. #P<0.05, versus PM2.5 group, n=3. |
PM2.5 Reduced the Expression of p-GSK-3β in HBECs
To assess the role of p-GSK-3β in HBECs treated with PM2.5, we examined the relative protein expression levels of GSK-3β and p-GSK-3β in HBECs. Western blot data revealed that the expression of p-GSK-3β did not change markedly after 24 h of PM2.5 (5–10 μg/mL) treatment, whereas the expression was significantly decreased after PM2.5 stimulation at 20 μg/mL compared with the control group (Figure 3A), which suggested that 24 h of exposure of HBECs to PM2.5 (20 μg/mL) led to a decrease in p-GSK-3β expression. Furthermore, Western blot data also revealed that the expression p-GSK-3β did not change markedly after 3 h of PM2.5 (20 μg/mL) treatment, but the expression was markedly decreased after 12 and 24 h of PM2.5 treatment, in a time-dependent manner (Figure 3B).
 |
Figure 3 PM2.5 reduced the expression of p-GSK-3β in HBECs. Abbreviations: PM2.5, particulate matter<2.5 μm. Notes: (A) Western blot analysis revealed that the expression of p-GSK-3β did not change markedly after 24 h of PM2.5 (5–10 μg/mL) treatment, whereas the expression was significantly decreased after PM2.5 stimulation at 20 μg/mL. (B) Western blot analysis showed that the expression of p-GSK-3β did not change markedly after 3 h of PM2.5 (20 μg/mL) treatment, but the expression was substantially decreased after 12 and 24 h of PM2.5 treatment. #P<0.05, compared to control group. n=3. |
GSK-3β Inhibitors Increased the Expression of p-GSK-3β in HBECs
To gain mechanistic insight into the effects of the GSK-3β inhibitors used, we examined the expression p-GSK-3β in HBECs. As expected, Western blot data showed that the GSK-3β inhibitors TDZD-8 and SB216763 significantly increased the expression of p-GSK-3β exposed to PM2.5. (Figure 4). In addition, the altered level of p-GSK-3β in the TDZD-8 group was stronger than that in the SB216763 group.
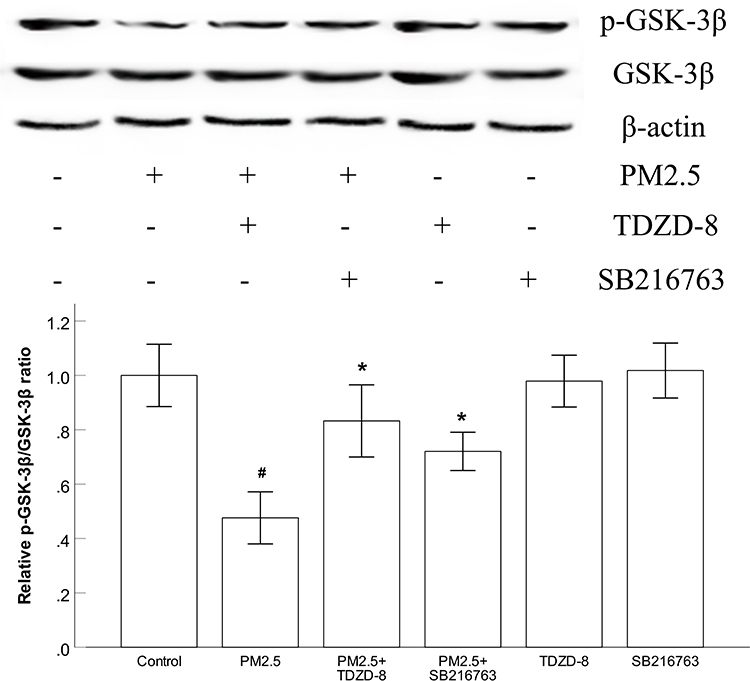 |
Figure 4 GSK-3β inhibitors increased the expression of p-GSK-3β in HBECs. Abbreviations: PM2.5, particulate matter<2.5 μm. Notes: (A) Western blot analysis showed that the GSK-3β inhibitors TDZD-8 and SB216763 significantly increased the expression of p-GSK-3β. #P<0.05 compared to the control group. *P<0.05, compared to PM2.5 group, n=3. |
GSK-3β Inhibitors Alleviated PM2.5-Activated JNK Pathways in HBECs
Since GSK-3β is an upstream regulator of the C-jun N-terminal kinase (JNK) signaling pathway, we explored the protein expression levels of JNK and p-JNK in HBECs treated with PM2.5. As shown in Figure 5, PM2.5 treatment significantly enhanced the ratio of p-JNK/JNK, while treatment with TDZD-8 and SB216763 markedly decreased the ratio of p-JNK/JNK induced by PM2.5. The altered ratios of p-JNK/JNK in the TDZD-8 groups were higher than those in the SB216763 groups.
