Back to Journals » OncoTargets and Therapy » Volume 9
GPR30 decreases with vascular aging and promotes vascular smooth muscle cells maintaining differentiated phenotype and suppressing migration via activation of ERK1/2
Authors Huang F, Yin J, Li K, Li Y, Qi H, Fang L, Yuan C, Liu W, Wang M, Li X
Received 25 January 2016
Accepted for publication 6 March 2016
Published 7 June 2016 Volume 2016:9 Pages 3415—3422
DOI https://doi.org/10.2147/OTT.S104972
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr William C. Cho
Fang Huang,1,2 Jianguo Yin,2 Keyu Li,1 Ying Li,1 Heng Qi,1 Li Fang,1 Cong Yuan,1 Weiwei Liu,1 Min Wang,2 Xiangping Li2
1Department of Cardiology, The First Hospital of Changsha, 2Department of Cardiology, The Second XiangYa Hospital of Central South University, Changsha, Hunan Province, People’s Republic of China
Abstract: Estrogen receptors, including classic nuclear receptors ERα, ERβ, and membrane receptor GPR30, are expressed in vascular tissues and exert protective actions in vascular diseases. But the expression pattern and functional roles of GPR30 in vascular smooth muscle cells (VSMCs) remain unclear. In this study, we found that ERα, ERβ, and GPR30 were decreased with VSMCs passaging in vitro or growing in vivo and activation of GPR30 promoted ERα expression. Then, we validated that activation of GPR30 significantly decreased migratory capability of VSMCs and suppressed ERα, whereas PDGF-BB (20 ng/mL) treatment caused increase of migration. And activation of GPR30 led to reduction of osteopontin and cellular retinol binding protein 1, enhancement of calponin and 3F8, and upregulation of total and phosphorylated ERK1/2 expression in VSMCs knocked down by GPR30, ERα, and ERβ or treated with PDGF-BB. These data suggest that GPR30 promotes VSMCs reducing migration and maintaining differentiated phenotype via activation of ERK1/2 pathway. Our findings provide novel mechanisms of GPR30 protection of VSMCs as well as a new target for prevention of vascular aging.
Keywords: vascular smooth muscle cell, estrogen receptor, G protein-coupled receptor 30, phenotype, migration
Introduction
The vascular smooth muscle cells (VSMCs) are highly specified cells in adults, possessing the contraction and adjustment of the blood vessel tension, maintain the blood pressure, and show a contractile phenotype with relatively low proliferative and migratory activity.1 However, in the development of age-related diseases, such as hypertension and atherosclerosis, the functions and properties of VSMCs will be affected by the pathological factors and manifest a synthetic phenotype, highly proliferative and migratory activity, contributing to arterial stiffening and thickening, calcification and plaque formation, and increasing the risk of cardiovascular events.2–4 Therefore, exploring the factors that play a critical role in controlling phenotype transformation and migration of VSMCs is helpful and necessary for development of novel therapeutic strategies for vascular diseases.
Estrogen can inhibit the endogenous aging of skin and reverse vascular calcification induced in vitro.5,6 The protective effects of estrogen on endothelium and vascular smooth muscle are dependent on its binding to the estrogen receptor.7 The loss of estrogen receptor will contribute to vascular aging and development of cardiovascular disease.8–10 Estrogen receptor ERα, ERβ, and GPR30 (a transmembrane estrogen-binding protein) have been identified in blood vessels of human and animal models.11,12 Experimental evidence suggests beneficial effects of estrogen on the vasculature, involving activation of ERα- and ERβ-mediated genomic effects in endothelial cells and VSMCs via the related signaling pathways.13,14 Estrogen is able to affect the functional genes regulating vascular tone as well as responding to vascular injury and atherosclerosis. Estrogen increases the expression of genes for vasodilatory enzymes, such as prostacyclin synthase and nitric oxide synthase. GPR30-mediated nongenomic effects are rapid responses and typically involve regulation of membrane bound and cytoplasmic regulatory proteins.15 In some cell types, ERα and ERβ correlate with caveolae and other signaling molecules to trigger GPR30-mediated second messengers and intracellular pathways, including MAPK and PI3K/Akt, and activation of ion channel fluxes.16 Although studies have shown that estrogen receptors (ERs) play a protective role in vascular aging, the expression pattern, interrelationship, and action mechanisms of GPR30, ERα, and ERβ in the protection of VSCMs from aging remain unclear.
