Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 15
Glucocorticoid Receptor α Mediates Roflumilast’s Ability to Restore Dexamethasone Sensitivity in COPD
Authors Reddy AT, Lakshmi SP, Banno A, Reddy RC
Received 7 September 2019
Accepted for publication 29 November 2019
Published 14 January 2020 Volume 2020:15 Pages 125—134
DOI https://doi.org/10.2147/COPD.S230188
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Richard Russell
Aravind T Reddy, 1, 2 Sowmya P Lakshmi, 1, 2 Asoka Banno, 1 Raju C Reddy 1, 2
1Department of Medicine, Division of Pulmonary, Allergy and Critical Care Medicine, University of Pittsburgh School of Medicine, Pittsburgh, PA 15213, USA; 2Veterans Affairs Pittsburgh Healthcare System, Pittsburgh, PA 15240, USA
Correspondence: Raju C Reddy
Department of Medicine, Division of Pulmonary, Allergy and Critical Care Medicine, University of Pittsburgh School of Medicine, 3459 Fifth Avenue, Pittsburgh, PA 15213, USA
Tel +1 412 360-6823
Fax +1 412 360-1919
Email [email protected]
Background: Glucocorticoids are commonly prescribed to treat inflammation of the respiratory system; however, they are mostly ineffective for controlling chronic obstructive pulmonary disease (COPD)-associated inflammation. This study aimed to elucidate the molecular mechanisms responsible for such glucocorticoid inefficacy in COPD, which may be instrumental to providing better patient outcomes. Roflumilast is a selective phosphodiesterase-4 (PDE4) inhibitor with anti-inflammatory properties in severe COPD patients who have a history of exacerbations. Roflumilast has a suggested ability to mitigate glucocorticoid resistance, but the mechanism is unknown.
Methods: To understand the mechanism that mediates roflumilast-induced restoration of glucocorticoid sensitivity in COPD, we tested the role of glucocorticoid receptor α (GRα). Roflumilast’s effects on GRα expression and transcriptional activity were assessed in bronchial epithelial cells from COPD patients.
Results: We found that both GRα expression and activity are downregulated in bronchial epithelial cells from COPD patients and that roflumilast stimulates both GRα mRNA synthesis and GRα’s transcriptional activity in COPD bronchial epithelial cells. We also demonstrate that roflumilast enhances dexamethasone’s ability to suppress pro-inflammatory mediator production, in a GRα-dependent manner.
Discussion: Our findings highlight the significance of roflumilast-induced GRα upregulation for COPD therapeutic strategies by revealing that roflumilast restores glucocorticoid sensitivity by sustaining GRα expression.
Keywords: nuclear hormone receptor, glucocorticoid resistance, phosphodiesterase-4 inhibitor; PDE4 inhibitor, airway inflammation, NF-κB, cAMP response element binding protein; CREB
Introduction
Chronic obstructive pulmonary disease (COPD) is a progressive lung disease characterized by persistent airflow limitation and impaired gas exchange and associated with chronic inflammation and tissue remodeling in the airways.1 It affects millions of people, and morbidity and mortality of COPD are expected to rise in the coming years, with its increasing prevalence.2 Current therapies are insufficient in preventing disease progression and exacerbations.3 Notably, glucocorticoids that effectively treat many inflammatory diseases including those of the respiratory system have proven inadequate in blocking COPD progression.4–6
Understanding of glucocorticoids’ failure to suppress inflammation in COPD4 remains largely incomplete. Among the mechanisms that could potentially account for such glucocorticoid resistance in COPD,7,8 a potential role of glucocorticoid receptor α (GRα) downregulation has yet to be assessed. Supporting this idea, chronic cigarette smoke exposure downregulated GRα expression in mice,9 and COPD patients who were smokers exhibited greater decrements in GRα expression vs. non-smokers than did smokers vs. non-smokers without COPD.9 These findings imply that restoring glucocorticoid receptor expression/function may recover glucocorticoid sensitivity and help enable effective COPD treatment.
Toward achieving this, we explored the effects and mechanism of action of roflumilast (Rofl), a drug indicated for treating severe COPD patients with a history of exacerbations, especially in those with chronic bronchitis.10–12 It is a selective phosphodiesterase-4 (PDE4) inhibitor that provides anti-inflammatory benefits in COPD. Rofl reduced LPS-induced release of various chemokines (CCL2, CCL3, CCL4, CXCL10) and tumor necrosis factor alpha (TNFα) from human lung macrophages in a concentration-dependent manner.13 It also decreased the numbers of pro-inflammatory cells and mediators in sputum of COPD patients in a placebo-controlled study.14 A study examining neutrophils from COPD patients ex vivo suggested that Rofl may restore glucocorticoid sensitivity, as measured by several glucocorticoid resistance markers.15 Post-hoc analysis of two clinical trials also revealed that COPD patients on inhaled corticosteroid were among those who benefited from the addition of Rofl and experienced a reduction in exacerbations.16
In this study, we investigated the relationship between GRα and glucocorticoid resistance in COPD, using human bronchial epithelial (HBE) cells. We found that GRα expression and activity were downregulated in HBE cells from COPD patients (COPD HBE cells), whereas pro-inflammatory NF-κB p65 activity was increased. We also observed that, in addition to stimulating GRα mRNA synthesis and inducing GRα transcriptional activity in COPD HBE cells, Rofl exerted a GRα-dependent additive enhancement of dexamethasone’s (Dex’s) anti-inflammatory action. Together, the results point to a molecular mechanism underlying glucocorticoid resistance, supporting a therapeutic strategy utilizing Rofl and informing the mechanism of Rofl-GRα interaction for effective glucocorticoid-based COPD therapy.
