Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Glycosaminoglycan-functionalized poly-lactide-co-glycolide nanoparticles: synthesis, characterization, cytocompatibility, and cellular uptake
Authors Lamichhane S, Arya N, Ojha N, Kohler E, Shastri VP
Received 30 August 2014
Accepted for publication 11 October 2014
Published 19 January 2015 Volume 2015:10(1) Pages 775—789
DOI https://doi.org/10.2147/IJN.S73508
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Surya P Lamichhane,1 Neha Arya,1,2 Nirdesh Ojha,3 Esther Kohler,1 V Prasad Shastri1,2,4
1Institute for Macromolecular Chemistry, University of Freiburg, Freiburg, 2Helmholtz Virtual Institute on “Multifunctional Biomaterials for Medicine”, 3Laboratory for Process Technology, Department of Microsystems Engineering, University of Freiburg, Freiburg, 4Centre for Biological Signaling Studies (BIOSS), University of Freiburg, Freiburg, Germany
Abstract: The efficient delivery of chemotherapeutics to the tumor via nanoparticle (NP)-based delivery systems remains a significant challenge. This is compounded by the fact that the tumor is highly dynamic and complex environment composed of a plurality of cell types and extracellular matrix. Since glycosaminoglycan (GAG) production is altered in many diseases (or pathologies), NPs bearing GAG moieties on the surface may confer some unique advantages in interrogating the tumor microenvironment. In order to explore this premise, in the study reported here poly-lactide-co-glycolide (PLGA) NPs in the range of 100–150 nm bearing various proteoglycans were synthesized by a single-step nanoprecipitation and characterized. The surface functionalization of the NPs with GAG moieties was verified using zeta potential measurements and X-ray photoelectron spectroscopy. To establish these GAG-bearing NPs as carriers of therapeutics, cellular toxicity assays were undertaken in lung epithelial adenocarcinoma (A549) cells, human pulmonary microvascular endothelial cells (HPMEC), and renal proximal tubular epithelial cells. In general NPs were well tolerated over a wide concentration range (100–600 µg/mL) by all cell types and were taken up to appreciable extents without any adverse cell response in A549 cells and HPMEC. Further, GAG-functionalized PLGA NPs were taken up to different extents in A459 cells and HPMEC. In both cell systems, the uptake of heparin-modified NPs was diminished by 50%–65% in comparison to that of unmodified PLGA. Interestingly, the uptake of chondroitin sulfate NPs was the highest in both cell systems with 40%–60% higher uptake when compared with that of PLGA, and this represented an almost twofold difference over heparin-modified NPs. These findings suggest that GAG modification can be explored as means of changing the uptake behavior of PLGA NPs and these NP systems have potential in cancer therapy.
Keywords: tumor microenvironment, nanocarriers, proteoglycans, polysaccharides, passive targeting
Introduction
Second only to heart disease, cancer accounts for nearly 13% of all deaths in the USA.1 In spite of significant advances in the detection of solid tumors and treatment options, the efficient delivery of anticancer agents to the target site still remains a primary challenge.2 Over the past decade, nanoparticle (NP)-based delivery systems have gained importance in drug-delivery applications and demonstrated potential in the tissue-targeted delivery of cancer therapeutics.3 Cancer therapeutics can be targeted either by passive or active targeting. “Passive targeting” utilizes the changes to permeability of vasculature associated with tumor environment, which promotes the diffusion and accumulation of macromolecules and drugs by “extravasation”, the enhanced permeation and retention effect to localize the therapy to the tumor environment.4 In this regard, NPs offer several advantages due to their size and ability to present cell- or tissue-specific moieties on their surface. However, following intravenous administration, the majority of the NPs end up in the liver and this has provided the impetus for developing strategies to enhance localization of therapy to tumors through active targeting.5,6 “Active targeting” is primarily based on ligand–receptor interactions between the receptors overexpressed by cancer cells and ligand-modified NPs that then promote affinity for cancer cells within solid tumor.7,8 This approach has many merits but presents a few challenges with regard to ensuring that the targeting ligand is expressed constitutively in all grades and stages of the tumor. Since NP biodistribution is influenced by protein adsorption, and extracellular matrix (ECM) polysaccharides have specific affinity for proteins, the functionalization of the NP surface with ECM polysaccharides or proteoglycans may present new avenues to tissue targeting, based on protein–ECM interactions.
The objective of the work reported here was therefore to synthesize and characterize poly-lactide-co-glycolide (PLGA) NPs bearing glycosaminoglycans (GAGs), namely heparin (H), hyaluronic acid (HA), chondroitin sulfate (CS), and dermatan sulfate (DS). As preliminary screening for establishing the safety of these GAG-modified NPs for injectable therapy, the toxicity of these in lung epithelial adenocarcinoma (A549), human pulmonary microvascular endothelial cells (HPMEC), and human renal proximal tubular epithelial cells (RPTEC) was studied by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay, lactate dehydrogenase (LDH) release, and induction of reactive oxygen species (ROS). In addition, to validate this system for the treatment of lung cancer, the uptake behavior of these NPs in A549 cells and HPMEC was evaluated.
Materials and methods
Chemicals
Chondroitin-4-sulfate sodium salt from bovine trachea, DS from procaine intestinal mucosa, HA sodium salt from Streptococcus equi, H sodium salt derived from bovine intestinal mucosa, and PLGA, 50:50 (number average molecular weight [Mw] 24,000–38,000 g/mol) were purchased from Sigma-Aldrich Co (St Louis, MO, USA). Acetone was procured from VWR International (Radnor, PA, USA) and tetrahydrofuran (THF) was procured from Carl Roth GmbH (Karlsruhe, Germany).
Nanoparticle preparation and characterization
GAG-functionalized NPs were synthesized by nanoprecipitation using a previously described single-step synthesis.9 Briefly, a 10 mg/mL solution of PLGA was prepared in a binary solvent system containing THF and acetone (9:1% v/v), and the aqueous phase containing the water-soluble polyelectrolyte GAG at the desired concentration of 0.45 mg/mL was prepared in in deionized water. NP formation was induced by the rapid addition of 1 mL of aqueous phase to an equal volume of organic phase. The resulting suspension was dialyzed against deionized water, which was replaced every 2 hours, for a total of 8–12 hours.
For preparation of fluorescently labeled NPs, 0.2 mg/mL of Rhodamine-B base (Sigma-Aldrich Co) was additionally introduced to the organic phase. NP size and polydispersity index (PDI) were measured by dynamic light scattering (DLS) using a Delsa™ Nano C (Beckman Coulter Inc, Brea, CA, USA) with a scattering angle of 165° at room temperature. At least three measurements (n=3) were taken and values reported as the average ± standard deviation (SD).
Zeta potential was measured in triplicate using the Delsa Nano C with a scattering angle of 15° at room temperature in deionized water, phosphate-buffered saline, and cell-culture medium, and the values reported as the average ± SD.
The morphology of the GAG-functionalized PLGA NPs was determined by transmission electron microscopy (TEM). Briefly, the NP suspension was stained with uranyl formate placed on TEM grid and then air dried before analyzing using a Carl Zeiss LEO 912 Omega microscope (Oberkochen, Germany).
Confirmation of surface functionalization by pH titration
The zeta potential of the GAG-modified NPs was determined as a function of pH using the Delsa Nano C equipped with a pH auto-titrator. Briefly, the zeta potential of the diluted NP suspension was measured at room temperature at pHs 2, 3, 5, and 7. The data were then used to calculate the isoelectric point (pIe) of the NP surface and this was compared with the pIe of the respective functional moiety.
X-ray photoelectron spectroscopy
X-ray photoelectron spectroscopy (XPS) was used to confirm the surface functionalization of the PLGA NPs with GAGs. The primary objective of the investigation was to identify the presence of H, HA, DS, and CS on the surface of the functionalized PLGA NPs. For this purpose, a wide-scan spectrum of each of the samples was obtained and corrected for background noise. The X-ray photoelectron spectra of the samples for carbon 1s (C1s) and oxygen 1s (O1s) were obtained at a higher resolution (narrow scan) for detailed analysis. Since the surfaces of the samples were nonconductive, the experimental binding energies were corrected for charge-induced shifts in binding energy using the charge correction factor. First the peaks were fitted for the O1s and the numerical value of the binding energy of the component peak was determined. This numerical value was then subtracted from the reference value of the O1s’ binding energy as found in the literature to obtain the charge correction factor.10 The X-ray photoelectron spectra for C1s were then de-convoluted using OriginPro software (v 9.8G; OriginLab Corporation, Northampton, MA, USA). The cumulative curve for each of the samples had an adjusted R2 value better than 0.995.
Cell-culture experiments
A549 cells were obtained from the Center for Biological Signaling Studies (BIOSS) Toolbox, University of Freiburg (Germany) and cultured in Dulbecco’s Modified Eagle’s Medium supplemented with 10% fetal bovine serum (Thermo Fisher Scientific, Waltham, MA, USA), and 1% penicillin/streptomycin (P/S) (100 U/mL; PAN-Biotech GmbH, Aidenbach, Germany). HPMEC were procured from ScienCell Research Laboratories (Carlsbad, CA, USA) and grown in endothelial-cell growth medium (ScienCell) supplemented with 5% fetal bovine serum and 1% P/S. RPTEC were purchased from Lonza Group (Basel, Switzerland) and were grown in renal-cell growth media (REBM basal medium, Lonza). All cells were maintained in a humidified incubator at 37°C with 5% CO2.
