Back to Journals » Journal of Blood Medicine » Volume 12
Glanzmann Thrombasthenia: Perspectives from Clinical Practice on Accurate Diagnosis and Optimal Treatment Strategies
Authors Mathews N, Rivard GE, Bonnefoy A
Received 11 March 2021
Accepted for publication 20 May 2021
Published 11 June 2021 Volume 2021:12 Pages 449—463
DOI https://doi.org/10.2147/JBM.S271744
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Martin H Bluth
Natalie Mathews,1 Georges-Etienne Rivard,2 Arnaud Bonnefoy2
1Division of Haematology/Oncology, Department of Paediatrics, The Hospital for Sick Children, Toronto, Ontario, Canada; 2Division of Hematology-Oncology, Department of Pediatrics, CHU Sainte-Justine, Université de Montréal, Montréal, Québec, H3T 1C5, Canada
Correspondence: Natalie Mathews
Division of Haematology/Oncology, Department of Paediatrics, The Hospital for Sick Children, 555 University Ave, Toronto, Ontario, M5G 1X8, Canada
Email [email protected]
Abstract: Glanzmann thrombasthenia (GT) is a rare autosomal recessive disorder of fibrinogen-mediated platelet aggregation due to a quantitative or qualitative deficit of the αIIbβ3 integrin at the platelet surface membrane resulting from mutation(s) in ITGA2B and/or ITGB3. Patients tend to present in early childhood with easy bruising and mucocutaneous bleeding. The diagnostic process requires consideration of more common disorders of haemostasis and coagulation prior to confirming the disorder with platelet light transmission aggregation, flow cytometry of CD41 and CD61 expression, and/or exon sequencing of ITGA2B and ITGB3. Antifibrinolytic therapy, recombinant activated factor VII, and platelet transfusions are the mainstay of therapy, although the latter may trigger formation of anti-platelet antibodies in GT patients and inadvertent platelet-refractory disease. The management of these patients therefore remains complex, particularly in the context of trauma, labour and delivery, and perioperative care. Bone marrow transplantation remains the sole curative option, although the venue of gene therapy is being increasingly explored as a future alternative for definitive treatment of GT.
Keywords: bleeding disorders, inherited platelet defects, platelet aggregation, ITGA2B, ITGB3, αIIbβ 3
Introduction
Glanzmann Thrombasthenia (GT) is a rare inherited bleeding disorder characterized by dysfunctional fibrinogen-mediated platelet aggregation due to decreased or dysfunctional αIIbβ3 integrin expression at the platelet surface membrane. This autosomal recessive condition affects approximately 1 in 1,000,000 people,1 though prevalence reaches up to 1 in 200,000 people2 in populations of increased consanguinity, including those coming from Pakistan, Iraqi Jewish groups, nomadic tribes of Jordan, South Indian Hindu communities, Roma camps within France, and the Canadian province of Newfoundland and Labrador.1–6
Historical Context
In 1918, Dr. Glanzmann, a Swiss pediatrician, coined the term “thrombasthenia,” or “weak platelets,” when describing a patient exhibiting purpura despite having platelets of normal quantity and appearance on peripheral smear.1,7–10 Curiously, he also noted absence of platelet clumping, a prolonged bleeding time, and inferior clot retraction.7 Forty-four years later, in 1962, Drs. Caen and Cousin demonstrated absence of platelet aggregation in response to ADP, adrenaline, thrombin, and collagen stimulation.1,11 In the 1970s, multiple teams12–15 studying the platelets of Glanzmann thrombasthenia patients identified a shared underlying deficiency of the membrane glycoprotein IIb/IIIa complex, now known as the αIIbβ3 integrin.
Role of αIIbβ3 Integrin
Wild-type platelets express approximately 50,000 copies of αIIbβ3 integrin at their surface membranes.16,17 The αIIb subunit, predominantly expressed within cells of the megakaryocytic lineage,18 is produced as a pro-peptide comprised of a heavy and light chain linked together by disulfide bridges.19–21 In addition to providing intramolecular stability, the integrity of the 674–687 disulfide bridge in particular has also been shown to be necessary for ultimate surface expression of the αIIbβ3 integrin.22 The αIIb subunit binds its β3 partner via calcium-depending bonds prior to undergoing post-translational modifications, including O- and N-glycosylation, within the endoplasmic reticulum and Golgi apparatus.20,21 Once integrated into the platelet membrane, each mature subunit within the final receptor complex contains a large globular extracellular domain, a single transmembrane domain, and a small cytoplasmic region that interacts with its neighboring cytoplasmic domain via salt bridge (Figure 1).20 The cytoplasmic domains also interact with other cytoplasmic and cytoskeletal proteins20,23 and facilitate both inside-out and outside-in signaling (Figure 2).20,24,25 In its resting state, the αIIbβ3 integrin receptor exists in a “bent” conformation whose extracellular domains are clasped and have a low affinity for binding ligands.1,26,27 A rise in the intracellular concentration of calcium leads to a talin-induced conformational change within the extracellular domains of both subunits, exposing their ligand binding sites.26–28 This inside-out activation allows ligand binding sites on each subunit to bind the same fibrinogen molecule, which in turns binds identical ligand binding sites on other platelets to establish a platelet plug.1 Meanwhile, fibrinogen binding also facilitates unclasping of the extracellular domains of the αIIb and β3 subunits26,27,29,30 and triggers protein kinase C-mediated cytoskeletal changes and platelet granule secretion, ultimately resulting in platelet spreading and fibrin clot stabilization.1 Clot formation and stabilization is further enhanced by β3’s ability to bind von Willebrand factor, fibronectin, and vitronectin.1,28,31 Furthermore, β3 promotes cleavage of Factor Xa, assisting with conversion of prothrombin to thrombin.8,32–34 β3 has also been shown to have a role in fibrin clot retraction that is independent from its ability to bind fibrinogen.8,10,35–37 Given the myriad of interactions that must occur for the αIIb and β3 subunits to reach the platelet membrane and promote platelet aggregation, one can imagine the many opportunities for this process to become disrupted. Each of these opportunities for error, in turn, implies the potential for countless theoretical GT-causing genetic variants.
 |
Figure 1 Schematic of αIIbβ3 integrin composed of αIIb and β3 subunits. The mature αIIb subunit contains extracellular heavy and light chains linked together via disulfide bridge. Both subunits contain extracellular, transmembrane, and cytoplasmic domains; the latter domains are linked via salt bridge. |
 |
Figure 2 Schematic of αIIbβ3 integrin undergoing inside-out and outside-in signaling. (A) Bent confirmation of αIIbβ3 integrin with intact salt bridge linking cytosolic domains of the subunits (low affinity for binding fibrinogen). (B) Binding of intracellular protein talin disrupts salt bridge and triggers separation of the cytosolic region of β3 from that of αIIb, resulting in a conformational change of the αIIbβ3 integrin into the upright position. In this position, fibrinogen is able to bind extracellular domains (high affinity for binding fibrinogen; inside-out signaling). (C) Fibrinogen, in turn, binds additional αIIbβ3 integrins to facilitate platelet aggregation, resulting in activation and recruitment of additional intracellular and cytosolic proteins, such as c-Src tyrosine kinase (c-Src), integrin-linked kinase (ILK), spleen tyrosine kinase (Syk), protein kinase C (PKC), and protein tyrosine phosphatase (PTP1B) and others, to facilitate processes including cytoskeletal reorganization for platelet spreading, clot stabilization, and clot retraction (outside-in signaling). |
Genetics