Materials and Methods
Cells and Treatments
HBE cells (Supplemental Table S1; normal HBE [NHBE] and diseased HBE [DHBE; also described as COPD HBE]) obtained from Lonza (Walkersville, MD, USA) were grown and maintained in bronchial epithelial cell medium (Lonza) supplemented with growth factors and hormones, according to the manufacturer’s instructions. These cells were cultured at 37°C in a humidified atmosphere of 5% CO2. Monolayer cultures at 90% confluence were deprived of growth factors prior to treatment.
Treatment with Dex (D4902; Sigma-Aldrich, St. Louis, MO, USA) and Rofl (15141; Cayman Chemical, Ann Arbor, MI, USA), GRα siRNA (SC35505; Santa Cruz Biotechnology, Santa Cruz, CA, USA) transfection, ELISA-based cytokine and chemokine measurement (DTA00D [TNFα] and D8000C [interleukin 8; IL-8]; R&D Systems, Minneapolis, MN, USA), and ELISA-based transcription factor-DNA binding assay (40096 [NFκB p65] and 45496 [GRα]; Active Motif, Carlsbad, CA, USA) have been described previously.17
Western Blotting
Protein concentrations were determined using the BCA Protein Assay kit (Pierce, Rockford, IL, USA). Western blotting was then performed as described.18 Primary antibodies against GRα (8992), Lamin B1 (20682), and β-Actin (1616) were purchased from Santa Cruz Biotechnology. The secondary antibodies, donkey anti-goat IRDye 680RD (926-68074) and goat anti-rabbit IRDye 800CW (925-32211), were obtained from LI-COR Biosciences (Lincoln, NE, USA). The infrared signal was detected with an Odyssey Infrared Imager (LI-COR Biosciences) and quantified by densitometric analysis.
RNA Isolation, Nascent RNA Capturing, and Real-Time PCR
RNA isolation, nascent RNA capturing, and real-time PCR were performed as described.19 Briefly, total RNA was isolated using the RNeasy Plus Mini Kit (Qiagen, Valencia, CA, USA) and nascent RNA transcripts were captured by the Click-iT Nascent RNA Capture Kit (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. A click reaction was performed using 5-EU-labelled mRNA and biotin azide; the mixture was incubated at room temperature for 30 min. Following overnight precipitation at −70°C, a biotin-labeled EU–mRNA-binding pull-down assay was performed using Dynabeads MyOne Streptavidin beads. The bead-bound mRNA was washed and used as a template for reverse transcription using iScript Advanced cDNA Synthesis Kit (Bio-Rad, Hercules, CA, USA). Quantitative real-time PCR was performed with specific primers for GRα and β-actin (Supplemental Table S2) and Fast SYBR Green Master Mix (Applied Biosystems, Foster City, CA, USA). Relative expression normalized to β-actin is presented.
DNA Affinity Precipitation
DNA affinity precipitation was performed as described.18 Briefly, Dynabeads M-280 Streptavidin (Invitrogen, Carlsbad, CA, USA) were mixed with biotinylated oligonucleotides containing the indicated cAMP response elements (CREs) (Supplemental Table S2) and incubated at room temperature for 30 min. Next, the biotinylated oligonucleotide-coupled beads were added to nuclear proteins and incubated for 1 h at room temperature with rotation. Samples were washed three times with cold binding and washing buffer. Bead-bound biotinylated oligonucleotide-protein complexes were eluted and subjected to Western blotting as described above with specific antibodies as indicated. An aliquot of the nuclear sample was set aside without streptavidin bead incubation to be used as an input control.
GRα Reporter Assay
To determine the activation of GRα by test compounds, we performed a GRα-specific luciferase reporter assay, using the GRα Reporter Assay System (IB00201; Indigo Biosciences, State College, PA, USA) according to the manufacturer’s instructions. Briefly, GRα reporter cells were cultured in Cell Recovery Medium and treated with various concentrations of Rofl, Dex, or combination of both (0–3 nM) for 24 h. At the end of the treatment, the culture medium was discarded, and Luciferase Detection Reagent was added to each well of the assay plate. The plate was then incubated for 5 min at room temperature, and luminescence was quantified with a luminometer.
Statistical Analysis
Data are presented as the mean ± SD. New England Journal of Medicine style was used to report P values. We determined the differences between experimental groups using an unpaired t-test or two-way analysis of variance followed by a Bonferroni multiple-comparison correction. To determine the EC50 from dose-response curves and drug additivity with the Bliss independence model, we used curve fitting with non-linear regression. For statistical analyses, we used GraphPad Prism 8.1.2 (GraphPad Software, La Jolla, CA, USA); differences with P values <0.05 were considered significant.
Results
COPD HBE Cells Show Decreased GRα Expression and Activity
To test the potential role of GRα in COPD pathophysiology, we determined its expression and activity levels in COPD patients. We found GRα protein expression was reduced in COPD HBE cells compared with NHBE cells (Figure 1A). Consistent with this decreased expression, GRα’s DNA-binding activity was lower in COPD HBE cells than in NHBE cells (Figure 1B), whereas the activity of pro-inflammatory NF-κB p65 was elevated (Figure 1B), pointing to a link between GRα downregulation and COPD-associated inflammation.