In this study, we firstly determined the expression profile of GPR30, ERα, and ERβ in VSMCs and arterial tissues, and then investigated the effects of GPR30 on the expression of ERα and ERβ in VSMCs, the migration and phenotypic transformation of VSMCs, and the underlying mechanisms. Our findings highlight that GPR30 plays a crucial role in ERs-mediated protective actions in VSMCs.
Materials and methods
Cell culture
Ethical approval was sought by the ethical committee of the First Hospital of Changsha. Primary VSMCs of rat carotid artery were purchased from PriCells (RAT-CELL-0009, Wuhan, People’s Republic of China), cultured in VSMCs basal medium (MED-0003, PriCells), and supplemented with special additives (SUP-003, PriCells), according to the manufacturer’s instructions, and incubated at 37°C in a humidified atmosphere with 5% CO2. Cells between passages 2 and 8 were used in the study.
Quantitative real-time reverse transcription polymerase chain reaction
Total RNA was extracted from VSMCs using TRIzol Reagent (Thermo Fisher Scientific, Waltham, MA, USA), according to the manufacturer’s instructions, and then the RNA was reverse transcribed using the PrimeScript RT Master Mix Perfect Real Time kit (TaKaRa, Dalian, People’s Republic of China) to obtain the cDNA. Using the cDNA as the template, a real-time PCR assay was performed using the following pairs of primers: ERα forward, 5′-CGCTTTTGAACCAGCAGG-3′ and ERα reverse, 5′-TTCCCGAGGCTTTGGTGTG-3′; ERβ forward, 5′-ATCTGTCCAGCCACGAATCA-3′ and ERβ reverse, 5′-ATTAGCACCTCCATCCAGCA-3′; GPR30 forward, 5′-AGCTCAGGCTGTATGTGGCG-3′ and GPR30 reverse, 5′-TGCTCCGTGCTGTCTGGTAT-3′; and β-actin forward, 5′-AGGCCCCTCTGAACCCTAAG-3′ and β-actin reverse, 5′-CCAGAGGCATACAGGGACAAC-3′. The 20 μL real-time PCR reaction included 0.5 μL of cDNA template, 0.25 μL of Primer F, 0.25 μL of Primer R, 10 μL of RNase-free dH2O, and 8 μL of 2.5× RealMasterMix (SYBR Green I). The reaction conditions included a predenaturation step at 95°C for 10 seconds, and 40 cycles of 95°C for 15 seconds and 60°C for 60 seconds. After the reaction, the data were subjected to statistical analysis.
Short hairpin RNA expression plasmids and stable transfection
The shRNA expression vectors against ERα, ERβ, and GPR30 were prepared by cloning of double-stranded oligonucleotides into the BamHI and HindIII sites in the pRNAT-U6.1/Neo vector (Thermo Fisher Scientific, Waltham, MA, USA). The sequences of shRNA against ERα, ERβ, and GPR30 were 5′-AAGTCGATTCCGCATGATGAA-3′, 5′-AAGTTCTAGCGAGCAGGTACA-3′, and 5′-CGAGCAGTATTACGATATC-3′, respectively. All of the constructs were confirmed by DNA sequencing. The VSMCs were transfected with lentivirus packaged pRNATU6.1-ERα-shRNA, -ERβ-shRNA, or GPR30-shRNA vector. The stable suppression of ERα, ERβ, or/and GPR30 in VSMCs was identified by Western blotting and quantitative real-time RT-PCR (qRT-PCR) assays. And the stable transfectants were selected and used for the following studies.