 |
Figure 5 GSK-3β inhibitors alleviated PM2.5-activated JNK pathways in HBECs. Abbreviations: PM2.5, particulate matter<2.5 μm. Notes: (A) Western blot analysis showed that the GSK-3β inhibitors TDZD-8 and SB216763 markedly decreased the PM2.5-induced ratio of p-JNK/JNK. #P<0.05, compared to control group. *P<0.05, compared to PM2.5 group, n=3. |
GSK-3β Inhibitors and JNK Inhibitors Suppressed PM2.5-Induced NF-κB Activation and IkBα Degradation in HBECs
NF-κB plays a critical role in the pathogenesis of airway inflammation, and GSK-3β can modulate the activity of NF-kB. To clarify the mechanism by which PM2.5 induces the expression of inflammatory cytokines, the effects of GSK-3β inhibitors on NF-κB activation and IkBα degradation were measured. As depicted in Figures 6A and B, stimulation with PM2.5 significantly enhanced the levels of phosphorylated NF-κB and IκBα and decreased the levels of IκBα in HBECs. However, treatment with TDZD-8 or SB216763 markedly diminished PM2.5-induced NF-κB activation and IkBα degradation, respectively. Moreover, the altered levels of phosphorylated NF-κB and IκBα in the TDZD-8 groups were greater than those in the SB216763 groups. Because the JNK signaling pathway is an upstream signaling pathway of NF-κB, we explored the level of phosphorylated NF-κB in HBECs treated with PM2.5 and the JNK inhibitor SP600125. As shown in Figure 6C, the JNK inhibitor SP600125 markedly abated the level of phosphorylated NF-κB induced by PM2.5.
 |
Figure 6 GSK-3β inhibitors and JNK inhibitors suppressed PM2.5-induced NF-κB activation and IkBα degradation in HBECs. Abbreviations: PM2.5, particulate matter<2.5 μm. Notes: (A) Western blot analysis revealed that the GSK-3β inhibitors TDZD-8 and SB216763 markedly diminished PM2.5-induced NF-κB phosphorylation. (B) Western blot analysis showed that TDZD-8 and SB216763 markedly suppressed PM2.5-induced IκBα phosphorylation and IkBα degradation, respectively. (C) The JNK inhibitor SP600125 markedly abated the level of phosphorylated NF-κB induced by PM2.5. #P<0.05, compared to control group. *P<0.05, compared to PM2.5 group, n=3. |
GSK-3β Inhibitors Blocked PM2.5-Induced Inflammatory Responses in HBECs
Consistent with the animal experiment results, the results of qRT–PCR and ELISA showed that PM2.5 increased the mRNA expression levels of the inflammatory cytokines HMGB1 and IL-6 and induced the secretion of HMGB1 and IL-6 in cell supernatants. To further assess the role of GSK-3β inhibitors in PM2.5-induced inflammatory responses in HBECs, HBECs were treated with the GSK-3β inhibitors TDZD-8 or SB216763. As shown in Figure 7A–C, TDZD-8 and SB216763 treatment significantly reversed PM2.5-induced outcomes at both the mRNA and protein levels. Furthermore, the altered levels of inflammation-related cytokines in the TDZD-8 groups were greater than those in the SB216763 groups.