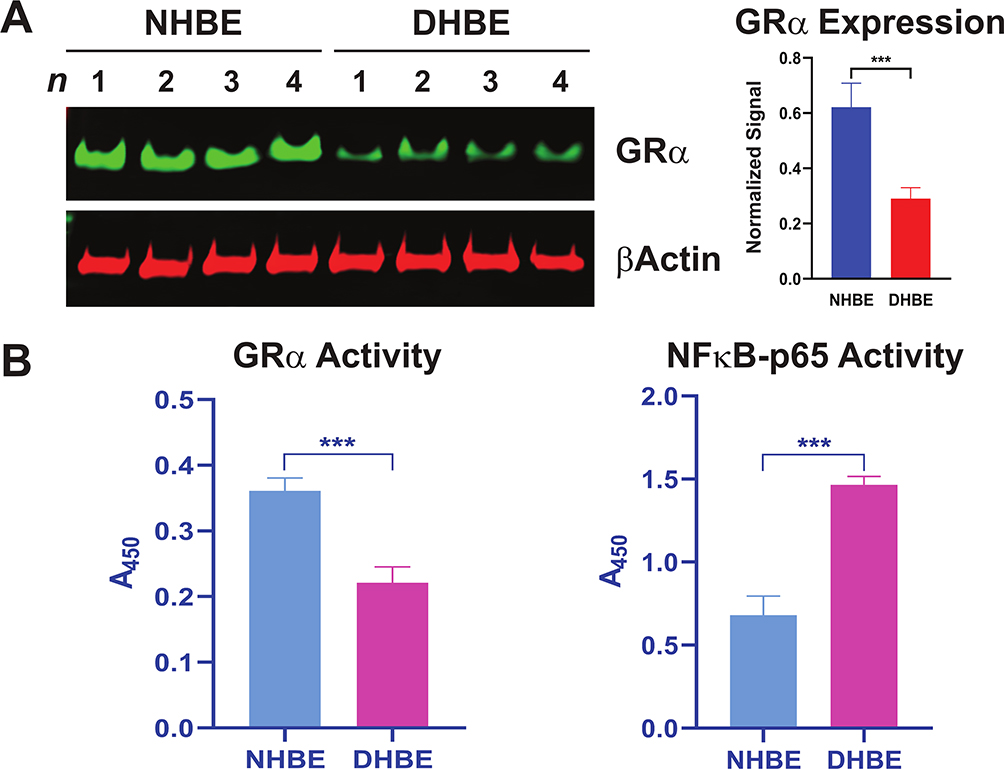 |
Figure 1 Reduced GRα expression and activity in COPD HBE cells. (A) GRα expression in DHBE (also described as COPD HBE in the text) and NHBE cells, determined by Western blotting and quantified by densitometric analysis. β-Actin served as a loading control. (B) DNA-binding activities of GRα and NF-κB p65 in NHBE and DHBE cells. Data are expressed as means ± SD. n = 4; ***P < 0.001. Abbreviations: DHBE, diseased human bronchial epithelial; GRα, glucocorticoid receptor α; HBE, human bronchial epithelial; NHBE, normal human bronchial epithelial. |
Rofl Induces GRα Gene Transcription in COPD HBE Cells
Rofl is an anti-inflammatory drug indicated for treatment to control disease exacerbations in severe COPD patients.10–12 It was suggested to restore glucocorticoid sensitivity in COPD patients,15,16 by unknown mechanisms. To explore the mechanism, we tested Rofl’s effects on GRα expression. Rofl enhanced GRα protein expression in a dose- and time-dependent manner (Figure 2A–B). We then tested the hypothesis that Rofl-induced upregulation of GRα expression reflects increased transcription by analyzing the dynamics of RNA synthesis and processing. Specifically, we measured nascent mRNA levels in COPD HBE cells treated with and without Rofl. We found that Rofl treatment increased the synthesis of nascent GRα mRNA (NR3C1 mRNA, which encodes GRα) at all observed time points (Figure 2C). Because enhanced mRNA stability could also increase protein expression, we tested whether Rofl altered decay of GRα mRNA by EU pulse-chase assay. Beyond the initial period, Rofl treatment failed to significantly alter GRα mRNA stability (Figure 2D). Together, these results indicate that Rofl promotes GRα expression at the transcriptional level, specifically by stimulating mRNA synthesis.
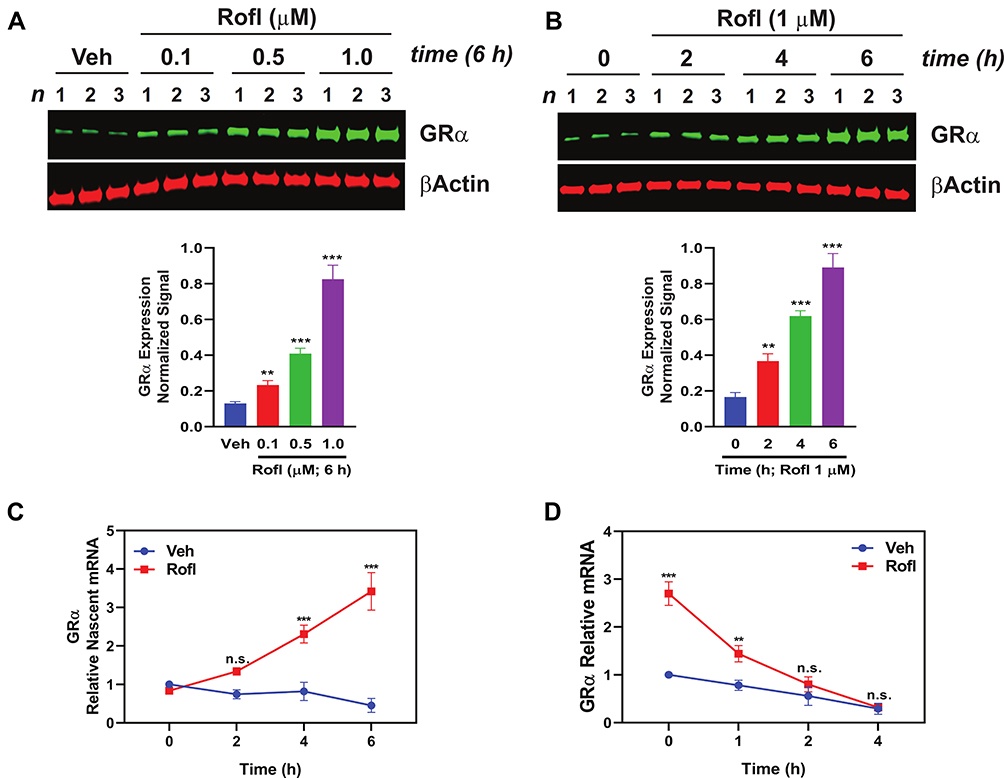 |
Figure 2 Rofl induces GRα expression in COPD HBE cells. (A-B) COPD HBE cells were treated with the indicated concentrations of Rofl for 6 h (A) or with 1 µM of Rofl for the indicated periods (B) and GRα levels determined by Western blotting in whole-cell extracts and quantified by densitometric analysis. β-Actin served as a loading control. (C) Time course of transcriptional response to Rofl. After COPD HBE cells were treated with 1 µM Rofl or Veh control for 6 h, nascent mRNA captured by Click-iT Nascent RNA Capture Kit. Relative mRNA levels of GRα were measured by real-time PCR. (D) Effects of Rofl vs. Veh control on GRα mRNA stability in COPD HBE cells were determined by incubation in growth medium containing 5-EU followed by incubation in growth medium without 5-EU for the indicated periods. After total mRNA isolation, labeled mRNA was captured and analyzed with Click-iT Nascent RNA Capture Kit. Data are expressed as means ± SD; n = 3. **P < 0.01, ***P < 0.001. Abbreviations: GRα, glucocorticoid receptor α; HBE, human bronchial epithelial; n.s., non-significant; Rofl, roflumilast; Veh, vehicle. |
Rofl Induces GRα Promoter Activation and GRα Transcriptional Activity
