Back to Journals » Clinical Ophthalmology » Volume 18
Glucocorticoid-Induced Ocular Hypertension and Glaucoma
Authors Harvey DH, Sugali CK, Mao W
Received 1 October 2023
Accepted for publication 22 January 2024
Published 16 February 2024 Volume 2024:18 Pages 481—505
DOI https://doi.org/10.2147/OPTH.S442749
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Devon Hori Harvey,1,2 Chenna Kesavulu Sugali,1,2 Weiming Mao1– 5
1Department of Ophthalmology, Indiana University School of Medicine, Indianapolis, IN, USA; 2Eugene and Marilyn Glick Eye Institute, Indiana University School of Medicine, Indianapolis, IN, USA; 3Department of Biochemistry & Molecular Biology, Indiana University School of Medicine, Indianapolis, IN, USA; 4Department of Pharmacology and Toxicology, Indiana University School of Medicine, Indianapolis, IN, USA; 5Stark Neurosciences Research Institute, Indiana University School of Medicine, Indianapolis, IN, USA
Correspondence: Weiming Mao, Department of Ophthalmology, Indiana University School of Medicine, RM305v, 1160 W. Michigan St, Indianapolis, IN, 46202, USA, Tel +1 317 278 0801, Email [email protected]
Abstract: Glucocorticoid (GC) therapy is indicated in many diseases, including ocular diseases. An important side-effect of GC therapy is GC-induced ocular hypertension (GIOHT), which may cause irreversible blindness known as GC-induced glaucoma (GIG). Here, we reviewed the pathological changes that contribute to GIOHT including in the trabecular meshwork and Schlemm’s canal at cellular and molecular levels. We also discussed the clinical aspects of GIOHT/GIG including disease prevalence, risk factors, the type of GCs, the route of GC administration, and management strategies.
Keywords: glucocorticoid, intraocular pressure, trabecular meshwork, aqueous humor outflow, mechanism, treatment
Introduction
Purpose and Method of Literature Search
Glaucoma is a leading cause of irreversible blindness worldwide.1 One of the complications of glucocorticoid (GC) therapy is glucocorticoid-induced ocular hypertension (GIOHT). If GIOHT is left untreated, some patients may develop glaucomatous retinal ganglion cell loss, which is a subtype of glaucoma: glucocorticoid-induced glaucoma (GIG).
The initial reports of GIOHT/GIG were published in the 1950s.2–6 Since then, there have been many studies attempting to understand the mechanism of GIOHT/GIG. This disease is important for scientists since GIOHT is frequently used as a disease model to study primary open-angle glaucoma. It is also important for clinicians since GC treatment is essential for many ocular and systemic diseases, and sometimes clinicians are in the dilemma of whether to continue GC treatment to control inflammation or to discontinue GC to preserve vision. We hope this article provides a quick overview of GIOHT/GIG with information from bench to bedside to both basic science researchers and clinicians.
The first part of this article reviews the basic-science aspects of the disease focusing on the potential molecular mechanisms. The second part of this article reviews the clinical aspects of the disease focusing on epidemiology as well as GC selection and administration.
Peer-reviewed literature was searched using the following terms in PubMed: (“glucocorticoid receptor”[tw] OR “steroid receptor”[tw] OR “corticosteroid receptor”[tw]) AND (“ocular hypertension”[tw] OR “glaucoma”); TM 11B-hydroxysteroid dehydrogenase glucocorticoid; 11B-hydroxysteroid dehydrogenase steroid responder; yap taz glucocorticoid receptor glaucoma; wnt signaling glucocorticoid receptor; tgfβ2 glucocorticoid receptor; cross linked actin network glucocorticoid; trabecular meshwork extracellular matrix glucocorticoid; glucocorticoid glaucoma distal; POAG and steroid-induced glaucoma: rimexolone steroid glaucoma; loteprednol steroid glaucoma; fluocinolone acetonide implant iop; retrobulbar injection steroid IOP; intravenous steroid iop; cutaneous steroid glaucoma; glaucoma Cushing syndrome; steroid-induced glaucoma treatment. Searches were conducted until August 2023.
Background
Intraocular Pressure (IOP)
The eye is filled with aqueous humor, and this fluid generates IOP. Intraocular pressure is determined by aqueous humor production (inflow), aqueous humor outflow, and episcleral vein pressure. Aqueous humor outflow has two pathways: the major pathway is the trabecular meshwork (TM) outflow pathway, also called the conventional outflow pathway (the pressure-dependent pathway); the minor pathway is the uveoscleral outflow pathway (the pressure-independent pathway).7 In GIG/GIOHT eyes, IOP elevation is primarily due to increased outflow resistance (or decreased outflow facility) in the TM outflow pathway.
In non-glaucomatous eyes, the normal IOP is approximately 16±5 mmHg. IOP higher than 21mmHg is usually considered OHT. Ocular hypertension can lead to glaucomatous optic nerve damage (glaucoma), although some individuals may have OHT for years or even throughout their life span without developing glaucoma. However, OHT is still the most important risk factor for developing glaucomatous optic nerve damage. Besides, GIOHT is not as “natural” as those who have “spontaneous” OHT. Also, individuals who develop GIOHT are susceptible to developing primary open-angle glaucoma.8,9 Therefore, people with GIOHT may be at higher risk of developing optic nerve damage.
The Anatomy of the TM and Schlemm’s Canal (SC)
As described previously, the majority of the aqueous humor drains through the TM outflow pathway. The tissue in this pathway includes the TM, Schlemm’s canal (SC), collector channels, aqueous veins, and episcleral veins. The conventional outflow pathway tissue can be further divided into the proximal outflow tissue and the distal outflow tissue. The proximal outflow tissue refers to the TM and inner wall of SC. The distal outflow tissue refers to the outer wall of SC, collector channels, aqueous veins, and episcleral veins.
The TM and SC are the key tissues of the TM outflow pathway. The TM can be divided into 3 regions, depending on their location: the uveoscleral meshwork, the corneoscleral meshwork, and the juxtacanalicular region (JCT).7,10,11 The uveoscleral meshwork is the most adjacent to the aqueous humor and is made of connective tissue trabeculae that create irregular openings through which aqueous humor passes.7 Adjoining the uveal meshwork is the corneoscleral meshwork, which consists of trabecular beams with elliptical openings that get progressively smaller as the aqueous humor flows outward.7 Next to the corneoscleral meshwork is the JCT, which consists of irregularly oriented fibrils and layers of TM cells that do not allow for aqueous to easily pass through.7 The SC has an inner wall and an outer wall. The inner wall of SC is a single layer of endothelium-like cells attached to the basal lamina and/or the JCT ECM, and there are tight junctions between these cells.7,12 The aqueous humor passes through this layer of cells via two types of pores: type I (transcellular) pores and type B (paracellular) pores.13 The JCT and the inner wall of SC contribute to the majority of the aqueous humor outflow resistance.14,15
Basic Science Review of GIG/GIOHT
The Mechanism of GCs
Glucocorticoids function through glucocorticoid receptors (GRs). There are two GR isoforms: GR⍺ and GRβ.16 Glucocorticoid receptor ⍺ is located primarily in the cytoplasm, bound to a large heterocomplex comprising several other chaperone proteins including heat shock proteins (HSP), the hop protein, p23, and immunophilins.17,18 After binding to GCs, the GC-GRα complex translocates into the nucleus, binds to glucocorticoid response elements (GRE), and regulates gene expression. The entire process is finely controlled and involves several regulatory proteins (see Figure 1).
 |
Figure 1 The glucocorticoid receptor signaling pathway. The cytosolic glucocorticoid receptor alpha (GRα) is associated with several chaperone proteins including heat-shock protein (HSP) 70, HOP, and HSP 90. After the recruitment of p23 and displacement of HSP 70 and HOP into the complex, the GRα achieves “client-maturation” and is able to bind to glucocorticoids (GCs). Immunophilins influence the affinity of GC binding with FKBP51 decreasing GC-GRα affinity and FKBP52 increasing GC-GRα affinity. These chaperone proteins facilitate the translocation of GC-GRα to the nucleus. The nuclear GC-GRα complex monomer, together with other transcription factors (TFs), functions as a transrepressor to inhibit gene expression. The GC-GR complex may also dimerize and function as a transactivator to increase gene expression. The GR beta (GRβ) is an alternatively spliced, dominant negative form of GRα. Created with Biorender.com. |
The GR binding proteins play an important role in the activation of the GR. Wang et al reported the structure of the GR loading complex using high-resolution cryo-electron microscopy and the role of these proteins.18 They described 4 stages of activation of the GR beginning with an association with hsp70 in an inactivated state. The Hop protein then loads the hsp70-GR receptor, with hsp90 creating the inactivated hsp70-GR-hop-hsp90 complex. After hsp90 ATP hydrolysis, hop and hsp70 dissociate and p23 associates, creating the activated, “client-maturation” GR-hsp90-p23 complex.18 This complex is further regulated by the association with immunophilins FKBP52 and FKBP51.19 The presence of FKBP52 leads to a higher affinity for GR-ligand binding and the presence of FKBP51 antagonizes FKBP52.19 It has been reported that higher levels of FKBP51 contribute to GC resistance.20 After the complex binds to the GC, the GR undergoes conformational change and nuclear translocation.21,22 Once in the nucleus, the GR⍺ acts as a transcription factor through either transactivation or transrepression.23 In the transactivation mode, the GR⍺ dimerizes and binds to the glucocorticoid response elements (GRE) at the promoter region of the target genes.24,25 In the transrepression mode, the GR⍺ remains as a monomer and interacts with other transcription factors (AP-1, NF-κB) resulting in the inhibition of target gene expression.24,26 The anti-inflammatory mechanism of GCs is complicated. Traditionally, it is believed that the inhibition of proinflammatory factors by GR transrepression plays a key role.27 However, studies have shown that the upregulation of anti-inflammatory factors by GR transactivation is also important.28
There have not been many reports on the role of GR-mediated transactivation/transrepression in GC-related ocular side effects, especially GIOHT/GIG. Recently, Patel and colleagues tried to dissect the GR transactivation and transrepression sub-pathways in GIOHT using the GRdim mice that have impaired GR transactivation but normal GR transrepression.23 They found that these GRdim mice did not develop GIOHT or GC-inducible changes in the TM after periocular DEX injections, which suggests that GIOHT is mediated through GR-transactivation.
Glucocorticoid receptor β is produced through alternative splicing of the GR gene NR3C1.29 In contrast to GR⍺ that has a fully functioning ligand-binding domain (LBD) at the c-terminus, the GRβ lacks this LBD domain. Although GRβ is unable to bind to GC, it is still able to dimerize with GR⍺, resulting in an inhibition of GR signaling by acting as a dominant negative transcription factor.29 Some studies showed that GRβ is decreased in the glaucomatous TM cells.30–32 Patel and colleagues overexpressed GRβ in the TM of DEX-induced OHT mice using the Ad5 adenovirus.33 They found that GRβ overexpression inhibited DEX-induced fibronectin, myocilin, and collagen 1 expression in the mouse TM, decreased DEX-induced OHT, and improved outflow facility. Therefore, it is hypothesized that GR⍺/GRβ ratio may determine an individual’s GC responsiveness.29
There are different mechanisms to regulate GR⍺/GRβ ratio in the cell, including GR alternative splicing, GR translocation, and GR polymorphisms.21,22,34–41
- Glucocorticoid receptor alternative splicing
As described previously, GR⍺ and GRβ originate from alternative splicing, and therefore the regulation of alternative splicing contributes to different GR⍺/GRβ ratios. Spliceosomes are protein complexes consisting of ribonucleoproteins and serine-arginine proteins (SRp).41 Jain and colleagues investigated the role of different SRps in GR splicing.38 They found that SRp30c and SRp40 overexpression decrease GR⍺/GRβ ratios and DEX-induced GR signaling activity in human TM cells. They also found that Bombesin, a peptide that decreases SRp20 while increasing SRp30c and SRp40 levels, decreases GRα/GRβ ratio, and suppresses DEX responses in human TM cells.38 In another study conducted by the same group, the authors found that a microbial-derived compound, thailanstatin, which regulates alternative splicing, increases GRβ (decreases GRα/GRβ ratio) and effectively attenuates the effect of DEX on human TM cells.39
To activate the GR signaling pathway, GRα needs to translocate from the cytoplasm to the nucleus. Zhang et al reported that the hsp90 inhibitor, 17-AAG, decreases GRβ nuclear translocation and increases its degradation in the cytoplasm, and therefore increases GRα/GRβ ratio.21 Since GRβ nuclear translocation requires FKBP51, they studied the effect of FK506 (an FKBP51 inhibitor) on GC-induced GR translocation in two transformed human TM cell lines (NTM5 and GTM3). In non-glaucomatous NTM5 cells, KF506 increased DEX-induced GRβ nuclear translocation. In contrast, such an increase of nuclear GRβ was not observed in glaucomatous GTM3 cells.22 These studies suggest that GRβ nuclear translocation may affect GR signaling activity in different TM cells and may contribute to differential GC responsiveness.21,22
Fingert et al studied 48 single nucleotide polymorphisms (SNPs) in three groups of subjects: POAG patients, GC responders, and normal (non-glaucomatous, non GC-responder) patients.35 These SNPs are associated with GR genes, FKBP genes, and SRp genes. They found that there was no significant difference in any of the SNPs among the three groups, suggesting that none of these SNPs are of high heritable risk in developing GIG or POAG.
In non-eye related studies, a biallelic polymorphism (Bcl1) in the GR gene NR3C1 was reported to be associated with GC responders compared to non-responders (in that study, responders/non-responders refer to disease treatment outcome, not IOP) in patients with inflammatory bowel disease.37 This polymorphism is associated with GC responsiveness in lymphocytes.34 However, Wang et al and Gerzenstein et al both found that there is no significant association between the Bcl1 polymorphism or other polymorphisms and the development of GIOHT, suggesting that they are unlikely to play a role in ocular GC responsiveness.36,40
Other Potential Regulatory Proteins of GR Signaling
Several other proteins may play a role in regulating GR signaling (Figure 2). The estrogen-related receptor β (ERRβ; also known as estrogen receptor-related 2) belongs to the orphan nuclear receptor family, a subgroup of the nuclear receptor superfamily. Trapp et al found that ERRβ inhibited the transcriptional activity of GRs in non-ocular cells.42 However, there is a lack of reports of such regulation in the TM.
 |
Figure 2 The regulation of glucocorticoid receptor signaling. Several proteins participate in the regulation of glucocorticoid receptor (GR) signaling. 1) The presence of GRβ, an alternatively spliced, dominant negative form GRα, inhibits GR signaling. 2) The estrogen-related receptor β (ERRβ) may inhibit GR transcriptional activity (in non-ocular cells). 3) P-glycoprotein actively transports glucocorticoids (GCs) out of the cell. 4) 11β-hydroxysteroid dehydrogenase 1/2 (11β-HSD 1/2) regulates the conversion of cortisol from cortisone and may contribute to increased GR signaling. |
LEM1 is a transporter protein found in the yeast, and it actively transports several molecules, including GCs, out of the cell.43,44 Kralli et al reported that the human gene Mrd1, also known as ABCB1, is functionally similar to LEM1 and produces a protein, P-glycoprotein, which actively transports GCs from the cell.43 Liu et al studied the effect of polymorphisms in the ABCB1 gene on the susceptibility of developing POAG.44 They found that two polymorphisms in the ABCB1 gene, 2677 G> T/A and 3435 C> T, are associated with susceptibility to POAG. Since POAG is closely related to GIOHT/GIG (over 90% POAG patients are GC-responders),45,46 ABCB1 may play a role in GIOHT/GIG.
Another protein that may play a role in the susceptibility of developing GIOHT/GIG is 11β-hydroxysteroid dehydrogenase (11β-HSD), which has two isozyme forms: 11β-hydroxysteroid dehydrogenase 1 (11β-HSD1) and 11β-hydroxysteroid dehydrogenase 2 (11β-HSD2). 11β-HSD1 converts cortisone to cortisol, and 11β-HSD2 converts cortisol to cortisone.47 It is known that high cortisol levels are a known risk factor for OHT and POAG.48–52 Carbenoloxone (CBX) is an inhibitor of 11β-HSD1 and was reported to lower IOP.47,53,54 Rauz et al showed that 11β-HSD1 is expressed in corneal endothelium and ciliary non-pigmented epithelium, but not in TM cells.47 However, they found that oral delivery of CBX lowered baseline IOP in healthy individuals. The authors hypothesized that 11β-HSD1 may contribute to GIOHT/GIG through several mechanisms including increasing cortisol in the aqueous humor.