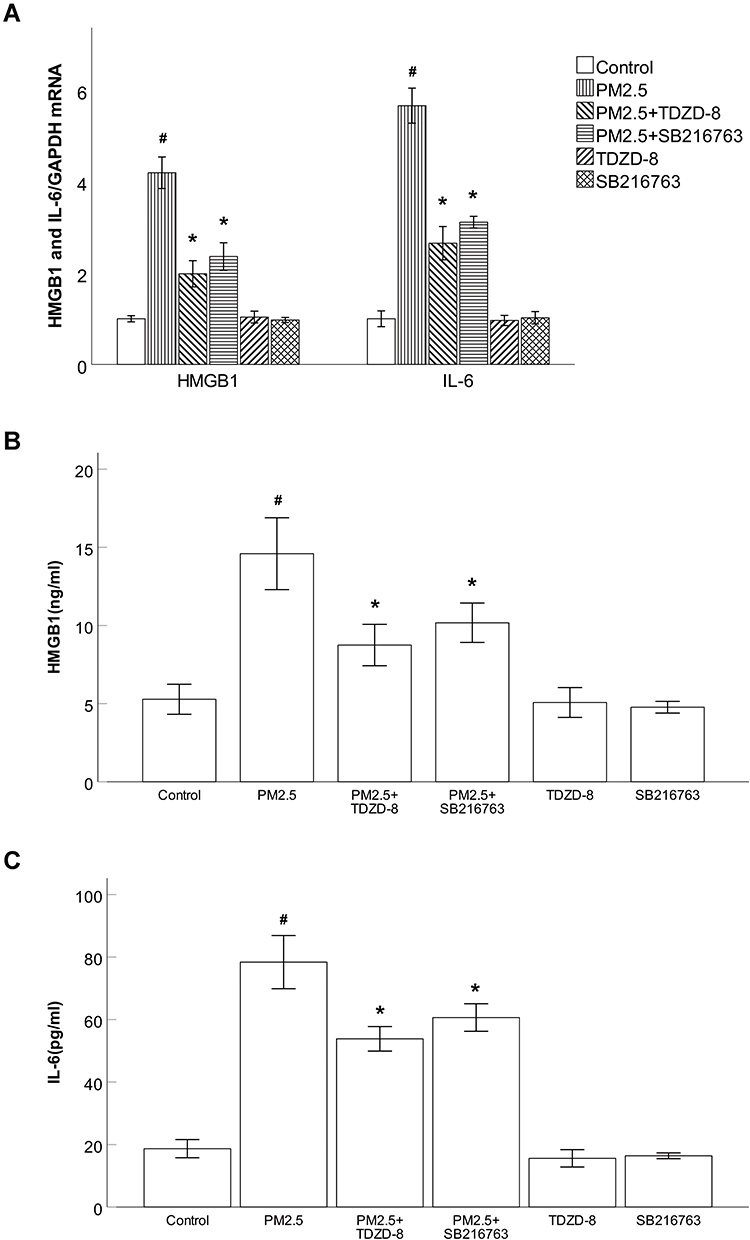 |
Figure 7 GSK-3β inhibitors blocked PM2.5-induced inflammatory responses in HBECs. Abbreviations: PM2.5, particulate matter<2.5 μm. Notes: (A) The results of qRT–PCR showed that the GSK-3β inhibitors TDZD-8 and SB216763 markedly attenuated the production of HMGB1 and IL-6 induced by PM2.5. (B) ELISA results showed that the GSK-3β inhibitors TDZD-8 and SB216763 markedly prevented the production of HMGB1 induced by PM2.5. (C) ELISA results showed that TDZD-8 and SB216763 markedly prevented the production of IL-6 induced by PM2.5. #P<0.05, compared to control group. *P<0.05, compared to PM2.5 group, n=3. |
Discussion
PM2.5 exposure has recently been reported to be associated with the development and progression of COPD. Here, we investigated the effects of GSK-3β inhibition on the PM2.5-associated airway inflammatory response in HBECs. We demonstrated that short-term exposure to PM2.5 induced an imbalance between inflammatory cytokines and anti-inflammatory cytokines and reduced p-GSK-3β expression in both mice and HBEC models. Reduced p-GSK-3β expression activated JNK pathways, caused NF-κB activation and IkB degradation, and then increased the levels of inflammatory cytokines in HBECs. Targeting GSK-3β inhibition may be a new strategy for the prevention or therapy of PM2.5-associated airway inflammation in COPD.