Western blotting
An amount of 1×107 VSMCs treated as indicated was lysed in cell lysate, and then centrifuged at 12,000× g for 20 minutes at 4°C. The supernatant was collected and denatured. Proteins were separated in 10% SDS-PAGE (dodecyl sulfate, sodium salt polyacrylamide gel electrophoresis) and blotted onto polyvinylidene difluoride membrane. The polyvinylidene difluoride membrane was treated with Tris Buffered Saline Tween containing 50 g/L skimmed milk at room temperature for 4 hours, followed by incubation with the primary antibodies anti-GPR30 (1:250, Abcam, Cambridge, MA, USA), anti-ERα (1:200, Abcam), ERβ (1:1,000, Abcam), anti-OPN (1:1,000, Abcam), anti-CRBP1 (1:200, Santa Cruz Biotechnology Inc., Dallas, TX, USA), anti-calponin (1:20,000, Abcam), anti-3F8 (1:500, Santa), Collage I/II (1:1,000, Abcam), anti-fibronectin (1:1,000, Abcam), anti-PI3K (1:500, Abcam), p-PI3K (1:500 dilution, Abcam), ERK1/2 (1:1,000, Abcam), p-ERK1/2 (1:500, Abcam), PKA (1:1,000, Abcam), p-PKA (1:2,500 dilution, Abcam), and anti-β-actin (1:2,500 dilution, ABZOOM BIOLABS Inc, Dallas, TX, USA), respectively, at 37°C for 1 hour. Membranes were rinsed and incubated for 1 hour with the correspondent peroxidase-conjugated secondary antibodies. Chemiluminescent detection was performed with the ECL kit (Pierce Chemical, Rockford, IL, USA). The amount of the protein of interest, expressed as arbitrary densitometric units, was normalized to the densitometric units of β-actin.
Immunocytochemistry analysis
Animal experiments were performed in strict accordance with the guide for the care and use of laboratory animals of Central South University. The rats were maintained under specific pathogen-free conditions in the Institute of Zoology, Xianya Hospital of Central South University. Six female rats were randomly divided into two groups and raised to 2 months and 18 months, respectively. Then, the rats were euthanized and the carotid artery tissues were excised and embedded in paraffin. The expressions of ERα, ERβ, and GPR30 in rat carotid artery tissues of 2- and 18-month-old rats were determined by immunocytochemistry. The root of the aorta was obtained from rat and fixed in 4% paraformaldehyde overnight. Tissue specimens were then cut at 5 μm thickness, and a standard immunostaining procedure was performed using antibodies against ERα, ERβ, and GPR30. The mean optical density value (D) and area (A) of brown particles in three visual fields of each section were calculated by the Leica Q550 image analysis system (Leica Microsystems, Wetzlar, Germany). The expression levels of target molecules in tissues were evaluated using the formula: integral density = D × A.
Cell migratory capacity analysis
VSMCs of passage 2 treated with G-1 (1 nM, Tocris Bioscience, Bristol, UK) for 48 hours or PDGF-BB (10 ng/mL) for 24 hours as indicated were plated at a final concentration of 2×103 cells per well in 96-well plates. The migratory capacity of cells was evaluated by transwell assay. In brief, migration experiments were performed in polycarbonate transwell inserts (8 μm pores, Corning Incorporated, Corning, NY, USA). Cells (1×106) in 200 μL culture medium were seeded in the upper chamber and cultured at 37°C for 6 hours. And the migrating cells in the bottom of the chamber were fixed and stained with crystal violet and photographed. The crystal violet was then eluted and the eluent of each group was measured by an ELISA reader (ELX-800 type, BioTek, Highland Park, FL, USA) to determine the optical density at 570 nm (OD570).
Statistical analysis
Data were expressed as mean ± standard deviation from at least three separate experiments. Statistical analysis was carried out using SPSS 15.0 software (SPSS Inc., Chicago, IL, USA). The difference between two groups was analyzed by the Student’s t-test. A value of P<0.05 was considered as statistically significant.
Results
ERα, ERβ, and GPR30 decrease in VSMCs with cells passaging or rat aging
To investigate whether the expression levels of ERs alter with VSMCs passaging in vitro or growing in vivo, the relative expression of ERα, ERβ, and GPR30 was compared among VSMCs of passages 2, 4, 6, 8, or between rat carotid artery tissues of 2- and 18-month-old rats. The quantitative RT-PCR and Western blot results showed that the ERα, ERβ, and GPR30 mRNAs and proteins were decreased with VSMCs passaging from P2 to P8 (Figure 1A–D). Then, we further found that the ERα, ERβ, and GPR30 proteins were more expressed in carotid artery tissues of 2-month-old rats compared with the 18-month-old rats (Figure 1E and F). These results demonstrate that estrogen receptors ERα, ERβ, and GPR30 were downregulated in VSMCs along with its passaging in vitro or growing in vivo.