The αIIb and β3 subunits are respectively encoded by ITGA2B (65 kbp) and ITGB3 (17 kbp), which are both found on chromosome 17 (17q21.31 and 17q21.32, respectively; OMIM # 607759 and OMIM #173470). ITGA2B and ITGB3 expression are not known to be coordinated,3,31 however, and mutations in either of these genes can lead to forms of GT that are phenotypically indistinguishable.1 GT inheritance is typically autosomal recessive and patients may exhibit homozygosity, particularly if consanguinity is present, or compound heterozygosity.8,20,31,38 ITGB3 mutations are more common, presumably due to its relatively larger coding region of 30 exons in comparison to the 15 exons comprising ITGA2B.1 As of February 2021, 475 ITGA2B and ITBG3 GT-causing mutations have been catalogued in the Glanzmann Thrombasthenia Database (https://glanzmann.mcw.edu/) and most commonly include nonsense, missense, and splice site variants.1,20 Large deletion and duplication mutations are rare.1,20 Mutations leading to a GT phenotype can be manifested by dysfunctional gene expression, protein folding, post-translational processing, trafficking to the platelet membrane, and ligand binding.1,8,39–43 In fact, missense mutations impeding such processes have helped researchers to identify functional coding sequences within ITGA2B and ITBG3.20,44–46 For example, ITGA2B c.818G>A disrupts the calcium-binding site with the β3 subunit and has been shown to result in lack of αIIbβ3 integrin expression at the platelet membrane.8,39–41 Alternatively, defects within αIIbβ3 integrin extracellular ligand binding sites result in a qualitative variant of GT in which quantities of αIIbβ3 integrin at the platelet membrane are otherwise intact. Such differences in αIIbβ3 integrin expression at the level of the platelet membrane are the basis for distinguishing clinical types of hereditary GT (Table 1). In Type I GT, platelets’ membrane expression of αIIbβ3 integrin is less than 5% of the wild-type quantity.47,48 Type I GT is most common, representing 62–78% of GT cases.1,3,4,20 Type II GT, in which 5–25% of normal of αIIbβ3 integrin expression is maintained,4,8,20,47,48 represents about 12–16% of the GT population.1,3,4,20 Type III represents a “variant” GT phenotype in which the αIIbβ3 integrin is present in sufficient quantities at the platelet membrane (ranging from 25% to 100% of reference levels),8,20,47,48 but is qualitatively dysfunctional, and represents 8–22% of affected patients.1,3,4,20 Mutations conferring a defective αIIbβ3 integrin result in varying clinical severities, but tend to involve ligand binding sites, such as ITGB3 c.719G>A, and inside-out signaling, such as ITGB3 c.2332T>C.8,45,49,50 Interestingly, gain-of-function GT-like cases have also been described involving compound heterozygous ITGA2B and ITBG3 mutations affecting membrane-adjacent residues resulting in auto-activation of αIIbβ3, reduced αIIbβ3 expression, and thrombocytopenia.1,51–53 In a minority of patients, no mutation may be found, suggesting unidentified causes of GT that could perhaps be attributed to non-sequenced promoter or intron regions, mutated proteins that may help facilitate αIIbβ3 integrin development and transport, or modulators affecting αIIbβ3 integrin expression, such as miRNA or epigenetic changes.20
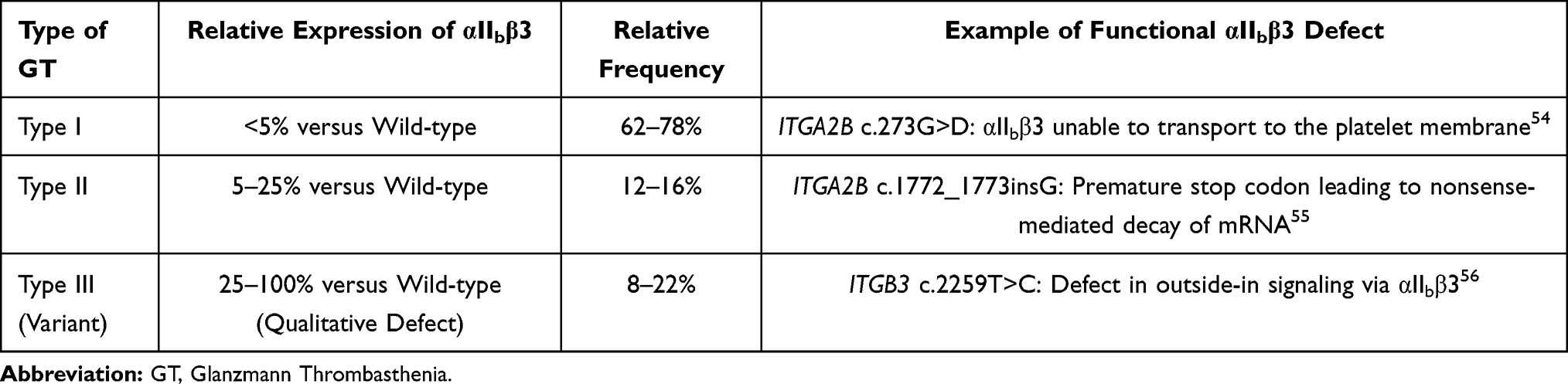 |
Table 1 Types of Glanzmann Thrombasthenia and Their Frequencies, with Examples of Genetic Mutations |
Acquired Glanzmann Thrombasthenia
Acquired GT is a disorder characterized by anti-αIIbβ3 complex-antibody-mediated platelet destruction.57,58 More common than its hereditary counterpart, acquired GT can manifest as primary immune thrombocytopenia (ITP) or occur secondary to autoimmune disorders, malignancies, or organ transplants.57–60 Certain medications, including the antimalarial quinine, antiarrhythmic quinidine, and various anticoagulants, including abciximab, have been identified as triggers for acquired GT.1,58
Clinical Presentation
Patients with hereditary GT tend to develop easy bruising and mucocutaneous bleeding symptoms (Table 2) early in life, with a mean age of diagnosis of 1 year of age and 15% of GT patients presenting beyond age 14 years.2,61 Males with GT may be diagnosed as a result of post-circumcision hemorrhage.8,62 Loss of primary teeth is another common source of bleeding during childhood.8 In rare cases, abnormal bleeding may not occur until adulthood, when a patient’s coagulation system is challenged by childbirth or another severe trauma.4,20 Bruising provoked by mild trauma is the most common symptom experienced, followed by mucocutaneous bleeding.4 Typically, bleeding symptoms are less severe than those seen in hemophilia patients,2,63 although more than two-thirds of patients require one or more platelet and/or red blood cell transfusions over their lifetime.20,64 Epistaxis is the most prevalent source of severe bleeding, affecting 60–80% of patients.4 This symptom is most prominent during childhood,4,65 when the nasal septum is most friable and also most likely to be subjected to the trauma of nose-picking.1
 |
Table 2 Clinical Presentations of Glanzmann Thrombasthenia |
The majority of females with GT experience heavy, prolonged, and/or more frequent menstrual bleeding.4 Gingival bleeding is also a source of concern, affecting up to 60% of patients, and may even result in iron deficiency anemia.4 This symptom may be remedied with improved oral hygiene. Gastrointestinal bleeding is more rare, affecting only 10–28% of patients,4,20 but may be particularly concerning in the presence of localized angiodysplasia.8 Intracranial hemorrhages, hemarthroses, hematuria, and organ bleeds have all been described in GT patients, but are exceedingly rare.1,4,8,20
Diagnosis
As with any patient presenting with easy bruising and/or mucocutaneous bleeding, it is important to take a detailed history of bleeding symptoms. Diagnostic bleeding scores, which quantify a given patient’s ongoing bleeding risk based on their historical symptoms and need for interventions, are generally useful for establishing a true bleeding tendency although no cut-off values have been established for GT.1,66 The ISTH/SSC bleeding assessment tool67 has, however, demonstrated an ability to identify patients with inherited platelet disorders, once von Willebrand disease has been ruled out. History should also include the presence of any past or present autoimmune or malignant diagnoses, as well as current symptoms that may point to an undetected underlying systemic condition that could trigger acquired GT. Past infections should also be queried, as mutations involving integrin regulatory proteins, including kindlin-3 and calcium and diacylglycerol-regulated guanine nucleotide exchange factor, can affect both platelet and leukocyte integrin function, resulting in a simultaneous immunodeficiency;58,68 these patients do not have GT. A medication history should include the use of antiplatelet medications, such as NSAIDs, and other anticoagulants that may suggest an alternative cause. Taking a family history and drawing a pedigree is encouraged, as it could provide critical information to help establish the presence of a familial bleeding disorder spanning multiple generations with a specific pattern of inheritance. When examining the patient, it is important to inspect the skin for signs of bruising as well as the mucocutaneous regions, including the nares, for evidence of bleeding. Additional areas of importance may be guided by history.
Preliminary investigations that are widely available and relatively inexpensive are necessary to help narrow the list of differential diagnoses (Figure 3), given the rarity of GT amongst haemostasis and coagulation disorders; for every one patient diagnosed with inherited GT, there are 1000 people diagnosed with von Willebrand disease,69 95 people diagnosed with immune thrombocytopenia,70 and 85 people diagnosed with haemophilia A.71 In terms of specific inherited platelet defects diagnosed per year, GT represents just 9.8% of this category.72 As Dr. Glanzmann discovered,7 complete blood count (CBC) and peripheral smear will typically reveal a normal quantity of platelets that are of a normal size and maintain a typical granular pattern, which helps differentiate GT from other platelet disorders, such as Bernard-Soulier and Grey Platelet Syndromes. In some cases, the CBC might also suggest iron deficiency anemia (IDA), which may be a consequence and/or a cause of abnormal bleeding, as arachidonic acid-induced platelet aggregation has been shown to improve following treatment of IDA.73,74 Additional CBC abnormalities would point towards a diagnosis other than GT. Coagulation studies, including INR, PTT, and fibrinogen, are normal in GT and help rule out coagulation factor deficiencies and fibrinogen disorders that could be alternative, more common causes of bleeding and abnormal platelet aggregation. Additionally, normal von Willebrand factor antigen and von Willebrand factor activity assays are expected and eliminate the possibility of von Willebrand disease.