Cross-Talk Between the GR Signaling Pathway and Other Signaling Pathways
The YAP/TAZ Pathway
The Yes-associated protein (YAP) and transcriptional coactivator with PDZ-binding motif (TAZ) are two proteins that play important roles in many biological processes, including adequate organ growth, through cellular signaling and regulation of gene expression.55 YAP/TAZ is commonly associated with the Hippo pathway, but there are other regulators of YAP/TAZ signaling, including mechanotransduction through cell structure, shape, and polarity, as well as intercellular communications.55,56 The YAP/TAZ pathway is also implicated in the development of several types of tumors.57,58
The Hippo pathway was originally discovered in Drosophila.59–63 In mammalian cells, when the Hippo pathway is on, MST1/2 forms a complex with the protein SAV1 and both proteins are phosphorylated and activated.55 Phospho-MST1/2 subsequently phosphorylates LATS 1/2 and MOB1A/B. Phospho-LATS 1/2-MOB1A/B complex phosphorylates YAP/TAZ leading to YAP/TAZ degradation. Without nuclear YAP/TAZ, gene expression is turned off. When the Hippo pathway is off, unphosphorylated MST1/2, SAV1, Mob1, and LATS1/2 lead to YAP/TAZ nuclear translocation where it binds to TEAD transcription factors and turns on gene expression.
Mechanotransduction is an important regulator of YAP/TAZ signaling, and part of this regulation is triggered by the changes in tension and conformation of F-actin.56 In brief, high-tension focal adhesions activate YAP/TAZ signaling, and low-tension focal adhesions inhibit YAP/TAZ signaling.64
There is an important cross-talk between GR signaling and YAP/TAZ (see Figure 3). Sorrentino and colleagues found that in breast cancer cells, the GR binds directly to the YAP promoter region and increases its expression.58 In addition, GC-induced fibronectin expression and actin reorganization increase YAP nuclear translocation in those cells.58 These effects are inhibited when the cells are co-treated with the GR antagonist RU486.57,58 Nakamura and colleagues found that GC-induced YAP activity may contribute to vocal folds fibrosis.65
 |
Figure 3 The cross-talk between glucocorticoid receptor and YAP/TAZ signaling pathways. The glucocorticoid receptor (GR) signaling pathway and the YAP/TAZ pathway cross-talk via 1) glucocorticoid (GC)-induced changes in ECM and actin cytoskeleton may lead to increased nuclear translocation of YAP; 2) the GRα-GC complex binds directly to the YAP gene promoter and increases its expression; 3) YAP/TAZ signaling inhibits GR signaling as TAZ prevents the binding of GRα to glucocorticoid response elements (GRE’s) which inhibits target gene transcription. |
Although GR signaling activates YAP/TAZ signaling, in contrast, YAP/TAZ signaling inhibits GR signaling. For example, Xu et al found that in hepatocytes, TAZ acts as an inhibitor of GR signaling by preventing its binding to the GRE of gluconeogenic genes since TAZ-knockout mice exhibit higher levels of glucose production and concentration in the blood.66
Recent studies have shown that YAP/TAZ signaling plays an important role in glaucoma-related ocular tissues including the lamina cribrosa,67 retinal capillary endothelial cells,68 and TM.69–71 The cross-talk between YAP/TAZ and GR signaling in the TM may contribute to GIOHT/GIG. Recently, Yoo and colleagues investigated the effect of statins, which are YAP/TAZ signaling inhibitors, on GC-treated TM cells.71 They found that Simvastatin significantly decreases nuclear YAP/TAZ and attenuates high extracellular matrix (ECM) stiffness induced by DEX. Their data suggested that Simvastatin may have therapeutic applications for GIOHT/GIG.71
The Wnt Pathway
The Wnt signaling pathway is evolutionarily conserved with a wide range of biological functions including the regulation of cell proliferation, cell fate determination, cell polarity during development, cell migration, apoptosis, and stem cell maintenance.72 Dysregulation of the Wnt signaling pathway has been reported in the pathogenesis of a variety of human diseases including developmental diseases, degenerative diseases, cancers,73 and glaucoma.74,75 The first member of the Wnt signaling pathway was identified in 1982,76 and since then, studies on Wnt signaling have been steadily increasing. The Wnt gene was originally derived from integrase-1 in mouse breast cancer and the wingless gene of Drosophila.76,77 Because the two genes and functional proteins are similar, researchers combined the terms as the Wnt gene.78
The Wnt signaling pathway includes the canonical and noncanonical pathways.72 The noncanonical Wnt pathways are independent of β-catenin-Tcell factor/lymphoid enhancer-binding factor (TCF/LEF). They include the Wnt/Ca2+ pathway, PCP pathway, and Rho/Rock pathway. The canonical Wnt pathway, also known as the Wnt/β-catenin pathway, involves the nuclear translocation of β-catenin and transcriptional regulation via TCF/LEF transcription factors.79 In the canonical Wnt pathway, in the absence of Wnt ligands, β-catenin undergoes degradation by the degradation complex formed by Axin2, adenomatous polyposis coli (APC), glycogen synthase kinase-3β (GSK3β), and casein kinase-1α (CKIα).80 When Wnt ligands bind to their transmembrane receptor Frizzled and co-receptor lipoprotein receptor-related protein 5/6 (LRP 5/6), the degradation complex disassembles, which allows cytosolic β-catenin to accumulate and translocate into the nucleus. Nuclear β-catenin associates with T-cell factors 1/3/4 (TCF1/3/4) or lymphoid enhancer binding factor 1(LEF-1). They bind to the TCF/LEF binding element and regulate gene expression.81
The Wnt pathway cross-talks with the GR pathway in several ways (Figure 4). The antagonists of the Wnt pathway such as dickkopf1 (DKK1)74 and sFRP182 are elevated in the glaucomatous TM. Overexpression of the two Wnt inhibitors induces OHT in human and/or mouse eyes.74,82 Mao and colleagues showed that there is a functional canonical Wnt signaling pathway in the TM and plays an important role in the regulation of IOP in the mouse eye.83
 |
Figure 4 The cross-talk between glucocorticoid receptor and Wnt signaling pathways. The glucocorticoid receptor (GR) signaling pathway and the Wnt signaling pathway cross-talk via 1) glucocorticoid (GC)-induced expression of DKK1 and SFRP1 inhibits the canonical Wnt signaling pathway in the TM; 2) canonical Wnt signaling inhibits GR signaling via unknown mechanisms); 3) GC-induced Wnt5a activates non-canonical Wnt signaling which is mediated by glypican-4. |
Bermudez and colleagues showed that the canonical Wnt pathway inhibitor DKK1 is elevated in the bovine TM after DEX treatment.46 More importantly, they found that DKK1 is more elevated in bovine responder TM cells compared to non-responder TM cells. In a recent study, Sugali and colleagues showed that the activation of canonical Wnt signaling attenuates DEX-induced GR signaling activity and pathological changes in the TM as well as prevents DEX-induced OHT in a mouse model.74 In contrast, inhibition of the canonical Wnt signaling enhances GR signaling. Ragunathan’s group also showed that DEX induces the expression of DKK1, DKK2, and SFRP1 in TM cells.84
Non-canonical Wnt signaling, in contrast, may contribute to glaucomatous changes. Yuan and colleagues reported the induction of Wnt5a by DEX, which contributes to cytoskeletal reorganization and CLAN formation via ROR2/RhoA/Rock signaling.85 Also, Rao’s group recently reported that glypican-4 mediates DEX and Wnt5a-induced changes in TM cell cultures.86
The TGFβ Pathway
TGFβ is a cytokine, which plays a key role in various biological processes including growth, differentiation, cell death, and migration.87–91 Three different isoforms have been identified in mammals: TGF-β1, TGF-β2, and TGF-β3.92 They bind to their transmembrane receptors TGFβRI and TGFβRII, which are serine/threonine kinases. Binding of TGFβ to its receptors activates TGFβ signaling via the phosphorylation of Smad2/3. Phospho-Smad2/3 and the co-Smad (Smad4; not phosphorylated) form a complex, and the complex translocates to the nucleus where it binds to the Smad binding element (SBE) to regulate gene transcription.93
Among different TGFβ proteins, TGFβ2 is one of the most well studied in glaucoma pathogenesis and plays an important role in the cross-talk to GR signaling (Figure 5). TGFβ2 levels are elevated in the aqueous humor and TM tissue of POAG patients.94,95 Experimentally, excessive TGFβ2 induces pathological changes including formation of CLANs, excessive synthesis and deposition of ECM proteins including fibronectin, Collagen I, IV, etc, in the TM as well as elevates IOP in perfusion cultured human eyes as well as in vivo rat and mouse eyes.95–99
 |
Figure 5 The TGFβ signaling pathway mediates the effect of the GR signaling pathway in the TM. Glucocorticoids (GCs) induce the expression of TGFβ2 which activates the TGFβ signaling in the TM, and this process seems to be SMAD3 dependent. |
Kasetti and colleagues showed that DEX elevates TGFβ signaling by inducing TGFβ2 expression in the TM.100 They also showed that Smad3 knock-out mice (with impaired TGFβ signaling) fail to develop ER stress or DEX-induced OHT. Their data suggested that DEX-mediated effects require/are via TGFβ signaling.
Glucocorticoid-Induced Pathological Changes in the TM, ECM, SC, and Distal Outflow Tissue
Glucocorticoid-Induced Changes in TM Cells
Cross-Linked Actin Networks (CLANs) in the TM
One of the unique features of TM cells (not seen in the inner wall of SC cells) is the formation of cross-linked actin networks (CLANs).101 Cross-linked actin networks are formed by F-actin and many actin-binding proteins.102–104 They consist of many small triangles and these triangles further form dome-shaped, three-dimensional structures frequently observed in the perinuclear region.101,105 CLANs are GC and TGFβ2-inducible in TM cells, and more importantly, glaucomatous TM cells and tissues form more CLANs.95,101,106–108 Other nonhuman TM cells, such as bovine83,109 and mouse110 TM cells, also form CLANs.
Although the detailed mechanisms by which CLANs form are still unclear, studies have shown that several signaling pathways participate in/affect CLAN formation. Besides GR and TGFβ signaling as described previously, Peters’ group showed that the activation of the integrin signaling pathway contributes to CLAN formation.104,111,112 Also, Yuan et al reported that DEX-induced noncanonical Wnt signaling increases CLAN formation.85
Multiple studies have investigated the potential intervention to prevent or reverse CLAN formation. Clark et al found that tetrahydrocortisol (THF), a cortisol metabolite, inhibits CLAN formation in GC-treated TM cells without blocking GR activity.107 Montecchi-Palmer and colleagues reported that the Rho-associated protein kinase (ROCK) inhibitor can completely or partially prevent CLAN formation and/or dissolve formed CLANs.105
However, the contribution of CLANs to OHT is still not clear. Recently, Peng and colleagues developed a stably transformed GTM cell line, which expresses the LifeAct-GFP fusion protein.113 This cell line enables live imaging of the actin cytoskeleton. Using this TM cell line, the authors showed that CLANs increase TM cell stiffness, inhibit TM cell phagocytosis, and decrease cells’ responsiveness to the actin depolymerization agent latrunculin B. The first two effects are also observed in primary human TM cells. The authors speculated that increased TM cell stiffness and decreased phagocytosis may contribute to increased outflow resistance and IOP.113 However, more research is needed to test this hypothesis.
Increased TM Cell Stiffness
Raghunathan and colleagues reported that after DEX treatment for 3 days, there is about 2-fold increase in primary human TM cell stiffness.84 It is still not entirely clear how DEX treatment elevates TM cell stiffness. Yuan et al and Fujimoto et al showed that DEX induces actin stress fibers85 and activates the ROCK pathway.114 Both events contribute to actin cytoskeletal contraction and reorganization, which could increase cell stiffness.115 Recently, Peng and colleagues used a vital actin dye SiR-actin to stain primary human TM cells for live imaging and atomic force microscopy.113 They showed that the TM cells with DEX-induced CLANs are stiffer than those treated with DEX but without CLANs, and the cells without CLANs are as soft as non-DEX treated control cells. As described previously, DEX-induced CLAN formation could be one of the contributors of elevated cell stiffness besides actin cytoskeleton contraction and other changes. How stiffer TM cells contribute to OHT is still not entirely clear, but many studies have shown that stiffer TM cells produce stiffer ECM and have compromised mechanosensory functions, which could increase outflow resistance and IOP.115–118
Decreased TM Cell Phagocytosis
Trabecular meshwork cells have the unique capability of phagocytosis, which enables them to clean up tissue/cell debris (eg, pigment, red blood cells, etc) and to maintain a clear pathway for aqueous humor drainage. Zhang and colleagues showed that DEX decreased TM cell phagocytosis and this decrease may contribute to OHT.32,119
Elevated Endoplasmic Reticulum (ER) Stress
Zode and colleagues showed that in DEX-induced OHT mouse eyes, ER stress markers such as GRP78, ATF4/6, and CHOP are elevated in the mouse anterior segment.120 Cotreatment with the ER stress inhibitor, 4-PBA, reduces DEX-induced ER stress markers and OHT. This study suggested that ER stress plays an important role in GIOHT/GIG.120
Cell Adhesion
Integrins are important cell-matrix adhesion proteins. Peters’ group has conducted extensive investigations on GC-induced integrin changes in the TM. They found that DEX activates αvβ3 integrins through conformational changes, which is likely via an inside-out process (ie, cytosolic changes activate αvβ3 from within), as well as increases αvβ3 integrin expression and affinity via the calcineurin/NFAT pathway.112,121,122 Besides, the integrin signaling pathway affects phagocytosis and CLAN formation in the TM.103,112,123,124
Glucocorticoid-Induced Changes in the TM ECM
The ECM is a key component of the TM outflow pathway. Glucocorticoids induce the accumulation of ECM proteins such as glycosaminoglycans, fibronectin, and collagens19,125,126 by increasing their production and/or decreasing metalloproteinases (MMPs, which degrade ECM).127–129 There are fingerprint-like materials at the JCT ECM underneath the inner wall of SC in GIOHT/GIG eyes.130 Overby’s group also reported an increase in the basement membrane of the inner wall endothelium of SC in GIG eyes.131
Besides changes in the amount of ECM, GCs make TM ECM stiffer, which may contribute to increased outflow resistance and GIOHT. Raghunathan and colleagues studied the effect of DEX on TM ECM.84 The authors treated TM cells with or without DEX and then removed the cells using a mild detergent. Using atomic force microscopy, they found that DEX increases the stiffness of the ECM around the treated cells. The short-term stiffness change induced by DEX can trigger changes in gene expression and lead to altered ECM deposition/composition in the long term, thus creating a feedforward loop perpetuating the glaucomatous changes.84 GCs may also inhibit MMP activation/activity by the induction of PAI-1, an inhibitor of tissue plasminogen activator (tPA), although there are limited reports.132 However, treatment with tPA was shown to lower IOP in GIOHT animal models. In a sheep GIOHT model, Gerometta and colleagues showed that intravitreal injection of tPA decreases IOP.133 Kumar and colleagues showed that the administration of tPA lowers IOP in a mouse GIOHT model.134 Gindina and colleagues observed that tPA does not exert its effect on MMP expression through enzymatic activity, but rather through a cytokine-mediated fashion as both enzymatically active and inactive tPA had similar effects on MMP expression after GC administration.135
Glucocorticoid-Induced Changes in the SC
Underwood and colleagues reported that DEX increases fluid flow resistance, tight junction formation and the associated protein ZO-1, as well as a reduction in interendothelial spaces.136 Glucocorticoids also induce actin cytoskeletal changes in SC inner wall cells, causing elevated cortical stiffness.137 Importantly, increased SC inner wall cell stiffness is also observed in POAG eyes.138 Therefore, high SC inner wall stiffness is likely to compromise pore formation and therefore to increase outflow resistance.137 Overby and colleagues found that in human donor eyes with long-term GC use, the SC basement membrane is more continuous and shows the morphology of a thick band.131
Glucocorticoid-Induced Pathological Changes in the Distal Outflow Tissue
Most of the GIOHT/GIG research focuses on the proximal outflow tissues (TM and SC). Recently, the distal outflow tissue has drawn increasing attention in glaucoma research. Rosenquist et al showed that the distal outflow tissue may contribute to about 25%–50% of the total outflow resistance.139 Gonzalez Jr. et al showed that the distal outflow tissue expresses contractile tissue markers using 2-photon imaging.140 McDonell and colleagues reported that endothelin-1 significantly elevates outflow resistance in perfusion cultured human donor eyes with the TM removed (thus these eyes only had distal outflow tissues), and this elevation could be reversed by co-treatment with a nitroxide donor compound.141 Chowdhury and colleague developed an approach to culture vascular distal outflow cells and showed that these cells are biologically different from TM cells.142 More importantly, a recent clinical study reported that one-eighth of the minimally invasive glaucoma surgery (surgical procedures that partially/completely remove the TM or open the TM from inside the eye) patients may still develop GIOHT,143 suggesting that the distal outflow tissue may also contribute to GIOHT/GIG.
