Back to Journals » International Journal of Nanomedicine » Volume 14
Glucose-responsive mesoporous silica nanoparticles to generation of hydrogen peroxide for synergistic cancer starvation and chemistry therapy
Authors Du X, Zhang T, Ma G, Gu X, Wang G, Li J
Received 25 November 2018
Accepted for publication 4 March 2019
Published 2 April 2019 Volume 2019:14 Pages 2233—2251
DOI https://doi.org/10.2147/IJN.S195900
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Mian Wang
Xiao Du,1 Tian Zhang,1 Guanglan Ma,1 Xiaochen Gu,2 Guangji Wang,3 Juan Li1
1Department of Pharmaceutics, School of Pharmacy, China Pharmaceutical University, Nanjing, , People’s Republic of China; 2College of Pharmacy, University of Manitoba, Winnipeg, MB, Canada; 3Center for New Drug Safety Evaluation and Research, China Pharmaceutical University, Nanjing, People’s Republic of China
Background: The combination of novel starving therapy with chemotherapy is one of the most promising strategies to achieve an effective antitumor activity.
Methods: Herein, we developed a multifunctional mesoporous silica nanoparticle (MSNs-GOx/PLL/HA) coated with poly (L-lysine) (PLL) and hyaluronic acid (HA) for co-delivery of glucose oxidase (GOx) and anticancer drug paclitaxel (PTX) for cancer treatment for the first time. Compared to single chemotherapy, introduction of GOx would not only selectively trigger the consumption of intracellular glucose, leading to the interruption of energy supply, but also elevat the endogenous H2O2 level, inducing stronger therapeutic effects.
Results: The novel drug delivery system possessed desirable particle diameter of 40 nm and exhibited a pH-sensitive drug release behavior. An in vitro cellular uptake study indicated that MSNs-GOx/PLL/HA nanoparticles effectively enhanced the cellular uptake of drug in an apparently CD44 receptor-dependent manner, and delivered more cargo into cytoplasm via endolysosomal escape effect in presence of PLL. The nanoplatform has also demonstrated amplified synergistic therapeutic effects for remarkable tumor inhibition in a xenograft animal tumor model.
Conclusion: Consequently, the developed synergistic starving-like/chemotherapy may provide a potential platform for next generation cancer therapy.
Keywords: combination therapy, glucose oxidase, hyaluronic acid, pH-sensitivity, nanomedicine
Introduction
Although chemotherapy has been a frontline component for current cancer treatment, its effectiveness is often limited due to undesired side effects on normal tissues. Thus, there is a critical need for improving the therapeutic efficacy of chemotherapy. Combination therapy is a promising strategy to achieve an effective antitumor activity, which could not only modulate different signaling pathways in cancer cells, but also sensitize cancer responsive to drugs, thereby reducing each drug dose to reduce side effects.1,2
Recently, cancer starvation therapy has been extensively investigated in cancer therapy. According to the Warburg effect, adenosine triphosphate (ATP) is preferentially produced by oxidative phosphorylation in normal cells with a regular microenvironment, while an enhancement of glycolysis to generate ATP is observed in tumor cells, which is a fast, but less efficient process.3 To compensate for this, proliferating cancer cells tend to uptake more glucose than normal tissues for producing more energy.4 Therefore, blocking the glucose supply or elevating the nutrients consumption in cancer cells have been proposed for cancer starvation therapy.5
Glucose oxidase (GOx) has drawn more attention for glucose biosensors, which could oxidize glucose into gluconic acid and H2O2.6 As we all know, H2O2 plays an active role in various physiological processes including cell growth, immune response, and senescence.7 According to a previous report, H2O2 at endogenous concentrations could cause malignant transformation of normal cells, but in turn would lead to cancerous cell death at high concentrations.8 Therefore, the application of GOx into tumor therapy would not only trigger the consumption of intracellular glucose, leading to the interruption of the energy supply, but also elevate the endogenous H2O2 level, inducing stronger intratumoral cytotoxicity.
Mesoporous silica nanoparticles (MSNs) have been demonstrated as one of the most promising drug carriers due to their uniform pore size, high surface area, large pore volume, high drug loading capability, remarkable biocompatibility and easy surface functionalization.9 Therefore, MSNs were employed for delivery of GOx and the chemotherapeutic agent may be an attractive choice. However, the traditional MSNs lacked cancer targeting ability, which may cause serious off-target effects and lead to obvious side effects.5 Thus, introduction of active targeting ligands or antibodies to the system is extremely demanding.10 Hyaluronic acid (HA), an anionic linear polysaccharide, has received extensive attention for its biocompatibility, biodegradability and non-immunogenicity, especially the high affinity to CD44 receptor.11,12 HA-modified nanocarriers are capable of simultaneously achieving passive targeting of solid tumors via enhanced permeation and retention (EPR) effect and active targeting through CD44-mediated endocytosis without additional targeting ligands.13,14
Therefore, in this study, MSNs were applied for the co-delivery of GOx and anticancer drug paclitaxel (PTX) to achieve synergistic therapeutic effects. Poly (L-lysine) (PLL) was coupled onto the surface of MSNs via pH-sensitive dynamic benzoic-imine covalent bond and served as “gatekeeper” to minimize the drug leakage. Additionally, PLL had more primary and secondary amines that would enable carriers to facilitate endosomal escape, due to the elevation of osmotic pressure of lysosome and endosome.15,16 Moreover, HA was coated at the surface of carriers to shield the positive charges of PLL to reduce hemolytic toxicity or cytotoxicity and to improve biocompatibility. As illustrated in Figure 1, the multifunctional nanoparticles (MSNs-GOx/PLL/HA) could enhance the permeation at solid tumor site via EPR effect and improved active targeting of cancer cells through CD44-mediated endocytosis. Once entering into the tumor cells, hyaluronidase (HAase) in the acid endo/lysosomes would actively de-shield HA from the carriers and expose the high positive charges of PLL, thereby leading to endolysosomal escape via the proton sponge effect to facilitate more drugs into cytoplasm.17,18 Meanwhile, GOx could oxidize intratumoral glucose for choking off the energy supply and elevate the toxic H2O2 level to further increase the therapeutic effect. Therefore, the combination of cancer starving therapy with chemotherapy might provide a potential platform for next generation cancer therapy.
 | Figure 1 Schematic illustration of MSNs-GOx/PLL/HA nanoparticles for synergistic tumor-targeted starvation and chemistry therapy. (A) The preparation processes of MSNs-GOx/PLL/HA nanoparticles. (B) The drug delivery of nanoparticles including steps of intravenous injection and EPR effect in blood circulation. (C) The cellular process containing the CD44-mediated cellular internalization, degradation of HA by HAase together with the exposure of PLL at HAase-rich tumor milieu, endosomal or lysosomal escape, cytoplasmic release of GOx and PTX and the intracellular reaction of GOx with glucose.Abbreviations: MSNs, mesoporous silica nanoparticles; GOx, glucose oxidase; PLL, poly (L-lysine); HA, hyaluronic acid; EPR, enhanced permeation and retention; HAase; hyaluronidase; PTX, paclitaxel. |
Materials and methods
Materials
GOx (100 units/mg) was applied by Sigma-Aldrich Co. (St Louis, MO, USA). HA (36 kDa) and PTX were purchased from Yew Biotechnology Co. Ltd. (Jiangsu, China). Triphosgene, N-ε-carbobenzyloxy-L-lysine (Lys), hexylamine, 4-(dimethylamino)-pyridine (DMAP), dicyclohexylcarbodiimide (DCC), tetrahydrofuran (THF), hexane, N,N-Dimethylformamide (DMF) were obtained from Energy Chemical Technology Co., Ltd (Shanghai, China). Hexadecyltrimethylammonium chloride (CTAC), tetraethyl orthosilicate (TEOS), triethanolamine (TEA) and 4-formylbenzoic acid were provided by Fuke Chemical Reagent Co. (Changsha, China). Methanol and ethanol were purchased from Han Bang Scientific Corporation (Huaian, Jiangsu, China).
HepG2 and MCF-7 cells were kindly provided by China Pharmaceutical University Center for Safety and Evaluation of New Drugs (Nanjing, Jiangsu, China). All cell experiments were permitted by the Ethical Committee of China Pharmaceutical University. Cells were cultured in DMEM with high glucose, containing 10% FBS, 100 U/mL penicillin and 100 μg/mL streptomycin. The cells were cultured in an incubator at 37°C in 5% CO2 atmosphere.