 |
Figure 3 Diagnostic algorithm for GT. *Consider proceeding to platelet light transmission aggregometry if suspicion for platelet defect remains high. **Consider genetic testing to identify specific mutation of ITGA2B and ITGB3 and/or flow cytometry to differentiate GT type. †Consider clot retraction assay (if available) and platelet light transmission aggregometry or genetic testing of ITGA2B and ITGB3 to make the diagnosis of GT Type III. ††Consider genetic testing to identify specific mutation of ITGA2B or ITGB3. Abbreviations: CBC, complete blood count; VWF Ag, von Willebrand factor antigen; VWF Act, von Willebrand factor activity; VWD, von Willebrand disease. |
Additional screening tests may be helpful prior to confirming the diagnosis of GT with platelet light transmission aggregometry, which requires a large volume of blood, 3–4 hours, and a specialized laboratory.75 A clot retraction assay can be performed using 1–2 mL of whole blood incubated overnight at 37 degrees Celsius, removing the newly formed clot, and quantifying the relative volume of serum within the remaining plasma sample that was extracted from the clot during the retraction process. The assay can also be performed using platelet rich plasma. Assuming a normal platelet quantity and adequate fibrinogen count and quality, an abnormal clot retraction is consistent with diagnosis of GT, as this assay specifically tests the outside-in signaling of the αIIbβ3 integrin.76,77 Furthermore, a result of no clot retract versus low-to-normal clot retraction can help differentiate between Types I and II GT, respectively.36 It should be noted, however, that this test has fallen out of favour, as it is only available is specialized laboratories and is no longer considered to be a required test in the diagnosis of GT.
Platelet function analyzer, or PFA 100, testing requires as little as 2 mL of blood and is a measure of primary hemostasis.75,78 By timing platelet plug formation over a membrane in the presence of stimulants (collagen and epinephrine or collagen and ADP) under high shear conditions, samples affected by platelet disorders and von Willebrand disease will be differentiated.78 While an abnormal PFA-100 is 100% sensitive for GT,75 it does not distinguish GT from severe von Willebrand disease or afibrinogenemia.1
Platelet light transmission aggregometry is a gold standard for establishing a diagnosis of GT.79 This test exposes a blood sample to various agonists to stimulate platelet plug formation and measures the subsequent transmission of light through the platelet suspension.80 Platelet aggregation in the presence of ristocetin, but not in the presence of ADP, collagen, thrombin, or adrenaline (<10% of reference values) is pathognomonic for the disorder.36,47,77 It may be necessary to repeat testing once to confirm the results or compare with a control sample in parallel, given the multiple pre-analytical and analytical variables that may affect the outcome of this investigation.1,80–83
Once Glanzmann thrombasthenia has been diagnosed, there are additional tests that are helpful for further characterization of the disorder. Flow cytometry with monoclonal antibody panels against CD41 and CD61 can quantify deficiencies in αIIb and β3 expression at the platelet membranes, respectively, and allow for identification of Type I (<5% expression) and Type II (5–25% expression) GT.84 Flow cytometry typically reveals at least 50% αIIb and β3 expression in Type III GT.36,84 Differentiating GT type is particularly helpful for risk stratifying patients who may develop alloimmunization following platelet transfusion (see Platelet Refractoriness and Alloimmunization). Furthermore, flow cytometry can also be used to identify specific antibodies against αIIbβ3 in cases of acquired GT.84,85 Genetic sequencing of ITGA2B and ITGB3 may also be performed to confirm the specific mutations involved. Multiple groups86–89 have recently developed high-throughput molecular diagnostic assays for patients with GT and other inherited bleeding disorders. It should be noted though that at this time, there does not seem to be a correlation between specific mutation and phenotype severity8 and even family members sharing similar mutations have been shown to have significant variability amongst their clinical outcomes.20
Management
Routine Follow-Up and Anticipatory Guidance
Any patient diagnosed with GT should be referred to a tertiary care centre with a haematologist experienced in treating patients with inherited bleeding disorders. This centre must be able to manage patients outside of regular clinic hours should severe bleeding occur. During regular clinic hours, the patient should have access to a multidisciplinary team, including a nursing coordinator, physiotherapist, social worker, and psychologist as necessary.2,90 Good oral hygiene and routine dental follow-up are also of the utmost importance.91 Patients with GT can and should receive routine vaccinations with the additional step of providing 15 minutes of applied pressure to the site to encourage proper hemostasis.90 Additional immunization against hepatitis A and B is encouraged, given heightened risk for exposure to blood products.90 Patients should receive adequate teaching about GT, including education on preventing bleeds, such as avoiding certain medications, including aspirin and NSAIDs, and high-impact physical activities. Patients should be counseled regarding recognition of bleeds and necessity of urgent medical intervention for prolonged or worrisome bleeding, as delay in treatment has been implicated in both hospital length of stay and overall treatment response.92,93 Any patient with a diagnosis of GT, or any clinically significant bleeding disorder, should wear a MedicAlert bracelet or similar piece of identification to flag their condition to emergency medical personnel in the event of incapacitation.
Pregnant women who have been diagnosed with GT, or whose partners have been diagnosed with GT, should be considered for prenatal testing if the parents of the fetus are consanguineous. Prenatal diagnosis is routinely confirmed by genetic testing,1 although flow cytometry has also been used.94 Pregnant women who are known to have GT should also be screened for anti- αIIbβ3 antibodies via monoclonal antibody-specific immobilization of platelet antigen (MAIPA) assay regularly throughout gestation,95 as these antibodies can cross the placenta and cause dangerously low platelet levels in the fetus.31,96,97 Mothers must be counseled about this risk and for this reason, it is important to test the newborn’s platelet count within the first few hours life8 if maternal antibodies have been detected. While mothers with GT are not expected to experience increased bleeding during the pregnancy itself, specific planning surrounding labour and delivery is indicated in order to prevent excessive bleeding in the intra- and post-partum periods (see Site-Specific considerations below).
General Approach to Bleeding
Minor bleeding episodes may be initially managed at home using manual compression and/or antifibrinolytic therapy. Depending on the bleeding site, manual compression may be achieved by applying pressure with gauze or, in the cases of epistaxis, pinching the soft cartilages of the nares closed using the index finger and thumb. Cold compresses are not recommended, as they have been shown to impair haemostasis and coagulation in patients with and without bleeding disorders.98,99 The use of oral antifibrinolytic therapy,31,47 including tranexamic acid (TXA) and aminocaproic acid, can be easily administered in the home setting and may be critical for stopping a bleed in its early stages. Antifibrinolytics should not be administered to patients experiencing gross hematuria, however.100,101 GT patients with bleeding that is refractory to these methods should seek emergency medical attention.
Patients with GT experiencing severe bleeding require some combination of three main treatment options to achieve haemostasis: antifibrinolytics, recombinant activated factor VII (rF7a), and platelet transfusion. DDAVP has also been used,47,102 although study results have been inconsistent1 and further investigation is needed in this area. In addition to providing haemostasis, the need for red blood cell transfusion must always be considered. These transfusions should be sourced from washed or frozen red blood cells in order to remove any residual platelets that could trigger alloimmunization.31 Unfortunately, the urgency of the situation may preclude the ability to wait for these products to become available and the risks and benefits of transfusing fresh non-washed pRBCs must be carefully weighed.
Antifibrinolytics, in addition to being available enterally, can be administered intravenously or topically using antifibrinolytic-soaked gauze or gel foam. Fibrin glue and topical thrombin are additional topical alternatives/complements to antifibrinolytics.
Platelet transfusion has been a mainstay of GT therapy for years. However, the benefit of providing functional platelets must be weighed against the risk of causing alloimmunization and rendering a patient refractory to subsequent platelet transfusions. GT Type I patients are at particular risk of alloimmunization against αIIb and β3 due to their inherent lack of self-expression of these antigens.20,31 Given the possibly fatal implications of platelet refractoriness, platelet transfusions should be reserved for life-threatening hemorrhages in the GT population.31 The use of platelet transfusions in girls and pre-menopausal women should be particularly avoided whenever possible given the added risk of transplacental antibody transfer causing future neonatal alloimmune thrombocytopenia.1,96 If and when choosing to transfuse platelets, they should be HLA-matched, leukocyte-reduced whenever possible to avoid HLA-alloimmunization.31 ABO compatibility offers an additional layer of protection31 and patients who have received any blood products, including only red blood cells, should be regularly screened for antibodies. Furthermore, there have even been some reports of antibody presence in non-transfused patients,20,31,103 which may have been infection-induced.