Cellular compatibility
The effect of the GAG-modified PLGA NPs on the proliferation of A549 cells, HPMEC, and RPTEC was determined using MTT assay. Since the formation of the insoluble formazan product requires mitochondrial esterase, this assay is frequently used to ascertain the metabolic status of cells. Briefly, cells were plated in 96-well plate (6×103 cells/well) and, 24 hours after plating, were treated with increasing concentrations of the functionalized NPs, with the MTT assay performed at the end of the 24 hours.
Briefly, the culture medium was replaced with 100 μL MTT solutions (0.25 mg/mL) and the plates were incubated for 3 hours at 37°C and in 5% CO2. Then, the MTT reagent from the wells was aspirated and 100 μL dimethyl sulfoxide (DMSO) was added to each well to dissolve the purple formazan crystals. The cell viability was assessed by absorbance at 550 nm using a microplate reader (BioTek Instruments, Inc, Winooski, VT, USA). The cell viability is represented as the percentage with respect to untreated samples (control) and represents an average of at least three independent experiments (n≥3).
Lactate dehydrogenase assay
The effect of the NPs on the integrity of the cell membrane was assayed using a LDH Cytotoxicity Assay Kit II (Abcam plc, Cambridge, England). The assay was performed as per the manufacturer’s instructions. LDH is released by cells in response to damage or loss of integrity of cell membrane and is an indicator cellular toxicity. Briefly, 4×104 cells/well were plated in a 96-well plate and treated with the functionalized PLGA NPs for 24 hours. Untreated cells were taken as the negative control and cells treated with lysis buffer were taken as the high control (total LDH in the cell). As a positive control, 1 μL of LDH was used to validate the assay. Following the incubation with NPs, the well plates were centrifuged at 600 g for 10 minutes and 10 μL of the medium was transferred to a fresh 96-well plate. The medium was then incubated with 100 μL of LDH reaction mixture for 15 minutes at room temperature and the absorbance was measured at 450 nm using the microplate reader (BioTek Instruments, Inc) with the reference wavelength at 650 nm. LDH was quantified using the following formula:
| (1) |
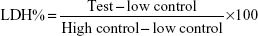
in which “low control” was the cells without any treatment and “high control” was the cells treated with lysis buffer (total LDH).
Induction of intracellular reactive oxygen species by glycosaminoglycan-modified nanoparticles
Intracellular ROS were determined using 2′,7′-dichlorofluorescein diacetate (DCFH-DA) (Abcam plc) using the protocol described by the supplier. DCFH-DA interacts with the intracellular ROS and gets converted to a fluorescent compound, dichlorofluorescein (DCF). The intensity of DCF is therefore directly proportional to intracellular ROS. Briefly, cells were plated in a 96-well plate (3×104 cells/well) and exposed to GAG-functionalized and un-functionalized PLGA NPs for 24 hours. Following this, the cells were washed twice with phosphate-buffered saline (PBS) and then incubated with a working solution of DCFH-DA (200 μL/well) at 37°C for 30 minutes. Fluorescence was then recorded at 485 nm excitation and 520 nm emission using the microplate reader (BioTek Instruments, Inc). Cells not exposed to NPs served as controls. Hydrogen peroxide (50 μM) was used to validate the assay for each run. The ROS were quantified using the following formula:
| (2) |
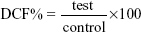
in which “control” was the cells without any treatment.
Cellular uptake of NPs
To investigate the cellular uptake of the functionalized PLGA NPs, cells were seeded in an eight-well chamber slide (2×104 cells/well). After 24 hours the cells were exposed to 200 μg/mL of surface-functionalized PLGA NPs and then incubated for 4 hours. The cells were then fixed with 4% paraformaldehyde and mounted with mounting media containing 4′,6-diamidino-2-phenylindole (DAPI; Vectashield®, Vector Laboratories Inc, Burlingame, CA, USA). The fluorescence images were then taken using a Carl Zeiss microscope.
Further, for flow cytometry (fluorescence-activated cell sorting [FACS]), 10×104 cells were seeded in a 12-well plate. Following overnight attachment, the cells were treated with 200 μg/mL of GAG-functionalized PLGA NPs for 4 hours. Before performing FACS, the samples were washed with PBS in order to remove non-internalized NPs from the cell surface. The uptake efficiency was determined with a FACS analyzer (Gallios™ Flow Cytometer, Beckman Coulter Inc) considering 10,000 counts per sample (n=3). The samples were analyzed using the Flowing Software (v 2.5.0; University of Turku, Turku, Finland).
Statistical analysis
The statistical analysis was performed using Student’s t-test and P-values <0.05 were considered statistically significant. Error bars represent SDs.
Results and discussion
Synthesis and characterization of GAG-functionalized PLGA NPs
Four GAGs commonly found in the ECM of tumor tissue – namely H, HA, CS, and DS – were selected for the surface modification of PLGA NPs due to their varied physicochemical characteristics and affinity for growth factors.11 The single-step nanoprecipitation method developed in our laboratory allows for the concomitant functionalization of NP during its formation and thus enables the rendering of a variety of biologically relevant information without resorting to multi-step synthesis for the generation of modified NPs. Moreover, this method enables the rapid production of NPs with the desired size and functional properties without the need for complex polymer backbone synthesis steps.9
The choice of acetone to THF ratio of 9:1 (v/v) as the binary solvent system to dissolve the polymer was based on an optimization study to get the target NP size of 100–150 nm. This size range is desirable as it is well-suited for cellular uptake,12 increased circulation time within the solid tumors, favors NP extravasation from vasculature, and reduces the likelihood of lung embolism.13,14 This latter attribute can augment the primary targeting modality to solid tumors, which is based on the phenomenon of enhanced permeation and retention effect.4
GAG-functionalized NPs were characterized in terms of their size and surface charge using DLS. NP size, determined using DLS, is plotted as a function of GAG concentration in Figure 1A. DLS measurements revealed that the particle sizes of H-, HA-, CS-, and DS-modified PLGA NPs in aqueous solutions were in the range of 100–160 nm with a narrow PDI of <0.2 in all cases except HA. The size of the GAG-functionalized NPs in aqueous solution was similar to the size of the non-functionalized PLGA NPs thereby demonstrating that the modification with GAGs yielded NPs with similar hydrodynamic radius. Furthermore, the increase in GAG concentration in the aqueous phase had no appreciable effect on NP size when the concentration was above 0.45 mg/mL except in the case of HA. It has been reported previously that aqueous-phase viscosity plays a crucial role in controlling NP size in the modified nanoprecipitation method and that there exits a linear correlation between aqueous-phase viscosity and NP size.9 Therefore, the unusual increase in size in the case of HA may be attributed to the greater viscosity of HA solutions at higher concentrations. Based on these observations, NPs produced in presence of a GAG concentration of 0.45 mg/mL were chosen for all the future studies as this particular concentration consistently resulted in NPs in the desired size range of 100–150 nm (Table S1).
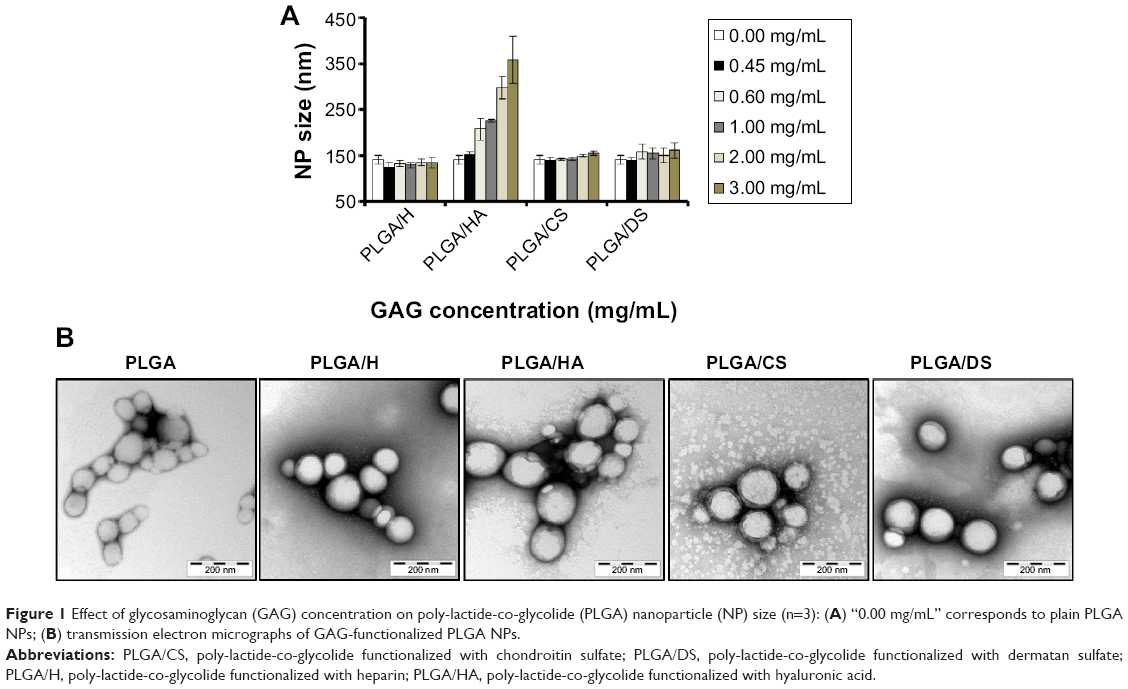 | Figure 1 Effect of glycosaminoglycan (GAG) concentration on poly-lactide-co-glycolide (PLGA) nanoparticle (NP) size (n=3): (A) “0.00 mg/mL” corresponds to plain PLGA NPs; (B) transmission electron micrographs of GAG-functionalized PLGA NPs. |
The morphology and size distribution of NPs were also investigated using TEM (Figure 1B and Table S2). TEM analysis confirmed that the GAG-modified NPs had a spherical morphology and the particles were homogeneously distributed. However, the size distribution as obtained from TEM (Table S2) was lower and in the range of 80–125 nm. This is not surprising as DLS always overestimates size as it is based on hydrodynamic radius, which is very sensitive to surface chemistry.