Clinical Science Review of GIG/GIOHT
Clinical Features of GIG/GIOHT
The clinical features of GIOHT/GIG are very similar to those of POAG. Unlike some POAG patients who do not show OHT (normal tension glaucoma), all GIOHT/GIG patients have elevated IOP. Depending on the responsiveness of these patients, their IOP may be significantly higher than that of POAG patients, which may lead to rapid disease progression.
Glucocorticoid Responsiveness and the Type of GCs
The type of GCs affects GC responsiveness (Table 1).31 Bojikian et al found that the incidence of GIOHT (defined as >50% increase in baseline IOP) following topical prednisolone post-cataract treatment to be 2.1% in non-glaucomatous eyes and 8.4% in glaucoma eyes.144 Rajendrababu et al analyzed GC response after the same treatment (topical prednisolone post-cataract treatment) and found that 3.2% (2.8% moderate and 0.4% high responders) non-glaucomatous patients had GIOHT (>6 mmHg increase in IOP).145 Badrinarayanan et al found that 28% of non-glaucomatous eyes developed OHT ≥21 mmHg after triamcinolone-acetonide injection, and 17% of non-glaucomatous eyes that received a DEX implant were GC responders (IOP ≥21mmHg).146 In a pediatric study, Krag et al found that 56% of children receiving systemic GCs had OHT (net increase in IOP ≥6 mmHg from baseline or a peak IOP ≥21 mmHg).147
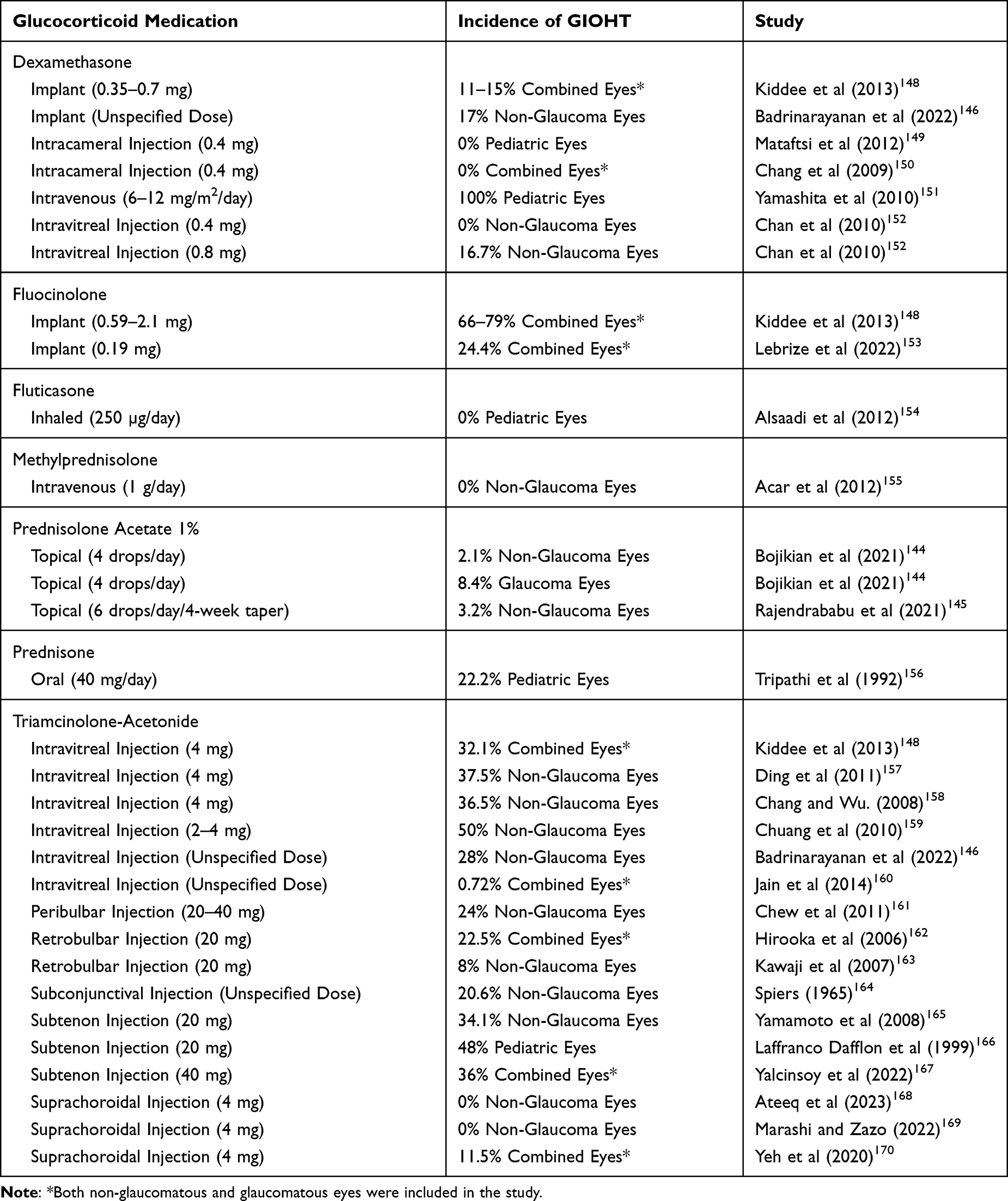 |
Table 1 A Comparison of the Incidence of Glucocorticoid-Induced Ocular Hypertension (GIOHT) of Different Glucocorticoids |
In 1975, Cantrill et al found that highly potent GCs are associated with a higher risk of GIOHT.171 In general, older GCs such as DEX, betamethasone, and prednisolone are more likely to induce OHT compared to newer GCs such as loteprednol etabonate, rimexolone, or difluprednate.172,173 Some studies showed that loteprednol etabonate is less likely to induce OHT (≥10 mm Hg from baseline) after short- or long-term use.174,175 Several characteristics of loteprednol etabonate make it an attractive alternative to older GCs: it is more rapidly metabolized after GR activation due to the replacement of a ketone group with an ester group at the carbon 20 position, and it is highly lipophilic with high affinity for the GR which allows for excellent penetration in the ocular tissue.175 In a study that compared loteprednol etabonate to prednisolone, Price et al found that loteprednol etabonate was not inferior to prednisolone in the prevention of DMEK graft rejection and with less chance to induce GIOHT.176
Raj et al compared the effect of prednisolone to rimexolone after corneal transplantation, and they found a significant decrease in the mean IOP elevation postoperatively after switching from prednisolone to rimexolone.177 Likewise, Chen et al reported a decrease in IOP over a 12 month follow-up for pediatric uveitis patients who were switched from DEX, prednisolone, or difluprednate to rimexolone.178 They also reported that rimexolone provided adequate treatment for pediatric anterior uveitis patients.178
In 2022, Kao et al studied the effects of difluprednate vs prednisolone after Ahmed glaucoma valve surgery.172 They observed that treatment success rates of the two GCs were comparable for 1-year post-operation. They also observed that the effort of the two GCs on IOP was similar. However, the patients receiving difluprednate required less additional glaucoma medications compared to those who received prednisolone.
These studies suggest that newer GCs may decrease the risk of GIOHT/GIG and have similar efficacy to older GCs.
Risk Factors of GIOHT/GIG
POAG and GC Responsiveness
Armaly and Becker first reported that POAG patients are at higher risk of developing GIOHT/GIG in the 1960s,5,6,179 suggesting that POAG patients are more likely to be GC responders. Their studies showed that about 40% of the general population are responders: among them, approximately 32% of showed a moderate response (6–15mmHg increase in IOP or IOP between 25–31mmHg) and approximately 4% showed a dramatic response (>15mmHg increase in IOP or IOP >31mmHg).180,181 In POAG-suspects and POAG patients, more than 90% are GC responders.5,179
Although new GCs have been developed to minimize GIOHT/GIG, POAG patients are still susceptible to GIOHT/GIG.182 A retrospective analysis reported a high risk of developing OHT in patients who received one or more intravitreal DEX implants.31,183 They also found that glaucoma patients who required dual or triple therapy have higher risk of developing GIOHT. Maier et al studied the incidence of IOP elevation in the patients who received Descemet’s membrane endothelial keratoplasty (DMEK) at 12 and 36 months post operation.184 Postoperative treatment for these patients included one GC eye drop 5 times daily that was weaned to 1 to 2 times daily over the course of 3 to 4 months, which was then continued for a full year postoperatively, unless an increase in IOP was identified.184 They found that the main risk factor for developing GIOHT was preexisting POAG.184 In addition, the mean IOP for preexisting glaucoma patients was significantly higher than that for non-glaucoma patients for the entire follow-up period.184
It is important to note that the family history of POAG also increases the risk of GIOHT/GIG, which was first reported by Armaly who found that 62% of offspring of glaucoma patients had an increase >6 mmHg in response to GC treatment.185 In another study, Bartlett et al found that after topical administration of GCs in healthy, first-degree offspring of POAG patients, 69% of them had an IOP increase of no less than 5mmHg.186 Mitchell et al found a significant association between the use of inhaled GC and developing OHT/GIG in patients with a family history of glaucoma.187 Although family history is involved, just like POAG, which is a multifactorial disease, GC responsiveness is also complex involving multiple genetic and environmental factors.188
The relationship between POAG and GIOHT/GIG was further demonstrated from a reversed perspective: patients who have a significant IOP elevation in response to GC treatment are more likely to develop POAG.8 Lewis et al studied patients with no previous glaucoma diagnosis, and found that 13% of high responders developed POAG over a period of 5 or more years.9 However, the same study showed that low responders had zero diagnoses of POAG during the same follow-up period.
All these studies show that GIOHT/GIG and POAG are closely related, and that is why GIOHT/GIG animal models are frequently used as POAG research models.
Age
Older age was considered a risk factor of GIOHT/GIG.6,179,188,189 Interestingly, recent studies have suggested that younger age is a risk, not older age.145 Choi et al performed a retrospective observational study involving 570 eyes of 455 adult patients who received intravitreal DEX injection and compared the rate of GIOHT based on approximate decade of life.182 They found that the greatest rate of GIOHT was in the youngest group (16–30 years) with 42.9% of patients developing an IOP elevation ≥10mmHg. The rate of IOP steadily decreased with each increasing decade of life, with the lowest rate of IOP elevation ≥10mmHg seen in patients 81–90 years at 9.1%.182 Malclès et al reported a similar finding in that patients with age ≤60 years were at increased risk.183 In addition, it was reported by Friedman et al that in eyes with uveitis, age <50 years increased the risk of IOP elevation following GC treatment.190
Pediatric studies have demonstrated that the risk of GIOHT/GIG is increased in young children.191,192 Lam et al studied the effect of GC eye drops in pediatric patients who received strabismus surgery.193 In that study, the patients were divided into two groups and received GC eye drops either 4 times daily or 2 times daily. The authors found that IOP elevation after GC treatment is age dependent: young children (<6 years old) showed greater net increase in IOP and less time to reach peak IOP. These studies suggest that caution should be taken with GC treatment in children and young adults and alternative options should be sought when appropriate.
Other Risk Factors
Connective tissue disorders,194 increased axial length,144,145,195 and type I Diabetes Mellitus196 have also been reported to increase the risk of developing GIOHT/GIG.
Methods of GC Administration
All methods of GC administration are associated with GIOHT/GIG, but the incident rate of each method is different (see Table 1). Common methods of GC administration include ocular, extraocular, and systemic administrations.31,189,197
Ocular administration includes topical, intraocular, and periocular routes. Extraocular administration includes inhaled and intranasal GC delivery. Systemic administration includes oral, intravenous, percutaneous, and excessive endogenous production. Studies show that GIOHT/GIG tends to occur more quickly with ocular administration, usually within weeks to months compared to systemic administration, which may occur after months or years of GC use.189,198
It is also important to understand that drug formulation may affect the risk of developing GIOHT/GIG as well. Hydrophilic compounds, such as GC phosphates, do not penetrate the cornea as well as lipophilic compounds, such as GC acetates. This may account for some of the differences in GC potency and the risk of IOP elevation.199,200
Topical Application
Topical GC use is more likely to lead to GIOHT/GIG than systemic use.188,189,201 Sihota et al found that 73.5% of GIG cases were caused by topical GC administration.202 Topical GC treatment is often used for various ocular diseases and as a post-operative/post-procedural regimen for ophthalmologic surgeries and laser treatments. Chronic GC users, most of whom have ocular inflammation such as uveitis and allergic conjunctivitis, are at high risk of developing GIOHT.197 Cessation of GC eye drops usually leads to pressure normalization within a few weeks,173,201 but some could last up to 18 months.31 As previously discussed, newer GCs may provide a better option in treating inflammation with decreased risk of IOP elevation compared to older GCs.
However, in very few cases, GIOHT persists even after GC cessation.203 We hypothesize that these patients may be at the preclinical stage of POAG (no IOP elevation), and there have already been pathological changes in their TM. The use of GCs accelerates these irreversible damages in the TM and brings these patients from preclinical POAG to clinical POAG with OHT.
Intraocular Injection
Intraocular administration most commonly refers to intravitreal injection but also includes intracameral injection and suprachoroidal injection. The use of intravitreal GCs has been continually increasing for retinal and choroidal diseases due to the proximity of GCs to the target tissue.204,205
Intravitreal injection may be a single injection of GCs or an implantation of a sustained-release device. Similar to topical administrations, drug solubility plays an important role. Hydrophilic GCs, such as DEX sodium phosphate, have a shorter half-life in the vitreous compared to hydrophobic GCs, such as triamcinolone acetonide.206,207 In addition, the status of the vitreous can influence drug bioavailability in the eye. Beer et al found that the elimination half-life of triamcinolone acetonide was decreased by approximately 83% in eyes with vitrectomy compared to that in non-vitrectomized eyes.208 Chin et al reported similar results, with a reduction of approximately 50% in triamcinolone acetonide half-life in vitrectomized eyes vs non-vitrectomized eyes.209
The most commonly used GCs for intravitreal injection are triamcinolone acetonide and DEX.31 Many studies have shown that on average, about 32.1% of the eyes developed GIOHT after 4mg triamcinolone acetonide injection.148 However, several other studies have reported the incidence of GIOHT after triamcinolone acetonide injection to be higher, with one study reporting up to 50%.157–159,210 In a United Kingdom national survey, the mean time between triamcinolone acetonide injection and reaching maximum IOP was 16 weeks although it could range from 1.5 weeks to 71 weeks.160 For DEX, one study found that 16.7% of the eyes developed GIOHT after 0.8mg DEX injection and that 0% of eyes developed GIOHT following 0.4mg DEX injection over a 4-week period.152
Implantable devices decrease the risk associated with repeated intravitreal injections and provide a sustained, low-dose GC treatment. This leads to less exposure of the TM to GCs and may decrease systemic side effects.211 Two GCs are currently available for implantable devices: fluocinolone acetonide (administered as a non-degradable implant) and DEX (administered as a degradable implant).