rF7a is an expensive47,104,105 but efficacious treatment for patients with GT experiencing moderate to severe bleeding. rF7a has a long shelf life at room temperature106,107 and is approved in several counties for use in patients with hemophilia, congenital factor VII deficiency, and GT. In particular, it is approved in the United States for use in GT patients who are refractory to platelets as well as in Europe for patients with GT who cannot receive platelets. Various studies have reported partial or better responses to rF7a for 67–93%104–106,108 of nonsurgical hemorrhages in GT patients, with or without the help of antifibrinolytics. Its efficacy has been found to be irrelevant to antibody presence.105 rF7a’s mechanism of action for GT patients is not entirely understood.104 However, it is thought that through activation of factor X, rF7a facilitates generation of thrombin, which can then bind and activate GT platelets via intact GP1b receptors.31,106,109,110
It is important to note that dosing of rF7a varies based on indication, and patients with GT typically require fewer total doses than hemophilia patients with inhibitors do.111 Patients with GT typically require 80–140 μg/kg intravenous every 2.5 hours or less until hemostasis is achieved.61,104 Continuous infusions of rF7a have only been rarely used and are not well studied.104
Site-Specific Considerations
As epistaxis is a major source of bleeding in children with GT, efforts should be made to prevent over-drying of the nasal mucosa in affected patients. These methods include use of humidifiers, saline nasal sprays and Vaseline gel. Some patients may find it helpful to sleep in a seated position when experiencing mild nasal or oral bleeding. Major epistaxis requires the expertise of an otolaryngologist. A 2010 retrospective study65 of 5 children with GT presenting on 63 occasions with epistaxis reported a hospitalization rate of 72%. Forty-two percent of these admissions required intensive care. While anterior nasal packing with or without topic hemostatic treatments were successful about one-third of the time, the administration of a bovine collagen matrix was deemed successful in just one-half of cases.
Oral cavity bleeding can commonly present in the setting of gingivitis. Daily flossing is therefore highly encouraged. Those with gingival bleeding may benefit from using antifibrinolytic therapy as a mouthwash.2,5,6 Children are also at risk of oral bleeding with the routine loss of primary teeth. Loose teeth can be treated with fibrin glue to help mitigate blood loss.3,112,113 Plastic splints can also be used to physically support hemostasis.114
Heavy uterine bleeding affects most women with GT. Efforts should be made to quantify this symptom using standardized scales, such as the Pictorial Blood Loss Assessment Chart.115,116 GT patients with heavy menses should be treated first line with antifibrinolytics. Patients who have refractory bleeding should be reviewed by a gynecologist and considered for oral contraceptive therapy or hormonal intrauterine devices, with or without adjunctive antifibrinolytics. Intravenous high-dose estrogen therapy over 1–2 days is an effective measure for these patients and should be administered in consultation with a gynecologist.35,117 It should be considered prior to administration of rF7a or platelet transfusion if bleeding is not life-threatening.
Bleeding associated with childbirth is a major concern in women with GT. Surprisingly, up to 50% of women with GT may not be diagnosed until facing the haemostatic challenges of labour and delivery.1,96 Epidural anesthesia is contraindicated in this population due to additional bleeding risks and an alternative pain management plan should be arranged beforehand.1,77 Women in labour for vaginal delivery or preoperative for caesarean section should be started on rF7a and antifibrinolytics in the presence of evidence of abnormal bleeding and may benefit from platelets as well. Platelets may continue to be required up to 7 days after delivery.8,118,119 Women experiencing postpartum hemorrhage should be managed with packed red blood cells and uterotonics.120
Perioperative Bleeding
The rate of perioperative bleeding in patients with inherited functional defects has been reported at 24.8%, with cardiovascular and urological surgeries bearing a particularly significant risk.121 The principles for preventing and managing surgical bleeds are similar to those for nonsurgical hemorrhages: patients tend to be managed with a combination of antifibrinolytics and rF7a, with or without platelets. One international retrospective review sponsored by Novo Nordisk Health Care AG involving 96 GT patients122 who underwent 101 surgeries in which rF7a and platelets were used, with or without antifibrinolytics, reported 100% efficacy in achieving hemostasis, regardless of alloimmunization status. This success rate was maintained for minor procedures when only rF7a and antifibrinolytics were given to patients with a history of antibodies and refractoriness. The same study found that this success rate decreased to 88.9% when rF7a was used without antifibrinolytics. The group that received platelets with or without antifibrinolytics had a success rate of just 67%. Another international survey in which ties to Novo Nordisk Health Care AG were disclosed reported 67% success in preventing surgical bleeding in 9 GT patients undergoing major operations and 92% of 25 GT patients undergoing minor procedures, with or without antifibrinolytics.105 rF7a dosing in surgical patients is 80–90 ug/kg intravenously immediately prior to surgery, with at least 2 repeated doses every 2–6 hours;123 some sources61,106,122 recommend dosing as high as 140 u/kg. Patients undergoing major operations are expected to require additional doses, with one review104 on rF7a use in surgical patients reporting median durations of treatment of 7 hours and 2 days for minor and major operations, respectively. In the post-operative period, haemostasis can be monitored clinically along with trending of hemoglobin.47
Platelet Refractoriness and Alloimmunization
Any GT patient presenting with refractoriness to platelet transfusion likely requires rF7a and should be considered for packed red blood cell transfusion. Anti-platelet antibodies, should always be suspected in the case of platelet refractoriness.8 The prevalence of alloimmunization, due either to anti- αIIbβ3 or anti-HLA antibodies, is as high at 30% in the general GT population.61 Patients who have developed HLA antibodies should be treated with HLA-compatible platelets. Alloimmunization against αIIbβ3 resulting in lack of response to all platelet transfusions, however, is a life-threatening complication for GT patients. Patients whose mutations prevent any αIIbβ3 expression at the platelet membrane are theoretically at the highest risk for developing anti- αIIbβ3 antibodies.31 Women seem to be more affected than men, although this risk may be due to the prevalence of transfusions for uterine bleeding.20,31 A relationship between number of past platelet transfusions and presence of platelet refractoriness in the GT population has not been established, however, and even residual platelets within red blood cell transfusions can trigger alloimmunization.31,124,125 When anti- αIIbβ3 antibodies are present, platelet transfusions in conjunction with antifibrinolytics have still been successful 71% of the time for non-surgical hemorrhages.106 However, rF7a has been shown to be effective in 91% of non-surgical bleeds, regardless of antibody status.106 rF7a has similarly been shown to be successful in treating 88% of surgical bleeds in antibody patients.122 Patients with antibodies have been shown to require more doses or rF7a than those without.106,122
Alloimmunization has been reported in up to 70% of pregnant women with GT.1,61 Pregnant mothers with GT who are immunized by fetal platelet antigens will produce antibodies that can later cross the placenta and induce life-threatening thrombocytopenia and hemorrhage in the fetus.31,96,97 Methods to lower antibody titre prenatally include plasma exchange, steroids, and intravenous immunoglobulin (IVIG),8,95,126,127 with IVIG dosing of 0.5–1g/kg per week associated with 97.3% success.95,128
Thrombosis Secondary to Recombinant Activated Factor VII
The rate of thromboses secondary to rF7a use in GT patients fortunately remains low. One literature review of rF7a use in GT patients found a thromboembolic event rate of 1.4% amongst 221 bleeding episodes.104 A review including data from the Glanzmann Thrombasthenia Registry found 5 cases of thromboembolism amongst 490 instances of rF7a use for nonsurgical bleeding and perioperative prophylaxis.129 Two of these cases occurred in patients over the age of 65 years, with one featuring additional risk factors of immobilization, surgery, and continuous rF7a infusion. Thromboembolic diagnoses reported in GT patients receiving rF7a within the literature include deep vein thrombosis, pulmonary embolism, ureteric obstruction, and intracardiac thrombi.105,130–132
Curative Therapy
Bone marrow transplantation has proven to be a curative option in several GT patients, including those with anti-platelet antibodies.31,133–139 In many cases, conditioning therapy has helped alleviate antibodies, which can otherwise threaten engraftment.50,117 Transplant has typically been performed in the pediatric population, with chronic graft versus host disease being a major morbidity.
Though still in the experimental stages, gene therapy has been explored as an option for treating GT. In 2011, Fang et al140 demonstrated increased αIIbβ3 expression in GT dog models who had been transfected with peripheral blood stem cells engineered to express human ITGA2B. Three years later, Sullivan et al141 generated induced pluripotent stem (iPS) cells from peripheral monocytes of 2 GT patients and transfected the iPS cells with αIIb cDNA at the AAVS1 locus, accompanied by a megakaryocyte-specific promoter. Thereafter, these patients exhibited αIIbβ3 platelet expression surpassing 50% and 70%.
Future Steps
The diagnosis and management of patients with GT continues to have many associated challenges. Diagnosis requires testing in specialized laboratories and further genetic testing, at least at this point, does little to help predict severity. However, by continuing to grow the Glanzmann Thrombasthenia Registry, our understanding of GT’s various mutations will become more developed. Currently, the mainstay of treatment remains supportive. However, as recognition of anti-platelet antibodies becomes more prevalent, the demand for curative options will certainly increase. To meet this demand, bone marrow transplant regimens will need to become more standardized for this population and recognized as a treatment option early in life. Moreover, the further development of gene therapy technology for GT patients will offer an alternative option to cure this otherwise lifelong disease.