Animals and tumor xenograft models
All animal experiments were carried out according to the guidelines of the Experimental Animal Center of China Pharmaceutical University and approved by the Ethics Committee of China Pharmaceutical University. Kunming SPF mice (male, 20±2 g) were provided by Qinglong Mountain Animal Center (Nanjing, China). To establish the tumor model, 0.2 mL of murine hepatic carcinoma cells (Heps, 5×106 cells/mice) were subcutaneously inoculated in the right armpit area of the mice.
Preparation of mesoporous silica nanoparticles (MSNs-GOx/PLL/HA)
Synthesis of benzaldehyde-functionalized MSNs (MSNs-CHO)
MSNs were prepared as described in previous study.19 Briefly, CTAC (2.00 g) and TEA, (0.16 g) were dispersed in 40 mL water and stirred at 95°C under intensive stirring for 1 hour. Subsequently, 1.5 mL of TEOS was added dropwise and the reaction took place for another 1 hour at 95°C. The product was obtained by centrifugation and washed with ethanol to remove the residual reactants. Afterwards, the template (CTAC) was removed in 1 wt% solution of sodium chloride (NaCl) in methanol for 3 hours, and the same procedure was repeated four times. Then the product was dried at 60°C in a vacuum. Subsequently, 0.50 g MSNs were dispersed in 10 mL DMSO solution, and 0.10 g 4-formylbenzoic acid, 0.02 g DMAP and 0.14 g DCC were added. The reaction proceeded for 24 hours, and product was recovered by centrifugation and dried in a vacuum to obtain MSNs-CHO.
Synthesis of MSNs-GOx
To prepare MSNs-GOx, 0.10 g MSNs-CHO was dispersed in 10 mL deionized water solution (pH 7.8), then 0.03 g GOx was added, and the drug delivery system was stirred at 4°C for 8 hours. The GOx could covalently graft onto the surface of MSNs through benzoic-imine linkage between the aldehyde group and amine group of GOx.6,20 Subsequently, the product was collected by centrifugation, dried in vacuum to obtain MSNs-GOx. The concentration of reacted GOx was determined by the BCA Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA) and the loading capacity of GOx was calculated by thermogravimetric analysis.21
Poly (L-lysine) surface modification (MSNs-GOx/PLL)
The synthesis route and characterization of PLL was shown in supplementaryinformation (
Lys(Z)-NCA (1.41 g, 4.58 mmol) was dissolved in 20 mL DMF and hexylamine (3.69 mg, 0.04 mmol) in 1 mL DMF was added. The reaction lasted for 48 hours at N2 atmosphere. Then the drug delivery system was poured into the mixture of cold methyl tert-butyl ether and hexane. The precipitate was filtered, dried in a vacuum to yield as PLL (Z).1H NMR (300 MHz, DMSO-d6, ppm): δ 0.82 (s, -CH3 in hexylamine), 4.98 (s, -CH2- in Z groups), 7.20–7.45 (m, -Ph in Z groups). FTIR peak cm:−1 1691 (C=O in the Z group), 1626 and 1536 (-NHCO-, amide bond of the PLL (Z) repeating unit) (
PLL (Z) (200 mg, 0.02 mmol) was dissolved in trifluoroacetic acid then HBr (307.45 mg, 3.80 mmol) was added. The reaction lasted for 2 hours, and then it was quenched with cold methyl tert-butyl ether. Then the crude solid product was filtered and washed three times to obtain the resulting solid PLL. The average molecular weight was detected by gel permeation chromatography (GPC). The analysis was taken with a Shimadzu GPC with Shimadzu RI and UV/Vis detection, and two 300 mm Waters ultrahydrogel GPC Columns using PEG standards. The dissolution solvent and mobile phase were composed of aqueous solution containing 0.2 M NaNO3 and 0.01 NaH2PO4 and the mobile phase at a flow rate of 1.0 mL/min. The average molecular weight (6400 Da) and polydispersity index (1.00) were calculated from the GPC retention time (20.58 minutes) in
The preparation process of MSNs-GOx/PLL was similar to that of MSNs-GOx. In brief, 0.10 g MSNs-GOx was dispersed in 10 mL deionized water (pH 7.8), then 0.03 g PLL was added, and the drug delivery system was further stirred for 12 hours at room temperature. Finally, the resulting MSNs-GOx/PLL particles were collected by centrifugation and dried via vacuum freeze-drying for further use.
Conjugating targeted ligand to MSNs-GOx/PLL and drug loading
In order to realize active tumor targeting of nanoparticles, HA was applied to coat the surface of the carriers by adding MSNs-GOx/PLL into the HA solution at a ratio of HA: MSNs-GOx/PLL=1:1 (w:w) and stirred for 8 hours. The final MSNs-GOx/PLL/HA particles were collected by centrifugation, washed with deionized water, and then dried via vacuum freeze-drying for further use.
To obtain PTX-loaded MSNs-GOx/PLL/HA, 0.10 g MSNs-CHO were dispersed in 10 mL PTX solution (10 mg/mL in DCM) and stirred for 12 hours at room temperature, and the drug delivery system was centrifuged and washed with ethanol twice, dried in a vacuum to obtain PTX-loaded MSNs. To prepare PTX-loaded MSNs-GOx, MSNs-GOx/PLL or MSNs-GOx/PLL/HA nanoparticles, blank MSNs were replaced by PTX-loaded MSNs and the rest of the procedure was the same as before. The coumarin (C6) labeled nanoparticles were prepared in a same way with PTX-loaded nanoparticles. Drug loading capability (DL) was calculated according to previous report.22
Characterization
The aldehyde group on the surface of MSNs was analyzed by the FTIR using a Thermo Scientific Nicolet 6700 FTIR spectrometer (Thermo Fisher Scientific, Waltham, MA, USA). Mesostructure ordering of MSNs and MSNs-CHO was determined by small-angle X-ray diffraction (SAXRD) on a Shimadzu XRD-6000 diffractometer (Kyoto, Japan). Nitrongen adsorption-desorption isotherms at 77 K were conducted on a Micrometitics Tristar 3000 system (Norcross, Georgia) to quantify the pore size distribution, surface area and pore volume of MSNs and MSNs-CHO. The morphology of MSNs, MSNs-CHO, MSNs-GOx, MSNs-GOx/PLL and MSNs-GOx/PLL/HA were examined using transmission electron microscopy (TEM). Thermogravimetric analysis (TGA) was performed under an N2 atmosphere with a heating rate of 10°C min−1 on a LINSEIS STA PT1600 (Selb, Germany) thermal analyzer. Scanning transmission electron microscopy (STEM) images and spectra of the MSNs-GOx/PLL/HA nanoparticles were obtained on JEOL JEM-2800 (Akishima, Tokyo, Japan) scanning transmission electron microscope with dual energy dispersive X-ray spectrometer (EDS) detectors at an electron beam energy of 200 kV. The size distribution and zeta potential of different mesoporous nanoparticles were investigated using a Malvern Zetasizer 3000 system (Malvern, UK). The structure of PLL and the intermediates were characterized by FT-IR and 1H-NMR spectra (Bruker, Karlsruhe,Germany).
Catalytic ability measurement
To investigate the catalytic ability of immobilized GOx, free GOx and MSNs-GOx/PLL/HA (GOx, 100 μg/mL) were incubated with glucose solutions (1000 μg/mL). At selected intervals (5 minutes, 30 minutes, 1 hour, 1.5, 2, 2.5 and 3 hours), samples were collected and the concentration of H2O2 and pH was detected by Hydrogen Peroxide AssayKit (Jiancheng, China) and a pH meter (pHS-25B, Dapu, Shanghai, China), respectively.
In vitro pH responsiveness and drug release profile
The release profile of PTX-loaded formulations was conducted in PBS buffer at different pH (pH 5.0, 6.5 or 7.4). In brief, PTX-loaded MSNs and MSNs-GOx/PLL/HA were introduced into a dialysis bag (MWCO: 1000 Da) and immersed in 50 mL of PBS containing 0.5% (w/v) Tween 80. The pH values were adjusted to 6.5 or 5.0 by adding diluted HCl in the solutions. The nanoparticles were kept at 37°C and shaken at a speed of 100 rpm. At predetermined times, a 1 mL sample was collected and PTX concentration measured by HPLC.