Confirmation of surface functionalization of poly-lactide-co-glycolide nanoparticles
One of the primary objectives of this study was to establish the suitability of GAG-functionalized NPs for injectable therapeutics. Establishing the presence of GAG on the NP surface was therefore a key step in this direction. To confirm the presence of GAG modification on the PLGA NPs, two characterization strategies were employed: zeta potential measurements and XPS. In general, all GAG-functionalized NPs showed a large negative zeta potential ranging from −30 to −40 mV in deionized water, which represented a significant decrease when compared with that of unmodified PLGA NPs which was found to be −21.66±1.50 mV (Figure 2A). Since pH is related to the surface chemistry of the NP, the pIe of NP’s surface can be used to confirm the nature of the ionizable moieties. A titration of zeta potential as a function of pH was undertaken (Figure 2B) and this revealed that the pIe values for each of the GAG-functionalized PLGA NPs closely matched the pKa of the dominant ionizable group associated with each respective GAG molecule (Table 1).
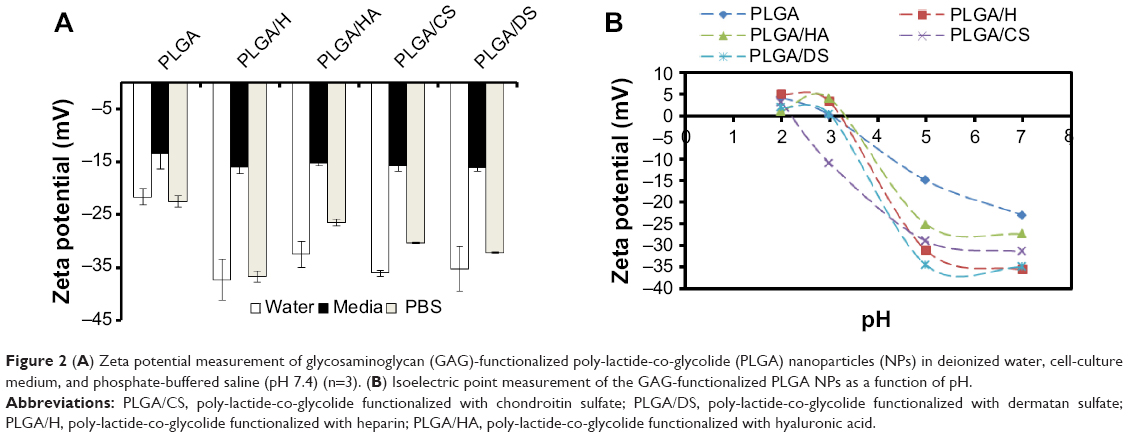 | Figure 2 (A) Zeta potential measurement of glycosaminoglycan (GAG)-functionalized poly-lactide-co-glycolide (PLGA) nanoparticles (NPs) in deionized water, cell-culture medium, and phosphate-buffered saline (pH 7.4) (n=3). (B) Isoelectric point measurement of the GAG-functionalized PLGA NPs as a function of pH. |
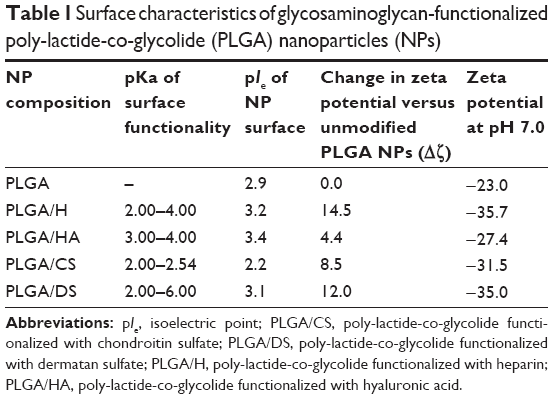 | Table 1 Surface characteristics of glycosaminoglycan-functionalized poly-lactide-co-glycolide (PLGA) nanoparticles (NPs) |
While the measurements strongly indicated the presence of GAG on the NP surface, direct confirmation of the surface chemistry was obtained using XPS. The processed C1S XPS spectra of PLGA (50:50) NPs (Figure 3A) showed three peaks: one at 289.1 eV, a second peak at 287.0 eV, and third peak at 285.1 eV corresponding to the carbon atoms in O-C=O, C-O-C, and C-H and C-C bonds, respectively (Table S3). In addition to these three peaks, the XPS spectrum of all the functionalized NPs had varying intensity in the peak around 285 eV and a shoulder at 285.5 eV. De-convolution of this peak confirmed the contribution from C-S and C-N bonds in the case of H, CS, and DS (Figure 3B, D, and E) and the contribution of C-N in the case of HA (Figure 3C). This provides direct evidence for the presence of a GAG-rich surface in these NPs (Table 2).
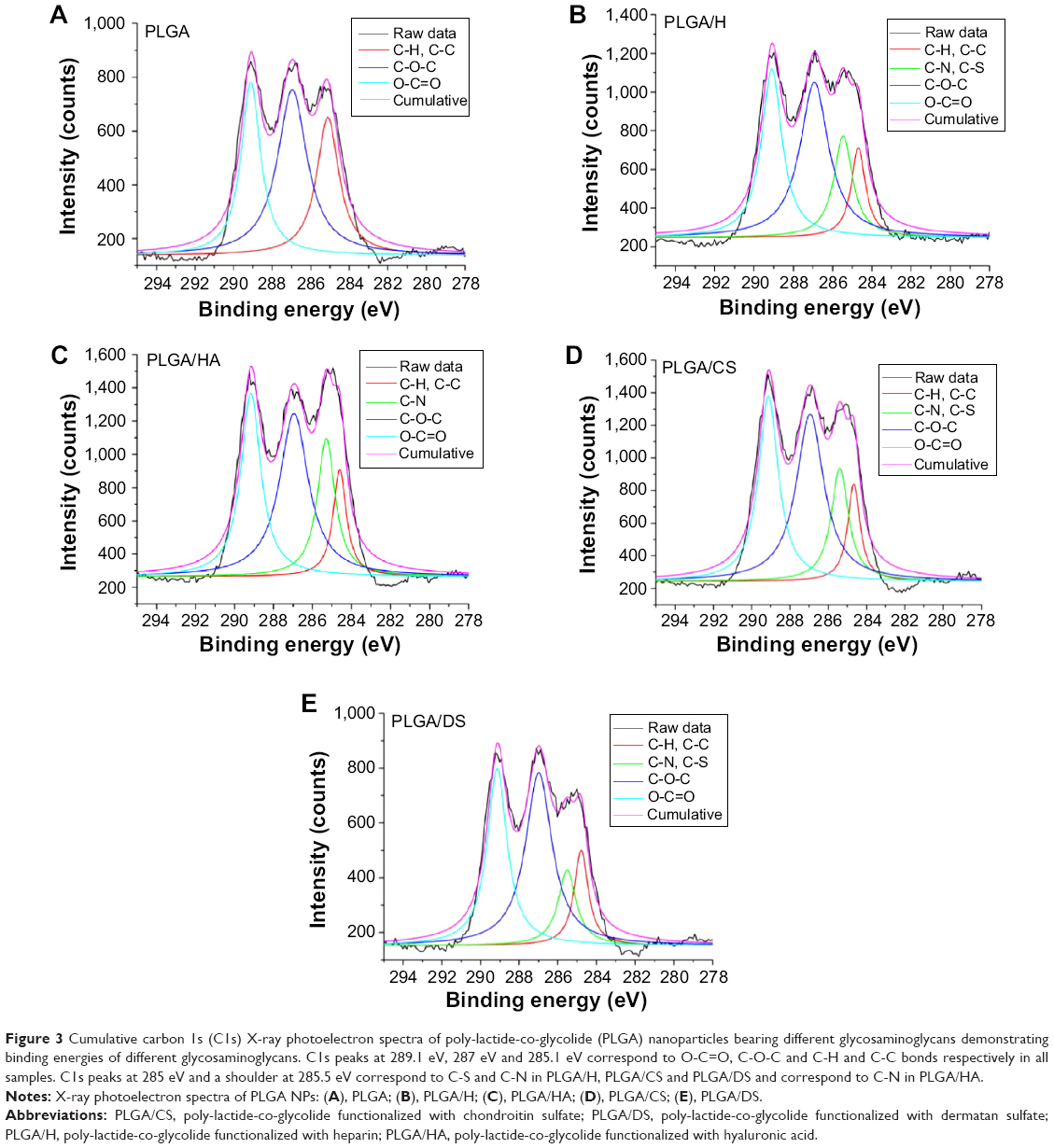 | Figure 3 Cumulative carbon 1s (C1s) X-ray photoelectron spectra of poly-lactide-co-glycolide (PLGA) nanoparticles bearing different glycosaminoglycans demonstrating binding energies of different glycosaminoglycans. C1s peaks at 289.1 eV, 287 eV and 285.1 eV correspond to O-C=O, C-O-C and C-H and C-C bonds respectively in all samples. C1s peaks at 285 eV and a shoulder at 285.5 eV correspond to C-S and C-N in PLGA/H, PLGA/CS and PLGA/DS and correspond to C-N in PLGA/HA. |
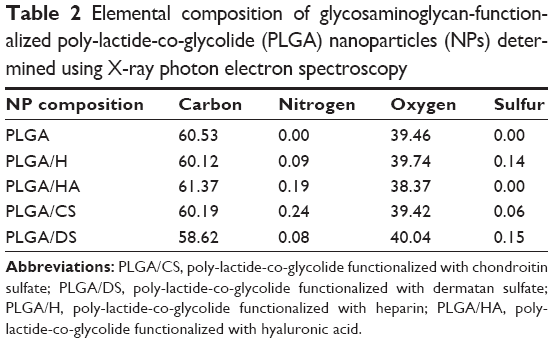 | Table 2 Elemental composition of glycosaminoglycan-functionalized poly-lactide-co-glycolide (PLGA) nanoparticles (NPs) determined using X-ray photon electron spectroscopy |
Cytotoxicity of glycosaminoglycan-functionalized poly-lactide-co-glycolide nanoparticles
The in vitro cytotoxicity of the GAG-functionalized PLGA NPs was assessed on three different cell types: A549 cells, HPMEC and RPTEC. The rationale for the selection of these cell types was based on our interest in exploiting these functionalized PLGA NPs for targeting solid tumors like lung cancer and also in addressing the pulmonary and renal toxicity of GAG-modified PLGA NPs. HPMEC were primarily chosen as a model system to address the pulmonary toxicity of these NPs, as these cells form a selective barrier responsible for the exchange of gases and regulation of various fluids and solutes across the blood and cellular compartments.15,16 Further, these cells have been reported to be prominent sites for the generation of ROS in the lungs during cellular injuries and inflammation.16,17 Additionally, RPTEC were chosen to address the renal toxicity of the functionalized NPs, as they have been commonly employed in understanding various renal diseases (eg, glomerulonephritis), injuries, inflammation, and toxicity.18 Also, one of the concerns with long-term use of NP-based therapeutics is renal toxicity.