Disclosure
The authors reported no conflicts of interest for this work.
References
1. Botero JP, Lee K, Branchford BR, et al. Glanzmann thrombasthenia: genetic basis and clinical correlates. Haematologica. 2020;105(4):888–894. doi:10.3324/haematol.2018.214239
2. Krause KA, Graham BC. Glanzmann Thrombasthenia. 2020. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2021. PMID: 30855858.
3. Nurden AT, Fiore M, Nurden P, Pillois X. Glanzmann thrombasthenia: a review of ITGA2B and ITGB3 defects with emphasis on variants, phenotypic variability, and mouse models. Blood. 2011;118(23):5996–6005. doi:10.1182/blood-2011-07-365635
4. George JN, Caen JP, Nurden AT. Glanzmann’s thrombasthenia: the spectrum of clinical disease. Blood. 1990;75(7):1383–1395. doi:10.1182/blood.V75.7.1383.1383
5. Lee A, Poon MC. Inherited platelet functional disorders: general principles and practical aspects of management. Transfus Apher Sci. 2018;57(4):494–501. doi:10.1016/j.transci.2018.07.010
6. Borhany M, Fatima H, Naz A, Patel H, Shamsi T. Pattern of bleeding and response to therapy in Glanzmann thrombasthenia. Haemophilia. 2012;18(6):e423–5. doi:10.1111/hae.12017
7. Glanzmann E. Hereditare hämorrhagische thrombasthenie. Ein Beitrag zur Pathologie der Blutplättchen. Jb Kinderheilk. 1918;88:133. German.
8. Bellucci S, Caen J. Molecular basis of Glanzmann’s thrombasthenia and current strategies in treatment. Blood Rev. 2002;16(3):193–202. doi:10.1016/s0268-960x(02)00030-9
9. Coller B. A brief history of ideas about platelets in health and disease. Platelets. 2007;170. doi:10.1016/B978-0-12-387837-3.00069-9
10. Stevens RF, Meyer S. Fanconi and Glanzmann: the men and their works. Br J Haematol. 2002;119(4):901–904. doi:10.1046/j.1365-2141.2002.03812.x
11. Caen J, Cousin C. [“In vivo” disorder of platelet adhesiveness in Willebrand’s disease and Glanzmann’s thrombasthenias. Trial interpretation]. Nouv Rev Fr Hematol. 1962;2:685–694. [Slovenian].
12. Phillips DR, Agin PP. Platelet membrane defects in Glanzmann’s thrombasthenia. Evidence for decreased amounts of two major glycoproteins. J Clin Invest. 1977;60(3):535–545. doi:10.1172/JCI108805
13. Kunicki TJ, Aster RH. Deletion of the platelet-specific alloantigen PlA1 from platelets in Glanzmann’s thrombasthenia. J Clin Invest. 1978;61(5):1225–1231. doi:10.1172/JCI109038
14. Hagen I, Solum NO. Further studies on the protein composition and surface structure of normal platelets and platelets from patients with Glanzmann’s thrombasthenia and Bernard-Soulier syndrome. Thromb Res. 1978;13(5):845–855. doi:10.1016/0049-3848(78)90189-5
15. Nurden AT, Caen JP. The different glycoprotein abnormalities in thrombasthenic and Bernard-Soulier platelets. Semin Hematol. 1979;16(3):234–250.
16. Nurden AT. Inherited abnormalities of platelets. Thromb Haemost. 1999;82(2):468–480. doi:10.1055/s-0037-1615867
17. Coller BS, Cheresh DA, Asch E, Seligsohn U. Platelet vitronectin receptor expression differentiates Iraqi-Jewish from Arab patients with Glanzmann thrombasthenia in Israel. Blood. 1991;77(1):75–83. doi:10.1182/blood.V77.1.75.75
18. Nurden P, Poujol C, Nurden AT. The evolution of megakaryocytes to platelets. Baillieres Clin Haematol. 1997;10(1):1–27. doi:10.1016/s0950-3536(97)80048-0
19. Mitchell WB, Li J, French DL, Coller BS. alphaIIbbeta3 biogenesis is controlled by engagement of alphaIIb in the calnexin cycle via the N15-linked glycan. Blood. 2006;107(7):2713–2719. doi:10.1182/blood-2005-07-2990
20. Nurden AT, Pillois X, Fiore M, et al. Expanding the mutation spectrum affecting αIIbβ3 integrin in Glanzmann thrombasthenia: screening of the ITGA2B and ITGB3 genes in a large international cohort. Hum Mutat. 2015;36(5):548–561. doi:10.1002/humu.22776
21. Carrell NA, Fitzgerald LA, Steiner B, Erickson HP, Phillips DR. Structure of human platelet membrane glycoproteins IIb and IIIa as determined by electron microscopy. J Biol Chem. 1985;260(3):1743–1749. doi:10.1016/S0021-9258(18)89656-9
22. González-Manchón C, Fernández-Pinel M, Arias-Salgado EG, et al. Molecular genetic analysis of a compound heterozygote for the glycoprotein (GP) IIb gene associated with Glanzmann’s thrombasthenia: disruption of the 674–687 disulfide bridge in GPIIb prevents surface exposure of GPIIb-IIIa complexes. Blood. 1999;93(3):866–875. doi:10.1182/blood.V93.3.866
23. Yang J, Ma YQ, Page RC, Misra S, Plow EF, Qin J. Structure of an integrin alphaIIb beta3 transmembrane-cytoplasmic heterocomplex provides insight into integrin activation. Proc Natl Acad Sci U S A. 2009;106(42):17729–17734. doi:10.1073/pnas.0909589106
24. Shattil SJ, Newman PJ. Integrins: dynamic scaffolds for adhesion and signaling in platelets. Blood. 2004;104(6):1606–1615. doi:10.1182/blood-2004-04-1257
25. Coller BS, Shattil SJ. The GPIIb/IIIa (integrin alphaIIbbeta3) odyssey: a technology-driven saga of a receptor with twists, turns, and even a bend. Blood. 2008;112(8):3011–3025. doi:10.1182/blood-2008-06-077891
26. Armstrong PC, Peter K. GPIIb/IIIa inhibitors: from bench to bedside and back to bench again. Thromb Haemost. 2012;107(5):808–814. doi:10.1160/TH11-10-0727
27. Zhu J, Negri A, Provasi D, Filizola M, Coller BS, Springer TA. Closed headpiece of integrin αIIbβ3 and its complex with an αIIbβ3-specific antagonist that does not induce opening. Blood. 2010;116(23):5050–5059. doi:10.1182/blood-2010-04-281154
28. Kieffer N, Phillips DR. Platelet membrane glycoproteins: functions in cellular interactions. Annu Rev Cell Biol. 1990;6:329–357. doi:10.1146/annurev.cb.06.110190.001553
29. Farrell DH, Thiagarajan P. Binding of recombinant fibrinogen mutants to platelets. J Biol Chem. 1994;269(1):226–231. doi:10.1016/S0021-9258(17)42338-6
30. Du X, Ginsberg MH. Integrin αIIbβ3 and platelet function. Thromb Haemost. 1997;78(1):96–100. doi:10.1055/s-0038-1657508
31. Fiore M, d’Oiron R, Pillois X, Alessi MC. Anti-α. Br J Haematol. 2018;181(2):173–182. doi:10.1111/bjh.15087
32. Gemmell CH, Sefton MV, Yeo EL. Platelet-derived microparticle formation involves glycoprotein IIb-IIIa. Inhibition by RGDS and a Glanzmann’s thrombasthenia defect. J Biol Chem. 1993;268(20):14586–14589. doi:10.1016/S0021-9258(18)82371-7
33. Reverter JC, Béguin S, Kessels H, Kumar R, Hemker HC, Coller BS. Inhibition of platelet-mediated, tissue factor-induced thrombin generation by the mouse/human chimeric 7E3 antibody. Potential implications for the effect of c7E3 Fab treatment on acute thrombosis and “clinical restenosis”. J Clin Invest. 1996;98(3):863–874. doi:10.1172/JCI118859
34. Byzova TV, Plow EF. Networking in the hemostatic system. Integrin alphaiibbeta3 binds prothrombin and influences its activation. J Biol Chem. 1997;272(43):27183–27188. doi:10.1074/jbc.272.43.27183
35. Solh T, Botsford A, Solh M. Glanzmann’s thrombasthenia: pathogenesis, diagnosis, and current and emerging treatment options. J Blood Med. 2015;6:219–227. doi:10.2147/JBM.S71319
36. Fiore M, Nurden AT, Nurden P, Seligsohn U. Clinical utility gene card for: Glanzmann thrombasthenia. Eur J Hum Genet. 2012;20(10). doi:10.1038/ejhg.2012.151