In a review of several studies, the incidence of significant IOP elevation after 3.5 mg DEX implant was 11%.148 This rate is less than that resulting from 0.59 mg fluocinolone acetonide implant (66%).148 Recently, a large study assessed the safety of DEX implants, and it reported that 26.5% of the subjects experienced a significant increase in IOP.212 Over 90% of these patients were managed with IOP-lowering eye drops and 0.5% required surgical treatment to lower their IOP.212 Similarly, in another study with patient data obtained from 4 European countries, it showed that in 1 year follow-up of patients who received fluocinolone acetonide implant, 24.4% of them developed significant IOP elevation.153
Intracameral injection (delivery to the anterior chamber) and suprachoroidal injection (delivery to the suprachoroidal space) are also used for GC administration.211 Mataftsi et al concluded that intracameral injection of preservative-free DEX did not increase the risk of GIOHT/GIG in pediatric patients after cataract surgery.149 Another study performed by Chang et al found that intracameral DEX injection at the end of cataract surgery significantly decreased the anterior chamber inflammation but did not increase the risk of GIOHT compared to the eyes that did not receive a DEX injection.150 Intracameral injections may avoid compliance issues, but also involve other risks that are not associated with topical treatment. Further studies should be conducted to understand the risk of developing GIOHT/GIG after intracameral GC treatment in the long term.
Suprachoroidal injection is primarily used to treat retina and choroid diseases, and triamcinolone acetonide is clinically available. Muya et al compared the pharmacokinetics and distribution of suprachoroidally injected triamcinolone acetonide to those of intravitreally injected triamcinolone acetonide using rabbit eyes.213 They found that there was a significantly higher exposure of retinal pigmented epithelium, choroid, and sclera tissues to triamcinolone acetonide in suprachoroidally injected eyes compared to intravitreally injected eyes. However, the exposure was similar in the retina between the two methods.213 Furthermore, the exposure of the lens, iris, and vitreous humor to triamcinolone acetonide was significantly decreased with minimal exposure in the aqueous humor in the suprachoroidal injection group.213 Recently, several other studies evaluated the efficacy of suprachoroidal injection of GCs in several retinal diseases including diabetic macular edema,168,169 uveitic macular edema,170,214,215 and retinal detachment secondary to Vogt-Koyanagi Harada disease.216 The results showed good efficacy with no significant increase in IOP. Therefore, suprachoroidal GC injection may be an alternative to intravitreal injection for treating retinal disease when GIOHT/GIG is a concern.
Periocular Injection
Periocular administration includes subconjunctival, subtenon, peribulbar, and retrobulbar injections. The concentration of GCs is higher in the anterior chamber using subconjunctival and subtenon injections. Peribulbar and retrobulbar injections lead to greater GC localization close to the retina and vitreous.197 The GCs injected subconjunctivally enter the eye through passive diffusion through the sclera.211 Subconjunctival injection is more efficient for ocular GC delivery compared to oral administration.217 However, some studies have shown that subconjunctival injection is less efficient since significant amounts of GCs leak through the injection site.218
Periocular injection of GCs also induces OHT. Some studies showed that 20.6% of scleritis patients developed OHT after subconjunctival injection of triamcinolone acetonide.164 Subtenon injection of GCs seems to cause high GIOHT incidence.219 Yamamoto et al reported GIOHT in 34.1% of the patients165 while Lafranco Dafflon et al reported GIOHT in 36% of the patients after subtenon GC injection.166 In a study of pediatric uveitis patients, Yalcinsoy et al observed GIOHT in 48% of the patients after subtenon GC injection.167 Chew et al evaluated the effect of peribulbar injection of triamcinolone acetonide, and the authors reported that 24% of the patients developed significant IOP elevation.161 For the retrobulbar GC injection, Hirooka et al retrospectively evaluated the incidence of GIOHT and found that the rate was 22.5%.162 However, Kawaji et al found that retrobulbar injection of triamcinolone acetonide caused GIOHT in only 8% of the patients.163
Inhalation and Intranasal Administration
In 1997, Garbe et al conducted a large case–control study showing that prolonged use (3 months or more) of inhaled GCs at high doses increased the risk of OHT and glaucoma.220 However, the authors did not have proper controls for POAG family history or high myopia, and there were concerns with the conclusions of that study and the true effect of inhaled GCs and the development of GIOHT/GIG.221 In 1999, Mitchell et al found a significant association between high doses of inhaled GCs and elevated IOP/glaucoma, but only in patients that had first-degree relatives with POAG.187
Since the publication of those studies, there have been conflicting reports. In 2017, Nath et al conducted a prospective observational study to investigate the prevalence of glaucoma in elderly patients receiving inhaled GCs.222 They found that the prevalence of glaucoma was 3.92% and the prevalence of OHT 16%. They also observed a dose-dependent relationship showing that 42.8% of the patients receiving high doses (501–1000 µg) of inhaled GCs daily developed OHT. Thus, the authors concluded that higher doses and longer usage of inhaled GCs are associated with GIOHT/GIG.222 In contrast, several studies have found no association between inhaled glucocorticoids and GIOHT/GIG.223–226 One such study was conducted by Gonzalez et al who conducted a case–control study for elderly patients who were taking inhaled GC and started on glaucoma treatment.227 They found that there was no dose-related association nor long-term use relationship between inhaled GC and GIOHT/GIG. Similarly, in a pediatric study conducted by Alsaadi et al, it was observed in children between 5 and 15 years of age there was no association between normal dose GC and the development of elevated IOP.154 In 2021, a comprehensive meta-analysis that included 18 studies and 31,665 subjects was conducted by Ishii et al to determine the association between inhaled GC and GIOHT/GIG.228 The results of their analysis showed no evidence that linked inhaled GC with ocular hypertension, glaucoma prevalence, or glaucoma incidence. Interestingly, they found there may be a reduced risk of glaucoma in patients who receive inhaled GC.
The result of the studies investigating the association between intranasal GC use and GIOHT/GIG is also inconsistent.220,225,229 Most of the studies conclude that there is no association between intranasal GC use and OHT. In 2019, Valenzuela et al conducted a meta-analysis including 10 randomized controlled trials and 2226 patients.230 They found there was no absolute risk of developing OHT when taking intranasal GCs. They also found there were zero cases of glaucoma reported within a year of follow-up. On the contrary, Bielory et al concluded that with increased availability of over-the-counter intranasal GCs, some individuals may develop elevated IOP, especially those with risk factors, and therefore should be monitored.231
The results of these studies suggest that there is a low risk of GIOHT/GIG with inhaled and intranasal GC treatment. However, IOP monitoring may be important for individuals taking high-dose or long-term inhaled or intranasal GCs, especially for those with risk factors such as disease and/or family history of POAG. A baseline IOP and IOPs at 1 and 3 months from the initiation of GC treatment are recommended for monitoring potential GIOHT/GIG.31
Oral Administration
In 1962, Bernstein and Schwartz reported that long-term use of oral GCs induced significant OHT.232 Other studies also showed an association between oral GC use and GIOHT/GIG.197,220,233 Tripathi et al reported that for every 10 mg of prednisone taken orally daily, there was a reciprocal increase of 1.4 mmHg in the mean IOP.156 Generally, oral use of GCs is less likely to develop GIOHT/GIG compared to topical use. However, one study showed that elderly patients who took 80 mg or more of hydrocortisone orally daily might have a similar risk of developing GIOHT/GIG as those receiving topical GCs.234
Intravenous Injection
Intravenous GCs are first-line treatment for several diseases including multiple sclerosis.235 Acar et al studied the effect of high dose intravenous methylprednisolone on IOP in multiple sclerosis patients with acute relapse.155 They found an initial significant increase in IOP during treatment and within 1-month post treatment. However, at two and three months after, IOP returned to normal levels and the patients showed no glaucomatous optic nerve damage.155 Another study by Yamashita et al investigated the effects of systemic DEX, including intravenous administration, on IOP in pediatric patients with acute lymphoblastic leukemia.151 They found that there was a significant rise in IOP in all patients with max IOP between treatment cycle 5 and 11. Thus, patients receiving intravenous GCs, especially those receiving long-term treatments, should be monitored for GIOHT/GIG.
Percutaneous GC Absorption
Cutaneous application of GCs may induce GIOHT/GIG.236–238 Most of these cases consisted of cutaneous application of GCs to the periorbital region. The skin of the eyelids/periorbital region is relatively thinner than other parts of the body, which facilitates GC penetration and absorption resulting in high concentrations of GCs in the eye. Takakuwa et al described a new clinical entity as “atopic glaucoma” which is glaucoma associated with atopic dermatitis.239 They found that 48 out of 62 eyes with atopic glaucoma had used GC ointments.239
However, Haeck et al conducted a retrospective study to determine the effect of cutaneous GC application on the risk of IOP elevation and glaucoma.240 They included 88 patients in their analysis, and found that 37 patients regularly applied GCs to the eyelids and periorbital regions. Among the 88 patients, only one developed transient OHT. Therefore, the authors concluded that there was no association between cutaneous topical GC and risk of GIOHT/GIG.240
Although the result of these studies is not consistent, the risk of GIOHT/GIG induced by cutaneous GC treatment seems to be lower than the other GC administration methods. However, for patients who use topical GC treatments regularly on the eyelid and periorbital region, IOP monitoring may be necessary.
Elevation of Endogenous GCs
Elevated IOP has been documented in diseases with high levels of endogenous GCs such as Cushing’s syndrome.241–244 In a recent case report, Habib et al described a male patient with OHT who was initially managed with close monitoring.244 Three months later, he was diagnosed with Cushing’s syndrome due to a microadenoma in the pituitary gland. After surgical removal of the microadenoma, his IOP returned to normal level. Therefore, the patient was diagnosed with GIOHT. For GIOHT patients resulting from elevated endogenous GCs, treatment of the underlying disease that leads to GC elevation should be initiated promptly to normalize IOP and prevent irreversible optic nerve damage.245
More importantly, as described previously, it is known that high cortisol levels are a known risk factor of POAG, while POAG is a risk factor of GIOHT/GIG. Therefore, high endogenous cortisol levels (which do not have to be as high as in diseases such as Cushing’s syndrome) are likely a risk factor of GIOHT/GIG.
Management of GIOHT/GIG
GIOHT may cause irreversible optic nerve damage and loss of vision (GIG). Thus, early diagnosis and intervention are necessary. General management of GIOHT/GIG includes the following: discontinuation of GC therapy if possible or switch to an alternative GC with less risk of inducing OHT, removing deposited GCs, as well as IOP-lowering eye drops and/or surgeries if required.246 For most patients, IOP will drop quickly as long as GCs are discontinued swiftly.246 For patients with chronic GIOHT/GIG, IOP may normalize after 1 to 4 weeks after GC discontinuation.247 One study showed that after GIOHT/GIG diagnosis, some patients required anti-glaucoma treatment for up to 18 months after discontinuation of GC use.202 For patients who experience OHT after intravitreal GC injection or GC-sustained release device implantation, removal of GCs or GC-sustained release devices may be needed to control their IOP.248 Also, Rezkallah et al showed that the XEN gel stent (Allergan Inc., CA), which is a drainage device between the anterior chamber and the subconjunctival space, successfully lowered IOP in 5 GIOHT patients caused by DEX implants.249 Furthermore, they found that it was safe to resume GC treatment with the implanted GC device and the concurrent use of the XEN gel stent.249 Medications and surgical treatments for GIOHT/GIG are practically identical to those for POAG, including IOP-lowering eye medicines, laser trabeculoplasty, drainage implant surgery, and filtering surgery.250 Several recent studies have shown that selective laser trabeculoplasty (SLT) is a safe and effective treatment for GIOHT/GIG.251–254 Davidson et al evaluated the efficacy and safety of SLT in patients who developed GIOHT due to the requirement of GC treatment after endothelial keratoplasty.252 They found that SLT may be a safe option to control IOP, which allows the continuation of GC treatment in patients at high risk of graft rejection.252
Patients with GIOHT/GIG should be monitored regularly and closely to determine their responses to treatment and to prevent further optic nerve damage. Goñi et al proposed the following method for IOP monitoring:255 1) a baseline IOP measurement and determination of glaucomatous damage; 2) risk factor assessment including IOP ≥ 15 mmHg, family history, previous episodes of GC-induced IOP elevation, glucocorticoid dosage, etc; 3) if IOP is >21 mmHg, then visual field testing as well as imaging of the optic nerve and retinal nerve fiber layer should be collected; treatment initiation can be delayed and a follow-up within 6 weeks is warranted; 4) if IOP is >25 mmHg, then visual field testing and imaging studies should be performed with initiation of first treatment using medical treatment or laser intervention. Follow-up should be conducted within 6 weeks, if IOP is >25 mmHg and the patient has received ≥2 medical therapies, then referral to a glaucoma specialist is warranted.
Potential novel therapies are under research, which target various aspects of the complex mechanism of GR signaling and/or GIOHT/GIG. For example, tPA lowered IOP in sheep and mouse GIOHT models by increasing the activity MMP’s and prevented, at least partially, GC-induced pathological changes in the TM.133–135,256,257 Using the same sheep model, Gerometta and colleagues showed a reduction in IOP after introducing a GC-inducible MMP1 gene expression vector into GIOHT sheep eyes.258 Patel and colleagues showed that the viral mediated expression of GRβ attenuated GC-induced OHT in mouse eyes.33 Sugali and colleagues also showed that viral mediated expression of Wnt3a to activate the Wnt pathway prevented GIOHT in the same mouse model.74 Further research should be conducted to better understand the complexities of the disease process and potential therapies that can be translated into clinical use.
Conclusion
Glucocorticoid-induced glaucoma is a complex secondary glaucoma that can lead to severe vision loss and even blindness. Glucocorticoids induce POAG-like changes the TM and SC. The GR signaling pathway is complicated with multiple regulators and it cross-talks with many other signaling pathways. Regardless of the method of GC administration and GC types, there is a risk for general population, especially those with POAG or family history of POAG, to develop GIOHT/GIG. The risk level also depends on many other factors. Early diagnosis and treatment of GIOHT/GIG are important to prevent optic nerve damage and to preserve visual function.
Funding
This work was supported by NEI R01EY026962 (W.M.), R01EY031700 (W.M.), R01EY031700-03S1 (W.M.), R21EY033929 (W.M.), BrightFocus Foundation G2023009S (W.M.), and a Challenge Grant from Research to Prevent Blindness (Department of Ophthalmology, Indiana University School of Medicine).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Sun Y, Chen A, Zou M, et al. Time trends, associations and prevalence of blindness and vision loss due to glaucoma: an analysis of observational data from the Global Burden of Disease Study 2017. BMJ Open. 2022;12(1):e053805. doi:10.1136/bmjopen-2021-053805
2. Thorne GW, Forsham PH, Frawley TF, et al. The clinical usefulness of ACTH and cortisone. N Engl J Med. 1950;242(21):824–834. doi:10.1056/NEJM195005252422105
3. F J. Cortisone et tension ocularire. Ann Ocul. 1954;187:805–816.
4. Covell LL. Glaucoma Induced by Systemic Steroid Therapy. Am J Ophthalmol. 1958;45(1):108–109. doi:10.1016/0002-9394(58)91403-X
5. Armaly MF. EFFECT OF CORTICOSTEROIDS ON INTRAOCULAR PRESSURE AND FLUID DYNAMICS. I. Arch Ophthalmol Chic. 1963;70:482–491. doi:10.1001/archopht.1963.00960050484010
6. Becker B, Mills DW. Corticosteroids and Intraocular Pressure. Arch Ophthalmol. 1963;70(4):500–507. doi:10.1001/archopht.1963.00960050502012
7. Llobet A, Gasull X, Gual A. Understanding Trabecular Meshwork Physiology: a Key to the Control of Intraocular Pressure? Physiology. 2003;18(5):205–209. doi:10.1152/nips.01443.2003
8. Kitazawa Y, Horie T. The Prognosis of Corticosteroid-Responsive Individuals. Arch Ophthalmol. 1981;99(5):819–823. doi:10.1001/archopht.1981.03930010819005
9. Lewis JM, Priddy T, Judd J, et al. Intraocular Pressure Response to Topical Dexamethasone as a Predictor for the Development of Primary Open-Angle Glaucoma. Am J Ophthalmol. 1988;106(5):607–612. doi:10.1016/0002-9394(88)90595-8
10. Bill A. Blood circulation and fluid dynamics in the eye. Physiol Rev. 1975;55(3):383–417. doi:10.1152/physrev.1975.55.3.383
11. Stamer WD, Clark AF. The many faces of the trabecular meshwork cell. Exp Eye Res. 2017;158:112–123. doi:10.1016/j.exer.2016.07.009
12. Stamer WD, Braakman ST, Zhou EH, et al. Biomechanics of Schlemm’s canal endothelium and intraocular pressure reduction. Prog Retin Eye Res. 2015;44:86–98. doi:10.1016/j.preteyeres.2014.08.002
13. Ethier CR, Coloma FM, Sit AJ, Johnson M. Two Pore Types in the Inner-Wall Endothelium of Schlemm’s Canal. Ophthalmol Vis. 1998;39(11):2041.