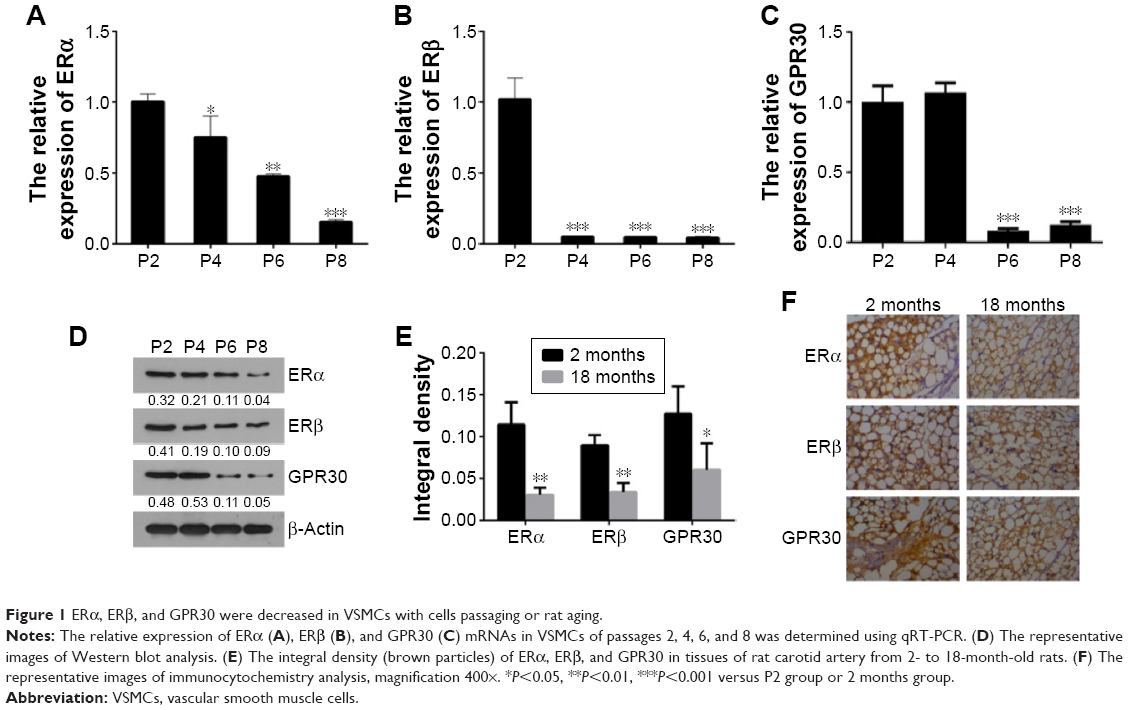 | Figure 1 ERα, ERβ, and GPR30 were decreased in VSMCs with cells passaging or rat aging. |
GPR30 has promotion actions on ERα expression but no clear effects on ERβ
To explore the influences of GPR30 on ERα and ERβ expression, the VSMCs were cotransfected with ERα + ERβ shRNAs or ERα + ERβ + GPR30 shRNAs. The quantitative RT-PCR and Western blot assays confirmed that the expression levels of ERα, ERβ mRNAs, or proteins were significantly suppressed in VSMCs cotransfected with ERα + ERβ shRNAs (Figure 2A–C). We then found that the ERα was less expressed in VSMCs cotransfected with ERα + ERβ + GPR30 shRNAs compared with ERα + ERβ shRNA group. Moreover, activation of GPR30 using its selective agonists G-1 could significantly enhance ERα mRNA levels in VSMCs cotransfected with ERα + ERβ + GPR30 shRNAs, even notably more than its expression in VSMCs cotransfected with ERα + ERβ shRNAs. However, neither inhibition nor activation of GPR30 had clear influence on ERβ (Figure 2D–F). These results indicate that activation of GPR30 may promote ERα expression, but it has no clear influence on the expression of ERβ.