The enhanced GRα expression we observed might reflect recruitment of a transcription factor to the GRα promoter, such as cAMP response element binding protein (CREB) that has been suggested to regulate GRα transcription in other cell types via direct binding to the GRα promoter.20,21 We thus tested whether Rofl promotes CREB binding to the CRE within the GRα promoter in COPD HBE cells by DNA-protein affinity assay. Rofl treatment increased the GRα CRE’s association with cellular CREB-containing complexes compared with those in vehicle (Veh)-treated controls (Figure 3A). As expected, the consensus CRE we used as a positive control recovered CREB, whereas non-specific oligonucleotides did not (Figure 3A). Mutations of the GRα CRE (Supplemental Table S2) abolished CREB binding (Figure 3A), indicating the specificity of GRα CRE-CREB interaction. These data suggest that Rofl promotes GRα expression by recruiting a CREB-dependent transcription complex that acts on the NR3C1 promoter.
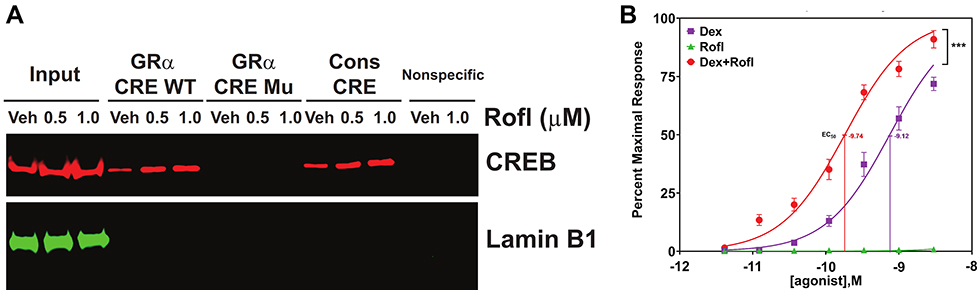 |
Figure 3 Rofl stimulates GRα promoter activity and induces GRα transcriptional activity. (A) Promoter activity. COPD HBE cells were treated with Rofl (0.5 or 1 µM) or Veh for 6 h, and then nuclear extracts were obtained and incubated with the indicated biotinylated double-stranded oligonucleotides corresponding to the following: WT or Mu GRα-CRE, the consensus CRE (positive control), or a nonspecific sequence (negative control). Bead-bound oligonucleotide-protein complexes were eluted and subjected to Western blotting to identify the presence of CREB. Western blotting for Lamin B1 was used as a control for non-specific interaction. Nuclear extracts without added nucleotides were loaded as input. (B) Transcriptional activity. GRα reporter cells were treated with the indicated concentrations (0–3 nM) of Rofl, Dex, or a combination of both for 24 h. GRα transcriptional activity, shown as the percent maximal response, was then measured using a GRα-specific reporter assay. Concentration-response curve fitting was performed by non-linear regression. Results were reproduced independently at least twice. Data are expressed as means ± SD; n = 3, ***P < 0.001. Abbreviations: CRE, cAMP response element; CREB, cAMP response element binding protein; Dex, dexamethasone; GRα, glucocorticoid receptor α; HBE, human bronchial epithelial; Mu, mutated; Rofl, roflumilast; Veh, vehicle; WT, wildtype. |
To assess the functional consequence of increased GRα expression, we compared GRα transcriptional activity in GRα reporter cells treated with Dex, Rofl, and both drugs combined. While having no effect on GRα transcriptional activity alone, Rolf significantly sensitized the transcriptional response to Dex, reducing its EC50 from 0.75 ± 0.16 to 0.18 ± 0.04 nM (Figure 3B). It also significantly increased the response of transcriptional activity to the maximal concentration of Rofl tested (Figure 3B).
Rofl Inhibits IL-8 and TNFα Production in COPD HBE Cells Additively with Dex in a GRα-Dependent Manner
Decreased cytokine and chemokine secretion is a key mechanism contributing to the anti-inflammatory actions of both Rofl and Dex.10,22,23 We tested for such actions in the HBE system and found concentration-dependent inhibition of the levels produced of the key inflammatory chemokine and cytokine, IL-8 and TNFα (Figure 4A–B), in Rofl- and Dex-treated COPD HBE cells. Consistent with its enhancing effects seen on GRα transcriptional activity (Figure 3B), the combination of Rofl and Dex gave greater suppression of IL-8 (Figure 4A) and TNFα (Figure 4B) release than either drug alone. Furthermore, we found that siRNA-mediated GRα knockdown (Figure 4E), but not scrambled control siRNA, blocked Rofl- and Dex-induced suppression of IL-8 and TNFα production (Figure 4C–D). Together, the results indicate that Rofl and Dex additively inhibited inflammatory cytokine and chemokine secretion in COPD HBE cells in a GRα-dependent fashion.