In the first set of experiments, the toxicity of surface-modified PLGA NPs as a function of concentration was determined by MTT assay (Figure 4). It was observed that with an increase in the NP concentration (functionalized as well as non-functionalized), there was a moderate decrease in cell viability. A similar decrease in the viability of A549 cells at higher concentrations has been reported by Grabowski et al in a polyvinyl alcohol/chitosan-coated PLGA NP system.19 Also, Chung et al have reported 5%–7% toxicity in NIH/3T3 fibroblast and SCC7 tumor cells following incubation with 100 μg/mL of PLGA NPs for 24 hours.20 The increase in toxicity at higher concentrations in all cases (including PLGA NPs) could be attributed to an increase in NP internalization by all cell types and not the surface moiety. The fact that the zeta potential of all four NPs decreased to values between −13.47 and −16.12 mV upon exposure to cell-culture medium (Figure 2A) is indicative of a partial neutralization of the surface charge following interaction with the serum proteins.21 Adsorbed serum proteins are expected to play a role in both cellular toxicity and uptake and this needs to be further investigated. Nevertheless, the results clearly indicate that the loss of cell viability was NP-concentration dependent as it was proportional to this variable rather than type of GAG, indicating that the surface functionalization was well tolerated by the cells.
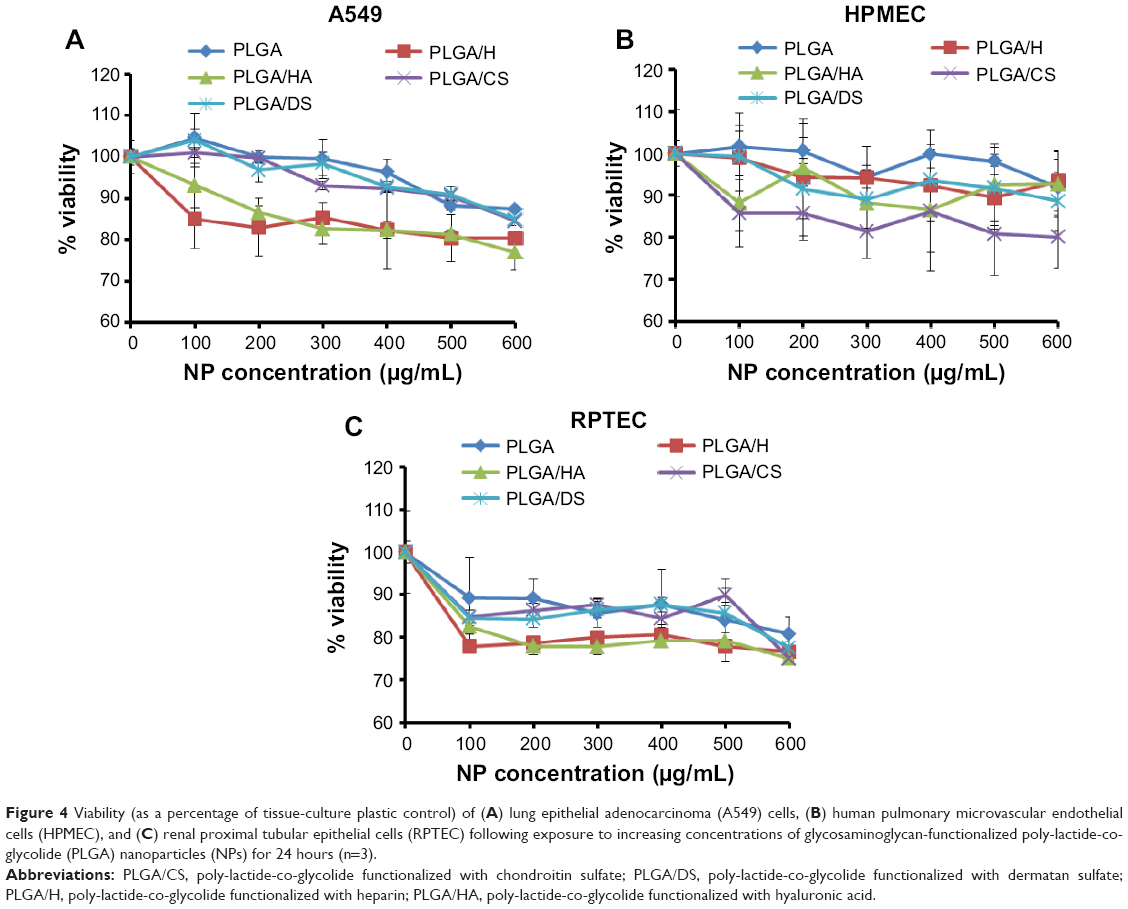 | Figure 4 Viability (as a percentage of tissue-culture plastic control) of (A) lung epithelial adenocarcinoma (A549) cells, (B) human pulmonary microvascular endothelial cells (HPMEC), and (C) renal proximal tubular epithelial cells (RPTEC) following exposure to increasing concentrations of glycosaminoglycan-functionalized poly-lactide-co-glycolide (PLGA) nanoparticles (NPs) for 24 hours (n=3). |
While cell viability provides insights into the general cytocompatibility of an NP system, secondary processes triggered by exposure to and ingestion of nanomaterials may determine long-term outcomes. In this regard, the release of intracellular LDH, which has been found to be associated with cellular necrosis,22 is a good indicator of cell-membrane integrity, and hence cellular toxicity induced by exposure to NPs. Assay for LDH in A549 cells, HPMEC, and RPTEC revealed that with increase in NP concentration, there was only a marginal increase in cellular toxicity (Figure 5). The increase in LDH was more prominent in HPMEC upon exposure to the GAG NPs, except in the case of DS-modified NPs, followed by A549 cells and RPTEC. Overall, the results indicated similar patterns of toxicity in all the experimental conditions and doses employed. The maximum cellular toxicity in all cells was observed at the highest NP concentration of 600 μg/mL, which in HPMEC was observed following exposure to PLGA/HA NPs and was 18.27%±5.02%, while the maximum cellular toxicity in A549 cells was similar (14.35%±3.50%) following exposure to PLGA/H NPs, and in RPTEC the maximum of 13.22±3.69 was seen in presence of PLGA/H NPs. The generally higher cytotoxicity of these NPs in A549 cells and HPMEC might be attributed to their higher proliferative capacity, oxidative stress, and higher metabolic activity, while the increased cytotoxicity observed in HPMEC in the presence of PLGA/HA could be attributed to enhanced uptake of functionalized PLGA/HA NPs by HPMEC, as is discussed later.