Cellular uptake
To investigate the cellular uptake, HepG2 cells were seeded in 24-well plates and cultured for 24 hours. Subsequently, cells were treated with C6-labeled formulations for 1, 2, 4, 8, 12, and 24 hours, and uptake assessed by flow cytometry (BD, Franklin Lakes, NJ, USA).
The real time observation of the cellular internalization process was carried out on confocal laser scanning microscopy (Carl Zeiss LSM 700, Carl Zeiss Meditec AG, Jena, Germany). Briefly, HepG2 cells were cultivated in a CLSM dish, and then incubated with C6 loaded MSNs-GOx/PLL/HA for 1, 2, 4, and 8 hours, respectively. Then, the original medium was discarded and washed with PBS to remove residual nanoparticles. Afterwards, cells were fixed with 4% paraformaldehyde and DAPI was applied to stain the nuclei, and visualized by confocal laser scanning microscopy (CLSM).
The endocytosis pathway and intracellular trafficking assay
To investigate the endocytosis pathway of MSNs-GOx/PLL/HA, HepG2 cells were treated with specific endocytosis inhibitors.23 In brief, cells were cultured in a CLSM dish and then treated with chlorpromazine hydrochloride (10 mg/mL, clathrin-mediated endocytosis inhibition), nystatin (15 μg/mL, caveolin-mediated endocytosis inhibition), amiloride (100 μg/mL, macropinocytosis inhibition) and sodium azide (3 μg/mL, ATP synthesis inhibition) for 1 hour. Subsequently, C6-labeled nanoparticles were added and incubated for another 4 hours. Then, cells were fixed with 4% paraformaldehyde, and nuclei were stained with DAPI and imaged by CLSM. For quantitative studies, cells were collected for further assessment by flow cytometry. In addition, the viability of cells incubated with the above inhibitors for 4 hours was also measured.
The intracellular transport of nanoparticles such as endoysosomal escape was investigated by CLSM. Briefly, HepG2 cells were cultured in CLSM dish, and then treated with C6-loaded nanoparticles for 1 hour, and washed with cold PBS to remove the residual nanoparticles. Subsequently, cells were further incubated with DMEM media for another 0, 2, or 4 hours and stained with Lyso-Tracker Red (Beyotime, Nanjing, China). After washing with PBS, samples were immediately visualized using CLSM.
Target efficacy of nanoparticles
The CD44 expression levels in HepG2 and MCF-7 cells were detected by flow cytometry. Briefly, cells were cultured in 24-well plates, and then washed with blocking buffer containing of 0.2% (v/v) FBS and 0.02% (v/v) sodium azide, and incubated with fluorescein isothiocyanate (FITC)-conjugated anti-mouse CD44 (20 mL, BD) and FITC-conjugated mouse IgG (20 mL, BD) at 4°C for 30 minutes. Subsequently, cells were treated with 200 μL of 4% paraformaldehyde, and analyzed by flow cytometry.
The CD44-mediated active tumor targeting effect was further verified by CLSM. In brief, HepG2 and MCF-7 cells were treated with C6-labeled MSNs-GOx/PLL/HA nanoparticles for 2 hours. Then, cells were fixed with 4% paraformaldehyde, and nuclei were stained with DAPI. For CD44 blocking experiments, cells were precultured with 10 mg/mL of HA in advance, and then incubated with MSNs-GOx/PLL/HA for 2 hours. Furthermore, the HA-coated nanoparticles were pretreated with HAase (1 mg/mL) for 2 hours, and then cells were incubated with the HAase-treated MSNs-GOx/PLL/HA nanoparticles for 2 hours. Cells were then treated following the same procedure as described above, and monitored using CLSM. Finally, the relative fluorescence intensity was also calculated.
Measurement of intracellular H2O2 concentration
The intracellular H2O2 concentration was detected by the BES-H2O2-Ac (Wako Pure Chemical Industries, Ltd, Osaka, Japan), a cell-permeable specific H2O2 fluorescent probe.24,25 The cells were cultured in 24-well plates, and then 50 µg/mL MSNs, MSNs-GOx, MSNs-GOx/PLL and MSNs-GOx/PLL/HA in glucose-free DMEM media were added into each well for 6 hours, respectively. The pure glucose-free DMEM media were also added as control. Then cells were washed with PBS and further incubated with 1 μM BES-H2O2-Ac for the detection of H2O2 in intracellular. Finally, the intracellular fluorescent intensity was monitored by flow cytometry.
To investigate the effect of glucose concentration on the amount of intracellular H2O2, cells were treated with MSNs-GOx/PLL/HA in different concentrations (50, 100, 200, 400, 800 µg/mL) of glucose-containing DMEM medium for 6 hours. The remaining preparation followed the same procedures as above.
The Hydrogen Peroxide Assay Kit (Beyotime) was applied for further quantitative detection of intracellular H2O2 level. The cells were cultured in 6-well plates, and then 50 µg/mL MSNs-GOx/PLL/HA in glucose-free DMEM or in different concentrations (50, 100, 200, 400, 800 µg/mL) of glucose-containing DMEM were added into each well. The pure glucose-free DMEM medium was also added as control. After 6 hours of incubation, cells were collected and quantified the intracellular H2O2 concentration following the manufacturer’s instructions.
In vitro synergistic antitumor assessment
An MTT assay was applied for evaluation of glucose on HepG2 cells proliferation. Briefly, cells were seeded in 96-well plates at a density of 5×103 cells/well and incubated for 24 hours. Then different concentrations of glucose-containing DMEM media were added and incubated for 24 hours. Subsequently, cells were incubated with 20 μL of MTT for 4 hours and 150 μL of DMSO. The absorbance was measured at 570 nm by a microplate reader. The viability of H2O2 against HepG2 cells was also evaluated. Moreover, the cytotoxicity of blank MSNs against HepG2 cells was also calculated.
In vitro synergistic antitumor effect of PTX-loaded MSNs, MSNs-GOx, MSNs-GOx/PLL and MSNs-GOx/PLL/HA was also studied in different concentrations of glucose-containing medium by MTT method. Briefly, 20 μg/mL samples in glucose-free DMEM media or 100 μg/mL glucose-containing DMEM media were added to each well and incubated with cells for 24 hours. The remaining procedures were similar to those described above. Additionally, in vitro antitumor efficacy of different concentration of PTX-loaded MSNs-GOx/PLL/HA in various glucose-containing DMEM media was also assessed.
Furthermore, in vitro observation of live/dead cells after different treatments was also studied. Briefly, cells were cultured in 24-well plates, and then incubated with MSNs, MSNs-GOx, MSNs-GOx/PLL and MSNs-GOx/PLL/HA (PTX, 2 μg/mL) in glucose-free DMEM media or 100 μg/mL glucose-containing DMEM media for 6 hours. Subsequently, cells were stained with calcein AM and propidium iodide (PI), according to Live and Dead Viability Assay Kit (KeyGEN Biotech, Nanjing, China), and captured by fluorescence microscope (Olympus Corporation, Osaka, Japan).
Cell apoptosis and mitochondrial membrane potential
The effect of nanoparticles on cell apoptosis was further investigated through the Hoechst and PI double staining method. Cells were cultured in 12-well plates, and then treated with different nanoparticles (PTX, 2 μg/mL) for 8 hours. The remaining procedure was in accordance with the Apoptosis and Necrosis Assay Kit (Beyotime) and imaged by fluorescence microscope.
The apoptosis-inducing capability of nanoparticles was further evaluated by Annexin V-FITC/PI Apoptosis Detection Kit (Beyotime). Briefly, cells were cultured in 12-well plates, and then treated with different nanoparticles (PTX, 2 μg/mL) for 8 hours. The following process was in accordance with the manufacturer’s protocol and monitored by flow cytometry. In addition, the apoptosis-inducing capability of MSNs-GOx/PLL/HA in different concentrations of glucose-containing media was also studied.