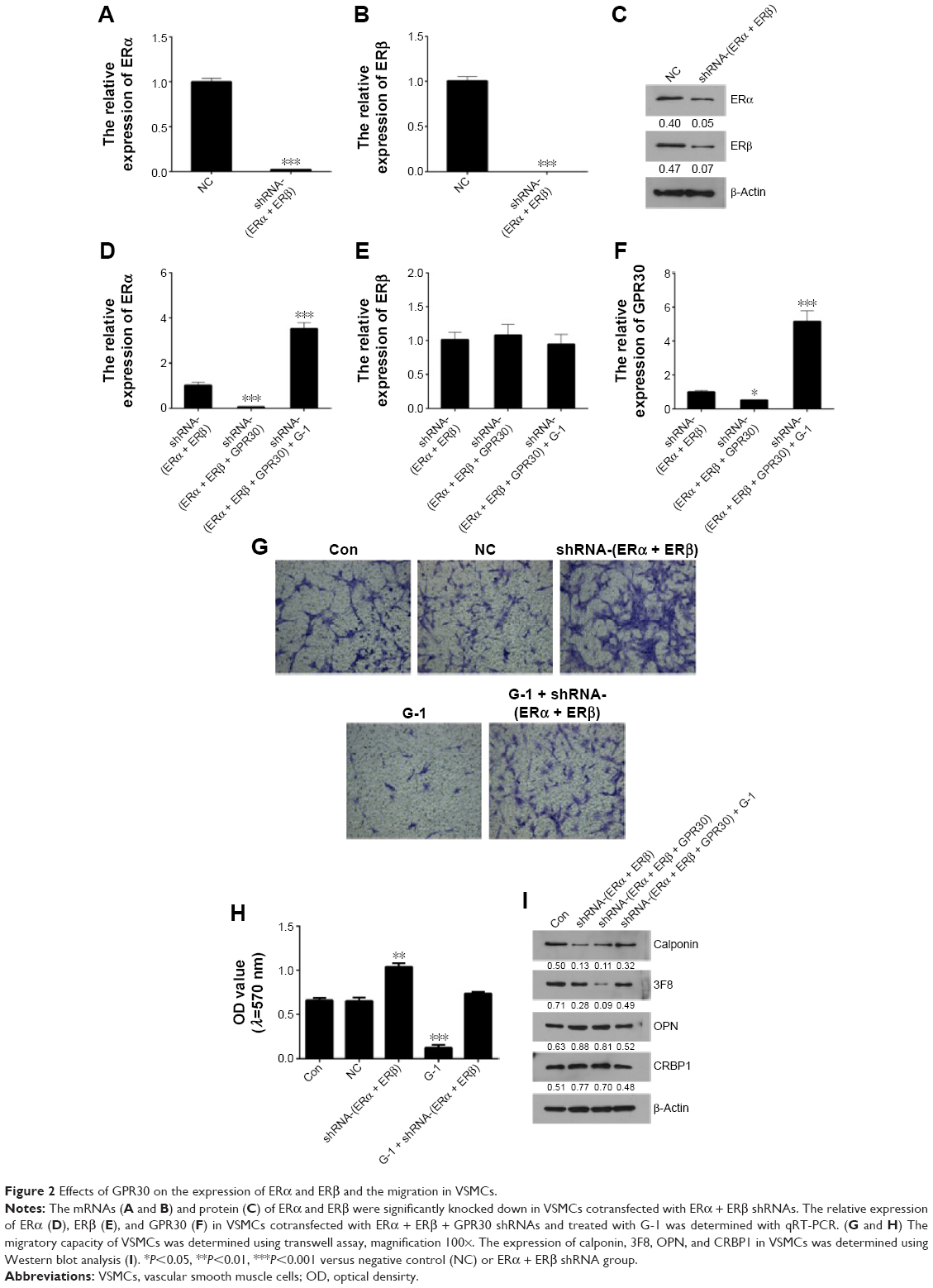 | Figure 2 Effects of GPR30 on the expression of ERα and ERβ and the migration in VSMCs. |
Activation of GPR30 promotes VSMCs maintaining differentiated phenotype and reducing migration
To elucidate the functional roles of GPR30 in VSMCs, the migration and phenotypic markers in VSMCs of passage 2 were determined using transwell assays and Western blot analysis. It was shown that inhibition of ERα and ERβ increased migration of VSMCs. However, activation of GPR30 using its selective agonists G-1 could decrease migration of VSMCs and force the enhanced migratory capability of VSMCs induced by inhibition of ERα and ERβ return to normal levels (Figure 2G and H). It suggests that ERα, ERβ, and GPR30 may play an inhibitory role in regulation of migratory capability in VSMCs. Then, the Western blot results illustrated that inhibition of ERα, ERβ, and/or GPR30 reduced the expression of differentiated phenotype markers (calponin and 3F8) and enhanced the expression of synthetic phenotype markers (OPN and CRBP1) in VSMCs, but activation of GPR30 in these VSMCs could partly resume the reduced expression of differentiated phenotypic markers (Figure 2I). It indicates that ERα, ERβ, and GPR30 may contribute to VSMCs maintaining a differentiated phenotype.