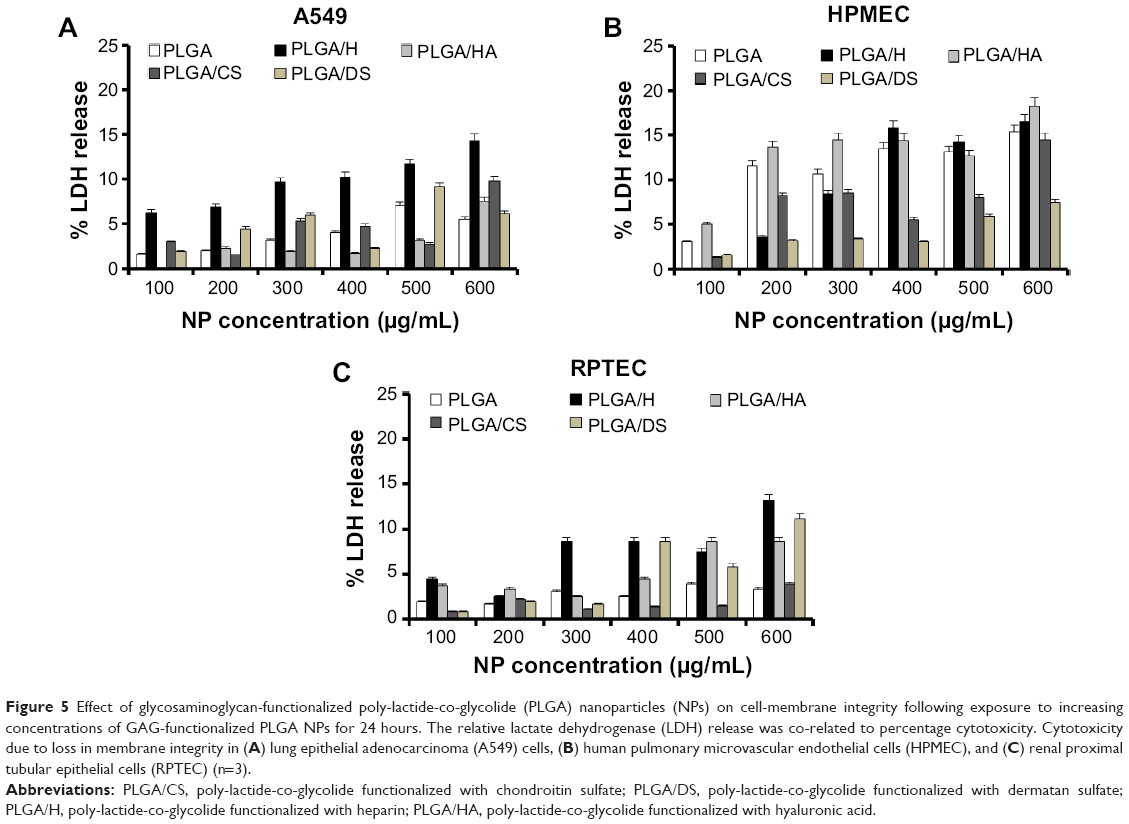 | Figure 5 Effect of glycosaminoglycan-functionalized poly-lactide-co-glycolide (PLGA) nanoparticles (NPs) on cell-membrane integrity following exposure to increasing concentrations of GAG-functionalized PLGA NPs for 24 hours. The relative lactate dehydrogenase (LDH) release was co-related to percentage cytotoxicity. Cytotoxicity due to loss in membrane integrity in (A) lung epithelial adenocarcinoma (A549) cells, (B) human pulmonary microvascular endothelial cells (HPMEC), and (C) renal proximal tubular epithelial cells (RPTEC) (n=3). |
It has been demonstrated in various mammalian cells that NP internalization induces an increased production of free oxygen radicals (also referred to as “ROS”) leading to cellular stress and inflammation.23 Therefore, as a third panel of cellular toxicity assessment in addition to MTT and LDH assays, ROS levels were determined (Figure 6). The induction of ROS in all the three cell types upon exposure to increasing concentrations of GAG-functionalized PLGA NPs was determined. It was observed that with an increase in GAG NP concentration, there was an increase in percentage DCF fluorescence indicative of an increase in ROS levels. This was consistent with what was already observed using the MTT and LDH assays. In A549 cells, the increase in ROS levels showed a dose dependency in all cases except in HA- and DS-functionalized NPs (Figure 6A). Interestingly, while PLGA/HA NPs triggered very low levels of ROS release, PLGA/DS NPs induced robust oxidative stress even at NP concentrations of 200 μg/mL. Similarly, in HPMEC, HA-modified NPs induced strong ROS levels even at a low concentration of 200 μg/mL (Figure 6B). The higher ROS levels in A549 cells on the other hand could be attributed to an increased state of oxidative stress in general in cancer cells due to the high degree of oncogenic transformation and increased metabolic activities.24–26 Our observation is corroborated by reports in the literature of an increase in production of ROS in both A549 cells and HPMEC after exposure to zinc oxide NPs.23,27
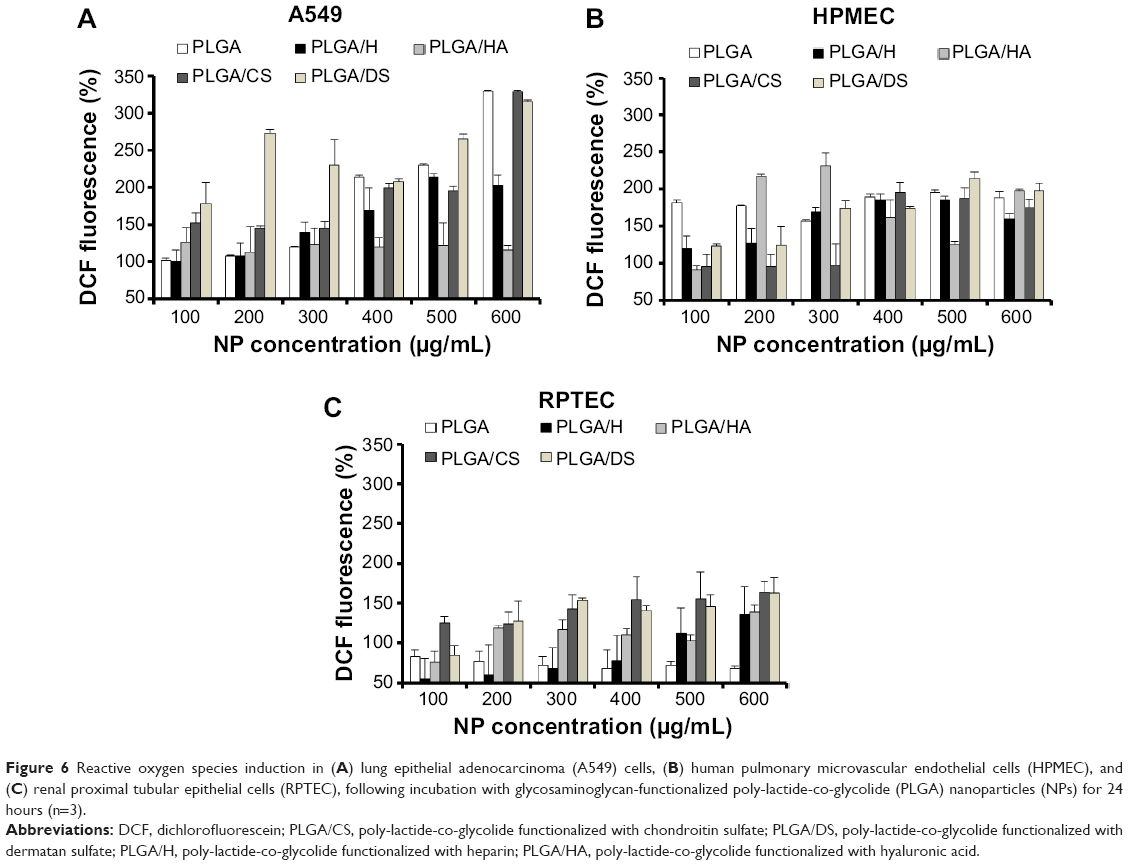 | Figure 6 Reactive oxygen species induction in (A) lung epithelial adenocarcinoma (A549) cells, (B) human pulmonary microvascular endothelial cells (HPMEC), and (C) renal proximal tubular epithelial cells (RPTEC), following incubation with glycosaminoglycan-functionalized poly-lactide-co-glycolide (PLGA) nanoparticles (NPs) for 24 hours (n=3). |
It is also well known that both A549 cells and HPMEC have an enhanced susceptibility toward ROS generation,24 and that lung microvascular endothelial cells are the primary sites for oxidative injury during the inflammation or injuries.16 Both of these factors might have played a role in the higher ROS production observed in these cells. Higher oxidative stress in A549 cells is advantageous, since it is known that ROS-generating agents like hydrogen peroxide28 and drug-delivery vehicles like carbon nanotubes and graphene oxide21 are potentiators for the enhanced toxicity of paclitaxel on cancer cells. In this regard, since GAG-functionalized PLGA NPs caused ROS production in A549 cells, they could be used as adjuvants for anticancer drugs like paclitaxel. In contrast, the ROS induction in RPTECs was significantly lower with a maximum of less than half of that with HPMECs and A549 cells, indicating a minimal level of oxidative stress in RPTEC as compared with A549 cells and HPMECs. This is an encouraging result, as renal toxicity is one of the concerns of long-term NP therapy.