The mitochondrial membrane potential (ΔΨm) was detected by the Mitochondrial Membrane potential Assay Kit with JC-1 (Beyotime). HepG2 cells (5×105 cells/well) dispersed in 6-well plates were incubated with different nanoparticles (PTX, 2 μg/mL) for 8 hours. The remaining procedure was in accordance with the kit manufacturer's protocol and analyzed by flow cytometry. Changes in mitochondrial membrane potential were analyzed by the following equations [22]:


Cell cycle analysis
Cell cycle distribution was studied by Cell Cycle Analysis Kit (Beyotime). HepG2 cells were cultured into 6-well plates, and then treated with different nanoparticles (PTX, 200 ng/mL) in 100 μg/mL glucose-containing DMEM media for 24 hours. Subsequently, cells were collected and fixed with 70% ethanol at −20°C for 12 hours, and re-suspended into 0.5 mL PI staining solution for 30 minutes at 37°C in dark. The other procedure was in accordance with the kit manufacturer's protocol and analyzed by flow cytometry.
In vivo pharmacokinetics
Sprague Dawley rats (180–220 g) were randomly divided into four groups (N=6) and intravenously administered with taxol, MSNs, MSNs-GOx and MSNs-GOx/PLL/HA nanoparticles with PTX at a dose of 2.5 mg/kg. At the time point of 0.083, 0.167, 0.25, 0.33, 0.5, 0.75, 1, 2, 4, 8, and 12 hours following injection, blood samples were collected and then transferred to a 10 mL centrifuge tube and extracted with 5 mL of methyl tert-butyl ether with 20 μL internal standard (diazepam, 500 ng/mL). The organic layer was collected, dried at 37°C, and the residue was then reconstituted with 100 μL methanol. 20 μL of the sample was injected into the HPLC system for PTX quantification. Pharmacokinetic parameters were obtained using a non-compartmental model by PK SOLVER (Nanjing, China).
In vivo biodistribution study
In vivo biodistribution of the preparations was studied in Kunming SPF mice. Tumor size was monitored with a Vernier caliper, and calculated as (a×b2)/2, where a and b are the major axis and minor axis. The mice were imaged at the predetermined time after intravenous injection DiR-labeled MSNs and MSNs-GOx/PLL/HA or free DiR, at DiR dose of 0.2 mg/kg. In the HA competition experiment, animals were pre-injected with 10 mg/kg of free HA 30 minutes before injected with MSNs-GOx/PLL/HA. At the established intervals, images were captured by an in vivo imaging system (DXS 4000PRO; Kodak, Rochester, NY, USA). After 24 hours of live imaging, mice were then sacrificed, and major organs as well as tumors were acquired and analyzed by the same method.
In vivo synergistic antitumor efficacy and security evaluation
Tumor-bearing mice were divided into five groups (N=6) and injected with different preparations every other day (total seven injections) that contained 7.5 mg/kg PTX. Saline was used as a negative control, and taxol as a positive control. The tumor size and body weight were recorded every day during the treatment period. At day 13, tumors and major organs, including heart, liver, spleen, lung, and kidney were also collected for histopathology analyses with H&E staining.
Tumor-bearing mice were randomly divided into five groups and administered with MSNs, MSNs-GOx, MSNs-GOx/PLL/HA (PTX, 7.5 mg/kg). Saline was used as a negative control, and taxol as a positive control. In 6 hours of post-injection, tumors were excised and weighted. Subsequently, hydrogen peroxide assay lysate was added for full homogenization, followed by centrifugation at 12,000 g for 5 minutes to obtain supernatant for further detection. The following process was in accordance with the manufacturer’s protocol of Hydrogen Peroxide Assay Kit (Beyotime).
Statistical analysis
Data are expressed as the mean ±SD. Differences between the groups were assessed by one-way ANOVA, and a P-value less than 0.05 was considered significant.
Results and discussion
Characterization of mesoporous silica nanoparticles
FTIR spectroscopy was used to confirm the synthesis of MSNs and MSNs-CHO. As shown in Figure 2A, the observed absorbance bands at 1,086 cm−1 was characteristic for the asymmetrical stretching vibration of Si-O-Si, and the absorption peak at 962 and 799 cm−1 were assigned to symmetric stretching vibration of Si-O-Si. The presence of a typical C=O stretching vibration of benzaldehyde at 1,692 cm−1 in MSNs-CHO confirmed the success of benzaldehyde functionalization. The new emerging peak at 676 cm−1 in MSNs-CHO was also a characteristic peak of the phenyl group. Then, the mesoporous characteristic of MSNs and MSNs-CHO was confirmed by SAXRD, N2 adsorption-desorption isotherms, and TEM analysis. The N2 adsorption-desorption isotherm revealed that MSNs possessed relatively high specific surface areas (463.95 m2/g) and well-defined pore sizes (2.51 nm) and pore volume (1.08 mL/g) (Figure 2C), which could also be confirmed by the absence of corresponding SAXRD peak(s) (Figure 2B).19 In comparison, MSNs-CHO had a slight decrease on specific surface area of (393.61 m2/g) and pore size (2.16 nm), and the pore volume was 0.982 mL/g. Moreover, the TEM results showed that the particle sizes of MSNs-CHO were all about 40 nm, which showed no obvious change from MSNs (Figure 2E), indicating that the success of benzaldehyde functionalization did not obviously alter the overall morphology of the nanoparticles.
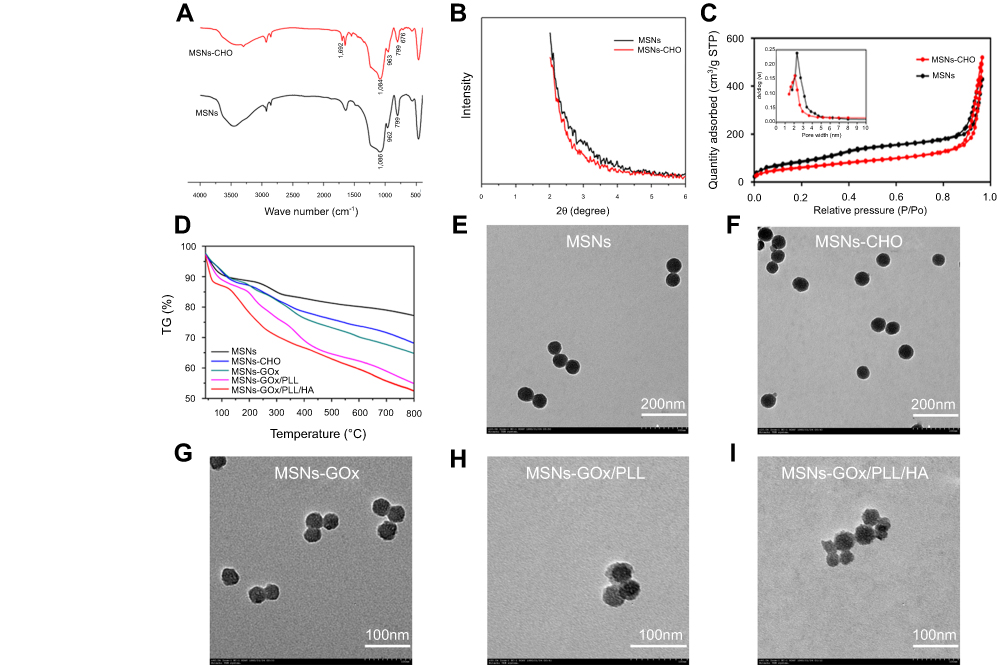 | Figure 2 (A) FTIR spectra of MSNs and MSNs-CHO. (B) SAXRD pattern of MSNs and MSNs-CHO. (C) Nitrogen adsorption-desorption isotherms and pore diameter distribution of MSNs and MSNs-CHO. (D) TGA curves of MSNs, MSNs-CHO, MSNs-GOx, MSNs-GOx/PLL and MSNs-GOx/PLL/HA. TEM of (E) MSNs (F), MSNs-CHO (G), MSNs-GOx (H), MSNs-GOx/PLL (I) and MSNs-GOx/PLL/HA.Abbreviations: MSNs, mesoporous silica nanoparticles; MSNs-CHO, benzaldehyde-functionalized mesoporous silica nanoparticles; GOx, glucose oxidase; PLL, poly (L-lysine); HA, hyaluronic acid; FTIR, Fourier transform infrared; SAXRD, small-angle X-ray diffraction; TGA, thermogravimetric analysis; TEM, transmission electron microscopy. |
Next, biofunctionalized PLL was also introduced onto the surfaces of the MSNs via benzoic-imine bond, which served as pH-responsive “gatekeepers” and endolysosomal escape materials. HA was coating the surfaces of the MSNs through electronic interaction. As shown in
Catalytic ability measurement
The concentration of reacted GOx was determined by the BCA Protein Assay Kit and the loading capacity of GOx was calculated as 7.63 wt%, and the result was in accordance with the analysis of TGA (Figure 2D).21 In addition, based on GOx-catalyzed decomposition reaction of glucose, we incubated free GOx and MSNs-GOx/PLL/HA with glucose solutions, and the results showed that free GOx and MSNs-GOx/PLL/HA could all effectively catalyze the conversion of glucose, leading to the quick decreasing in pH and the increasing of H2O2 concentration, which indicated the high catalytic capability of GOx (Figure 3A and B).27 Therefore, it concluded that GOx could still keep its activity after immobilization.