GPR30 protection of VSMCs involving activation of ERK1/2
To reveal whether ERK1/2 pathway participates in GPR30 regulating the phenotype of VSMCs, the VSMCs of passage 2 were treated with PDGF-BB or G-1 and the expression levels of phenotype markers were determined. As shown in Figure 3, PDGF-BB (20 ng/mL) induced reduced calponin and 3F8 and enhanced OPN, CRBP1, collagen I/II, and fibronectin in VSMCs in a time-dependent manner. It confirms that PDGF-BB can promote the conversion of VSMCs from a contractile phenotype into a synthetic phenotype and stimulate production of vascular fibrosis- and calcification-related factors in VSMCs. It is shown that PDGF-BB-induced reduced calponin and 3F8 expression, enhanced OPN and CRBP1 expression, and elevated migratory capability in VSMCs could be partly reversed by activation of GPR30 using G-1. We then found that activation of GPR30 could also partly recover PDGF-BB-induced decreased total and phosphorylated ERK1/2 expression in VSMCs. These data indicate that GPR30 protection of VSMCs from suffering pathological changes induced by PDGF-BB is possibly involved in upregulation of expression and activity of ERK1/2.
 | Figure 3 The protective actions of GPR30 on VSMCs. |
Discussion
Estrogen receptors play an important regulatory role in the reproductive, cardiovascular, nervous, immune system, and so on. ERs can be divided into the classic nuclear receptors, including ERα and ERβ, which induce a genomic effect by transcription of specific target genes, and the membrane receptor GPR30, which mediates a rapid nongenomic effect by a second messenger system.17 GPR30 is located in the surface of cell membrane, apparato reticulare, and endoplasmic reticulum.18 It is widely distributed in human heart, lung, liver, ovary, breast, brain tissues, and so on and expressed in carotid, thoracic, mesenteric, and pulmonary arterial vessels.19,20 Epidemiological investigations and experimental studies have revealed that estrogen has protective effects on vascular system against aging via binding of ERs. The alteration of ERs with age in VSMCs may affect its response to estrogen.21 It is reported that GPR30 expression was significantly decreased in the aorta of aged female Lewis rats and adult male rats, causing reduced vasorelaxation to estradiol and G-1.8 Furthermore, this decline has nothing to do with estrogen reduction. Therefore, it is necessary to reveal the expression alternation of GPR30 in vascular tissues, including VSMCs with aging. In the present study, we found that the expression levels of ERα, ERβ, and GPR30 were decreased with VSMCs passaging from passages 2 to 8 and less expressed in carotid artery tissues of 18-month-old rats than the 12-month-old rats. These results demonstrate that estrogen receptor ERα, ERβ, and GPR30 may diminish in VSMCs along with its passaging in vitro or growing in vivo, implying that estrogen receptors may decline with vascular aging.
To reveal the effects of GPR30 on ERα or ERβ expression in VSMCs, we firstly established a cell model of VSMCs in which ERα and ERβ were effectively knocked down. Then we discovered that further knockdown of GPR30 in this cell model would lead to less ERα expression. Accordingly, activation of GPR30 using its selective agonists G-1 caused enhancement of ERα expression. But neither inhibition nor activation of GPR30 had a clear influence on ERβ. Studies indicate that a potential crosstalk or cooperation may exist among GPR30, ERα, aryl hydrocarbon receptor, and ErbB family signaling pathways.22,23 GPR30 may function in conjunction with ERα to assemble a signal complex essential for rapid estrogen signaling.16 Our results suggest that activation of GPR30 signaling pathway may promote ERα expression, possibly resulting in accentuation of the estrogen-induced ERα signaling and effects in VSMCs.