14. Mäepea O, Bill A. The pressures in the episcleral veins, Schlemm’s canal and the trabecular meshwork in monkeys: effects of changes in intraocular pressure. Exp Eye Res. 1989;49(4):645–663. doi:10.1016/S0014-4835(89)80060-0
15. Overby DR, Stamer WD, Johnson M. The changing paradigm of outflow resistance generation: towards synergistic models of the JCT and inner wall endothelium. Exp Eye Res. 2009;88(4):656–670. doi:10.1016/j.exer.2008.11.033
16. Encío IJ, Detera-Wadleigh SD. The genomic structure of the human glucocorticoid receptor. J Biol Chem. 1991;266(11):7182–7188.
17. Wordinger RJ, Clark AF. Effects of glucocorticoids on the trabecular meshwork: towards a better understanding of glaucoma. Prog Retin Eye Res. 1999;18(5):629–667. doi:10.1016/S1350-9462(98)00035-4
18. Wang RYR, Noddings CM, Kirschke E, Myasnikov AG, Johnson JL, Agard DA. Structure of Hsp90−Hsp70−Hop−GR reveals the Hsp90 client-loading mechanism. Nature. 2022;601(7893):460–464. doi:10.1038/s41586-021-04252-1
19. Cox MB, Johnson JL. The Role of p23, Hop, Immunophilins, and Other Co-chaperones in Regulating Hsp90 Function. In: Calderwood SK, Prince TL editors. Molecular Chaperones: Methods and Protocols. Methods in Molecular Biology. Humana Press; 2011:45–66. doi:10.1007/978-1-61779-295-3_4
20. Smith DF, Toft DO. Minireview: the Intersection of Steroid Receptors with Molecular Chaperones: observations and Questions. Mol Endocrinol. 2008;22(10):2229–2240. doi:10.1210/me.2008-0089
21. Zhang X, Clark AF, Yorio T. Heat Shock Protein 90 Is an Essential Molecular Chaperone for Nuclear Transport of Glucocorticoid Receptor β. Invest Ophthalmol Vis Sci. 2006;47(2):700–708. doi:10.1167/iovs.05-0697
22. Zhang X, Clark AF, Yorio T. FK506-Binding Protein 51 Regulates Nuclear Transport of the Glucocorticoid Receptor β and Glucocorticoid Responsiveness. Invest Ophthalmol Vis Sci. 2008;49(3):1037–1047. doi:10.1167/iovs.07-1279
23. Patel GC, Millar JC, Clark AF. Glucocorticoid Receptor Transactivation Is Required for Glucocorticoid-Induced Ocular Hypertension and Glaucoma. Invest Ophthalmol Vis Sci. 2019;60(6):1967–1978. doi:10.1167/iovs.18-26383
24. Bledsoe RK, Montana VG, Stanley TB, et al. Crystal Structure of the Glucocorticoid Receptor Ligand Binding Domain Reveals a Novel Mode of Receptor Dimerization and Coactivator Recognition. Cell. 2002;110(1):93–105. doi:10.1016/S0092-8674(02)00817-6
25. Caamaño CA, Morano MI, Dalman FC, Pratt WB, Akil H. A Conserved Proline in the hsp90 Binding Region of the Glucocorticoid Receptor Is Required for hsp90 Heterocomplex Stabilization and Receptor Signaling*. J Biol Chem. 1998;273(32):20473–20480. doi:10.1074/jbc.273.32.20473
26. Reichardt HM, Kaestner KH, Tuckermann J, et al. DNA Binding of the Glucocorticoid Receptor Is Not Essential for Survival. Cell. 1998;93(4):531–541. doi:10.1016/S0092-8674(00)81183-6
27. Sundahl N, Bridelance J, Libert C, De Bosscher K, Beck IM. Selective glucocorticoid receptor modulation: new directions with non-steroidal scaffolds. Pharmacol Ther. 2015;152:28–41. doi:10.1016/j.pharmthera.2015.05.001
28. Hübner S, Dejager L, Libert C, Tuckermann JP. The glucocorticoid receptor in inflammatory processes: transrepression is not enough. Biol Chem. 2015;396(11):1223–1231. doi:10.1515/hsz-2015-0106
29. Jain A, Wordinger RJ, Yorio T, Clark AF. Role of the alternatively spliced glucocorticoid receptor isoform GRβ in steroid responsiveness and glaucoma. J Ocul Pharmacol Ther off J Assoc Ocul Pharmacol Ther. 2014;30(2–3):121–127. doi:10.1089/jop.2013.0239
30. Zhang X, Clark AF, Yorio T. Regulation of Glucocorticoid Responsiveness in Glaucomatous Trabecular Meshwork Cells by Glucocorticoid Receptor-β. Invest Ophthalmol Vis Sci. 2005;46(12):4607–4616. doi:10.1167/iovs.05-0571
31. Roberti G, Oddone F, Agnifili L, et al. Steroid-induced glaucoma: epidemiology, pathophysiology, and clinical management. Surv Ophthalmol. 2020;65(4):458–472. doi:10.1016/j.survophthal.2020.01.002
32. Zhang X, Ognibene CM, Clark AF, Yorio T. Dexamethasone inhibition of trabecular meshwork cell phagocytosis and its modulation by glucocorticoid receptor β. Exp Eye Res. 2007;84(2):275–284. doi:10.1016/j.exer.2006.09.022
33. Patel GC, Liu Y, Millar JC, Clark AF. Glucocorticoid receptor GRβ regulates glucocorticoid-induced ocular hypertension in mice. Sci Rep. 2018;8:862. doi:10.1038/s41598-018-19262-9
34. Cuzzoni E, De Iudicibus S, Bartoli F, Ventura A, Decorti G. Association between BclI polymorphism in the NR3C1 gene and in vitro individual variations in lymphocyte responses to methylprednisolone. Br J Clin Pharmacol. 2012;73(4):651–655. doi:10.1111/j.1365-2125.2011.04130.x
35. Fingert JH, Alward WL, Wang K, Yorio T, Clark AF. Assessment of SNPs associated with the human glucocorticoid receptor in primary open-angle glaucoma and steroid responders. Mol Vis. 2010;16:596–601.
36. Gerzenstein SM, Pletcher MT, Cervino ACL, et al. Glucocorticoid Receptor Polymorphisms and Intraocular Pressure Response to Intravitreal Triamcinolone Acetonide. Ophthalmic Genet. 2008;29(4):166–170. doi:10.1080/13816810802320217
37. Iudicibus SD, Stocco G, Martelossi S, et al. Genetic predictors of glucocorticoid response in pediatric patients with inflammatory bowel diseases. J Clin Gastroenterol. 2011;45(1). doi:10.1097/MCG.0b013e3181e8ae93
38. Jain A, Wordinger RJ, Yorio T, Clark AF. Spliceosome Protein (SRp) Regulation of Glucocorticoid Receptor Isoforms and Glucocorticoid Response in Human Trabecular Meshwork Cells. Invest Ophthalmol Vis Sci. 2012;53(2):857–866. doi:10.1167/iovs.11-8497
39. Jain A, Liu X, Wordinger RJ, Yorio T, Cheng YQ, Clark AF. Effects of Thailanstatins on Glucocorticoid Response in Trabecular Meshwork and Steroid-Induced Glaucoma. Invest Ophthalmol Vis Sci. 2013;54(5):3137–3142. doi:10.1167/iovs.12-11480
40. Wang XQ, Duan ZX, He XG, Zhou XY. Clinical relevance of the glucocorticoid receptor gene polymorphisms in glucocorticoid−induced ocular hypertension and primary open angle glaucoma. Int J Ophthalmol. 2015;8(1):169–173. doi:10.3980/j.issn.2222-3959.2015.01.30
41. Will CL, Lührmann R. Spliceosome Structure and Function. Cold Spring Harb Perspect Biol. 2011;3(7):a003707. doi:10.1101/cshperspect.a003707
42. Trapp T, Holsboer F. Nuclear Orphan Receptor as a Repressor of Glucocorticoid Receptor Transcriptional Activity (*). J Biol Chem. 1996;271(17):9879–9882. doi:10.1074/jbc.271.17.9879
43. Kralli A, Bohen SP, Yamamoto KR. LEM1, an ATP-binding-cassette transporter, selectively modulates the biological potency of steroid hormones. Proc Natl Acad Sci. 1995;92(10):4701–4705. doi:10.1073/pnas.92.10.4701
44. Liu H, Yang ZK, Li Y, Zhang WJ, Wang YT, Duan XC. ABCB1 variants confer susceptibility to primary open-angle glaucoma and predict individual differences to latanoprost treatment. Biomed Pharmacother. 2016;80:115–120. doi:10.1016/j.biopha.2016.02.028
45. Becker B, Hahn KA. Topical Corticosteroids and Heredity in Primary Open-Angle Glaucoma*. Am J Ophthalmol. 1964;57(4):543–551. doi:10.1016/0002-9394(64)92500-0
46. Bermudez JY, Webber HC, Brown B, Braun TA, Clark AF, Mao W. A Comparison of Gene Expression Profiles between Glucocorticoid Responder and Non-Responder Bovine Trabecular Meshwork Cells Using RNA Sequencing. PLoS One. 2017;12(1):e0169671. doi:10.1371/journal.pone.0169671
47. Rauz S, Walker EA, Shackleton CHL, Hewison M, Murray PI, Stewart PM. Expression and Putative Role of 11β-Hydroxysteroid Dehydrogenase Isozymes within the Human Eye. Invest Ophthalmol Vis Sci. 2001;42(9):2037–2042.
48. McCarty GR, Schwartz B. Increased plasma noncortisol glucocorticoid activity in open-angle glaucoma. Invest Ophthalmol Vis Sci. 1991;32(5):1600–1608.
49. Ray S, Mehra KS, Misra S, Singh R. Plasma cortisol in glaucoma. Ann Ophthalmol. 1977;9(9):1151–1154.
50. Rozsíval P, Hampl R, Obenberger J, Stárka L, Rehák S. Aqueous humour and plasma cortisol levels in glaucoma and cataract patients. Curr Eye Res. 1981;1(7):391–396. doi:10.3109/02713688109019976
51. Schwartz B, McCarty G, Rosner B. Increased plasma free cortisol in ocular hypertension and open angle glaucoma. Arch Ophthalmol Chic IL 1960. 1987;105(8):1060–1065. doi:10.1001/archopht.1987.01060080062029
52. Schwartz B, Seddon JM. Increased plasma cortisol levels in ocular hypertension. Arch Ophthalmol Chic IL 1960. 1981;99(10):1791–1794. doi:10.1001/archopht.1981.03930020665008
53. Choi KJ, Na YJ, Park SB, Jung WH, Sung HR, Kim KY. Carbenoxolone prevents chemical eye ischemia-reperfusion-induced cell death via 11β-hydroxysteroid dehydrogenase type 1 inhibition. Pharmacol Res. 2017;123:62–72. doi:10.1016/j.phrs.2017.07.002
54. Choi KJ, Na YJ, Jung WH, et al. Protective effect of a novel selective 11β-HSD1 inhibitor on eye ischemia-reperfusion induced glaucoma. Biochem Pharmacol. 2019;169:113632. doi:10.1016/j.bcp.2019.113632
55. Piccolo S, Dupont S, Cordenonsi M. The Biology of YAP/TAZ: hippo Signaling and Beyond. Physiol Rev. 2014;94(4):1287–1312. doi:10.1152/physrev.00005.2014
56. Totaro A, Panciera T, Piccolo S. YAP/TAZ upstream signals and downstream responses. Nat Cell Biol. 2018;20(8):888–899. doi:10.1038/s41556-018-0142-z
57. Ruggeri N REGULATION OF YAP BY GLUCOCORTICOIDS. Doctoral Thesis. Università degli studi di Trieste; 2015. Available from: https://www.openstarts.units.it/handle/10077/11122.
58. Sorrentino G, Ruggeri N, Zannini A, et al. Glucocorticoid receptor signalling activates YAP in breast cancer. Nat Commun. 2017;8:14073. doi:10.1038/ncomms14073
59. Harvey KF, Pfleger CM, Hariharan IK. The Drosophila Mst ortholog, hippo, restricts growth and cell proliferation and promotes apoptosis. Cell. 2003;114(4):457–467. doi:10.1016/s0092-8674(03)00557-9
60. Jia J, Zhang W, Wang B, Trinko R, Jiang J. The Drosophila Ste20 family kinase dMST functions as a tumor suppressor by restricting cell proliferation and promoting apoptosis. Genes Dev. 2003;17(20):2514–2519. doi:10.1101/gad.1134003
61. Pantalacci S, Tapon N, Léopold P. The Salvador partner Hippo promotes apoptosis and cell-cycle exit in Drosophila. Nat Cell Biol. 2003;5(10):921–927. doi:10.1038/ncb1051
62. Udan RS, Kango-Singh M, Nolo R, Tao C, Halder G. Hippo promotes proliferation arrest and apoptosis in the Salvador/Warts pathway. Nat Cell Biol. 2003;5(10):914–920. doi:10.1038/ncb1050
63. Wu S, Huang J, Dong J, Pan D. hippo encodes a Ste-20 family protein kinase that restricts cell proliferation and promotes apoptosis in conjunction with salvador and warts. Cell. 2003;114(4):445–456. doi:10.1016/s0092-8674(03)00549-x
64. Ibar C, Kirichenko E, Keepers B, Enners E, Fleisch K, Irvine KD. Tension-dependent regulation of mammalian Hippo signaling through LIMD1. J Cell Sci. 2018;131(5):jcs214700. doi:10.1242/jcs.214700
65. Nakamura R, Bing R, Doyle CP, Garabedian MJ, Branski RC. Glucocorticoids activate Yes-associated protein in human vocal fold fibroblasts. Exp Cell Res. 2021;405(2):112681. doi:10.1016/j.yexcr.2021.112681
66. Xu S, Liu Y, Hu R, et al. TAZ inhibits GR and coordinates hepatic glucose homeostasis in normal physiologic states. eLife. 2020:e57462. doi:10.1101/2020.05.12.091264
67. Murphy R, Irnaten M, Hopkins A, et al. Matrix Mechanotransduction via Yes-Associated Protein in Human Lamina Cribrosa Cells in Glaucoma. Invest Ophthalmol Vis Sci. 2022;63(1):16. doi:10.1167/iovs.63.1.16
68. Gu X, Ge L, Ren B, et al. Glucocorticoids Promote Extracellular Matrix Component Remodeling by Activating YAP in Human Retinal Capillary Endothelial Cells. Front Cell Dev Biol. 2021;9. doi:10.3389/fcell.2021.738341
69. Li H, Raghunathan V, Stamer WD, Ganapathy PS, Herberg S. Extracellular Matrix Stiffness and TGFβ2 Regulate YAP/TAZ Activity in Human Trabecular Meshwork Cells. Front Cell Dev Biol. 2022;10: 844342.
70. Liu Z, Li S, Qian X, Li L, Zhang H, Liu Z. RhoA/ROCK-YAP/TAZ Axis Regulates the Fibrotic Activity in Dexamethasone-Treated Human Trabecular Meshwork Cells. Front Mol Biosci. 2021;8: 728514.