Taken together, the results from the three toxicity screens indicate only a low–moderate level of cellular toxicity following exposure to GAG-functionalized PLGA NPs, over a wide concentration range.
Cellular uptake of glycosaminoglycan-functionalized poly-lactide-co-glycolide nanoparticles
Since the objective of the present study was to develop an NP platform to deliver therapeutics to lung tumors, preliminary studies were carried out to ascertain the suitability of GAG NPs for intracellular delivery in A549 cells and HPMEC. It was observed that GAG-modified PLGA NPs were differentially taken up by both the cell types, however, the trend of cellular uptake was identical in both cell systems (Figures 7, 8, S1, and S2). Both the cell types showed higher uptake of CS- and HA-coated NPs in comparison to unmodified PLGA NPs, with PLGA/CS NPs showing a 40%–65% higher uptake than unmodified NPs. However, in contrast, a marked 50%–65% reduction in uptake of H-modified NPs, in comparison to unmodified NPs, was observed in both cell systems. Interestingly, this represented a twofold increase in comparison to NPs modified with CS. It is possible that the higher uptake of PLGA/CS NPs in A549 cells and HPMEC is favored by the expression of cluster of differentiation (CD) 44 in both cell types, which is a known binding site for CS.29–32 The reasoning behind the possibility of CD44 playing a role in mediating NP uptake in these cells is further bolstered by the observation that NPs modified with HA, which is known to bind to CD44, are also taken up at relatively higher levels in both cells. Additionally, in the case of HPMEC, the enhanced uptake observed with HA NPs may also be due to specific receptors for HA – that is, the HA receptor for endocytosis (HARE), also referred to as “Stabilin-2” (encoded by the gene STAB2) and “FEEL-2”32 – that are expressed by endothelial cells.
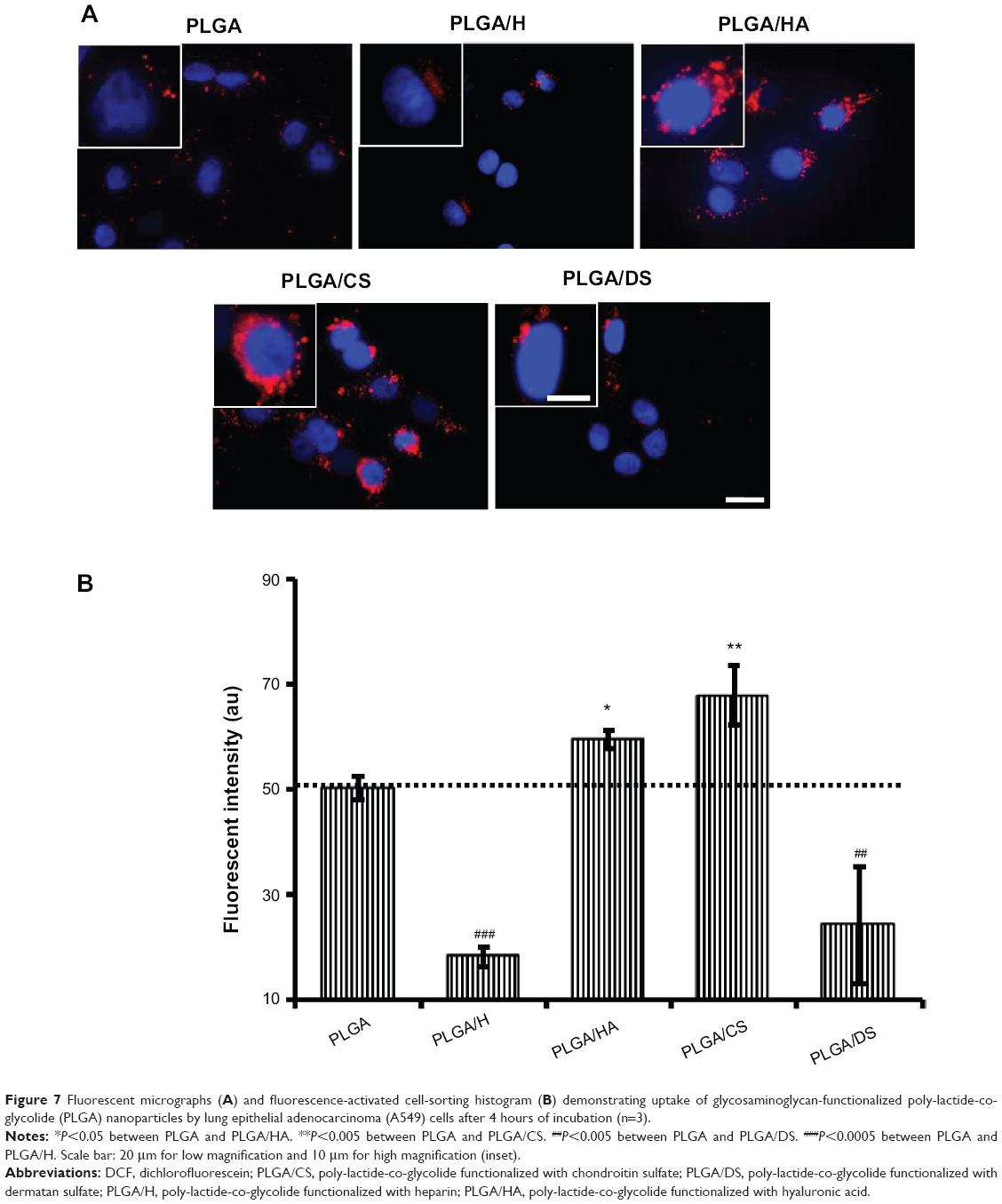 | Figure 7 Fluorescent micrographs (A) and fluorescence-activated cell-sorting histogram (B) demonstrating uptake of glycosaminoglycan-functionalized poly-lactide-co-glycolide (PLGA) nanoparticles by lung epithelial adenocarcinoma (A549) cells after 4 hours of incubation (n=3). |
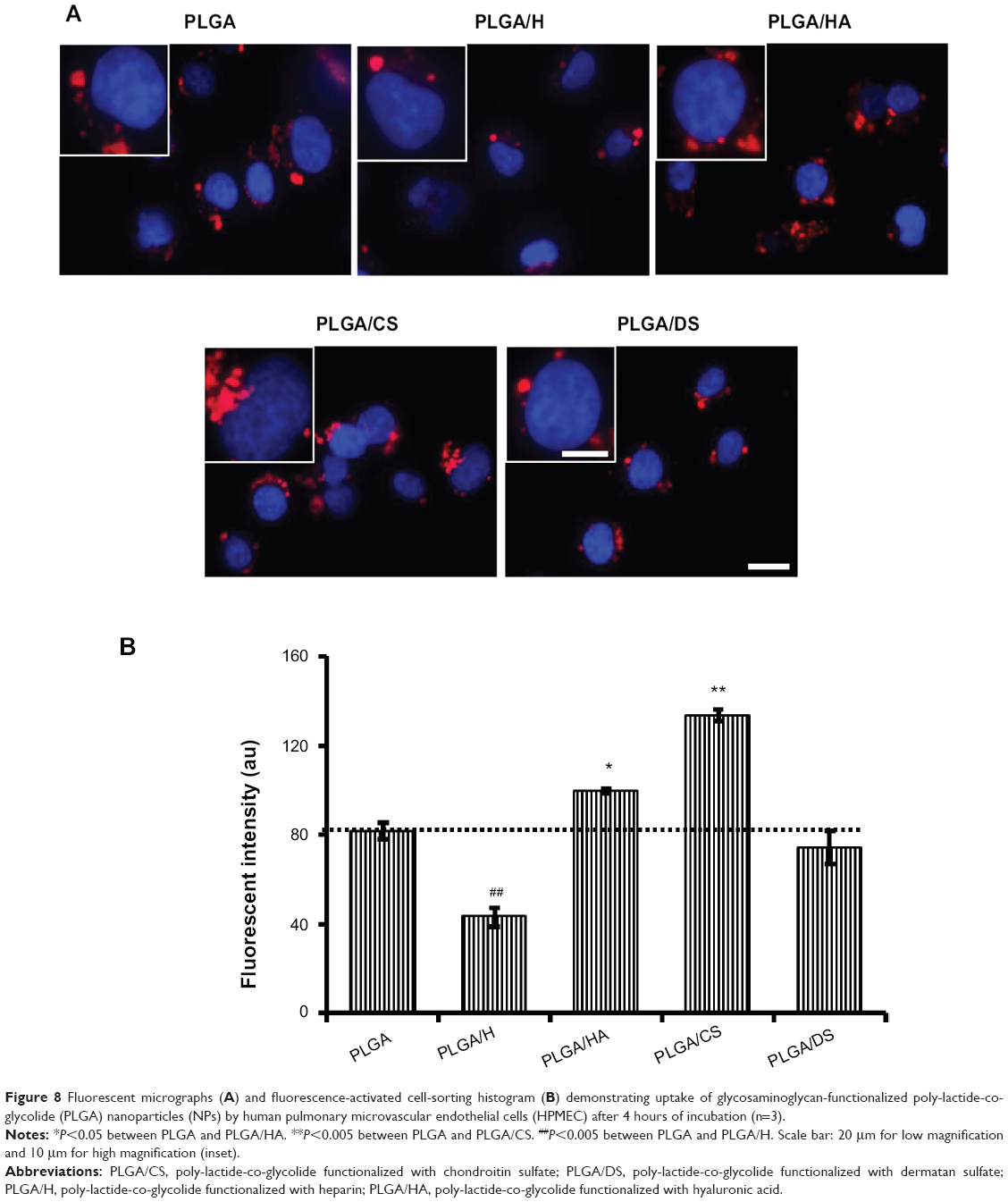 | Figure 8 Fluorescent micrographs (A) and fluorescence-activated cell-sorting histogram (B) demonstrating uptake of glycosaminoglycan-functionalized poly-lactide-co-glycolide (PLGA) nanoparticles (NPs) by human pulmonary microvascular endothelial cells (HPMEC) after 4 hours of incubation (n=3). |
In analyzing these findings, it is important to note that the surface concentration of the GAGs can play an important role in altering cellular interactions and uptake. Further studies need to be carried out to develop semi-empirical relationships between GAG functionalization and cellular uptake. Nevertheless, these results taken in sum suggest that GAGs can play a role in controlling NP uptake, and provide confirmation for the hypothesis of this study. While the exact pathway of uptake needs to be further elucidated using several cell systems, the fact that the NPs were mostly concentrated in the perinuclear region is indicative of a predominantly endocytic uptake pathway.13,33,34 The findings of this study therefore indicate that GAG-functionalized PLGA NPs have potential as nanocarriers for the delivery of cancer therapeutics.