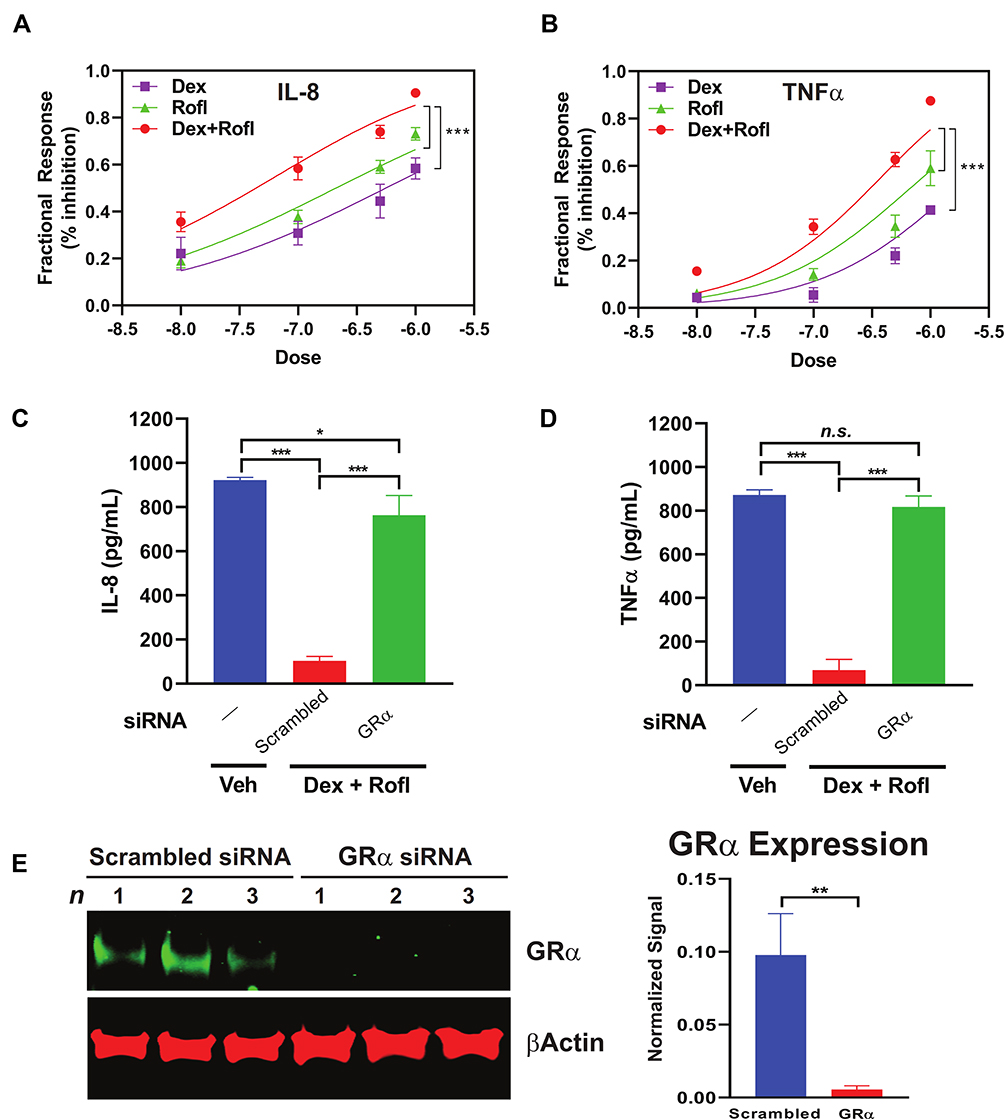 |
Figure 4 Rofl inhibits IL-8 and TNFα production in COPD HBE cells additively with Dex in a GRα-dependent manner. (A-B) COPD HBE cells were treated with Rofl, Dex, or combined Rofl and Dex at indicated concentrations for 6 h. Culture medium was then collected, and IL-8 (A) and TNFα (B) levels in medium were determined by ELISA. Inhibitory effects of treatments are shown as fractional response. Curves were fitted by non-linear regression and the Bliss independence model. (C-D) COPD HBE cells received 24 h-transfection with scrambled or GRα siRNA and then were treated with Veh or combined Rofl and Dex (1 µM each) for 6 h. IL-8 (C) and TNFα (D) levels in culture medium were determined by ELISA. Data are expressed as means ± SD; n = 3. *P < 0.05, ***P < 0.001. (E) GRα expression in siRNA-transfected COPD HBE cells was determined by Western blotting and quantified by densitometric analysis. β-Actin served as a loading control. **P < 0.01.Abbreviations: Dex, dexamethasone; GRα, glucocorticoid receptor α; HBE, human bronchial epithelial; IL-8, interleukin 8; n.s., non-significant; Rofl, roflumilast; TNFα, tumor necrosis factor alpha. |
Discussion
Despite their mainstay role in management of COPD-associated exacerbations, glucocorticoids provide only modest clinical benefits for most patients.24–26 New strategies to enhance their efficacy to control lung inflammation in COPD could thus aid clinical management and outcomes. Here we provide the first evidence, to our knowledge, that GRα downregulation contributes to glucocorticoid resistance in COPD HBE cells. Furthermore, we found that Rofl stimulates GRα mRNA synthesis and thereby augments Dex’s anti-inflammatory effects. These findings show that increased GRα expression can reverse glucocorticoid resistance and also clearly reveal a new mechanistic basis of Rofl’s therapeutic actions in COPD,15,16,27 as GRα knockdown blocked the cytokine-inhibitory action of Rofl combined with Dex.