GSK-3β is a serine-threonine protein kinase that plays a key role in regulating the inflammatory response by converging several signaling pathways, but its role in the regulation of PM2.5-related airway inflammation is not clear. An epidemiological study showed that PM2.5 exposure led to airway inflammation in COPD.13,14 Long-term inflammatory mediators, including TNF-α, IL-1β, and IL-6, are strongly associated with the processes in chronic inflammatory diseases.15 Moreover, HMGB1 is implicated as a late inflammatory mediator that can irritate the release of many inflammatory mediators, such as IL-1β, IL-6, and TNF-α.16 IL-10 plays an anti-inflammatory role by reducing the production of inflammatory mediators. A study reported that GSK-3β inhibitors prevented mouse lung inflammation and injury caused by bleomycin.17 Consistently, our present research also observed that PM2.5 increased the levels of the inflammatory cytokines HMGB1 and IL-6 and decreased the expression of the anti-inflammatory cytokines IL-10 and p-GSK-3β in mice. Inflammatory cytokine and p-GSK-3β expression also displayed the same trend in HBECs exposed to PM2.5. Intervention with the GSK-3β inhibitors TDZD-8 and SB216763 significantly suppressed the PM2.5-induced inflammatory response in HBECs, and TDZD-8 showed a stronger anti-inflammatory effect than B216763, which suggested that inhibition of GSK-3β activity led to potent anti-inflammatory effects in cell models caused by PM2.5.
What are the molecular mechanisms by which GSK-3β inhibitors decrease the inflammatory response caused by PM2.5? GSK3β phosphorylation at Ser9 will cause its inactivation.18 We clearly showed that GSK-3β inhibitors increased the expression of p-GSK-3β in HBECs exposed to PM2.5. Growing evidence has demonstrated an association between GSK-3β and NF-κB activation. NF-κB is an important transcription factor because it promotes the secretion of inflammatory cytokines in HBECs after external stimulation. Active GSK-3β increases NF-kB phosphorylation and promotes the phosphorylation and degradation of the inhibitory protein κB (IκB) and then enhances the expression of inflammatory genes.19 In contrast, GSK-3β inhibitors decreased the activation of NF-κB, which in turn caused a decreased formation of inflammatory mediators.20,21 Consistent with previous reports, our results indicated that inhibitors of GSK-3β can suppress NF-κB activation and IkB degradation in HBECs after PM2.5 treatment.
Previous studies have demonstrated that inactivation of GSK-3β regulates the phosphorylation of JNK in normal bronchial epithelial cells activated by quercetin.6 JNK is a serine/threonine protein kinase that can regulate the development of inflammation. Very recently, a study indicated that the activation of JNK promoted the activation of NF-κB, further increasing the release of inflammatory factors.22 Moreover, the JNK inhibitor SP600125 inhibited PM2.5-induced expression of the inflammatory mediator IL-6 in human osteoarthritis synovial fibroblasts.23 As predicted, in our study, we also found that GSK-3β inhibitors inhibited the ratio of p-JNK/JNK in HBECs caused by PM2.5. Therefore, we showed that PM2.5 induced JNK phosphorylation and NF-kB activation via GSK-3β in HBECs, promoting the development of airway inflammation.