71. Yoo H, Singh A, Li H, et al. Simvastatin Attenuates Glucocorticoid-Induced Human Trabecular Meshwork Cell Dysfunction via YAP/TAZ Inactivation. Curr Eye Res. 2023;1(2):1–14. doi:10.1080/02713683.2023.2206067
72. Logan CY, Nusse R. The Wnt Signaling Pathway in Development and Disease. Annu Rev Cell Dev Biol. 2004;20(1):781–810. doi:10.1146/annurev.cellbio.20.010403.113126
73. Clevers H. Wnt/β-Catenin Signaling in Development and Disease. Cell. 2006;127(3):469–480. doi:10.1016/j.cell.2006.10.018
74. Sugali CK, Rayana NP, Dai J, et al. The Canonical Wnt Signaling Pathway Inhibits the Glucocorticoid Receptor Signaling Pathway in the Trabecular Meshwork. Am J Pathol. 2021;191(6):1020–1035. doi:10.1016/j.ajpath.2021.02.018
75. Webber HC, Bermudez JY, Millar JC, Mao W, Clark AF. The Role of Wnt/β-Catenin Signaling and K-Cadherin in the Regulation of Intraocular Pressure. Invest Ophthalmol Vis Sci. 2018;59(3):1454–1466. doi:10.1167/iovs.17-21964
76. Nusse R, Varmus HE. Many tumors induced by the mouse mammary tumor virus contain a provirus integrated in the same region of the host genome. Cell. 1982;31(1):99–109. doi:10.1016/0092-8674(82)90409-3
77. Sharma RP, Chopra VL. Effect of the wingless (wg1) mutation on wing and haltere development in Drosophila melanogaster. Dev Biol. 1976;48(2):461–465. doi:10.1016/0012-1606(76)90108-1
78. Nusse R, Brown A, Papkoff J, et al. A new nomenclature for int-1 and related genes: the Wnt gene family. Cell. 1991;64(2):231. doi:10.1016/0092-8674(91)90633-A
79. Komiya Y, Habas R. Wnt signal transduction pathways. Organogenesis. 2008;4(2):68–75.
80. He X, Semenov M, Tamai K, Zeng X. LDL receptor-related proteins 5 and 6 in Wnt/β-catenin signaling:Arrows point the way. Development. 2004;131(8):1663–1677. doi:10.1242/dev.01117
81. MacDonald BT, Tamai K, He X. Wnt/β-catenin signaling: components, mechanisms, and diseases. Dev Cell. 2009;17(1):9–26. doi:10.1016/j.devcel.2009.06.016
82. Wang WH, McNatt LG, Pang IH, et al. Increased expression of the WNT antagonist sFRP-1 in glaucoma elevates intraocular pressure. J Clin Invest. 2008;118(3):1056–1064. doi:10.1172/JCI33871
83. Mao W, Liu Y, Mody A, Montecchi-Palmer M, Wordinger RJ, Clark AF. Characterization of a spontaneously immortalized bovine trabecular meshwork cell line. Exp Eye Res. 2012;105:53–59. doi:10.1016/j.exer.2012.10.007
84. Raghunathan VK, Morgan JT, Park SA, et al. Dexamethasone Stiffens Trabecular Meshwork, Trabecular Meshwork Cells, and Matrix. Invest Ophthalmol Vis Sci. 2015;56(8):4447–4459. doi:10.1167/iovs.15-16739
85. Yuan Y, Call MK, Yuan Y, et al. Dexamethasone Induces Cross-Linked Actin Networks in Trabecular Meshwork Cells Through Noncanonical Wnt Signaling. Invest Ophthalmol Vis Sci. 2013;54(10):6502–6509. doi:10.1167/iovs.13-12447
86. Maddala R, Eldawy C, Bachman W, Soderblom EJ, Rao PV. Glypican-4 regulated actin cytoskeletal reorganization in glucocorticoid treated trabecular meshwork cells and involvement of Wnt/PCP signaling. J Cell Physiol. 2023;238(3):631–646. doi:10.1002/jcp.30953
87. Barrett CSX, Millena AC, Khan SA. TGF-β Effects on Prostate Cancer Cell Migration and Invasion Require FosB. Prostate. 2017;77(1):72–81. doi:10.1002/pros.23250
88. Clark DA, Coker R. Transforming growth factor-beta (TGF-beta). Int J Biochem Cell Biol. 1998;30(3):293–298. doi:10.1016/s1357-2725(97)00128-3
89. Duenker N. Transforming growth factor-beta (TGF-beta) and programmed cell death in the vertebrate retina. Int Rev Cytol. 2005;245:17–43. doi:10.1016/S0074-7696(05)45002-0
90. Gao T, Cao Y, Hu M, Du Y. The activation of TGF-β signaling promotes cell migration and invasion of ectopic endometrium by targeting NRP2. Reprod Biol. 2022;22(4):100697. doi:10.1016/j.repbio.2022.100697
91. Zhang Y, Alexander PB, Wang XF. TGF-β Family Signaling in the Control of Cell Proliferation and Survival. Cold Spring Harb Perspect Biol. 2017;9(4):a022145. doi:10.1101/cshperspect.a022145
92. Lawrence DA. Transforming growth factor-beta: a general review. Eur Cytokine Netw. 1996;7(3):363–374.
93. Itoh S, Itoh F, Goumans MJ, ten Dijke P. Signaling of transforming growth factor-β family members through Smad proteins. Eur J Biochem. 2000;267(24):6954–6967. doi:10.1046/j.1432-1327.2000.01828.x
94. Tripathi RC, Li J, Chan WF, Tripathi BJ. Aqueous humor in glaucomatous eyes contains an increased level of TGF-beta 2. Exp Eye Res. 1994;59(6):723–727. doi:10.1006/exer.1994.1158
95. Wordinger RJ, Sharma T, Clark AF. The Role of TGF-β2 and Bone Morphogenetic Proteins in the Trabecular Meshwork and Glaucoma. J Ocul Pharmacol Ther. 2014;30(2–3):154–162. doi:10.1089/jop.2013.0220
96. Gottanka J, Chan D, Eichhorn M, Lütjen-Drecoll E, Ethier CR. Effects of TGF-beta2 in perfused human eyes. Invest Ophthalmol Vis Sci. 2004;45(1):153–158. doi:10.1167/iovs.03-0796
97. Montecchi-Palmer M, Bermudez JY, Webber HC, Patel GC, Clark AF, Mao W. TGFβ2 Induces the Formation of Cross-Linked Actin Networks (CLANs) in Human Trabecular Meshwork Cells Through the Smad and Non-Smad Dependent Pathways. Invest Ophthalmol Vis Sci. 2017;58(2):1288–1295. doi:10.1167/iovs.16-19672
98. Robertson JV, Golesic E, Gauldie J, West-Mays JA. Ocular Gene Transfer of Active TGF-β Induces Changes in Anterior Segment Morphology and Elevated IOP in Rats. Invest Ophthalmol Vis Sci. 2010;51(1):308–318. doi:10.1167/iovs.09-3380
99. Uemura T, Sakai O, Sakamoto Y. Transgenic mice expressing Transforming growth factor-β2 increased intraocular pressure. Invest Ophthalmol Vis Sci. 2019;60(9):3773.
100. Kasetti RB, Maddineni P, Patel PD, Searby C, Sheffield VC, Zode GS. Transforming growth factor β2 (TGFβ2) signaling plays a key role in glucocorticoid-induced ocular hypertension. J Biol Chem. 2018;293(25):9854–9868. doi:10.1074/jbc.RA118.002540
101. Clark AF, Wilson K, McCartney MD, Miggans ST, Kunkle M, Howe W. Glucocorticoid-induced formation of cross-linked actin networks in cultured human trabecular meshwork cells. Invest Ophthalmol Vis Sci. 1994;35(1):281–294.
102. Bermudez JY, Webber HC. Two-Dimensional Differential In-Gel Electrophoresis (2D-DIGE) Reveals Proteins Associated with Cross-Linked Actin Networks in Human Trabecular Meshwork Cells. J Clin Exp Ophthalmol. 2016;07(04). doi:10.4172/2155-9570.1000584
103. Filla MS, Woods A, Kaufman PL, Peters DM. Beta1 and beta3 integrins cooperate to induce syndecan-4-containing cross-linked actin networks in human trabecular meshwork cells. Invest Ophthalmol Vis Sci. 2006;47(5):1956–1967. doi:10.1167/iovs.05-0626
104. Filla MS, Schwinn K, Sheibani M. Distinct β1 and β3 integrin pathways converge to regulate cross-linked actin network (CLAN) formation in human trabecular meshwork (HTM) cells. Invest Ophthalmol Vis Sci. 2009;50(12):5723–5731. doi:10.1167/iovs.08-3215
105. Bermudez JY, Montecchi-Palmer M, Mao W, Clark AF. Cross-linked actin networks (CLANs) in glaucoma. Exp Eye Res. 2017;159:16–22. doi:10.1016/j.exer.2017.02.010
106. Wilson K, McCartney MD, Miggans ST, Clark AF. Dexamethasone induced ultrastructural changes in cultured human trabecular meshwork cells. Curr Eye Res. 1993;12(9):783–793. doi:10.3109/02713689309020383
107. Clark AF, Lane D, Wilson K, Miggans ST, McCartney MD. Inhibition of dexamethasone-induced cytoskeletal changes in cultured human trabecular meshwork cells by tetrahydrocortisol. Invest Ophthalmol Vis Sci. 1996;37(5):805–813.
108. O’Reilly S, Pollock N, Currie L, Paraoan L, Clark AF, Grierson I. Inducers of Cross-Linked Actin Networks in Trabecular Meshwork Cells. Invest Ophthalmol Vis Sci. 2011;52(10):7316–7324. doi:10.1167/iovs.10-6692
109. Wade NC, Grierson I, O’Reilly S, et al. Cross-linked actin networks (CLANs) in bovine trabecular meshwork cells. Exp Eye Res. 2009;89(5):648–659. doi:10.1016/j.exer.2009.06.006
110. Mao W, Liu Y, Wordinger RJ, Clark AF. A Magnetic Bead-Based Method for Mouse Trabecular Meshwork Cell Isolation. Invest Ophthalmol Vis Sci. 2013;54(5):3600–3606. doi:10.1167/iovs.13-12033
111. Clark R, Nosie A, Walker T, et al. Comparative Genomic and Proteomic Analysis of Cytoskeletal Changes in Dexamethasone-Treated Trabecular Meshwork Cells. Mol Cell Proteomics MCP. 2013;12(1):194–206. doi:10.1074/mcp.M112.019745
112. Filla MS, Schwinn MK, Nosie AK, Clark RW, Peters DM. Dexamethasone-Associated Cross-Linked Actin Network Formation in Human Trabecular Meshwork Cells Involves β3 Integrin Signaling. Invest Ophthalmol Vis Sci. 2011;52(6):2952–2959. doi:10.1167/iovs.10-6618
113. Peng M, Rayana NP, Dai J, et al. Cross-linked actin networks (CLANs) affect stiffness and/or actin dynamics in transgenic transformed and primary human trabecular meshwork cells. Exp Eye Res. 2022;220:109097. doi:10.1016/j.exer.2022.109097
114. Fujimoto T, Inoue T, Kameda T, et al. Involvement of RhoA/Rho-associated kinase signal transduction pathway in dexamethasone-induced alterations in aqueous outflow. Invest Ophthalmol Vis Sci. 2012;53(11):7097–7108. doi:10.1167/iovs.12-9989
115. Wang K, Read AT, Sulchek T, Ethier CR. Trabecular meshwork stiffness in glaucoma. Exp Eye Res. 2017;158:3–12. doi:10.1016/j.exer.2016.07.011
116. Raghunathan VK, Benoit J, Kasetti R, et al. Glaucomatous cell derived matrices differentially modulate non-glaucomatous trabecular meshwork cellular behavior. Acta Biomater. 2018;71:444–459. doi:10.1016/j.actbio.2018.02.037
117. Last JA, Pan T, Ding Y, et al. Elastic modulus determination of normal and glaucomatous human trabecular meshwork. Invest Ophthalmol Vis Sci. 2011;52(5):2147–2152. doi:10.1167/iovs.10-6342
118. Sherwood JM, Stamer WD, Overby DR. A model of the oscillatory mechanical forces in the conventional outflow pathway. J R Soc Interface. 2019;16(150):20180652. doi:10.1098/rsif.2018.0652
119. Matsumoto Y, Johnson DH. Dexamethasone decreases phagocytosis by human trabecular meshwork cells in situ. Invest Ophthalmol Vis Sci. 1997;38(9):1902–1907.
120. Zode GS, Sharma AB, Lin X, et al. Ocular-specific ER stress reduction rescues glaucoma in murine glucocorticoid-induced glaucoma. J Clin Invest. 2014;124(5):1956–1965. doi:10.1172/JCI69774
121. Faralli JA, Gagen D, Filla MS, Crotti TN, Peters DM. Dexamethasone increases αvβ3 integrin expression and affinity through a calcineurin/NFAT pathway. Biochim Biophys Acta. 2013;1833(12):3306–3313. doi:10.1016/j.bbamcr.2013.09.020
122. Gagen D, Faralli JA, Filla MS, Peters DM. The role of integrins in the trabecular meshwork. J Ocul Pharmacol Ther off J Assoc Ocul Pharmacol Ther. 2014;30(2–3):110–120. doi:10.1089/jop.2013.0176
123. Filla MS, Schwinn MK, Sheibani N, Kaufman PL, Peters DM. Regulation of cross-linked actin network (CLAN) formation in human trabecular meshwork (HTM) cells by convergence of distinct beta1 and beta3 integrin pathways. Invest Ophthalmol Vis Sci. 2009;50(12):5723–5731. doi:10.1167/iovs.08-3215
124. Gagen D, Filla MS, Clark R, Liton P, Peters DM. Activated αvβ3 integrin regulates αvβ5 integrin-mediated phagocytosis in trabecular meshwork cells. Invest Ophthalmol Vis Sci. 2013;54(7):5000–5011. doi:10.1167/iovs.13-12084
125. Overby DR, Clark AF. Animal Models of Glucocorticoid-Induced Glaucoma. Exp Eye Res. 2015;141:15–22. doi:10.1016/j.exer.2015.06.002
126. Tektas OY, Lütjen-Drecoll E. Structural changes of the trabecular meshwork in different kinds of glaucoma. Exp Eye Res. 2009;88(4):769–775. doi:10.1016/j.exer.2008.11.025
127. el-Shabrawi Y, Eckhardt M, Berghold A, et al. Synthesis pattern of matrix metalloproteinases (MMPs) and inhibitors (TIMPs) in human explant organ cultures after treatment with latanoprost and dexamethasone. Eye Lond Engl. 2000;14(Pt 3A):375–383. doi:10.1038/eye.2000.92
128. Mohd Nasir NA, Agarwal R, Krasilnikova A, Abdul Kadir SH S, Iezhitsa I. Effect of dexamethasone on the expression of MMPs, adenosine A1 receptors and NFKB by human trabecular meshwork cells. J Basic Clin Physiol Pharmacol. 2020;31(6). doi:10.1515/jbcpp-2019-0373
129. Mohd Nasir NA, Agarwal R, Krasilnikova A, Abdul Kadir SH S, Iezhitsa I. Effect of trans-resveratrol on dexamethasone-induced changes in the expression of MMPs by human trabecular meshwork cells: involvement of adenosine A1 receptors and NFkB. Eur J Pharmacol. 2020;887:173431. doi:10.1016/j.ejphar.2020.173431
130. Rohen JW, Linnér E, Witmer R. Electron microscopic studies on the trabecular meshwork in two cases of corticosteroid-glaucoma. Exp Eye Res. 1973;17(1):19–31. doi:10.1016/0014-4835(73)90164-4
131. Overby DR, Bertrand J, Tektas OY, et al. Ultrastructural Changes Associated With Dexamethasone-Induced Ocular Hypertension in Mice. Invest Ophthalmol Vis Sci. 2014;55(8):4922–4933. doi:10.1167/iovs.14-14429
132. Dibas A, Clark AF, Yorio T. Increased expression of glucocorticoid receptor B induces a novel transforming growth factor beta-2 isoform in trabecular meshwork cells. Invest Ophthalmol Vis Sci. 2016;57(12):86.
133. Gerometta R, Kumar S, Shah S, Alvarez L, Candia O, Danias J. Reduction of Steroid-Induced Intraocular Pressure Elevation in Sheep by Tissue Plasminogen Activator. Invest Ophthalmol Vis Sci. 2013;54(13):7903–7909. doi:10.1167/iovs.13-12801
134. Kumar S, Shah S, Tang HM, Smith M, Borrás T, Danias J. Tissue plasminogen activator in trabecular meshwork attenuates steroid induced outflow resistance in mice. PLoS One. 2013;8(8):e72447. doi:10.1371/journal.pone.0072447
135. Gindina S, Barron AO, Hu Y, Dimopoulos A, Danias J. Tissue plasminogen activator rescues steroid-induced outflow facility reduction via non-enzymatic action. Mol Vis. 2021;27:691–705.