Consistent with our data, Marwick et al found GRα protein expression was decreased in peripheral lung parenchyma of COPD patients compared to that in non-COPD patients (smokers and non-smokers).9 Similarly, HBE cells of COPD patients exhibited lower GRα transcript levels than those from non-COPD individuals.28 Extending those findings, we found that Rofl enhances GRα mRNA synthesis to yield sustained elevation of GRα protein expression and thereby sensitizes and enhances HBE cells’ responses to the anti-inflammatory actions of Dex. Our finding that Rofl upregulates GRα expression by increasing transcription rate, and not by stabilizing mRNA, agrees with prior findings in other cell types.29,30 Our data strongly indicate that altered GRα expression and function contribute mechanistically to glucocorticoid sensitivity/resistance in COPD.
Our findings that Rofl restores glucocorticoid sensitivity support Rofl’s therapeutic value for treating glucocorticoid-refractory patients. Prior findings agree, as Rofl’s active metabolite Rofl N-oxide and Dex were found to work synergistically/additively to suppress release of certain inflammation markers from neutrophils15 and HBE cells27 of COPD patients. Combined Rofl N-oxide and Dex also reduced corticosteroid resistance biomarkers.15 Our mechanistic findings that Rofl action is GRα mediated in the COPD context merits assessment in clinical trials, as certain other pathway-specific compounds found capable of reversing glucocorticoid resistance are being investigated or in clinical trials.7 This is important, since, although inhaled corticosteroids are well-tolerated, most COPD patients are insensitive to their ameliorative effects.24–26 Rofl is effective in reducing exacerbations, but associated with several significant adverse effects.31–33 Thus, combination therapy may yield the greatest therapeutic outcome; it may enhance patients’ responsiveness to glucocorticoids while minimizing or avoiding undesired off-target effects of Rofl treatment by using the minimal sensitizing dose. Such strategy should be comprehensively assessed in future studies.
Airway epithelium is the immediate target of inhaled toxicants, including those in cigarette smoke, and therefore plays a central role in COPD development and progression. It is also the primary site of glucocorticoids’ anti-inflammatory actions. Thus, our findings that GRα expression in HBE cells is required for Rofl-mediated recovery of glucocorticoid sensitivity highlight the potential of airway epithelial GRα not only as an attractive therapeutic target but also as a useful biomarker for glucocorticoid resistance in COPD. In this connection, the development of a diagnostic genomic tool to predict clinical response to glucocorticoid therapy in asthma has been proposed.34 A similar approach that assesses GRα expression level to identify subsets of patients who would likely benefit from glucocorticoid treatment as well as the addition of Rofl can be applied to COPD. Rofl is currently indicated for patients suffering severe COPD with chronic bronchitis and a high risk of severe exacerbations.23 Rofl, administered in combination with standard therapies, reduced incidence of moderate/severe exacerbations and hospitalization in this patient population.23 Extending our present study and assessing GRα expression in a clinical trial with COPD patients with and without chronic bronchitis may provide further evidence for Rofl’s therapeutic benefits in COPD.
Previously, glucocorticoid treatment was found to reverse the reduction in GRα mRNA levels seen in COPD patients.28 In contrast, others found that Dex downregulated GRα mRNA and GRα protein expression in BEAS-2B HBE cell line, implying secondary glucocorticoid resistance.35 These apparent discrepancies may be attributable to different experimental system/conditions; the former finding concerned GRα expression in primary bronchial epithelial cells from COPD patients receiving glucocorticoids, whereas the latter observation was made using a cell line treated in vitro with glucocorticoids. It is currently unknown when glucocorticoid resistance evolves during the course of COPD pathogenesis, a question beyond the scope of this study that warrants investigation. Nevertheless, clear understanding of such timing, including whether or not glucocorticoid insensitivity is induced by glucocorticoid treatment itself, will be key to designing and developing the most efficacious and potent therapies. Clinical tests of GRα levels, if developed, could also serve as an assessment tool to monitor glucocorticoids’ efficacy and patients’ responses during their therapy.
By inhibiting PDE4 hydrolytic activity, Rofl treatment increases intracellular cAMP and cAMP signaling, leading to various cellular and physiological outcomes.10,23 In alveolar macrophages from COPD patients ex vivo, Rofl induced CREB activation and its nuclear localization.36 In ovarian cancer cells, Rofl augmented the cAMP/PKA/CREB pathway and thereby restored their cisplatin sensitivity.37,38 Our finding that Rofl promoted CREB binding to the CRE within the GRα promoter are consistent with those results. Further assessment of the role of the cAMP/PKA/CREB axis in Rofl’s actions, including whether and how such signaling is altered, will inform about the molecular mechanism of GRα downregulation and thereby glucocorticoid resistance in COPD. CREB expression and activation in lungs are impaired with age, as they were reduced in adult compared to young mice, and further decreased in old vs. adult animals.39 As COPD is considered a disease of accelerated lung aging,40,41 our findings imply that any such impairment may also conceivably contribute to COPD-associated GRα downregulation.
Glucocorticoid resistance occurs in inflammatory conditions other than COPD.7,42 Thus, our findings on GRα’s role in glucocorticoid resistance and Rofl’s ability to restore glucocorticoid sensitivity in COPD may prove relevant to other diseases associated with chronic inflammation, a worthy topic for future study.
Conclusion
In conclusion, our study highlights Rofl’s therapeutic value for the treatment of glucocorticoid-refractory patients by showing that decreased GRα expression mediates glucocorticoid resistance and that Rofl blocks such GRα downregulation and thereby restores glucocorticoid sensitivity.