Conclusion
PLGA NPs functionalized with H, HA, CS, and DS GAGs were synthesized using a single-step modified nanoprecipitation process. The presence of GAG on the NP surface was verified by pIe measurements of the NP surface and XPS analysis. The cytocompatibility of GAG-modified PLGA NPs was assessed in three cell systems: A549 cells, HPMEC, and RPTEC, using MTT assay, LDH release, and induction of ROS. Overall, GAG-functionalized NPs showed very good cytocompatibility and did not invoke any adverse response in any of the cell systems investigated. Moreover, the particles underwent cellular internalization to differing extents in A549 cells and HPMEC and offer a potential platform for introducing a bias in the cellular uptake of PLGA NPs.
Based on these findings, GAG-functionalized PLGA NPs are promising delivery vehicles to the tumor microenvironment. However, to fully exploit the targeting potential of these NPs, their uptake mechanism in various cell systems needs to be fully understood and their biodistribution in the absence and presence of a tumor burden needs to be evaluated.
Acknowledgments
The authors would like to thank Dr Ralf Thomann, University of Freiburg, for TEM. This work was funded by the INTERREG IV Upper Rhine program (A21 NANO@MATRIX), the Excellence Initiative of the German Federal and State Governments Grant EXC 294, and the Helmholtz Virtual Institute on Multifunctional Biomaterials for Medicine.
Disclosure
The authors declare no conflicts of interest in this work.
References
Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63(1):11–30. | ||
Torchilin VP, Lukyanov AN. Peptide and protein drug delivery to and into tumors: challenges and solutions. Drug Discov Today. 2003;8(6):259–266. | ||
Liu Y, Miyoshi H, Nakamura M. Nanomedicine for drug delivery and imaging: a promising avenue for cancer therapy and diagnosis using targeted functional nanoparticles. Int J Cancer. 2007;120(12):2527–2537. | ||
Torchilin V. Tumor delivery of macromolecular drugs based on the EPR effect. Adv Drug Delivery Rev. 2011;63(3):131–135. | ||
Hans ML, Lowman AM. Biodegradable nanoparticles for drug delivery and targeting. Curr Opin Solid State Mater Sci. 2002;6(4):319–327. | ||
Davis ME, Chen ZG, Shin DM. Nanoparticle therapeutics: an emerging treatment modality for cancer. Nat Rev Drug Discov. 2008;7(9):771–782. | ||
Huwyler J, Drewe J, Krähenbühl S. Tumor targeting using liposomal antineoplastic drugs. Int J Nanomed. 2008;3(1):21–29. | ||
van Vlerken LE, Vyas TK, Amiji MM. Poly(ethylene glycol)-modified nanocarriers for tumor-targeted and intracellular delivery. Pharm Res. 2007;24(8):1405–1414. | ||
Sussman EM, Clarke MB Jr, Shastri VP. Single-step process to produce surface-functionalized polymeric nanoparticles. Langmuir. 2007;23(24):12275–12279. | ||
Oswald S, Baunack S. Application of XPS and factor analysis for non-conducting materials. Surf Interface Anal. 1997;25(12):942–947. | ||
Sasisekharan R, Shriver Z, Venkataraman G, Narayanasami U. Roles of heparan-sulphate glycosaminoglycans in cancer. Nat Rev Cancer. 2002;2(7):521–528. | ||
Suh KY, Yang JM, Khademhosseini A, et al. Characterization of chemisorbed hyaluronic acid directly immobilized on solid substrates. J Biomed Mater Res Part B Appl Biomater. 2005;72(2):292–298. | ||
Schipper ML, Iyer G, Koh AL, et al. Particle size, surface coating, and PEGylation influence the biodistribution of quantum dots in living mice. Small. 2009;5(1):126–134. | ||
Kulkarni S, Feng SS. Effects of particle size and surface modification on cellular uptake and biodistribution of polymeric nanoparticles for drug delivery. Pharm Res. 2013;30(10):2512–2522. | ||
Brigham KL. Oxidant stress and adult respiratory distress syndrome. Eur Respir J. 1990;11:482s–484s. | ||
Kelly J, Moore T, Babal P, Diwan A, Stevens T, Thompson W. Pulmonary microvascular and macrovascular endothelial cells: differential regulation of Ca2+ and permeability. Am J Physiol. 1998;274(5):L810–L819. | ||
Moore TM, Chetham PM, Kelly JJ, Stevens T. Signal transduction and regulation of lung endothelial cell permeability. Interaction between calcium and cAMP. Am J Physiol. 1998;275(2):L203–L222. | ||