 | Figure 3 (A) The generated H2O2 concentration and pH value (B) arising from the reaction between free GOx and MSNs-GOx/PLL/HA in glucose medium. In vitro PTX release profiles from MSNs (C) and MSNs-GOx/PLL/HA (D) at different pH values via a stepwise acidification method. (E) Cellular uptake of C6-labeled different nanoparticles. Results are expressed as mean fluorescence values determined by flow cytometry after 1, 2, 4, 8, 12 and 24 hour of incubation. Significant difference from MSNs-C6: ***P<0.001. (F) Real time confocal microscopy images of HepG2 after incubation with C6 labeled MSNs-GOx/PLL/HA.Abbreviations: GOx, glucose oxidase; MSNs, mesoporous silica nanoparticles; MSNs-CHO, benzaldehyde-functionalized mesoporous silica nanoparticles; PLL, poly (L-lysine); HA, hyaluronic acid; C6, coumarin. |
pH-dependent drug release
An in vitro release experiment was conducted to study whether MSNs-GOx/PLL/HA would respond well to the pH change and serve as a pH-responsive system for efficient drug delivery. For comparison, the release of PTX from uncoated MSNs was also measured. As illustrated in Figure 3C, MSNs showed an obvious burst release with more than 55% PTX release at pH 7.4 within 12 hours. The premature release of cargo may result in off-target toxicity to normal bystander cells. In contrast, MSNs-GOx/PLL/HA was more stable in a simulated body fluid (pH 7.4), and the drug release amount was less than 15% within 24 hours, indicating an effectively controlled capability by the supported polymers layer under physiological conditions (Figure 3D). However, drug release from MSNs-GOx/PLL/HA showed an obvious increase by a stepwise addition of diluted HCl into the medium, and acidification of the medium to pH 6.5 (simulating tumor microenvironment) and 5.0 (simulating the endosomal-lysosomal drug delivery system in tumor cell) gave rise to an increased release to 42% and 78%, respectively. This phenomenon could be attributed to pH-induced cleavage of benzoic-imine bond and consequently elimination of the surface modification polymer material on the pore outlets. The above results demonstrated that MSNs-GOx/PLL/HA presented a typical pH-dependent drug release behavior, which would facilitate selective drug release within tumor cells.
Cellular uptake and endocytosis pathway
Efficient cellular internalization of nanoparticles is important for drug delivery and efficient therapy.28 As displayed in Figure 3E, the mean fluorescence intensity between MSNs and MSNs-GOx showed no obvious difference. In contrast, the signal of MSNs-GOx/PLL and MSNs-GOx/PLL/HA was significantly increased, and fluorescence intensity amounted to 3.1- and 4.9-fold higher than that of MSNs preparation. Two factors might be responsible for the above results.On the one hand, MSNs-GOx/PLL with a highly positive charged surface could be beneficial for negatively charged membrane absorptions. On the other hand, the CD44-mediated endocytosis based on the active targeting effect of HA played an important role on enhanced uptake of nanoparticles compared to the electronic adsorptive endocytosis.29 In addition, CLSM results illustrated that nanoparticles could be effective uptake into cells, and fluorescence intensity increased with incubation time, suggesting a time-dependent cellular uptake (Figure 3F).
Subsequently, the endocytosis pathway of MSNs-GOx/PLL/HA was also studied. As shown in Figure 4A, cell viabilities were all above 90% after treatment with different inhibitors, implying the safety of inhibitors on cells. In addition, NaN3 reduced cellular uptake of nanoparticles by about 42% (Figure 4B), while chlorpromazine and nystatin lowered the cellular uptake by about 33% and 30%. The qualitative results from CLSM (Figure 4C) were in accordance with the analysis by flow cytometry. Therefore, these results illustrated that the internalization of MSNs-GOx/PLL/HA was an energy-dependent process, and clathrin-mediated endocytosis as well as caveolin-mediated endocytosis all played an important role in the uptake of nanoparticles.
 | Figure 4 (A) Viability of HepG2 cells treated with different inhibitors. (B) Effects of inhibitors on endocytosis in HepG2 cells. Significant difference from control: **P<0.01, ***P<0.001. (C) Confocal microscope images of HepG2 cells after pre-incubated with different inhibitors. Scale bar: 10 μm. (D) Confocal microscope images of HepG2 cells for intracellular delivery of C6-labeled MSNs, MSNs-GOx, MSNs-GOx/PLL and MSNs-GOx/PLL/HA for different times. The late endosomes and lysosomes were stained by Lyso-Tracker red. 1: overlay of 1, 2 and 3; 2: green fluorescent of C6-labeled nanoparticles; 3: red fluorescent of endo/lysosomes; 4: bright field of cells; 1 hour: incubation with nanoparticles for 1 hour, followed by imaged by CLSM; 1 +2 hours: incubation with nanoparticles for 1 hour, followed by washing and further incubation for 2 hours; 1 +4 hours: incubation with nanoparticles for 1 hour, followed by washing and further incubation for 4 hours; Scale bar: 10 μm.Abbreviations: GOx, glucose oxidase; MSNs, mesoporous silica nanoparticles; MSNs-CHO, benzaldehyde-functionalized mesoporous silica nanoparticles; PLL, poly (L-lysine); HA, hyaluronic acid; C6, coumarin; CLSM, confocal laser scanning microscopy. |
Intracellular trafficking
The endolysosomal degradation pathway was considered as a major obstacle for drug delivery, and it reported that extracellular cargo processed by clathrin-mediated endocytosis was then trafficked to the endolysosomal drug delivery system.30–32 Thus, CLSM was applied to monitor intracellular trafficking and endosomal escape of nanoparticles. As shown in Figure 4D, after 1 hour of incubation, colocalization of green C6-labeled nanoparticles and red Lyso-Tracker was observed in all groups, illustrating that nanoparticles could entrap endosomes at an early stage of cellular uptake.17 However, the merged yellow signal showed no significant change in MSNs and MSNs-GOx treatment after further 4 hours of incubation. In contrast, the merged signal in MSNs-GOx/PLL and MSNs-GOx/PLL/HA groups was significantly reduced after 2 hours incubation and a minimal amount of colocalization was observed after further 4 hours, indicating the endosomal escape of both nanoparticles through the proton sponge effect of PLL. Accordingly, the results confirmed that MSNs-GOx/PLL/HA nanoparticles would facilitate endosomal escape through the proton sponge effect, which was beneficial for efficient drug delivery.33
Target efficacy of nanoparticles
To investigate the affinity of MSNs-GOx/PLL/HA for CD44 receptor on cancer cells, CD44 expression levels on HepG2 and MCF-7 cells were studied. The result confirmed that HepG2 cells highly overexpressed CD44 receptor, while MCF-7 cells showed a low level of CD44 (Figure 5A). Thereafter, the CD44-mediated internalization was verified by CLSM. As shown in Figure 5B, when cells were treated with MSNs-GOx/PLL/HA for 2 hours, a visible green fluorescence was detected in HepG2 cells, while an insignificant C6 signal was observed in MCF-7 cells. Moreover, to further study the interaction between CD44 and HA, two cell lines were pretreated with excess HA to block the CD44 receptor on cell membrane. The results showed that fluorescence exhibited significant decrease in HepG2 cells compared to that without treatment with HA, while no obvious change was detected in MCF-7 cells, which further confirmed that MSNs-GOx/PLL/HA could be taken up by tumor cells via CD44-mediated internalization. In addition, we also incubated MSNs-GOx/PLL/HA with HAase to degrade HA shell of nanoparticles, followed by study of the cellular uptake. The fluorescence showed a dramatic reduction compared to that of untreated cells, and then the green signal was further quantified by CLSM (Figure 5C and D). These results all indicated that a CD44-mediated endocytosis pathway driven by CD44-HA affinity played a vital role in the uptake of MSNs-GOx/PLL/HA nanoparticles.