The alteration of VSMC features represents a critical event in the pathobiology of arterial wall since it contributes to the vascular remodeling and decline of function with aging and favors the progression of atherosclerosis.24 ERs are possibly involved in regulation of phenotypic transformation and migration in VSMCs.25–27 Our results confirmed that activation of GPR30 could partly resume the loss of ERα- and ERβ-mediated increase of migration in VSMCs. Inhibition of ERα, ERβ, and/or GPR30 reduced calponin and 3F8 and enhanced OPN and CRBP1 expression in VSMCs; however, activation of GPR30 partly recovered the differentiated phenotypic markers expressed in VSMCs. Then, we further validated that activation of GPR30 could inhibit PDGF-BB promotion of VSMCs migration and dedifferentiation. The potential metabolism is involved in upregulation of expression and activity of ERK1/2. Our results suggest that GPR30 plays a suppressor in regulation of migratory capability of VSMCs and contributes to VSMCs maintaining a differentiated phenotype via activation of ERK1/2 pathway. GPR30 activation may cause the dissociation of Gα-GTPase from the heterotrimeric Gαβγ complex. Dissociated Gβγ subunit will activate membrane associated matrix metalloproteinases with subsequent transient activation of mitogen-activated protein kinase ERK-1/2.28,29 And Gα-GTPase may act on membrane-associated adenylyl cyclase, generating the second messenger cAMP, which in turn inhibits ERK-1/2 activity via a cAMP-dependent protein kinase mechanism.30 In addition, in some breast cancer cells, silencing of GPR30 function had no effects on estradiol-induced cAMP elevation and ERK activation. Although GPR30 has been identified in the vasculature, its action mechanisms need further investigation.
In conclusion, we found that ERα, ERβ, and GPR30 were decreased with VSMCs passaging in vitro or growing in vivo, and activation of GPR30 promoted ERα expression. The results confirm that GPR30 promotes VSMCs maintaining differentiated phenotype and reducing migration through activation of ERK1/2 pathway. Our findings provide novel mechanisms of GPR30 protection of VSMCs as well as a target for prevention of vascular aging.
Disclosure
The authors report no conflicts of interest in this work.
References
Thyberg J, Nilsson J, Palmberg L, Sjolund M. Adult human arterial smooth muscle cells in primary culture. Modulation from contractile to synthetic phenotype. Cell Tissue Res. 1985;239(1):69–74. | ||
Fukuda N, Hu WY, Satoh C, et al. Contribution of synthetic phenotype on the enhanced angiotensin II-generating system in vascular smooth muscle cells from spontaneously hypertensive rats. J Hypertens. 1999;17(8):1099–1107. | ||
Sreejayan N, Yang X. Isolation and functional studies of rat aortic smooth muscle cells. Methods Mol Med. 2007;139:283–292. | ||
Devlin AM, Clark JS, Reid JL, Dominiczak AF. DNA synthesis and apoptosis in smooth muscle cells from a model of genetic hypertension. Hypertension. 2000;36(1):110–115. | ||
Quatresooz P, Pierard-Franchimont C, Gaspard U, Piérard GE. Skin climacteric aging and hormone replacement therapy. J Cosmet Dermatol. 2006;5(1):3–8. | ||
Osako MK, Nakagami H, Koibuchi N, et al. Estrogen inhibits vascular calcification via vascular RANKL system: common mechanism of osteoporosis and vascular calcification. Circ Res. 2010;107(4):466–475. | ||
Zhao J, Imbrie GA, Baur WE, et al. Estrogen receptor-mediated regulation of microRNA inhibits proliferation of vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2013;33(2):257–265. | ||
Lindsey SH, da Silva AS, Silva MS, Chappell MC. Reduced vasorelaxation to estradiol and G-1 in aged female and adult male rats is associated with GPR30 downregulation. Am J Physiol Endocrinol Metab. 2013;305(1):E113–E118. | ||
Post WS, Goldschmidt-Clermont PJ, Wilhide CC, et al. Methylation of the estrogen receptor gene is associated with aging and atherosclerosis in the cardiovascular system. Cardiovasc Res. 1999;43(4):985–991. | ||