136. Underwood JL, Murphy CG, Chen J, et al. Glucocorticoids regulate transendothelial fluid flow resistance and formation of intercellular junctions. Am J Physiol. 1999;277(2):C330–342. doi:10.1152/ajpcell.1999.277.2.C330
137. Vahabikashi A, Park CY, Perkumas K, et al. Probe Sensitivity to Cortical versus Intracellular Cytoskeletal Network Stiffness. Biophys J. 2019;116(3):518–529. doi:10.1016/j.bpj.2018.12.021
138. Overby DR, Zhou EH, Vargas-Pinto R, et al. Altered mechanobiology of Schlemm’s canal endothelial cells in glaucoma. Proc Natl Acad Sci U S A. 2014;111(38):13876–13881. doi:10.1073/pnas.1410602111
139. Rosenquist R, Epstein D, Melamed S, Johnson M, Grant WM. Outflow resistance of enucleated human eyes at two different perfusion pressures and different extents of trabeculotomy. Curr Eye Res. 1989;8(12):1233–1240. doi:10.3109/02713688909013902
140. Gonzalez JM, Ko MK, Hong YK, Weigert R, Tan JCH. Deep tissue analysis of distal aqueous drainage structures and contractile features. Sci Rep. 2017;7(1):17071. doi:10.1038/s41598-017-16897-y
141. McDonnell F, Dismuke WM, Overby DR, Stamer WD. Pharmacological regulation of outflow resistance distal to Schlemm’s canal. Am J Physiol Cell Physiol. 2018;315(1):C44–C51. doi:10.1152/ajpcell.00024.2018
142. Roy Chowdhury U, Fautsch MP. Isolation and Culture of Vascular Distal Outflow Pathway (VDOP) Cells From Human Donor Eyes. Curr Protoc. 2022;2(8):e528. doi:10.1002/cpz1.528
143. Abtahi M, Rudnisky CJ, Nazarali S, Damji KF. Incidence of steroid response in microinvasive glaucoma surgery with trabecular microbypass stent and ab interno trabeculectomy. Can J Ophthalmol. 2022;57(3):167–174. doi:10.1016/j.jcjo.2021.04.008
144. Bojikian KD, Nobrega P, Roldan A, Forrest SL, Tsukikawa M, Chen PP. Incidence of and Risk Factors for Steroid Response After Cataract Surgery in Patients With and Without Glaucoma. J Glaucoma. 2021;30(4):e159–e163. doi:10.1097/IJG.0000000000001785
145. Rajendrababu S, Pallamparthy S, Arunachalam A, et al. Incidence and risk factors for postoperative intraocular pressure response to topical prednisolone eye drops in patients undergoing phacoemulsification. Int Ophthalmol. 2021;41(12):3999–4007. doi:10.1007/s10792-021-01972-1
146. Badrinarayanan L, Rishi P, George R, Isaac N, Rishi E. for the Sankara Nethralaya Vitreo-Retinal group (SNVR). Incidence, Risk Factors, Treatment, and Outcome of Ocular Hypertension following Intravitreal Steroid Injections: a Comparative Study. Ophthalmologica. 2022;245(5):431–438. doi:10.1159/000522504
147. Krag S, Larsen D, Albertsen BK, Glerup M. Risk of ocular hypertension in children treated with systemic glucocorticoid. Acta Ophthalmol (Copenh). 2021;99(8):e1430–e1434. doi:10.1111/aos.14820
148. Kiddee W, Trope GE, Sheng L, et al. Intraocular Pressure Monitoring Post Intravitreal Steroids: a Systematic Review. Surv Ophthalmol. 2013;58(4):291–310. doi:10.1016/j.survophthal.2012.08.003
149. Mataftsi A, Dabbagh A, Moore W, Nischal KK. Evaluation of whether intracameral dexamethasone predisposes to glaucoma after pediatric cataract surgery. J Cataract Refract Surg. 2012;38(10):1719. doi:10.1016/j.jcrs.2012.05.034
150. Chang DT, Herceg MC, Bilonick RA, Camejo L, Schuman JS, Noecker RJ. Intracameral dexamethasone reduces inflammation on the first postoperative day after cataract surgery in eyes with and without glaucoma. Clin Ophthalmol Auckl NZ. 2009;3:345–355.
151. Yamashita T, Kodama Y, Tanaka M, Yamakiri K, Kawano Y, Sakamoto T. Steroid-induced glaucoma in children with acute lymphoblastic leukemia: a possible complication. J Glaucoma. 2010;19(3):188–190. doi:10.1097/IJG.0b013e3181af321d
152. Chan CKM, Mohamed S, Lee VYW, Lai TYY, Shanmugam MP, Lam DSC. Intravitreal dexamethasone for diabetic macular edema: a pilot study. Ophthalmic Surg Lasers Imaging off J Int Soc Imaging Eye. 2010;41(1):26–30. doi:10.3928/15428877-20091230-05
153. Lebrize S, Arnould L, Bourredjem A, et al. Intraocular Pressure Changes After Intravitreal Fluocinolone Acetonide Implant: results from Four European Countries. Ophthalmol Ther. 2022;11(3):1217–1229. doi:10.1007/s40123-022-00504-z
154. Alsaadi M, Osuagwu U, Almubrad T. Effects of inhaled fluticasone on intraocular pressure and central corneal thickness in asthmatic children without a family history of glaucoma. Middle East Afr J Ophthalmol. 2012;19(3):314–319. doi:10.4103/0974-9233.97936
155. Acar M, Gedizlioglu M, Koskderelioglu A, Ozturk F, Kilinc S, Talay N. Effect of High-Dose Intravenous Methyl-prednisolone Treatment on Intraocular Pressure in Multiple Sclerosis Patients with Relapse. Eur Neurol. 2012;68(1):20–22. doi:10.1159/000337615
156. Tripathi RC, Kirschner BS, Kipp M, et al. Corticosteroid treatment for inflammatory bowel disease in pediatric patients increases intraocular pressure. Gastroenterology. 1992;102(6):1957–1961. doi:10.1016/0016-5085(92)90319-T
157. Ding X, Li J, Hu X, Yu S, Pan J, Tang S. PROSPECTIVE STUDY OF INTRAVITREAL TRIAMCINOLONE ACETONIDE VERSUS BEVACIZUMAB FOR MACULAR EDEMA SECONDARY TO CENTRAL RETINAL VEIN OCCLUSION. RETINA. 2011;31(5):838. doi:10.1097/IAE.0b013e3181f4420d
158. Chang YC, Wu WC. Elevation of Intraocular Pressure After Intravitreal Injection of Triamcinolone Acetonide in Taiwanese Patients. Kaohsiung J Med Sci. 2008;24(2):72–77. doi:10.1016/S1607-551X(08)70100-1
159. Chuang LH, Yeung L, Wang NK, Chen HSL, Ku WC, Lai CC. Secondary Ocular Hypertension After Intravitreal Injection with 2 mg or 4 mg of Triamcinolone in Retinal Vein Occlusion. J Ocul Pharmacol Ther. 2010;26(4):325–328. doi:10.1089/jop.2010.0039
160. Jain S, Thompson JR, Foot B, Tatham A, Eke T. Severe intraocular pressure rise following intravitreal triamcinolone: a national survey to estimate incidence and describe case profiles. Eye. 2014;28(4):399–401. doi:10.1038/eye.2013.306
161. Chew EY, Glassman AR, Beck RW, et al. Ocular side effects associated with peribulbar injections of triamcinolone acetonide for diabetic macular edema. Retina (Philadelphia, Pa). 2011;31(2):284–289. doi:10.1097/IAE.0b013e3181f049a8
162. Hirooka K, Shiraga F, Tanaka S, Baba T, Mandai H. Risk Factors for Elevated Intraocular Pressure After Trans-Tenon Retrobulbar Injections of Triamcinolone. Jpn J Ophthalmol. 2006;50(3):235–238. doi:10.1007/s10384-005-0306-9
163. Kawaji T, Takano A, Inomata Y, et al. Trans-Tenon’s retrobulbar triamcinolone acetonide injection for macular oedema related to branch retinal vein occlusion. Br J Ophthalmol. 2008;92(1):81–83. doi:10.1136/bjo.2007.124578
164. Spiers F. Topical Steroids and Intraocular Pressure. Acta Ophthalmol (Copenh). 1965;43(5):735–745. doi:10.1111/j.1755-3768.1965.tb00346.x
165. Yamamoto Y, Komatsu T, Koura Y, Nishino K, Fukushima A, Ueno H. Intraocular pressure elevation after intravitreal or posterior sub-Tenon triamcinolone acetonide injection. Can J Ophthalmol. 2008;43(1):42–47. doi:10.3129/i07-186
166. Lafranco Dafflon M, Tran VT, Guex-Crosier Y, Herbort CP. Posterior sub-Tenon’s steroid injections for the treatment of posterior ocular inflammation: indications, efficacy and side effects. Graefes Arch Clin Exp Ophthalmol. 1999;237(4):289–295. doi:10.1007/s004170050235
167. Yalcinsoy KO, Ozdal PC, Sen E, Elgin U. Intraocular Pressure Elevation After Posterior Subtenon Triamcinolone Acetonide Injection in Pediatric Non-Infectious Uveitis. Beyoglu Eye J. 2022;7(4):298–303. doi:10.14744/bej.2022.97752
168. Ateeq A, Majid S, Ahmed Memon N, Hayat N, Qadeem Somroo A, Fattah Memon A. Suprachoroidal injection of triamcinolone acetonide for management of resistant diabetic macular edema (SCTA): original article. J Pak Med Assoc. 2023;73(2):239–244. doi:10.47391/JPMA.2239
169. Marashi A, Zazo A. Suprachoroidal injection of triamcinolone acetonide using a custom-made needle to treat diabetic macular edema post pars plana vitrectomy: a case series. J Int Med Res. 2022;50(4):03000605221089807. doi:10.1177/03000605221089807
170. Yeh S, Khurana RN, Shah M, et al. Efficacy and Safety of Suprachoroidal CLS-TA for Macular Edema Secondary to Noninfectious Uveitis: Phase 3 Randomized Trial. Ophthalmology. 2020;127(7):948–955. doi:10.1016/j.ophtha.2020.01.006
171. Cantrill HL, Palmberg PF, Zink HA, Waltman SR, Podos SM, Becker B. Comparison of in Vitro Potency of Corticosteroids with Ability to Raise Intraocular Pressure. Am J Ophthalmol. 1975;79(6):1012–1017. doi:10.1016/0002-9394(75)90687-X
172. Kao BW, Fong CW, Yu Y, Ying GS, Gedde SJ, Han Y. Surgical Outcomes of Ahmed Glaucoma Valve Implantation with Postoperative Use of Prednisolone Acetate versus Difluprednate. Ophthalmol Glaucoma. 2022;5(5):468–475. doi:10.1016/j.ogla.2022.03.003
173. Tripathi RC, Parapuram SK, Tripathi BJ, Zhong Y, Chalam KV. Corticosteroids and Glaucoma Risk. Drugs Aging. 1999;15(6):439–450. doi:10.2165/00002512-199915060-00004
174. Korenfeld M, Nichols KK, Goldberg D, et al. Safety of KPI-121 Ophthalmic Suspension 0.25% in Patients With Dry Eye Disease: a Pooled Analysis of 4 Multicenter, Randomized, Vehicle-Controlled Studies. Cornea. 2021;40(5):564. doi:10.1097/ICO.0000000000002452
175. Sheppard JD, Comstock TL, Cavet ME. Impact of the Topical Ophthalmic Corticosteroid Loteprednol Etabonate on Intraocular Pressure. Adv Ther. 2016;33(4):532–552. doi:10.1007/s12325-016-0315-8
176. Price MO, Feng MT, Scanameo A, Price FWJ. Loteprednol Etabonate 0.5% Gel Vs. Prednisolone Acetate 1% Solution After Descemet Membrane Endothelial Keratoplasty: prospective Randomized Trial. Cornea. 2015;34(8):853. doi:10.1097/ICO.0000000000000475
177. Raj A, Salvador-Culla B, Anwar H, Sykakis E, Figueiredo MS, Figueiredo FC. Long-term Outcomes on de novo Ocular Hypertensive Response to Topical Corticosteroids After Corneal Transplantation. Cornea. 2020;39(1):45. doi:10.1097/ICO.0000000000002142
178. Chen YH, Gepstein R, Sharief L, et al. Outcome and risk of ocular complications of managing children with chronic anterior uveitis with topical rimexolone 1%. Int Ophthalmol. 2020;40(5):1061–1068. doi:10.1007/s10792-020-01358-9
179. Armaly MF. Effect of Corticosteroids on Intraocular Pressure and Fluid Dynamics: II.he Effect of Dexamethasone in the Glaucomatous Eye. Arch Ophthalmol. 1963;70(4):492–499. doi:10.1001/archopht.1963.00960050494011
180. Becker B. Intraocular pressure response to topical corticosteroids. Invest Ophthalmol. 1965;4:198–205.
181. Feroze KB, Zeppieri M, Khazaeni L. Steroid Induced Glaucoma. StatPearls Publishing; 2023. http://www.ncbi.nlm.nih.gov/books/NBK430903/.