 | Figure 5 (A) The CD44 expression levels of HepG2 and MCF-7 cells were determined by flow cytometry. (B) The CD44-specific uptake in HepG2 and MCF-7 cells and (C) Z-line fluorescence intensity of C6 were determined by confocal laser scanning microscopy (Scale bar: 10 μm). The distances are indicated by the green lines in (B). (D) Relative fluorescence intensity based on (C); I: HepG2 (MSNs-GOx/PLL/HA), II: HepG2 (blocked), III: HepG2 (HAase), IV: MCF-7 (MSNs-GOx/PLL/HA), V: MCF-7 (blocked), VI: MCF-7 (HAase).Abbreviations: MSNs, mesoporous silica nanoparticles; GOx, glucose oxidase; PLL, poly (L-lysine); HA, hyaluronic acid; HAase, hyaluronidase; C6, coumarin. |
Measurement of intracellular H2O2 concentration
To demonstrate MSNs-GOx/PLL/HA could elevate the endogenous H2O2 level, a cell-permeable specific H2O2 fluorescent probe (BES-H2O2-Ac) was used to assess H2O2 generation on HepG2 cells.24,25 As shown in Figure 6A, a larger amount of H2O2 was released from GOx-loaded nanoparticles, indicating that GOx could oxidize intracellular glucose to produce H2O2. Meanwhile, MSNs-GOx/PLL/HA treatment showed a higher fluorescence intensity than other groups, which might be attributed to the active effect of the HA shell on increasing the intracellular accumulation of GOx. Subsequently, to further study the effect of glucose concentration on intracellular H2O2 release, cells were incubated with MSNs-GOx/PLL/HA in various levels of glucose-containing medium (Figure 6B). As expected, intracellular H2O2 level was elevated with increased glucose concentrations, suggesting a glucose-dependent H2O2 release. In addition, quantitative determination of H2O2 in cells was also studied and the results were in agreement with flow cytometry analysis (Figure 6C and D). Therefore, we could infer that MSNs-GOx/PLL/HA would effectively trigger the consumption of intracellular glucose, leading to interruption of the energy supply, and also elevate the endogenous H2O2 level, inducing stronger intratumoral cytotoxicity.
 | Figure 6 (A) Flow cytometry analysis of intracellular H2O2 generation (using the BES-H2O2-Ac probe) in HepG2 cells after being treated with different preparations. (B) The analysis of intracellular H2O2 generation after incubation with MSNs-GOx/PLL/HA in different concentrations (50, 100, 200, 400, 800 μg/mL) of glucose-containing DMEM media. (C) The quantitative determination of H2O2 level in HepG2 cells after MSNs-GOx/PLL/HA treatment in glucose-free DMEM media. (D) The quantitative determination of H2O2 level in HepG2 cells after incubation with MSNs-GOx/PLL/HA in different concentrations (50, 100, 200, 400, 800 μg/mL) of glucose-containing DMEM media.Abbreviations: MSNs, mesoporous silica nanoparticles; GOx, glucose oxidase; PLL, poly (L-lysine); HA, hyaluronic acid. |
In vitro synergistic antitumor efficacy
To confirm the synergistic antitumor efficacy caused by GOx-mediated consumption of intracellular glucose and the elevation of the endogenous H2O2 level as well as the combination with chemotherapy, a series of cell viability experiments were performed by different methods. As presented in Figure 7A, during the tumor growth, glucose serves as an energy supplier to promote cancerous cell proliferation, while a high concentration of H2O2 would lead to significant cell death (Figure 7B). Thus, the starvation-like therapy via GOx-triggered decomposition of glucose into toxic H2O2 was a feasible strategy, which would produce a much stronger anticancer effect than conventional starving therapy that only blocked the glucose supply. Subsequently, the good safety of blank MSNs was confirmed by MTT assay (
 | Figure 7 Viabilities of HepG2 cells after 24 hours of incubation with different concentrations of glucose (A) and H2O2 (B). (C) The cell viability after different treatments in the absence and presence of 100 mg/mL glucose. (D) The cell viability after 24 hours of incubation with MSNs-GOx/PLL/HA (20 or 40 μg/mL) in different concentrations (0, 50, 100, 200 μg/mL) of glucose-containing DMEM media. Significant difference from control: *P<0.05, **P<0.01, ***P<0.001. (E) Fluorescent images of live and dead HepG2 cells after different treatments in glucose-free DMEM media and 100 μg/mL glucose-containing DMEM media and stained by calcein AM and PI, respectively.Abbreviations: MSNs, mesoporous silica nanoparticles; GOx, glucose oxidase; PLL, poly (L-lysine); HA, hyaluronic acid; AM, acetoxymethyl; PI, propidium iodide. |
Cell apoptosis and mitochondrial membrane potential
To further demonstrate synergistic antitumor efficacy, the apoptosis-inducing capability of different nanoparticles was assessed by the Hoechst and PI double staining method, and morphological changes in cell nuclei were captured by fluorescence microscope. The blue fluorescent Hoechst 33,342 was a cell permeable nucleic acid dye usually used to identify chromatin condensation and fragmentation by staining the condensed nuclei of apoptotic cells. The red fluorescent PI was a cell impairment DNA-binding dye, which would stain cells as the plasma membrane permeability increased and plasma membrane integrity lost.34 Therefore, cells in late apoptosis or necrosis would show pink fluorescence after staining. As observed in Figure 8A, the nuclei of untreated cells appeared to homogeneous fluorescence with no evidence of segmentation and fragmentation after Hoechst staining, while cell nuclei became severely fragmented after being treated with different preparations, illustrating that the nuclei were segmented into dense nuclear parts and then distributed into apoptotic bodies.35 The PI staining for late apoptotic cells only displayed the homogeneous fluorescence, which was consistent with the cell death process.29 Moreover, the quantitative analysis of apoptosis was assessed by the Annexin V-FITC/PI method. The apoptotic rate of the control cells could be negligible, and the percentage of early and late apoptotic cells of the taxol group was 19%. After treatment with GOx modified nanoparticles, the apoptotic rate was clearly increased, especially MSNs-GOx/PLL/HA group (Figure 8B). Meanwhile, the apoptotic effect was further improved with increased glucose concentrations (Figure 8C). Thus, the above results all directly confirmed that GOx played an irreplaceable role in enhancing synergistic antineoplastic therapy.
 | Figure 8 (A) The morphological changes of HepG2 cells detected with dual staining of Hoechst 33,342/PI and captured by fluorescence microscope (magnification 200x). Arrows indicate (1) viable cells with normal nuclei; (2) live cells with apoptotic nuclei; (3) dead cells with normal nuclei; and (4) dead cells with apoptotic nuclei. (B) Apoptosis rate of HepG2 cells was determined by Annexin V-FITC/PI staining. (C) Apoptosis rate of HepG2 cells was analyzed by Annexin V-FITC/PI staining after treatment with MSNs-GOx/PLL/HA in in different concentrations (50, 100, 200, 400, 800 μg/mL) of glucose. (D) Change in mitochondrial membrane potential ΔΨm of HepG2 cells treated with various preparations for 8 hours analyzed by flow cytometry. Significant difference from control: *P<0.05, **P<0.01, ***P<0.001. (E) Effects of treatment with different preparations on the cell cycle of HepG2 cells. (F) The percent of cell cycle distribution after treatment with various formulations for 24 hours. (G) Plasma concentration-time curves of taxol and PTX-loaded nanoparticles in rats after intravenous administration of 2.5 mg/kg PTX (mean ±SD, N=6).Abbreviations: PI, propidium iodide; FITC, fluorescein isothiocyanate; MSNs, mesoporous silica nanoparticles; GOx, glucose oxidase; PLL, poly (L-lysine); HA, hyaluronic acid. |
Moreover, mitochondrial membrane potential (ΔΨm) is considered an important parameter of mitochondria function and the loss of ΔΨm is regarded as a hallmark of cell apoptosis.36 Therefore, the apoptotic effect was further confirmed by morphological membrane potential changes. As shown in Figure 8D, compared with negative control, relative ΔΨm value of the MSNs-GOx/PLL/HA group presented a significant decrease, and results were in agreement with cell apoptotic analysis.