Wynne FL, Payne JA, Cain AE, Reckelhoff JF, Khalil RA. Age-related reduction in estrogen receptor-mediated mechanisms of vascular relaxation in female spontaneously hypertensive rats. Hypertension. 2004;43(2):405–412. | ||
Lindner V, Kim SK, Karas RH, Kuiper GG, Gustafsson JA, Mendelsohn ME. Increased expression of estrogen receptor-beta mRNA in male blood vessels after vascular injury. Circ Res. 1998;83(2):224–229. | ||
Karas RH, Patterson BL, Mendelsohn ME. Human vascular smooth muscle cells contain functional estrogen receptor. Circulation. 1994;89(5):1943–1950. | ||
Stirone C, Boroujerdi A, Duckles SP, Krause DN. Estrogen receptor activation of phosphoinositide-3 kinase, akt, and nitric oxide signaling in cerebral blood vessels: rapid and long-term effects. Mol Pharmacol. 2005;67(1):105–113. | ||
Filardo EJ, Thomas P. Minireview: G protein-coupled estrogen receptor-1, GPER-1: its mechanism of action and role in female reproductive cancer, renal and vascular physiology. Endocrinology. 2012;153(7):2953–2962. | ||
Simoncini T, Mannella P, Fornari L, Caruso A, Varone G, Genazzani AR. Genomic and non-genomic effects of estrogens on endothelial cells. Steroids. 2004;69(8–9):537–542. | ||
Hammes SR, Levin ER. Extranuclear steroid receptors: nature and actions. Endocr Rev. 2007;28(7):726–741. | ||
Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science. 2005;307(5715):1625–1630. | ||
Koganti S, Snyder R, Gumaste U, Karamyan VT, Thekkumkara T. 2-Methoxyestradiol binding of GPR30 down-regulates angiotensin AT(1) receptor. Eur J Pharmacol. 2014;723:131–140. | ||
Notas G, Kampa M, Pelekanou V, Castanas E. Interplay of estrogen receptors and GPR30 for the regulation of early membrane initiated transcriptional effects: a pharmacological approach. Steroids. 2012;77(10):943–950. | ||
Reslan OM, Yin Z, do Nascimento GR, Khalil RA. Subtype-specific estrogen receptor-mediated vasodilator activity in the cephalic, thoracic, and abdominal vasculature of female rat. J Cardiovasc Pharmacol. 2013;62(1):26–40. | ||
Bowling MR, Xing D, Kapadia A, et al. Estrogen effects on vascular inflammation are age dependent: role of estrogen receptors. Arterioscler Thromb Vasc Biol. 2014;34(7):1477–1485. | ||
Tarnow P, Tralau T, Luch A. G protein-coupled receptor 30 ligand G-1 increases aryl hydrocarbon receptor signalling by inhibition of tubulin assembly and cell cycle arrest in human MCF-7 cells. Arch Toxicol. Epub 2015 Oct 16. | ||
Ruan SQ, Wang SW, Wang ZH, Zhang SZ. Regulation of HRG-beta1-induced proliferation, migration and invasion of MCF-7 cells by upregulation of GPR30 expression. Mol Med Rep. 2012;6(1):131–138. | ||
Orlandi A, Bochaton-Piallat ML, Gabbiani G, Spagnoli LG. Aging, smooth muscle cells and vascular pathobiology: implications for atherosclerosis. Atherosclerosis. 2006;188(2):221–230. | ||
Huang X, Jin Y, Zhou D, Xu G, Huang J, Shen L. IQGAP1 modulates the proliferation and migration of vascular smooth muscle cells in response to estrogen. Int J Mol Med. 2015;35(5):1460–1466. | ||
Zheng S, Chen X, Hong S, et al. 17β-Estradiol inhibits vascular smooth muscle cell migration via up-regulation of striatin protein. Gynecol Endocrinol. 2015;31(8):618–624. | ||
Gros R, Ding Q, Davis M, et al. Delineating the receptor mechanisms underlying the rapid vascular contractile effects of aldosterone and estradiol. Can J Physiol Pharmacol. 2011;89(9):655–663. | ||
Prenzel N, Zwick E, Daub H, et al. EGF receptor transactivation by G-protein-coupled receptors requires metalloproteinase cleavage of proHB-EGF. Nature. 1999;402(6764):884–888. | ||
Filardo EJ, Thomas P. GPR30: a seven-transmembrane-spanning estrogen receptor that triggers EGF release. Trends Endocrinol Metab. 2005;16(8):362–367. | ||
Filardo EJ, Quinn JA, Frackelton AR Jr, Bland KI. Estrogen action via the G protein-coupled receptor, GPR30: stimulation of adenylyl cyclase and cAMP-mediated attenuation of the epidermal growth factor receptor-to-MAPK signaling axis. Mol Endocrinol. 2002;16(1):70–84. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.