37. Ward CM, Kestin AS, Newman PJ. A Leu262Pro mutation in the integrin beta(3) subunit results in an alpha(IIb)-beta(3) complex that binds fibrin but not fibrinogen. Blood. 2000;96(1):161–169. doi:10.1182/blood.V96.1.161
38. Ruan J, Peyruchaud O, Alberio L, et al. Double heterozygosity of the GPIIb gene in a Swiss patient with Glanzmann’s thrombasthenia. Br J Haematol. 1998;102(4):918–925. doi:10.1046/j.1365-2141.1998.00852.x
39. French DL. The molecular genetics of Glanzmann’s thrombasthenia. Platelets. 1998;9(1):5–20. doi:10.1080/09537109876951
40. Schlegel N, Gayet O, Morel-Kopp MC, et al. The molecular genetic basis of Glanzmann’s thrombasthenia in a gypsy population in France: identification of a new mutation on the alpha IIb gene. Blood. 1995;86(3):977–982. doi:10.1182/blood.V86.3.977.977
41. Vinciguerra C, Bordet JC, Beaune G, Grenier C, Dechavanne M, Négrier C. Description of 10 new mutations in platelet glycoprotein IIb (alphaIIb) and glycoprotein IIIa (beta3) genes. Platelets. 2001;12(8):486–495. doi:10.1080/095371001317126383
42. Fullard J, Murphy R, O’Neill S, Moran N, Ottridge B, Fitzgerald DJ. A Val193Met mutation in GPIIIa results in a GPIIb/IIIa receptor with a constitutively high affinity for a small ligand. Br J Haematol. 2001;115(1):131–139. doi:10.1046/j.1365-2141.2001.03075.x
43. Tadokoro S, Tomiyama Y, Honda S, et al. Missense mutations in the β3 subunit have a different impact on the expression and function between αIIbβ3 and αvβ3. Blood. 2002;99(3):931–938. doi:10.1182/blood.v99.3.931
44. Loftus JC, O’Toole TE, Plow EF, Glass A, Frelinger AL, Ginsberg MH. A beta 3 integrin mutation abolishes ligand binding and alters divalent cation-dependent conformation. Science. 1990;249(4971):915–918. doi:10.1126/science.2392682
45. Bajt ML, Ginsberg MH, Frelinger AL, Berndt MC, Loftus JC. A spontaneous mutation of integrin alpha IIb beta 3 (platelet glycoprotein IIb-IIIa) helps define a ligand binding site. J Biol Chem. 1992;267(6):3789–3794. doi:10.1016/S0021-9258(19)50595-6
46. Chen YP, Djaffar I, Pidard D, et al. Ser-752–>Pro mutation in the cytoplasmic domain of integrin beta 3 subunit and defective activation of platelet integrin alpha IIb beta 3 (glycoprotein IIb-IIIa) in a variant of Glanzmann thrombasthenia. Proc Natl Acad Sci U S A. 1992;89(21):10169–10173. doi:10.1073/pnas.89.21.10169
47. Ganapule A, Jain P, Abubacker FN, et al. Surgical procedures in patients with Glanzmann’s thrombasthenia: case series and literature review. Blood Coagul Fibrinolysis. 2017;28(2):171–175. doi:10.1097/MBC.0000000000000524
48. Casati V, D’Angelo A, Barbato L, et al. Perioperative management of a heterozygous carrier of Glanzmann’s thrombasthenia submitted to coronary artery bypass grafting with cardiopulmonary bypass. Anesth Analg. 2006;103(2):309–311. doi:10.1213/01.ane.0000226087.11062.cd
49. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. doi:10.1038/gim.2015.30
50. Nurden AT. Acquired antibodies to αIIbβ3 in Glanzmann thrombasthenia: from transfusion and pregnancy to bone marrow transplants and beyond. Transfus Med Rev. 2018;32(3):155–164. doi:10.1016/j.tmrv.2018.05.002
51. Kashiwagi H, Kunishima S, Kiyomizu K, et al. Demonstration of novel gain-of-function mutations of αIIbβ3: association with macrothrombocytopenia and glanzmann thrombasthenia-like phenotype. Mol Genet Genomic Med. 2013;1(2):77–86. doi:10.1002/mgg3.9
52. Jayo A, Conde I, Lastres P, et al. L718P mutation in the membrane-proximal cytoplasmic tail of beta 3 promotes abnormal alpha IIb beta 3 clustering and lipid microdomain coalescence, and associates with a thrombasthenia-like phenotype. Haematologica. 2010;95(7):1158–1166. doi:10.3324/haematol.2009.018572
53. Gresele P, Falcinelli E, Giannini S, et al. Dominant inheritance of a novel integrin beta3 mutation associated with a hereditary macrothrombocytopenia and platelet dysfunction in two Italian families. Haematologica. 2009;94(5):663–669. doi:10.3324/haematol.2008.002246
54. Poncz M, Rifat S, Coller BS, et al. Glanzmann thrombasthenia secondary to a Gly273–>Asp mutation adjacent to the first calcium-binding domain of platelet glycoprotein IIb. J Clin Invest. 1994;93(1):172–179. doi:10.1172/JCI116942
55. Losonczy G, Rosenberg N, Boda Z, et al. Three novel mutations in the glycoprotein IIb gene in a patient with type II Glanzmann thrombasthenia. Haematologica. 2007;92(5):698–701. doi:10.3324/haematol.10847
56. Nurden P, Poujol C, Winckler J, Combrié R, Caen JP, Nurden AT. A Ser752–>Pro substitution in the cytoplasmic domain of beta3 in a Glanzmann thrombasthenia variant fails to prevent interactions between the alphaIIbbeta3 integrin and the platelet granule pool of fibrinogen. Br J Haematol. 2002;118(4):1143–1151. doi:10.1046/j.1365-2141.2002.03758.x
57. Sharma P, Kar R, Bhargava R, Ranjan R, Mishra PC, Saxena R. Acquired platelet dysfunction in 109 patients from a tertiary care referral hospital. Clin Appl Thromb Hemost. 2011;17(1):88–93. doi:10.1177/1076029610379397
58. Nurden AT. Acquired Glanzmann thrombasthenia: from antibodies to anti-platelet drugs. Blood Rev. 2019;36:10–22. doi:10.1016/j.blre.2019.03.004
59. Cines DB, Bussel JB, Liebman HA, Luning Prak ET. The ITP syndrome: pathogenic and clinical diversity. Blood. 2009;113(26):6511–6521. doi:10.1182/blood-2009-01-129155
60. Linge P, Fortin PR, Lood C, Bengtsson AA, Boilard E. The non-haemostatic role of platelets in systemic lupus erythematosus. Nat Rev Rheumatol. 2018;14(4):195–213. doi:10.1038/nrrheum.2018.38
61. Poon MC, Di Minno G, d’Oiron R, Zotz R. New insights into the treatment of Glanzmann thrombasthenia. Transfus Med Rev. 2016;30(2):92–99. doi:10.1016/j.tmrv.2016.01.001
62. Awidi AS. Increased incidence of Glanzmann’s thrombasthenia in Jordan as compared with Scandinavia. Scand J Haematol. 1983;30(3):218–222. doi:10.1111/j.1600-0609.1983.tb01477.x
63. Iqbal I, Farhan S, Ahmed N. Glanzmann thrombasthenia: a clinicopathological profile. J Coll Physicians Surg Pak. 2016;26(8):647–650.
64. Franchini M, Favaloro EJ, Lippi G. Glanzmann thrombasthenia: an update. Clin Chim Acta. 2010;411(1–2):1–6. doi:10.1016/j.cca.2009.10.016
65. Rosas RR, Kurth MH, Sidman J. Treatment and outcomes for epistaxis in children with Glanzmann’s thrombasthenia. Laryngoscope. 2010;120(12):2374–2377. doi:10.1002/lary.21034
66. Lowe GC, Lordkipanidzé M, Watson SP; group UGs. Utility of the ISTH bleeding assessment tool in predicting platelet defects in participants with suspected inherited platelet function disorders. J Thromb Haemost. 2013;11(9):1663–1668. doi:10.1111/jth.12332
67. Gresele P, Orsini S, Noris P, et al. Validation of the ISTH/SSC bleeding assessment tool for inherited platelet disorders: a communication from the platelet physiology SSC. J Thromb Haemost. 2020;18(3):732–739. doi:10.1111/jth.14683
68. Rognoni E, Ruppert R, Fässler R. The kindlin family: functions, signaling properties and implications for human disease. J Cell Sci. 2016;129(1):17–27. doi:10.1242/jcs.161190
69. Lillicrap D, James P. Von Willebrand Disease: An Introduction for the Primary Care Physician. Montreal, QC, Canada: World Federation of Hemophilia; 2009.