Additionally, to confirm whether the two signaling pathways of JNK and NF-kB were functionally connected or separately induced by PM2.5, we suppressed JNK expression through the JNK inhibitor SP600125. In accordance with previous studies, our results showed that pretreatment with SP600125 markedly abated the level of phosphorylated NF-κB induced by PM2.5, indicating that JNK plays an essential role in PM2.5-induced NF-kB activation and the inflammatory response. However, the level of phosphorylated NF-κB reduced by SP60025 was still higher than that in the GSK-3β inhibitor group. These data suggested that in addition to JNK, NF-κB might also be regulated by other signaling pathways.
Limitations
Our study has certain limitations. In our PM2.5-induced chronic model in vivo, a GSK-3β inhibitor (TDZD-8 or SB216763) was not administered to confirm the direct effect of GSK-3β in a PM2.5-related mouse model. However, this will be the focus of our next study.
Conclusion
In summary, the results of this study suggested beneficial effects of GSK-3β inhibitors in attenuating inflammatory responses in PM2.5-induced HBEC models. The mechanism is involved in the suppression of JNK phosphorylation, the inhibition of NF-κB activation, and the prevention of NF-κB-dependent proinflammatory gene expression. Thus, inhibition of GSK-3β may be a potential preventive and therapeutic strategy against PM2.5-associated airway inflammation in COPD. Further studies are required to verify these results.
Funding
This study was supported by the Natural Science Foundation of Guangdong, China (2019A1515011430 and 2020A1515010264), the Science and Technology Program of Guangzhou, China (202002030080), and the National Natural Science Foundation of China (81900044).
Disclosure
The authors report no conflicts of interest in this work.
References
1. The L. Air pollution: a major threat to lung health. Lancet. 2019;393(10183):1774. doi:10.1016/S0140-6736(19)30992-4
2. Gao N, Xu W, Ji J, et al. Lung function and systemic inflammation associated with short-term air pollution exposure in chronic obstructive pulmonary disease patients in Beijing, China. Environ Health. 2020;19(1):12. doi:10.1186/s12940-020-0568-1
3. Vogelmeier CF, Criner GJ, Martinez FJ, et al. Global Strategy for the Diagnosis, Management and Prevention of Chronic Obstructive Lung Disease 2017 Report: GOLD Executive Summary. Respirology. 2017;22(3):575–601.
4. Wang Y, Jia M, Yan X, et al. Increased neutrophil gelatinase-associated lipocalin (NGAL) promotes airway remodelling in chronic obstructive pulmonary disease. Clin Sci (Lond). 2017;131(11):1147–1159. doi:10.1042/CS20170096
5. Kaidanovich-Beilin O, Woodgett JR. GSK-3: functional insights from cell biology and animal models. Front Mol Neurosci. 2011;4:40. doi:10.3389/fnmol.2011.00040
6. Lee KH, Yoo CG. Simultaneous inactivation of GSK-3beta suppresses quercetin-induced apoptosis by inhibiting the JNK pathway. Am J Physiol Lung Cell Mol Physiol. 2013;304(11):L782–9. doi:10.1152/ajplung.00348.2012
7. Peng Z, Zha L, Yang M, Li Y, Guo X, Feng Z. Effects of ghrelin on pGSK-3beta and beta-catenin expression when protects against neuropathic pain behavior in rats challenged with chronic constriction injury. Sci Rep. 2019;9(1):14664. doi:10.1038/s41598-019-51140-w
8. Zhang H, Sha J, Feng X, et al. Dexmedetomidine ameliorates LPS induced acute lung injury via GSK-3beta/STAT3-NF-kappaB signaling pathway in rats. Int Immunopharmacol. 2019;74:105717. doi:10.1016/j.intimp.2019.105717