182. Choi W, Bae HW, Choi EY, et al. Age as a risk factor for steroid-induced ocular hypertension in the non-paediatric population. Br J Ophthalmol. 2020;104(10):1423–1429. doi:10.1136/bjophthalmol-2019-314559
183. Malclès A, Dot C, Voirin N, et al. SAFETY OF INTRAVITREAL DEXAMETHASONE IMPLANT (OZURDEX): the SAFODEX study. Incidence and Risk Factors of Ocular Hypertension. RETINA. 2017;37(7):1352. doi:10.1097/IAE.0000000000001369
184. Maier AKB, Pilger D, Gundlach E, Winterhalter S, Torun N. Long-term Results of Intraocular Pressure Elevation and Post-DMEK Glaucoma After Descemet Membrane Endothelial Keratoplasty. Cornea. 2021;40(1):26. doi:10.1097/ICO.0000000000002363
185. Armaly MF. Inheritance of Dexamethasone Hypertension and Glaucoma. Arch Ophthalmol. 1967;77(6):747–751. doi:10.1001/archopht.1967.00980020749006
186. Bartlett JD, Woolley TW, Adams CM. Identification of High Intraocular Pressure Responders to Topical Ophthalmic Corticosteroids. J Ocul Pharmacol Ther. 1993;9(1):35–45. doi:10.1089/jop.1993.9.35
187. Mitchell P, Cumming RG, Mackey DA. Inhaled corticosteroids, family history, and risk of glaucoma. Ophthalmology. 1999;106(12):2301–2306. doi:10.1016/S0161-6420(99)90530-4
188. Razeghinejad MR, Katz LJ. Steroid-Induced Iatrogenic Glaucoma. Ophthalmic Res. 2012;47(2):66–80. doi:10.1159/000328630
189. Jones RI, Rhee DJ. Corticosteroid-induced ocular hypertension and glaucoma: a brief review and update of the literature. Curr Opin Ophthalmol. 2006;17(2):163. doi:10.1097/01.icu.0000193079.55240.18
190. Friedman DS, Holbrook JT, Ansari H, et al. Risk of Elevated Intraocular Pressure and Glaucoma in Patients with Uveitis: results of the Multicenter Uveitis Steroid Treatment Trial. Ophthalmology. 2013;120(8):1571–1579. doi:10.1016/j.ophtha.2013.01.025
191. Biedner BZ, David R, Grudsky A, Sachs U. Intraocular pressure response to corticosteroids in children. Br J Ophthalmol. 1980;64(6):430–431. doi:10.1136/bjo.64.6.430
192. Hutcheson KA. Steroid-induced glaucoma in an infant. J Am Assoc Pediatr Ophthalmol Strabismus. 2007;11(5):522–523. doi:10.1016/j.jaapos.2007.03.008
193. Lam DS, Fan DS, Ng JS, Yu CB, Wong CY, Cheung AY. Ocular hypertensive and anti-inflammatory responses to different dosages of topical dexamethasone in children: a randomized trial. Clin Experiment Ophthalmol. 2005;33(3):252–258. doi:10.1111/j.1442-9071.2005.01022.x
194. Gaston H, Absolon MJ, Thurtle OA, Sattar MA. Steroid responsiveness in connective tissue diseases. Br J Ophthalmol. 1983;67(7):487–490. doi:10.1136/bjo.67.7.487
195. Podos SM, Becker B, Ross Morton W. High Myopia And Primary Open-Angle Glaucoma. Am J Ophthalmol. 1966;62(6):1039–1043. doi:10.1016/0002-9394(66)92551-7
196. Becker B, Bresnick G, Chevrette L, Kolker AE, Oaks MC, Cibis A. Intraocular Pressure and Its Response to Topical Corticosteroids in Diabetes. Arch Ophthalmol. 1966;76(4):477–483. doi:10.1001/archopht.1966.03850010479003
197. Hámor A, Markó R, Rák T, Csutak A. A szteroidterápia hatása az intraocularis nyomásra. Orv Hetil. 2022;163(34):1345–1352. doi:10.1556/650.2022.32548
198. Dibas A, Yorio T. Glucocorticoid Therapy and ocular hypertension. Eur J Pharmacol. 2016;787:57–71. doi:10.1016/j.ejphar.2016.06.018
199. Agrahari V, Mandal A, Agrahari V, et al. A COMPREHENSIVE INSIGHT ON OCULAR PHARMACOKINETICS. Drug Deliv Transl Res. 2016;6(6):735–754. doi:10.1007/s13346-016-0339-2
200. Benedetti MS, Whomsley R, Poggesi I, et al. Drug metabolism and pharmacokinetics. Drug Metab Rev. 2009;41(3):344–390. doi:10.1080/10837450902891295
201. Razeghinejad MR, Myers JS, Katz LJ. Iatrogenic Glaucoma Secondary to Medications. Am J Med. 2011;124(1):20–25. doi:10.1016/j.amjmed.2010.08.011
202. Sihota R, Konkal VL, Dada T, Agarwal HC, Singh R. Prospective, long-term evaluation of steroid-induced glaucoma. Eye. 2008;22(1):26–30. doi:10.1038/sj.eye.6702474
203. Phulke S, Kaushik S, Kaur S. Steroid-induced Glaucoma: an Avoidable Irreversible Blindness. J Curr Glaucoma Pract. 2017;11(2):67–72. doi:10.5005/jp-journals-l0028-1226
204. Cunningham MA, Edelman JL, Kaushal S. Intravitreal Steroids for Macular Edema: the Past, the Present, and the Future. Surv Ophthalmol. 2008;53(2):139–149. doi:10.1016/j.survophthal.2007.12.005
205. Muftuoglu IK, Tokuc EO, Sümer F, Karabas VL. Evaluation of retinal inflammatory biomarkers after intravitreal steroid implant and Ranibizumab injection in diabetic macular edema. Eur J Ophthalmol. 2022;32(3):1627–1635. doi:10.1177/11206721211029465
206. Graham RO, Peyman GA. Intravitreal injection of dexamethasone. Treatment of experimentally induced endophthalmitis. Arch Ophthalmol Chic IL 1960. 1974;92(2):149–154. doi:10.1001/archopht.1974.01010010155016
207. Mansoor S, Kuppermann BD, Kenney MC. Intraocular sustained-release delivery systems for triamcinolone acetonide. Pharm Res. 2009;26(4):770–784. doi:10.1007/s11095-008-9812-z
208. Beer PM, Bakri SJ, Singh RJ, Liu W, Peters GB, Miller M. Intraocular concentration and pharmacokinetics of triamcinolone acetonide after a single intravitreal injection. Ophthalmology. 2003;110(4):681–686. doi:10.1016/S0161-6420(02)01969-3
209. Chin HS, Park TS, Moon YS, Oh JH. Difference in clearance of intravitreal triamcinolone acetonide between vitrectomized and nonvitrectomized eyes. Retina (Philadelphia, Pa). 2005;25(5):556–560. doi:10.1097/00006982-200507000-00002
210. Ansari EA, Ali N. Intraocular Pressure Following Intravitreal Injection of Triamcinolone Acetonide. Open Ophthalmol J. 2008;2:119–122. doi:10.2174/1874364100802010119
211. Gaballa SA, Kompella UB, Elgarhy O, et al. Corticosteroids in ophthalmology: drug delivery innovations, pharmacology, clinical applications, and future perspectives. Drug Deliv Transl Res. 2021;11(3):866–893. doi:10.1007/s13346-020-00843-z
212. Rajesh B, Zarranz-Ventura J, Fung AT, et al. Safety of 6000 intravitreal dexamethasone implants. Br J Ophthalmol. 2020;104(1):39–46. doi:10.1136/bjophthalmol-2019-313991
213. Muya L, Kansara V, Cavet ME, Ciulla T. Suprachoroidal Injection of Triamcinolone Acetonide Suspension: ocular Pharmacokinetics and Distribution in Rabbits Demonstrates High and Durable Levels in the Chorioretina. J Ocul Pharmacol Ther. 2022;38(6):459–467. doi:10.1089/jop.2021.0090
214. Fung S, Syed YY. Suprachoroidal Space Triamcinolone Acetonide: a Review in Uveitic Macular Edema. Drugs. 2022;82(13):1403–1410. doi:10.1007/s40265-022-01763-7
215. Price KW, Albini TA, Yeh S. Suprachoroidal Injection of Triamcinolone— review of a Novel Treatment for Macular Edema Caused by Noninfectious Uveitis. US Ophthalmic Rev. 2020;13(2):76–79. doi:10.17925/usor.2020.13.2.76
216. Tabl AA, Elsayed MA, Tabl MA. Suprachoroidal triamcinolone acetonide injection: a novel therapy for serous retinal detachment due to Vogt-Koyanagi Harada disease. Eur J Ophthalmol. 2022;32(6):3482–3488. doi:10.1177/11206721221085420
217. Weijtens O, Schoemaker RC, Lentjes EGWM, Fphtm R, Cohen AF, van MJC. Dexamethasone concentration in the subretinal fluid after a subconjunctival injection, a peribulbar injection, or an oral dose. Ophthalmology. 2000;107(10):1932–1938. doi:10.1016/S0161-6420(00)00344-4
218. Conrad JM, Robinson JR. Mechanisms of anterior segment absorption of pilocarpine following subconjunctival injection in albino rabbits. J Pharm Sci. 1980;69(8):875–884. doi:10.1002/jps.2600690806
219. McGhee CNJ, Dean S, Danesh-Meyer H. Locally administered ocular corticosteroids: benefits and risks. Drug Saf. 2002;25(1):33–55. doi:10.2165/00002018-200225010-00004
220. Garbe E, LeLorier J, Boivin JF, Suissa S. Inhaled and nasal glucocorticoids and the risks of ocular hypertension or open-angle glaucoma. JAMA. 1997;277(9):722–727.
221. Macris N. Glucocorticoid use and risks of ocular hypertension and glaucoma. JAMA. 1997;277(24):1929. doi:10.1001/jama.277.24.1929b
222. Nath T, Roy SS, Kumar H, Agrawal R, Kumar S, Satsangi SK. Prevalence of Steroid-Induced Cataract and Glaucoma in Chronic Obstructive Pulmonary Disease Patients Attending a Tertiary Care Center in India. Asia-Pac J Ophthalmol. 2017;6(1):28. doi:10.22608/APO.201616
223. Marcus MW, Müskens RPHM, Ramdas WD, et al. Corticosteroids and Open-Angle Glaucoma in the Elderly. Drugs Aging. 2012;29(12):963–970. doi:10.1007/s40266-012-0029-9
224. Duh MS, Walker AM, Lindmark B, Laties AM. Association between intraocular pressure and budesonide inhalation therapy in asthmatic patients. Ann Allergy Asthma Immunol. 2000;85(5):356–361. doi:10.1016/S1081-1206(10)62545-8
225. Johnson LN, Soni CR, Johnson MAJ, Madsen RW. Short-term use of inhaled and intranasal corticosteroids is not associated with glaucoma progression on optical coherence tomography. Eur J Ophthalmol. 2012;22(5):695–700. doi:10.5301/ejo.5000141
226. Ahmadi N, Snidvongs K, Kalish L, et al. Intranasal corticosteroids do not affect intraocular pressure or lens opacity: a systematic review of controlled trials. Rhinol J. 2015;53(4):290–302. doi:10.4193/Rhino15.020
227. Gonzalez AV, Li G, Suissa S, Ernst P. Risk of glaucoma in elderly patients treated with inhaled corticosteroids for chronic airflow obstruction. Pulm Pharmacol Ther. 2010;23(2):65–70. doi:10.1016/j.pupt.2009.10.014
228. Ishii M, Horita N, Takeuchi M, et al. Inhaled Corticosteroid and Secondary Glaucoma: a Meta-analysis of 18 Studies. Allergy Asthma Immunol Res. 2021;13(3):435–449. doi:10.4168/aair.2021.13.3.435
229. Opatowsky I, Feldman RM, Gross R, Feldman ST. Intraocular Pressure Elevation Associated with Inhalation and Nasal Corticosteroids. Ophthalmology. 1995;102(2):177–179. doi:10.1016/S0161-6420(95)31039-1
230. Valenzuela CV, Liu JC, Vila PM, Simon L, Doering M, Lieu JEC. Intranasal Corticosteroids Do Not Lead to Ocular Changes: a Systematic Review and Meta-analysis. Laryngoscope. 2019;129(1):6–12. doi:10.1002/lary.27209
231. Bielory B, Bielory L. Over-The-counter migration of steroid use: impact on the eye. Curr Opin Allergy Clin Immunol. 2014;14(5):471–476. doi:10.1097/ACI.0000000000000099
232. Bernstein HN, Schwartz B. Effects of long-term systemic steroids on ocular pressure and tonographic values. Arch Ophthalmol Chic IL 1960. 1962;68:742–753. doi:10.1001/archopht.1962.00960030746009
233. Chadha V, Cruickshank I, Swingler R, Sanders R. Advanced glaucomatous visual loss and oral steroids. BMJ. 2008;337:a670. doi:10.1136/bmj.a670
234. Garbe E, LeLorier J, Boivin JF, Suissa S. Risk of ocular hypertension or open-angle glaucoma in elderly patients on oral glucocorticoids. Lancet Lond Engl. 1997;350(9083):979–982. doi:10.1016/S0140-6736(97)03392-8
235. Wiendl H, Gold R, Berger T, et al. Multiple Sclerosis Therapy Consensus Group (MSTCG): position statement on disease-modifying therapies for multiple sclerosis (white paper). Ther Adv Neurol Disord. 2021;14:17562864211039648. doi:10.1177/17562864211039648
236. Garrott HM, Walland MJ. Glaucoma from topical corticosteroids to the eyelids. Clin Experiment Ophthalmol. 2004;32(2):224–226. doi:10.1111/j.1442-9071.2004.00787.x
237. Sahni D, Darley CR, Hawk JLM. Glaucoma induced by periorbital topical steroid use − A rare complication. Clin Exp Dermatol. 2004;29(6):617–619. doi:10.1111/j.1365-2230.2004.01610.x
238. Daniel BS, Orchard D. Ocular side−effects of topical corticosteroids: what a dermatologist needs to know. Australas J Dermatol. 2015;56(3):164–169. doi:10.1111/ajd.12292
239. Takakuwa K, Hamanaka T, Mori K, et al. Atopic Glaucoma: clinical and Pathophysiological Analysis. J Glaucoma. 2015;24(9):662. doi:10.1097/IJG.0000000000000069
240. Haeck IM, Rouwen TJ, Timmer-de Mik L, de Bruin-Weller MS, Bruijnzeel-Koomen CA. Topical corticosteroids in atopic dermatitis and the risk of glaucoma and cataracts. J Am Acad Dermatol. 2011;64(2):275–281. doi:10.1016/j.jaad.2010.01.035
241. Huschle OK, Jonas JB, Koniszewski G, Buchfelder M, Fahlbusch R. Glaucoma in central hypothalamic-hypophyseal Cushing syndrome. Fortschritte Ophthalmol Z Dtsch Ophthalmol Ges. 1990;87(5):453–456.
242. Virevialle C, Brasnu E, Fior R, Baudouin C. Open-angle glaucoma secondary to Cushing syndrome related to an adrenal adenoma: case report. J Fr Ophtalmol. 2014;37(10):e169. doi:10.1016/j.jfo.2014.01.018
243. Griffin S, Boyce T, Edmunds B, Hills W, Grafe M, Tehrani S. Endogenous hypercortisolism inducing reversible ocular hypertension. Am J Ophthalmol Case Rep. 2019;16:100573. doi:10.1016/j.ajoc.2019.100573
244. Habib SN, Lin Z, Puvanachandra N. Ocular hypertension secondary to high endogenous steroid load in Cushing’s disease. BMJ Case Rep. 2019;12(1):bcr-2018–226738. doi:10.1136/bcr-2018-226738
245. Ma Y, Chen Z, Ma Z, et al. Increased Risk of Ocular Hypertension in Patients With Cushing’s Disease. J Glaucoma. 2022;31(12):941. doi:10.1097/IJG.0000000000002113
246. Horan T, Salim S Characteristics and Management of Steroid-Induced Glaucoma. American Academy of Ophthalmology; 2021. Available from: https://www.aao.org/eyenet/article/management-of-steroid-induced-glaucoma.
247. Weinreb RN, Polansky JR, Kramer SG, Baxter JD. Acute effects of dexamethasone on intraocular pressure in glaucoma. Invest Ophthalmol Vis Sci. 1985;26(2):170–175.
248. Agrawal S, Agrawal J, Agrawal TP. Management of intractable glaucoma following intravitreal triamcinolone acetonide. Am J Ophthalmol. 2005;139(3):575–576. doi:10.1016/j.ajo.2004.10.025
249. Rezkallah A, Mathis T, Denis P, Kodjikian L. XEN Gel Stent to Treat Intraocular Hypertension After Dexamethasone-Implant Intravitreal Injections: 5 Cases. J Glaucoma. 2019;28(1):e5. doi:10.1097/IJG.0000000000001092
250. Steroid-Induced Glaucoma - EyeWiki. EyeWiki; 2023. Available from: https://eyewiki.aao.org/Steroid-Induced_Glaucoma.
251. Zhou Y, Pruet CM, Fang C, Khanna CL. Selective laser trabeculoplasty in steroid-induced and uveitic glaucoma. Can J Ophthalmol. 2022;57(4):277–283. doi:10.1016/j.jcjo.2021.05.006
252. Davidson M, Berkowitz E, Roberts H, Wanas A, Myerscough J. Selective Laser Trabeculoplasty for Steroid-Induced Ocular Hypertension following Endothelial Keratoplasty. Curr Eye Res. 2022;47(10):1362–1365. doi:10.1080/02713683.2022.2088800
253. AlObaida I, Al Owaifeer AM, Alotaibi H, Alsafi A, Ali Aljasim L. Outcomes of selective laser trabeculoplasty in corticosteroid-induced ocular hypertension and glaucoma. Eur J Ophthalmol. 2022;32(3):1525–1529. doi:10.1177/11206721211023310
254. Xiao J, Zhao C, Liang A, Zhang M, Cheng G. Efficacy and Safety of High-Energy Selective Laser Trabeculoplasty for Steroid-Induced Glaucoma in Patients with Quiescent Uveitis. Ocul Immunol Inflamm. 2021;29(4):766–770. doi:10.1080/09273948.2019.1687730
255. Goñi FJ, Stalmans I, Denis P, et al. Elevated Intraocular Pressure After Intravitreal Steroid Injection in Diabetic Macular Edema: monitoring and Management. Ophthalmol Ther. 2016;5(1):47–61. doi:10.1007/s40123-016-0052-8
256. Hu Y, Barron AO, Gindina S, et al. Investigations on the Role of the Fibrinolytic Pathway on Outflow Facility Regulation. Invest Ophthalmol Vis Sci. 2019;60(5):1571–1580. doi:10.1167/iovs.18-25698
257. Gindina S, Hu Y, Barron AO, Qureshi Z, Danias J. Tissue plasminogen activator attenuates outflow facility reduction in mouse model of juvenile open angle glaucoma. Exp Eye Res. 2020;199:108179. doi:10.1016/j.exer.2020.108179
258. Gerometta R, Spiga MG, Borrás T, Candia OA. Treatment of sheep steroid-induced ocular hypertension with a glucocorticoid-inducible MMP1 gene therapy virus. Invest Ophthalmol Vis Sci. 2010;51(6):3042–3048. doi:10.1167/iovs.09-4920
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.