Abbreviations
COPD, chronic obstructive pulmonary disease; CRE, cAMP response element; CREB, cAMP response element binding protein; Dex, dexamethasone; DHBE, diseased human bronchial epithelial; GRα, glucocorticoid receptor α; HBE, human bronchial epithelial; IL-8, interleukin 8; Mu, mutated; NHBE, normal human bronchial epithelial; PDE4, phosphodiesterase-4; Rofl, roflumilast; TNFα, tumor necrosis factor alpha; Veh, vehicle; WT, wildtype.
Acknowledgments
This work was supported by a merit review award from the United States Department of Veterans Affairs (VA) and National Institutes of Health (NIH) grant HL137842 (to RCR).
Disclaimer
The contents in this article do not represent the views of the United States Department of Veterans Affairs or the United States Government.
Disclosure
Dr. Raju C Reddy reports grants from NIH R01 and VA Merit, during the conduct of the study. The authors report no other conflicts of interest in this work.
References
1. Vogelmeier CF, Criner GJ, Martinez FJ, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive lung disease 2017 Report. GOLD executive summary. Am J Respir Crit Care Med. 2017;195:557–582. doi:10.1164/rccm.201701-0218PP
2. Vestbo J, Hurd SS, Agusti AG, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med. 2013;187:347–365. doi:10.1164/rccm.201204-0596PP
3. Lakshmi SP, Reddy AT, Reddy RC. Emerging pharmaceutical therapies for COPD. Int J Chron Obstruct Pulmon Dis. 2017;12:2141–2156. doi:10.2147/COPD.S121416
4. Adcock IM, Ito K. Glucocorticoid pathways in chronic obstructive pulmonary disease therapy. Proc Am Thorac Soc. 2005;2:313–319. discussion 40-1. doi:10.1513/pats.200504-035SR.
5. Burge PS, Calverley PM, Jones PW, Spencer S, Anderson JA, Maslen TK. Randomised, double blind, placebo controlled study of fluticasone propionate in patients with moderate to severe chronic obstructive pulmonary disease: the ISOLDE trial. BMJ. 2000;320:1297–1303. doi:10.1136/bmj.320.7245.1297
6. Calverley P, Pauwels R, Vestbo J, et al. long-acting beta2 agonists study g. Combined salmeterol and fluticasone in the treatment of chronic obstructive pulmonary disease: a randomised controlled trial. Lancet. 2003;361:449–456. doi:10.1016/S0140-6736(03)12459-2
7. Barnes PJ, Adcock IM. Glucocorticoid resistance in inflammatory diseases. Lancet. 2009;373:1905–1917. doi:10.1016/S0140-6736(09)60326-3
8. Gross KL, Lu NZ, Cidlowski JA. Molecular mechanisms regulating glucocorticoid sensitivity and resistance. Mol Cell Endocrinol. 2009;300:7–16. doi:10.1016/j.mce.2008.10.001
9. Marwick JA, Caramori G, Stevenson CS, et al. Inhibition of PI3Kdelta restores glucocorticoid function in smoking-induced airway inflammation in mice. Am J Respir Crit Care Med. 2009;179:542–548. doi:10.1164/rccm.200810-1570OC
10. Kawamatawong T. Roles of roflumilast, a selective phosphodiesterase 4 inhibitor, in airway diseases. J Thorac Dis. 2017;9:1144–1154. doi:10.21037/jtd
11. Wedzicha JA, Rabe KF, Martinez FJ, et al. Efficacy of roflumilast in the COPD frequent exacerbator phenotype. Chest. 2013;143:1302–1311. doi:10.1378/chest.12-1489
12. Singh D, Agusti A, Anzueto A, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive lung disease: the GOLD science committee report 2019. Eur Respir J. 2019;53. doi:10.1183/13993003.00164-2019.
13. Buenestado A, Grassin-Delyle S, Guitard F, et al. Roflumilast inhibits the release of chemokines and TNF-alpha from human lung macrophages stimulated with lipopolysaccharide. Br J Pharmacol. 2012;165:1877–1890. doi:10.1111/j.1476-5381.2011.01667.x
14. Grootendorst DC, Gauw SA, Verhoosel RM, et al. Reduction in sputum neutrophil and eosinophil numbers by the PDE4 inhibitor roflumilast in patients with COPD. Thorax. 2007;62:1081–1087. doi:10.1136/thx.2006.075937
15. Milara J, Lluch J, Almudever P, Freire J, Xiaozhong Q, Cortijo J. Roflumilast N-oxide reverses corticosteroid resistance in neutrophils from patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2014;134:314–322. doi:10.1016/j.jaci.2014.02.001
16. Rennard SI, Calverley PM, Goehring UM, Bredenbroker D, Martinez FJ. Reduction of exacerbations by the PDE4 inhibitor roflumilast–the importance of defining different subsets of patients with COPD. Respir Res. 2011;12:18. doi:10.1186/1465-9921-12-18
17. Reddy AT, Lakshmi SP, Banno A, Reddy RC. Role of GPx3 in PPARgamma-induced protection against COPD-associated oxidative stress. Free Radic Biol Med. 2018;126:350–357. doi:10.1016/j.freeradbiomed.2018.08.014
18. Lakshmi SP, Reddy AT, Banno A, Reddy RC. Airway epithelial cell peroxisome proliferator-activated receptor gamma regulates inflammation and mucin expression in allergic airway disease. J Immunol. 2018;201:1775–1783. doi:10.4049/jimmunol.1800649
19. Lakshmi SP, Reddy AT, Reddy RC. Transforming growth factor beta suppresses peroxisome proliferator-activated receptor gamma expression via both SMAD binding and novel TGF-beta inhibitory elements. Biochem J. 2017;474:1531–1546. doi:10.1042/BCJ20160943