van Kooten C, van der Linde X, Woltman AM, van Es LA, Daha MR. Synergistic effect of interleukin-1 and CD40L on the activation of human renal tubular epithelial cells. Kidney Int. 1999;56(1):41–51. | ||
Grabowski N, Hillaireau H, Vergnaud J, et al. Toxicity of surface-modified PLGA nanoparticles toward lung alveolar epithelial cells. Int J Pharm. 2013;454(2):686–694. | ||
Chung YI, Kim JC, Kim YH, et al. The effect of surface functionalization of PLGA nanoparticles by heparin- or chitosan-conjugated Pluronic on tumor targeting. J Control Release. 2010;143(3):374–382. | ||
Arya N, Arora A, Vasu K, Sood AK, Katti DS. Combination of single walled carbon nanotubes/graphene oxide with paclitaxel: a reactive oxygen species mediated synergism for treatment of lung cancer. Nanoscale. 2013;5(7):2818–2829. | ||
Lewinski N, Colvin V, Drezek R. Cytotoxicity of nanoparticles. Small. 2008;4(1):26–49. | ||
Vandebriel RJ, De Jong WH. A review of mammalian toxicity of ZnO nanoparticles. Nanotechnol Sci Appl. 2012;5:61–71. | ||
Sosa V, Moliné T, Somoza R, Paciucci R, Kondoh H, Lleonart ME. Oxidative stress and cancer: An overview. Ageing Res Rev. 2013;12(1):376–390. | ||
Benz CC, Yau C. Ageing, oxidative stress and cancer: paradigms in parallax. Nat Rev Cancer. 2008;8(11):875–879. | ||
Valko M, Rhodes CJ, Moncol J, Izakovic M, Mazur M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem Biol Interact. 2006;160(1):1–40. | ||
Apopa P, Qian Y, Shao R, et al. Iron oxide nanoparticles induce human microvascular endothelial cell permeability through reactive oxygen species production and microtubule remodeling. Part Fibre Toxicol. 2009;6:1. | ||
Alexandre J, Batteux F, Nicco C, et al. Accumulation of hydrogen peroxide is an early and crucial step for paclitaxel-induced cancer cell death both in vitro and in vivo. Int J Cancer. 2006;119(1):41–48. | ||
Hiscox S, Jiang W. Regulation of endothelial CD44 expression and endothelium-tumour cell interactions by hepatocyte growth factor/scatter factor. Biochem Biophys Res Commun. 1997;233(1):1–5. | ||
Griffioen AW, Coenen MJ, Damen CA, et al. CD44 is involved in tumor angiogenesis; an activation antigen on human endothelial cells. Blood. 1997;90(3):1150–1159. | ||
Penno MB, August JT, Baylin SB, et al. Expression of CD44 in human lung tumors. Cancer Res. 1994;54(5):1381–1387. | ||
Harris EN, Kyosseva SV, Weigel JA, Weigel PH. Expression, processing, and glycosaminoglycan binding activity of the recombinant human 315-kDa hyaluronic acid receptor for endocytosis (HARE). J Biol Chem. 2007;282(5):2785–2797. | ||
Kim SH, Jeong JH, Chun KW, Park TG. Target-specific cellular uptake of PLGA nanoparticles coated with poly(L-lysine)-poly(ethylene glycol)-folate conjugate. Langmuir. 2005;21(19):8852–8857. | ||
Lai SK, Hida K, Man ST, et al. Privileged delivery of polymer nanoparticles to the perinuclear region of live cells via a non-clathrin, non-degradative pathway. Biomaterials. 2007;28(18):2876–2884. |
Supplementary materials
 | Table S1 Size distribution of glycosaminoglycan (GAG)-functionalized poly-lactide-co-glycolide (PLGA) nanoparticles (NPs) (0.45 mg/mL of GAG) as determined by dynamic light scattering |
 | Table S2 Size distribution of glycosaminoglycan (GAG)-functionalized poly-lactide-co-glycolide (PLGA) nanoparticles (NPs) (0.45 mg/mL of GAG) as determined by transmission electron microscopy |
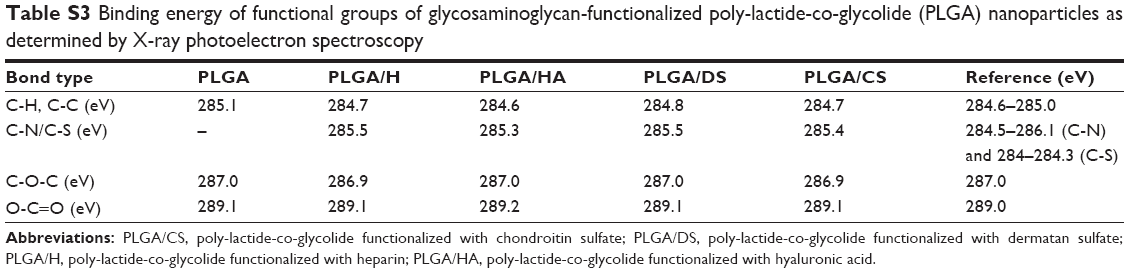 | Table S3 Binding energy of functional groups of glycosaminoglycan-functionalized poly-lactide-co-glycolide (PLGA) nanoparticles as determined by X-ray photoelectron spectroscopy |
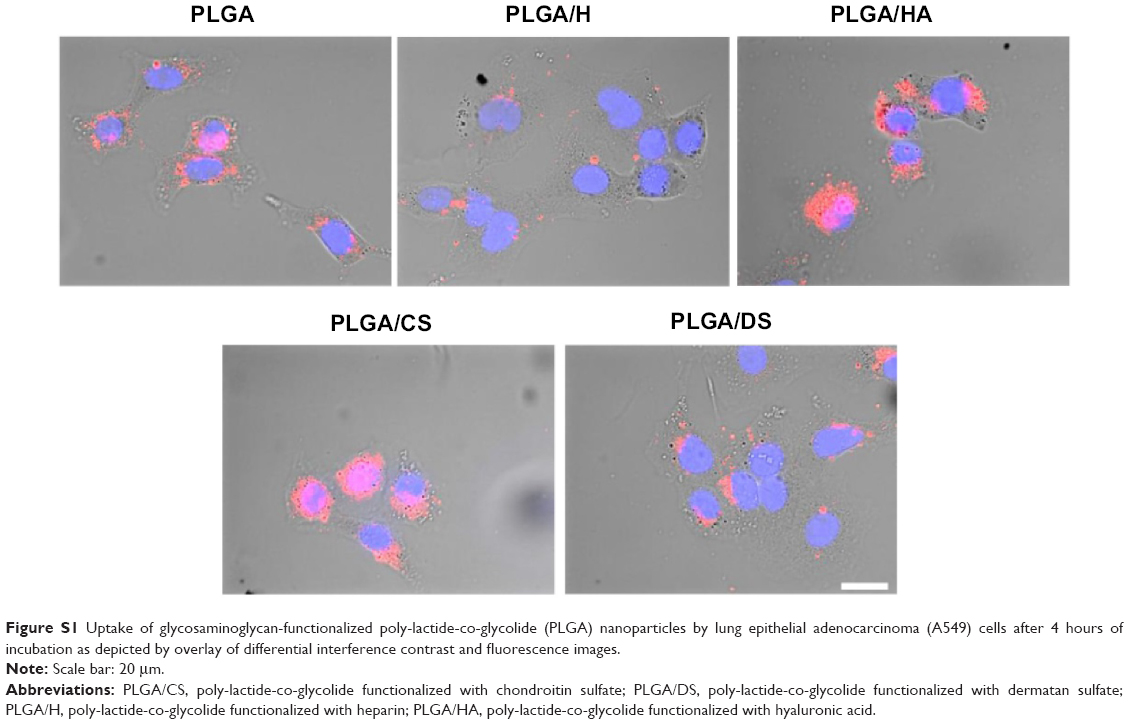 | Figure S1 Uptake of glycosaminoglycan-functionalized poly-lactide-co-glycolide (PLGA) nanoparticles by lung epithelial adenocarcinoma (A549) cells after 4 hours of incubation as depicted by overlay of differential interference contrast and fluorescence images. |
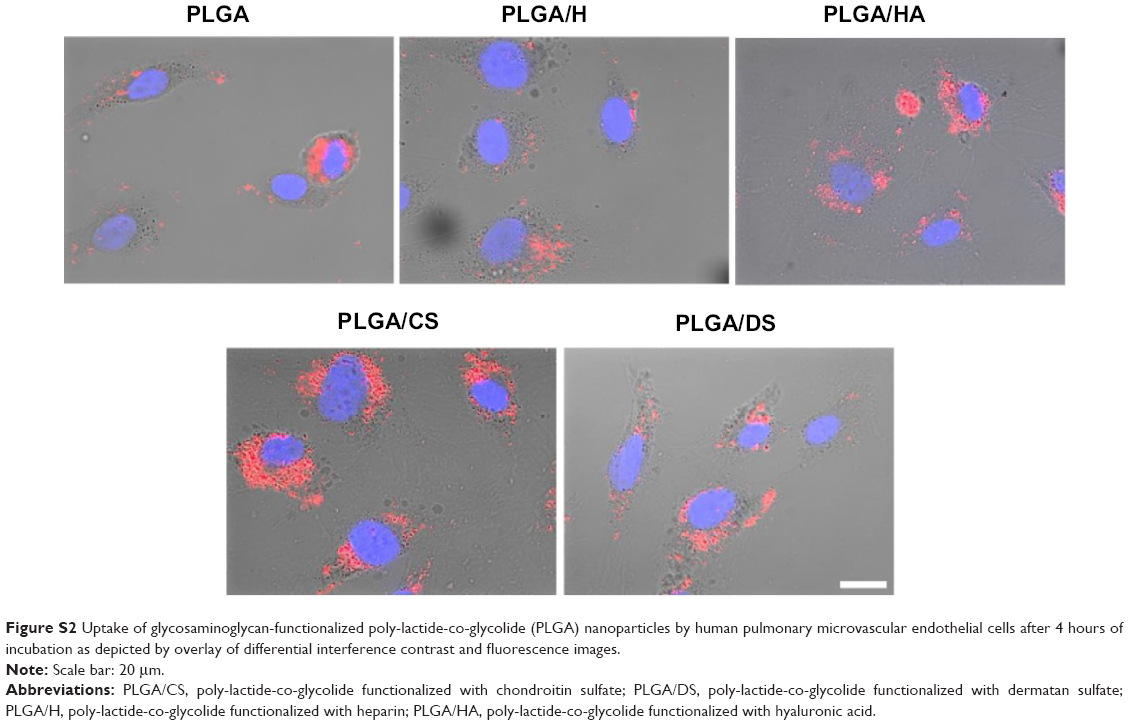 | Figure S2 Uptake of glycosaminoglycan-functionalized poly-lactide-co-glycolide (PLGA) nanoparticles by human pulmonary microvascular endothelial cells after 4 hours of incubation as depicted by overlay of differential interference contrast and fluorescence images. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.