9. Wang H, Brown J, Martin M. Glycogen synthase kinase 3: a point of convergence for the host inflammatory response. Cytokine. 2011;53(2):130–140. doi:10.1016/j.cyto.2010.10.009
10. Ngkelo A, Hoffmann RF, Durham AL, et al. Glycogen synthase kinase-3beta modulation of glucocorticoid responsiveness in COPD. Am J Physiol Lung Cell Mol Physiol. 2015;309(10):L1112–23. doi:10.1152/ajplung.00077.2015
11. Hummler SC, Rong M, Chen S, Hehre D, Alapati D, Wu S. Targeting glycogen synthase kinase-3beta to prevent hyperoxia-induced lung injury in neonatal rats. Am J Respir Cell Mol Biol. 2013;48(5):578–588. doi:10.1165/rcmb.2012-0383OC
12. Ferraro M, Gjomarkaj M, Siena L, Di Vincenzo S, Pace E. Formoterol and fluticasone propionate combination improves histone deacetylation and anti-inflammatory activities in bronchial epithelial cells exposed to cigarette smoke. Biochim Biophys Acta Mol Basis Dis. 2017;1863(7):1718–1727. doi:10.1016/j.bbadis.2017.05.003
13. Zou W, Wang X, Hong W, et al. PM2.5 Induces the Expression of Inflammatory Cytokines via the Wnt5a/Ror2 Pathway in Human Bronchial Epithelial Cells. Int J Chron Obstruct Pulmon Dis. 2020;15:2653–2662. doi:10.2147/COPD.S270762
14. Zhao J, Li M, Wang Z, et al. Role of PM2.5 in the development and progression of COPD and its mechanisms. Respir Res. 2019;20(1):120. doi:10.1186/s12931-019-1081-3
15. Lv H, Liu Q, Wen Z, Feng H, Deng X, Ci X. Xanthohumol ameliorates lipopolysaccharide (LPS)-induced acute lung injury via induction of AMPK/GSK3beta-Nrf2 signal axis. Redox Biol. 2017;12:311–324. doi:10.1016/j.redox.2017.03.001
16. Chen B, Na F, Yang H, et al. Ethyl pyruvate alleviates radiation-induced lung injury in mice. Biomed Pharmacother. 2017;92:468–478. doi:10.1016/j.biopha.2017.05.111
17. Gurrieri C, Piazza F, Gnoato M, et al. 3-(2,4-dichlorophenyl)-4-(1-methyl-1H-indol-3- yl)-1H-pyrrole-2,5-dione (SB216763), a glycogen synthase kinase-3 inhibitor, displays therapeutic properties in a mouse model of pulmonary inflammation and fibrosis. J Pharmacol Exp Ther. 2010;332(3):785–794. doi:10.1124/jpet.109.153049
18. Paul J, Naskar K, Chowdhury S, Chakraborti T, De T. TLR mediated GSK3beta activation suppresses CREB mediated IL-10 production to induce a protective immune response against murine visceral leishmaniasis. Biochimie. 2014;107(Pt):
19. Gong R, Ge Y, Chen S, et al. Glycogen synthase kinase 3beta: a novel marker and modulator of inflammatory injury in chronic renal allograft disease. Am J Transplant. 2008;8(9):1852–1863. doi:10.1111/j.1600-6143.2008.02319.x
20. Jin H, Yang X, Zhao K, Zhao L, Chen C, Yu J. Glycogen synthase kinase-3 beta inhibitors protect against the acute lung injuries resulting from acute necrotizing pancreatitis. Acta Cir Bras. 2019;34(6):e201900609. doi:10.1590/s0102-865020190060000009
21. Ma B, Hottiger MO. Crosstalk between Wnt/beta-Catenin and NF-kappaB Signaling Pathway during Inflammation. Front Immunol. 2016;7:378. doi:10.3389/fimmu.2016.00378
22. Yu T, Dong D, Guan J, Sun J, Guo M, Wang Q. Alprostadil attenuates LPS-induced cardiomyocyte injury by inhibiting the Wnt5a/JNK/NF-kappaB pathway. Herz. 2020;45(Suppl 1):130–138. doi:10.1007/s00059-019-4837-0
23. Liu JF, Chi MC, Lin CY, et al. PM2.5 facilitates IL-6 production in human osteoarthritis synovial fibroblasts via ASK1 activation. J Cell Physiol. 2021;236(3):2205–2213. doi:10.1002/jcp.30009
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.