20. Govindan MV. Recruitment of cAMP-response element-binding protein and histone deacetylase has opposite effects on glucocorticoid receptor gene transcription. J Biol Chem. 2010;285:4489–4510. doi:10.1074/jbc.M109.072728
21. Penuelas I, Encio IJ, Lopez-Moratalla N, Santiago E. cAMP activates transcription of the human glucocorticoid receptor gene promoter. J Steroid Biochem Mol Biol. 1998;67:89–94. doi:10.1016/S0960-0760(98)00097-1
22. Barnes PJ. Corticosteroid resistance in patients with asthma and chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2013;131:636–645. doi:10.1016/j.jaci.2012.12.1564
23. Wedzicha JA, Calverley PM, Rabe KF. Roflumilast: a review of its use in the treatment of COPD. Int J Chron Obstruct Pulmon Dis. 2016;11:81–90. doi:10.2147/COPD.S89849
24. Bourbeau J, Christodoulopoulos P, Maltais F, Yamauchi Y, Olivenstein R, Hamid Q. Effect of salmeterol/fluticasone propionate on airway inflammation in COPD: a randomised controlled trial. Thorax. 2007;62:938–943. doi:10.1136/thx.2006.071068
25. Culpitt SV, Maziak W, Loukidis S, Nightingale JA, Matthews JL, Barnes PJ. Effect of high dose inhaled steroid on cells, cytokines, and proteases in induced sputum in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1999;160:1635–1639. doi:10.1164/ajrccm.160.5.9811058
26. Keatings VM, Jatakanon A, Worsdell YM, Barnes PJ. Effects of inhaled and oral glucocorticoids on inflammatory indices in asthma and COPD. Am J Respir Crit Care Med. 1997;155:542–548. doi:10.1164/ajrccm.155.2.9032192
27. Milara J, Morell A, Ballester B, et al. Roflumilast improves corticosteroid resistance COPD bronchial epithelial cells stimulated with toll like receptor 3 agonist. Respir Res. 2015;16:12. doi:10.1186/s12931-015-0179-5
28. Korn SH, Thunnissen FB, Wesseling GJ, Arends JW, Wouters EF. Glucocorticoid receptor mRNA levels in bronchial epithelial cells of patients with COPD: influence of glucocorticoids. Respir Med. 1998;92:1102–1109. doi:10.1016/S0954-6111(98)90402-4
29. Dong Y, Poellinger L, Gustafsson JA, Okret S. Regulation of glucocorticoid receptor expression: evidence for transcriptional and posttranslational mechanisms. Mol Endocrinol. 1988;2:1256–1264. doi:10.1210/mend-2-12-1256
30. Rosewicz S, McDonald AR, Maddux BA, Goldfine ID, Miesfeld RL, Logsdon CD. Mechanism of glucocorticoid receptor down-regulation by glucocorticoids. J Biol Chem. 1988;263:2581–2584.
31. Calverley PM, Sanchez-Toril F, McIvor A, Teichmann P, Bredenbroeker D, Fabbri LM. Effect of 1-year treatment with roflumilast in severe chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2007;176:154–161. doi:10.1164/rccm.200610-1563OC
32. Fabbri LM, Calverley PM, Izquierdo-Alonso JL, et al. M, groups Ms. Roflumilast in moderate-to-severe chronic obstructive pulmonary disease treated with longacting bronchodilators: two randomised clinical trials. Lancet. 2009;374:695–703. doi:10.1016/S0140-6736(09)61252-6
33. Giembycz MA. Phosphodiesterase-4: selective and dual-specificity inhibitors for the therapy of chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2005;2:326–333. discussion 40-1. doi:10.1513/pats.200504-041SR.
34. Hakonarson H, Bjornsdottir US, Halapi E, et al. Profiling of genes expressed in peripheral blood mononuclear cells predicts glucocorticoid sensitivity in asthma patients. Proc Natl Acad Sci U S A. 2005;102:14789–14794. doi:10.1073/pnas.0409904102
35. Pujols L, Mullol J, Perez M, et al. Expression of the human glucocorticoid receptor alpha and beta isoforms in human respiratory epithelial cells and their regulation by dexamethasone. Am J Respir Cell Mol Biol. 2001;24:49–57. doi:10.1165/ajrcmb.24.1.4024
36. Lea S, Metryka A, Li J, et al. The modulatory effects of the PDE4 inhibitors CHF6001 and roflumilast in alveolar macrophages and lung tissue from COPD patients. Cytokine. 2019;123:154739. doi:10.1016/j.cyto.2019.154739
37. Gong S, Chen Y, Meng F, et al. Roflumilast enhances cisplatin-sensitivity and reverses cisplatin-resistance of ovarian cancer cells via cAMP/PKA/CREB-FtMt signalling axis. Cell Prolif. 2018;51:e12474. doi:10.1111/cpr.12474
38. Gong S, Chen Y, Meng F, Zhang Y, Wu H, Wu F. Roflumilast restores cAMP/PKA/CREB signaling axis for FtMt-mediated tumor inhibition of ovarian cancer. Oncotarget. 2017;8:112341–112353. doi:10.18632/oncotarget.22866
39. Rolewska P, Simm A, Silber RE, Bartling B. Reduced expression level of the cyclic adenosine monophosphate response element-binding protein contributes to lung aging. Am J Respir Cell Mol Biol. 2014;50:201–211. doi:10.1165/rcmb.2013-0057OC
40. Ito K, Barnes PJ. COPD as a disease of accelerated lung aging. Chest. 2009;135:173–180. doi:10.1378/chest.08-1419
41. MacNee W. Is chronic obstructive pulmonary disease an accelerated aging disease? Ann Am Thorac Soc. 2016;13(Suppl 5):S429–S37. doi:10.1513/AnnalsATS.201602-124AW
42. Rodriguez JM, Monsalves-Alvarez M, Henriquez S, Llanos MN, Troncoso R. Glucocorticoid resistance in chronic diseases. Steroids. 2016;115:182–192. doi:10.1016/j.steroids.2016.09.010
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.