70. Michel M. Immune thrombocytopenic purpura: epidemiology and implications for patients. Eur J Haematol Suppl. 2009;71:3–7. doi:10.1111/j.1600-0609.2008.01206.x
71. Iorio A, Stonebraker JS, Chambost H, et al. Establishing the prevalence and prevalence at birth of hemophilia in males: a meta-analytic approach using national registries. Ann Intern Med. 2019;171(8):540–546. doi:10.7326/m19-1208
72. Gresele P, Harrison P, Bury L, et al. Diagnosis of suspected inherited platelet function disorders: results of a worldwide survey. J Thromb Haemost. 2014;12(9):1562–1569. doi:10.1111/jth.12650
73. Kürekçi AE, Atay AA, Sarící SU, Zeybek C, Köseoğlu V, Ozcan O. Effect of iron therapy on the whole blood platelet aggregation in infants with iron deficiency anemia. Thromb Res. 2000;97(5):281–285. doi:10.1016/s0049-3848(99)00150-4
74. Akay OM, Akin E, Mutlu FS, Gulbas Z. Effect of iron therapy on platelet function among iron-deficient women with unexplained menorrhagia. Pathophysiol Haemost Thromb. 2008;36(2):80–83. doi:10.1159/000173726
75. Moenen F, Vries MJA, Nelemans PJ, et al. Screening for platelet function disorders with multiplate and platelet function analyzer. Platelets. 2019;30(1):81–87. doi:10.1080/09537104.2017.1371290
76. McCabe White M, Jennings LK. Platelet Protocols: Research and Clinical Laboratory Procedures. Academic Press; 1999.
77. Bolton-Maggs PH, Chalmers EA, Collins PW, et al. A review of inherited platelet disorders with guidelines for their management on behalf of the UKHCDO. Br J Haematol. 2006;135(5):603–633. doi:10.1111/j.1365-2141.2006.06343.x
78. Favaloro EJ. Clinical utility of the PFA-100. Semin Thromb Hemost. 2008;34(8):709–733. doi:10.1055/s-0029-1145254
79. Mezzano D, Quiroga T, Pereira J. The level of laboratory testing required for diagnosis or exclusion of a platelet function disorder using platelet aggregation and secretion assays. Semin Thromb Hemost. 2009;35(2):242–254. doi:10.1055/s-0029-1220785
80. Cattaneo M, Lecchi A, Zighetti ML, Lussana F. Platelet aggregation studies: autologous platelet-poor plasma inhibits platelet aggregation when added to platelet-rich plasma to normalize platelet count. Haematologica. 2007;92(5):694–697. doi:10.3324/haematol.10999
81. Dacie SJV, Lewis SM. Practical Haematology.
82. Cattaneo M. Inherited platelet-based bleeding disorders. J Thromb Haemost. 2003;1(7):1628–1636. doi:10.1046/j.1538-7836.2003.00266.x
83. Philp R. Methods of Testing Proposed Anti Thrombotic Drugs. CRC Press; 1981.
84. Mutreja D, Sharma RK, Purohit A, Aggarwal M, Saxena R. Evaluation of platelet surface glycoproteins in patients with Glanzmann thrombasthenia: association with bleeding symptoms. Indian J Med Res. 2017;145(5):629–634. doi:10.4103/ijmr.IJMR_718_14
85. Giannini S, Mezzasoma AM, Guglielmini G, Rossi R, Falcinelli E, Gresele P. A new case of acquired Glanzmann’s thrombasthenia: diagnostic value of flow cytometry. Cytometry B Clin Cytom. 2008;74(3):194–199. doi:10.1002/cyto.b.20396
86. Owaidah T, Saleh M, Baz B, et al. Molecular yield of targeted sequencing for Glanzmann thrombasthenia patients. NPJ Genom Med. 2019;4:4. doi:10.1038/s41525-019-0079-6
87. Bastida JM, Lozano ML, Benito R, et al. Introducing high-throughput sequencing into mainstream genetic diagnosis practice in inherited platelet disorders. Haematologica. 2018;103(1):148–162. doi:10.3324/haematol.2017.171132
88. Downes K, Megy K, Duarte D, et al. Diagnostic high-throughput sequencing of 2396 patients with bleeding, thrombotic, and platelet disorders. Blood. 2019;134(23):2082–2091. doi:10.1182/blood.2018891192
89. Simeoni I, Stephens JC, Hu F, et al. A high-throughput sequencing test for diagnosing inherited bleeding, thrombotic, and platelet disorders. Blood. 2016;127(23):2791–2803. doi:10.1182/blood-2015-12-688267
90. Amesse C, Baillargeon L, Bissonnette D, et al. Glanzmann Thrombasthenia: An Inherited Bleeding Disorder.
91. Mehta DN, Bhatia R. Dental considerations in the management of Glanzmann’s thrombasthenia. Int J Clin Pediatr Dent. 2010;3(1):51–56. doi:10.5005/jp-journals-10005-1054
92. Almeida AM, Khair K, Hann I, Liesner R. The use of recombinant factor VIIa in children with inherited platelet function disorders. Br J Haematol. 2003;121(3):477–481. doi:10.1046/j.1365-2141.2003.04286.x
93. Jayakrishnan TT, Limonnik V, Shah D, Mewawalla P. Glanzmann’s thrombasthenia: how listening to the patient is sometimes the simple key to good medicine! Case Rep Med. 2020;2020:4862987. doi:10.1155/2020/4862987
94. Kulkarni B, Ghosh K, Shetty S. Second trimester prenatal diagnosis in Glanzmann’s thrombasthenia. Haemophilia. 2016;22(2):e99–e100. doi:10.1111/hae.12865
95. Soni P, Mantri S, Prabhudesai A, Patil R, Shanmukhaiah C, Shetty S. Triple jeopardy: a case of Glanzmann’s thrombasthenia with anti-GPIIb-IIIa antibodies and HPA incompatibility resulting in stillbirth. Thromb Res. 2019;181:141–144. doi:10.1016/j.thromres.2019.07.022
96. Siddiq S, Clark A, Mumford A. A systematic review of the management and outcomes of pregnancy in Glanzmann thrombasthenia. Haemophilia. 2011;17(5):e858–69. doi:10.1111/j.1365-2516.2011.02516.x
97. Léticée N, Kaplan C, Lémery D. Pregnancy in mother with Glanzmann’s thrombasthenia and isoantibody against GPIIb-IIIa: is there a foetal risk? Eur J Obstet Gynecol Reprod Biol. 2005;121(2):139–142. doi:10.1016/j.ejogrb.2005.02.011
98. Forsyth AL, Zourikian N, Valentino LA, Rivard GE. The effect of cooling on coagulation and haemostasis: should “Ice” be part of treatment of acute haemarthrosis in haemophilia? Haemophilia. 2012;18(6):843–850. doi:10.1111/j.1365-2516.2012.02918.x
99. Forsyth AL, Zourikian N, Rivard GE, Valentino LA. An ‘ice age’ concept? The use of ice in the treatment of acute haemarthrosis in haemophilia. Haemophilia. 2013;19(6):e393–6. doi:10.1111/hae.12265
100. Levy JH, Koster A, Quinones QJ, Milling TJ, Key NS. Antifibrinolytic therapy and perioperative considerations. Anesthesiology. 2018;128(3):657–670. doi:10.1097/ALN.0000000000001997
101. Tengborn L, Blombäck M, Berntorp E. Tranexamic acid–an old drug still going strong and making a revival. Thromb Res. 2015;135(2):231–242. doi:10.1016/j.thromres.2014.11.012
102. Lombardo VT, Sottilotta G. Recombinant activated factor VII combined with desmopressin in preventing bleeding from dental extraction in a patient with Glanzmann’s thrombasthenia. Clin Appl Thromb Hemost. 2006;12(1):115–116. doi:10.1177/107602960601200120
103. Ghosh K, Kulkarni B, Shetty S, Nair S. Antiplatelet antibodies in cases of Glanzmann’s thrombasthenia with and without a history of multiple platelet transfusion. Indian J Hum Genet. 2009;15(1):23–27. doi:10.4103/0971-6866.50866
104. Rajpurkar M, Chitlur M, Recht M, Cooper DL. Use of recombinant activated factor VII in patients with Glanzmann’s thrombasthenia: a review of the literature. Haemophilia. 2014;20(4):464–471. doi:10.1111/hae.12473
105. Poon MC, D’Oiron R, Von Depka M, et al. Prophylactic and therapeutic recombinant factor VIIa administration to patients with Glanzmann’s thrombasthenia: results of an international survey. J Thromb Haemost. 2004;2(7):1096–1103. doi:10.1111/j.1538-7836.2004.00767.x
106. Di Minno G, Zotz RB, d’Oiron R, et al. The international, prospective Glanzmann thrombasthenia registry: treatment modalities and outcomes of non-surgical bleeding episodes in patients with Glanzmann thrombasthenia. Haematologica. 2015;100(8):1031–1037. doi:10.3324/haematol.2014.121475
107. Dubick MA, Dorfman R, Klemcke HG, et al. Stability of activated recombinant human factor VII (rFVIIa) after 28 days storage at 24° C and 43° C. Blood. 2008;112:4074. doi:10.1182/blood.V112.11.4074.4074
108. Lak M, Scharling B, Blemings A, et al. Evaluation of rFVIIa (NovoSeven) in Glanzmann patients with thromboelastogram. Haemophilia. 2008;14(1):103–110. doi:10.1111/j.1365-2516.2007.01592.x
109. Lisman T, Moschatsis S, Adelmeijer J, Nieuwenhuis HK, De Groot PG. Recombinant factor VIIa enhances deposition of platelets with congenital or acquired alpha IIb beta 3 deficiency to endothelial cell matrix and collagen under conditions of flow via tissue factor-independent thrombin generation. Blood. 2003;101(5):1864–1870. doi:10.1182/blood-2002-09-2761
110. Poon MC. Clinical use of recombinant human activated factor VII (rFVIIa) in the prevention and treatment of bleeding episodes in patients with Glanzmann’s thrombasthenia. Vasc Health Risk Manag. 2007;3(5):655–664.