Cell cycle analysis
As reported that the main mechanism of PTX involves inhibition of the microtubules depolymerization leading to formation of stable microtubules and inhibition of spindle formation during late G2 phase and M phase of the cell cycle.37 Therefore, the cell cycle progression of HepG2 cells after being treated with different preparations was further evaluated by monitoring DNA content through flow cytometry. As shown in Figure 8E, all the formulations induced the arrest of cell growth at different levels, with MSNs-GOx/PLL/HA exhibiting a superior efficacious G2/M phase arrest, which was in accordance with the in vitro apoptotic results (Figure 8F).
In vivo pharmacokinetics
The mean plasma concentration versus time profiles were plotted (Figure 8G) and corresponding pharmacokinetic parameters were summarized in
In vivo tumor targeting and biodistribution
To demonstrate tumor targeting ability, mice were administrated with different preparations, and monitored whole-body fluorescence biodistribution by an in vivo imaging method. As shown in Figure 9A, intense fluorescence signal was rapidly detected in liver 1 hour post-injection, demonstrating that nanoparticles could be rapidly distributed and metabolized by the liver.38 Moreover, as time progressed, MSNs-GOx/PLL/HA could efficiently accumulate at the tumor site, and the fluorescence signal was maintained for up to 24 hours. By comparison, almost no signal was detected in the tumor site 12 hours post-injection with free DiR. To further verify the active targeting effect of HA shell of nanoparticles, excess free HA solution was pre-injected for competitively blocking the CD44 receptors on the tumor cells, followed by injection with MSNs-GOx/PLL/HA. As expected, the DiR signal showed a significant reduction at the tumor site, indicating that the HA shell not only improved the biostability of nanoparticles in circulatory system, but also endowed with the active tumor targeting ability.39 Subsequently, the mice were sacrificed, and tumors as well as major organs were excised for ex vivo imaging, which further verified the in vivo biodistribution results (Figure 9B and C). Therefore, these results confirmed that MSNs-GOx/PLL/HA was a promising platform to increase the accumulation of cargo in tumor sites.
 | Figure 9 (A) In vivo fluorescence imaging of Heps tumor-bearing mice after intravenous injection with free DiR (I), DiR-loaded MSNs (II), MSNs-GOx/PLL/HA (III) and MSNs-GOx/PLL/HA with pre-injection of the free HA (10 mg/kg) for 1 hour (IV). (B) Fluorescence images of major organs and tumor ex vivo 24 hours post-injection, including heart, liver, spleen, lung, kidney, and tumor. (C) Fluorescence intensity of the DiR signals in different tissues harvested from the mice at 24 hours post-injection. (D) Tumor growth curves. (E) Body weight changes. (F) Average tumor weight after the study. (G) The change of H2O2 concentrations in Heps tumors at 6 hours after injection with different preparation. Data were shown as mean ±SD, (N=6). Significant difference from control: *P<0.05, **P<0.01, ***P<0.001. (H) Representative images of the H&E-stained tumor sections after treatment with different nanoparticles. Black arrows indicate examples of positive tumor suppression with H&E staining. The images were taken at 200× magnification. Abbreviations: MSNs, mesoporous silica nanoparticles; GOx, glucose oxidase; PLL, poly (L-lysine); HA, hyaluronic acid. |
In vivo synergistic antitumor efficacy and security evaluation
In vivo synergistic cancer starvation and chemistry therapy of the drug-loaded nanoparticles was further evaluated in Heps tumor bearing mice with saline as negative control and taxol as positive control. As shown in Figure 9D, tumor volume of saline group increased by 20-fold compared to that on the first day of treatment. Meanwhile, PTX-loaded MSNs exhibited improved tumor suppression in comparison to saline, while there was no significant difference from the taxol group. In comparison, significant delays in tumor growth were detected in drug and GOx co-delivery of MSNs-GOx and MSNs-GOx/PLL/HA, especially the MSNs-GOx/PLL/HA group. Furthermore, MSNs-GOx and MSNs-GOx/PLL/HA (Figure 9G) all caused a drastic increase in intratumoral H2O2 concentration, which further confirmed that GOx-modified nanoparticles could consume the intratumoral glucose for H2O2 generation, leading to a significantly synergistic effect in shrinking tumors (
Moreover, at the end of experiment, average tumor weight from the saline group was 2.03 g, while that from MSNs-GOx/PLL/HA group was only 0.21 g (Figure 9F); there were significant differences among the two groups. In contrast, taxol and MSNs seemed to be less effective in inhibiting the tumor growth, which could be attributed to the limited intratumoral H2O2 concentration that lead to a relatively low therapy efficacy. Body weight was another evaluation parameter to check for adverse effects of the testing preparations.22 As shown in Figure 9E, mice in the saline or nanoparticles groups all showed no weight loss for the duration of the study, while taxol treatment showed a significant reduction, demonstrating a satisfactory safety profile for MSNs-GOx/PLL/HA nanoparticles.
Furthermore, at the end of the experiments, the antitumor efficiency was also assessed through histological analysis of tumor tissues by H&E staining method. As shown in Figure 9H, all the GOx containing therapeutic groups showed signs of tumor growth inhibition, especially the MSNs-GOx/PLL/HA treatment, which presented the maximum cell toxicity and the highest level of tumor necrosis.29,38 The patterns of cell apoptosis in tumor tissues were in agreement with the results of the tumor inhibition study. Moreover, pathological changes of major organs were also analyzed. In comparison with saline and taxol, the nanoparticles groups did not induce inflammatory response, cell degeneration, or necrosis in major organs (
Conclusion
In summary, a glucose-responsive MSN was successfully developed and reported for the first time. The novel nanocarriers demonstrated improved cellular accumulation and enhanced tumor targeting specificity via the CD44-mediated internalization. Importantly, the GOx-loaded nanoparticle could effectively decompose the intratumoral glucose to produce toxic H2O2 to inhibit cancerous cells proliferation and further block the energy supply, achieving strong starvation-like antitumor effect. Furthermore, chemotherapy drug was further enhancing the apoptosis capability and maximized the synergistic therapeutic efficacy both in vitro and in vivo. Therefore, combining cancer-targeted starvation with chemistry therapy might provide a potential platform for cancer treatment.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 81373363); Key New Drug Innovation Project from the Ministry of Science and Technology of the People’s Republic of China (No. 2009ZX09310-004); the National Major Scientific and Technological Special Project for ‘‘Significant New Drugs Development” during the Twelfth Five-year Plan Period (2015ZX09501001); Postgraduate Research and Innovation Plan Project in Jiangsu Province (KYLX16_1178), and a Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Ruttala HB, Chitrapriya N, Kaliraj K, et al. Facile construction of bioreducible crosslinked polypeptide micelles for enhanced cancer combination therapy. Acta Biomater. 2017;63:135–149. doi:10.1016/j.actbio.2017.09.002
2. Hu Q, Sun W, Wang C, Gu Z. Recent advances of cocktail chemotherapy by combination drug delivery systems. Adv Drug Deliv Rev. 2016;98:19–34. doi:10.1016/j.addr.2015.10.022
3. Gomes AS, Ramos H, Soares J, Saraiva L. p53 and glucose metabolism: an orchestra to be directed in cancer therapy. Pharmacol Res. 2018;131:75–86. doi:10.1016/j.phrs.2018.03.015
4. DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008;7(1):11–20. doi:10.1016/j.cmet.2007.10.002
5. Zhang C, Ni D, Liu Y, Yao H, Bu W, Shi J. Magnesium silicide nanoparticles as a deoxygenation agent for cancer starvation therapy. Nat Nanotechnol. 2017;12(4):378–386. doi:10.1038/nnano.2016.280
6. Khan AY, Noronha SB, Bandyopadhyaya R. Glucose oxidase enzyme immobilized porous silica for improved performance of a glucose biosensor. Biochem Eng J. 2014;91:78–85. doi:10.1016/j.bej.2014.07.011
7. Chen Q, Liang C, Sun X, et al. H2O2-responsive liposomal nanoprobe for photoacoustic inflammation imaging and tumor theranostics via in vivo chromogenic assay. Proc Natl Acad Sci USA. 2017;114(21):5343–5348. doi:10.1073/pnas.1701976114
8. Lopez-Lazaro M. Dual role of hydrogen peroxide in cancer: possible relevance to cancer chemoprevention and therapy. Cancer Lett. 2007;252(1):1–8. doi:10.1016/j.canlet.2006.10.029
9. Aznar E, Oroval M, Pascual L, Murguia JR, Martinez-Manez R, Sancenon F. Gated materials for on-command release of guest molecules. Chem Rev. 2016;116(2):561–718. doi:10.1021/acs.chemrev.5b00456
10. Bildstein L, Dubernet C, Couvreur P. Prodrug-based intracellular delivery of anticancer agents. Adv Drug Deliv Rev. 2011;63(1–2):3–23. doi:10.1016/j.addr.2010.12.005
11. Yang X, Li Y, Li M, Zhang L, Feng L, Zhang N. Hyaluronic acid-coated nanostructured lipid carriers for targeting paclitaxel to cancer. Cancer Lett. 2013;334(2):338–345. doi:10.1016/j.canlet.2012.07.002
12. Qiu L, Li Z, Qiao M, et al. Self-assembled pH-responsive hyaluronic acid-g-poly(L-histidine) copolymer micelles for targeted intracellular delivery of doxorubicin. Acta Biomater. 2014;10(5):2024–2035. doi:10.1016/j.actbio.2013.12.025
13. Yang JA, Kong WH, Sung DK, et al. Hyaluronic acid-tumor necrosis factor-related apoptosis-inducing ligand conjugate for targeted treatment of liver fibrosis. Acta Biomater. 2015;12:174–182. doi:10.1016/j.actbio.2014.10.002
14. Du X, Yin S, Wang Y, Gu X, Wang G, Li J. Hyaluronic acid-functionalized half-generation of sectorial dendrimers for anticancer drug delivery and enhanced biocompatibility. Carbohydr Polym. 2018;202:513–522. doi:10.1016/j.carbpol.2018.09.015
15. Noh I, Kim HO, Choi J, et al. Co-delivery of paclitaxel and gemcitabine via CD44-targeting nanocarriers as a prodrug with synergistic antitumor activity against human biliary cancer. Biomaterials. 2015;53:763–774. doi:10.1016/j.biomaterials.2015.03.006
16. Sun X, Liu C, Liu D, Li P, Zhang N. Novel biomimetic vectors with endosomal-escape agent enhancing gene transfection efficiency. Int J Pharm. 2012;425(1–2):62–72. doi:10.1016/j.ijpharm.2012.01.010
17. Yin T, Liu J, Zhao Z, et al. Smart nanoparticles with a detachable outer shell for maximized synergistic antitumor efficacy of therapeutics with varying physicochemical properties. J Control Release. 2016;243:54–68. doi:10.1016/j.jconrel.2016.09.036
18. Jiang T, Mo R, Bellotti A, Zhou J, Gu Z. Gel–liposome-mediated co-delivery of anticancer membrane-associated proteins and small-molecule drugs for enhanced therapeutic efficacy. Adv Funct Mater. 2014;24(16):2295–2304. doi:10.1002/adfm.v24.16
19. Pan L, He Q, Liu J, et al. Nuclear-targeted drug delivery of TAT peptide-conjugated monodisperse mesoporous silica nanoparticles. J Am Chem Soc. 2012;134(13):5722–5725. doi:10.1021/ja211035w
20. Cheng J, Liu Q, Shuhendler AJ, Rauth AM, Wu XY. Optimizing the design and in vitro evaluation of bioreactive glucose oxidase-microspheres for enhanced cytotoxicity against multidrug resistant breast cancer cells. Colloids Surf B Biointerfaces. 2015;130:164–172. doi:10.1016/j.colsurfb.2015.04.002
21. Li SY, Cheng H, Xie BR, et al. Cancer cell membrane camouflaged cascade bioreactor for cancer targeted starvation and photodynamic therapy. ACS Nano. 2017;11(7):7006–7018. doi:10.1021/acsnano.7b02533
22. Du X, Yin S, Zhou F, et al. Reduction-sensitive mixed micelles for selective intracellular drug delivery to tumor cells and reversal of multidrug resistance. Int J Pharm. 2018;550(1–2):1–13. doi:10.1016/j.ijpharm.2018.08.019
23. Xiao L, Xiong X, Sun X, et al. Role of cellular uptake in the reversal of multidrug resistance by PEG-b-PLA polymeric micelles. Biomaterials. 2011;32(22):5148–5157. doi:10.1016/j.biomaterials.2011.03.071
24. Maeda H, Fukuyasu Y, Yoshida S, et al. Fluorescent probes for hydrogen peroxide based on a non-oxidative mechanism. Angew Chem Int Ed Engl. 2004;43(18):2389–2391. doi:10.1002/anie.200452381
25. Kanzaki H, Shinohara F, Kajiya M, Fukaya S, Miyamoto Y, Nakamura Y. Nuclear nrf2 induction by protein transduction attenuates osteoclastogenesis. Free Radic Biol Med. 2014;77:239–248. doi:10.1016/j.freeradbiomed.2014.09.006
26. Fan W, Lu N, Huang P, et al. Glucose-responsive sequential generation of hydrogen peroxide and nitric oxide for synergistic cancer starving-like/gas therapy. Angew Chem Int Ed Engl. 2017;56(5):1229–1233. doi:10.1002/anie.201610682
27. Zhang R, Feng L, Dong Z, et al. Glucose & oxygen exhausting liposomes for combined cancer starvation and hypoxia-activated therapy. Biomaterials. 2018;162:123–131. doi:10.1016/j.biomaterials.2018.02.004
28. Zhang X, Li F, Guo S, et al. Biofunctionalized polymer-lipid supported mesoporous silica nanoparticles for release of chemotherapeutics in multidrug resistant cancer cells. Biomaterials. 2014;35(11):3650–3665. doi:10.1016/j.biomaterials.2014.01.013
29. Assanhou AG, Li W, Zhang L, et al. Reversal of multidrug resistance by co-delivery of paclitaxel and lonidamine using a TPGS and hyaluronic acid dual-functionalized liposome for cancer treatment. Biomaterials. 2015;73:284–295. doi:10.1016/j.biomaterials.2015.09.022
30. Aied A, Greiser U, Pandit A, Wang W. Polymer gene delivery: overcoming the obstacles. Drug Discov Today. 2013;18(21–22):1090–1098. doi:10.1016/j.drudis.2013.06.014
31. Bitoque DB, Am RDC, Silva GA. Insights on the intracellular trafficking of PDMAEMA gene therapy vectors. Mater Sci Eng C Mater Biol Appl. 2018;93:277–288.
32. Zhang X, Sun N, Zheng M, Kim KM. Clathrin-mediated endocytosis is responsible for the lysosomal degradation of dopamine D3 receptor. Biochem Biophys Res Commun. 2016;476(4):245–251. doi:10.1016/j.bbrc.2016.05.104
33. Wan Y, Moyle PM, Toth I. Endosome escape strategies for improving the efficacy of oligonucleotide delivery systems. Curr Med Chem. 2015;22(29):3326–3346.
34. Syed Abdul Rahman SN, Abdul Wahab N, Abd MSN. In vitro morphological assessment of apoptosis induced by antiproliferative constituents from the rhizomes of curcuma zedoaria. Evid Based Complement Alternat Med. 2013;2013:5–20. doi:10.1155/2013/257108
35. Ziegler U, Groscurth P. Morphological features of cell death. News Physiol Sci. 2004;19:124–128.
36. Bu H, He X, Zhang Z, Yin Q, Yu H, Li Y. A TPGS-incorporating nanoemulsion of paclitaxel circumvents drug resistance in breast cancer. Int J Pharm. 2014;471(1–2):206–213. doi:10.1016/j.ijpharm.2014.05.039
37. Wang AT, Liang DS, Liu YJ, Qi XR. Roles of ligand and TPGS of micelles in regulating internalization, penetration and accumulation against sensitive or resistant tumor and therapy for multidrug resistant tumors. Biomaterials. 2015;53:160–172. doi:10.1016/j.biomaterials.2015.02.077
38. Yin S, Huai J, Chen X, et al. Intracellular delivery and antitumor effects of a redox-responsive polymeric paclitaxel conjugate based on hyaluronic acid. Acta Biomater. 2015;26:274–285. doi:10.1016/j.actbio.2015.08.029
39. Dong Q, Zhang H, Han Y, et al. Tumor environment differentiated “nanodepot” programmed for site-specific drug shuttling and combinative therapy on metastatic cancer. J Control Release. 2018;283:59–75. doi:10.1016/j.jconrel.2018.05.027
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.