111. Birschmann I, Klamroth R, Eichler H, Schenk J, Kirchmaier CM, Halimeh S. Results of the WIRK prospective, non-interventional observational study of recombinant activated factor VII (rFVIIa) in patients with congenital haemophilia with inhibitors and other bleeding disorders. Haemophilia. 2013;19(5):679–685. doi:10.1111/hae.12156
112. Rakocz M, Lavie G, Martinowitz U. Glanzmann’s thrombasthenia: the use of autologous fibrin glue in tooth extractions. ASDC J Dent Child. 1995;62(2):129–131.
113. Franco R, Miranda M, Di Renzo L, De Lorenzo A, Barlattani A, Bollero P. Glanzmann’s thrombastenia: the role of tranexamic acid in oral surgery. Case Rep Dent. 2018;2018:9370212. doi:10.1155/2018/9370212
114. Nurden AT. Glanzmann thrombasthenia. Orphanet J Rare Dis. 2006;1:10. doi:10.1186/1750-1172-1-10
115. Herman MC, Mak N, Geomini PM, et al. Is the Pictorial Blood Loss Assessment Chart (PBAC) score associated with treatment outcome after endometrial ablation for heavy menstrual bleeding? A cohort study. BJOG. 2017;124(2):277–282. doi:10.1111/1471-0528.14434
116. Higham JM, O’Brien PM, Shaw RW. Assessment of menstrual blood loss using a pictorial chart. Br J Obstet Gynaecol. 1990;97(8):734–739. doi:10.1111/j.1471-0528.1990.tb16249.x
117. Nurden AT, Pillois X, Wilcox DA. Glanzmann thrombasthenia: state of the art and future directions. Semin Thromb Hemost. 2013;39(6):642–655. doi:10.1055/s-0033-1353393
118. Sherer DM, Lerner R. Glanzmann’s thrombasthenia in pregnancy: a case and review of the literature. Am J Perinatol. 1999;16(6):297–301. doi:10.1055/s-2007-993875
119. Boval B, Bellucci S, Boyer-Neumann C. Glanzmann’s thrombasthenia and pregnancy: clinical observations and management of four affected women Congress of the International Society of Thrombosis and Hemostasis. Thromb Haemost. 2001;P1154.
120. Punt MC, Schuitema PCE, Bloemenkamp KWM, Kremer Hovinga ICL, van Galen KPM. Menstrual and obstetrical bleeding in women with inherited platelet receptor defects-A systematic review. Haemophilia. 2020;26(2):216–227. doi:10.1111/hae.13927
121. Orsini S, Noris P, Bury L, et al. Bleeding risk of surgery and its prevention in patients with inherited platelet disorders. Haematologica. 2017;102(7):1192–1203. doi:10.3324/haematol.2016.160754
122. Poon MC, d’Oiron R, Zotz RB, et al. The international, prospective Glanzmann thrombasthenia registry: treatment and outcomes in surgical intervention. Haematologica. 2015;100(8):1038–1044. doi:10.3324/haematol.2014.121384
123. Nordisk N, Inc. NovoSeven® RT, coagulation factor VIIa (Recombinant) [package insert]. Plainsboro, NJ; 2019.
124. Santoro C, Rago A, Biondo F, et al. Prevalence of allo-immunization anti-HLA and anti-integrin αIIbβ3 in Glanzmann thromboasthenia patients. Haemophilia. 2010;16(5):805–812. doi:10.1111/j.1365-2516.2010.02230.x
125. Laurian Y, Tisseron-Maury B, Bibi Triki T, Kaplan C, Gaudelus J. Red blood cell transfusion in patients with type 1 Glanzmann’s thrombasthenia. J Thromb Haemost. 2005;3(10):2346–2347. doi:10.1111/j.1538-7836.2005.01579.x
126. Vivier M, Treisser A, Naett M, et al. [Glanzmann’s thrombasthenia and pregnancy. Contribution of plasma exchange before scheduled cesarean section]. J Gynecol Obstet Biol Reprod. 1989;18(4):507–513. [French].
127. Winkelhorst D, Murphy MF, Greinacher A, et al. Antenatal management in fetal and neonatal alloimmune thrombocytopenia: a systematic review. Blood. 2017;129(11):1538–1547. doi:10.1182/blood-2016-10-739656
128. Rayment R, Brunskill SJ, Soothill PW, Roberts DJ, Bussel JB, Murphy MF. Antenatal interventions for fetomaternal alloimmune thrombocytopenia. Cochrane Database Syst Rev. 2011;5:CD004226. doi:10.1002/14651858.CD004226.pub3
129. Poon MC. The Use of recombinant activated factor VII in patients with Glanzmann’s thrombasthenia. Thromb Haemost. 2021;121(3):332–340. doi:10.1055/s-0040-1718373
130. Browning L, Dogra S, Bourbonnais J The use of recombinant factor VIIa use in a patient with Glanzmann’s Thrombasthenia and diffuse alveolar hemorrhage.
131. Wertz D, Boveroux P, Péters P, Lenelle J, Franssen C. Surgical resection of a sphenoid wing meningioma in a patient with Glanzmann thrombasthenia. Acta Anaesthesiol Belg. 2011;62(2):83–86.
132. Phillips R, Richards M. Venous thrombosis in Glanzmann’s thrombasthenia. Haemophilia. 2007;13(6):758–759. doi:10.1111/j.1365-2516.2007.01555.x
133. Bellucci S, Damaj G, Boval B, et al. Bone marrow transplantation in severe Glanzmann’s thrombasthenia with antiplatelet alloimmunization. Bone Marrow Transplant. 2000;25(3):327–330. doi:10.1038/sj.bmt.1702139
134. Bellucci S, Devergie A, Gluckman E, et al. Complete correction of Glanzmann’s thrombasthenia by allogeneic bone-marrow transplantation. Br J Haematol. 1985;59(4):635–641. doi:10.1111/j.1365-2141.1985.tb07358.x
135. Flood VH, Johnson FL, Boshkov LK, et al. Sustained engraftment post bone marrow transplant despite anti-platelet antibodies in Glanzmann thrombasthenia. Pediatr Blood Cancer. 2005;45(7):971–975. doi:10.1002/pbc.20365
136. Ishaqi MK, El-Hayek M, Gassas A, et al. Allogeneic stem cell transplantation for Glanzmann thrombasthenia. Pediatr Blood Cancer. 2009;52(5):682–683. doi:10.1002/pbc.21888
137. Kitko CL, Levine JE, Matthews DC, Carpenter PA. Successful unrelated donor cord blood transplantation for Glanzmann’s thrombasthenia. Pediatr Transplant. 2011;15(3):e42–6. doi:10.1111/j.1399-3046.2009.01251.x
138. Wiegering V, Sauer K, Winkler B, Eyrich M, Schlegel PG. Indication for allogeneic stem cell transplantation in Glanzmann’s thrombasthenia. Hamostaseologie. 2013;33(4):305–312. doi:10.5482/HAMO-12-08-0014
139. Ramzi M, Dehghani M, Haghighat S, Nejad HH. Stem cell transplant in severe Glanzmann thrombasthenia in an adult patient. Exp Clin Transplant. 2016;14(6):688–690. doi:10.6002/ect.2014.0165
140. Fang J, Jensen ES, Boudreaux MK, et al. Platelet gene therapy improves hemostatic function for integrin alphaIIbbeta3-deficient dogs. Proc Natl Acad Sci U S A. 2011;108(23):9583–9588. doi:10.1073/pnas.1016394108
141. Sullivan SK, Mills JA, Koukouritaki SB, et al. High-level transgene expression in induced pluripotent stem cell-derived megakaryocytes: correction of Glanzmann thrombasthenia. Blood. 2014;123(5):753–757. doi:10.1182/blood-2013-10-530725
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.