")
Back to Journals » International Journal of Nanomedicine » Volume 9 » Issue 1
Ginkgo biloba: a natural reducing agent for the synthesis of cytocompatible graphene
Authors Gurunathan S , Han JW, Park JH, Eppakayala V, Kim J
Received 25 August 2013
Accepted for publication 11 November 2013
Published 7 January 2014 Volume 2014:9(1) Pages 363—377
DOI https://doi.org/10.2147/IJN.S53538
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Sangiliyandi Gurunathan, Jae Woong Han, Jung Hyun Park, Vasuki Eppakayala, Jin-Hoi Kim
Department of Animal Biotechnology, Konkuk University, Seoul, South Korea
Background: Graphene is a novel two-dimensional planar nanocomposite material consisting of rings of carbon atoms with a hexagonal lattice structure. Graphene exhibits unique physical, chemical, mechanical, electrical, elasticity, and cytocompatible properties that lead to many potential biomedical applications. Nevertheless, the water-insoluble property of graphene restricts its application in various aspects of biomedical fields. Therefore, the objective of this work was to find a novel biological approach for an efficient method to synthesize water-soluble and cytocompatible graphene using Ginkgo biloba extract (GbE) as a reducing and stabilizing agent. In addition, we investigated the biocompatibility effects of graphene in MDA-MB-231 human breast cancer cells.
Materials and methods: Synthesized graphene oxide (GO) and GbE-reduced GO (Gb-rGO) were characterized using various sequences of techniques: ultraviolet-visible (UV-vis) spectroscopy, Fourier-transform infrared spectroscopy (FTIR), dynamic light scattering (DLS), scanning electron microscopy (SEM), atomic force microscopy (AFM), and Raman spectroscopy. Biocompatibility of GO and Gb-rGO was assessed in human breast cancer cells using a series of assays, including cell viability, apoptosis, and alkaline phosphatase (ALP) activity.
Results: The successful synthesis of graphene was confirmed by UV-vis spectroscopy and FTIR. DLS analysis was performed to determine the average size of GO and Gb-rGO. X-ray diffraction studies confirmed the crystalline nature of graphene. SEM was used to investigate the surface morphologies of GO and Gb-rGO. AFM was employed to investigate the morphologies of prepared graphene and the height profile of GO and Gb-rGO. The formation of defects in Gb-rGO was confirmed by Raman spectroscopy. The biocompatibility of the prepared GO and Gb-rGO was investigated using a water-soluble tetrazolium 8 assay on human breast cancer cells. GO exhibited a dose-dependent toxicity, whereas Gb-rGO-treated cells showed significant biocompatibility and increased ALP activity compared to GO.
Conclusion: In this work, a nontoxic natural reducing agent of GbE was used to prepare soluble graphene. The as-prepared Gb-rGO showed significant biocompatibility with human cancer cells. This simple, cost-effective, and green procedure offers an alternative route for large-scale production of rGO, and could be used for various biomedical applications, such as tissue engineering, drug delivery, biosensing, and molecular imaging.
Keywords: alkaline phosphatase activity, atomic force microscopy, biocompatibility, cell viability, graphene, Fourier-transform infrared spectroscopy, scanning electron microscopy, Raman spectroscopy, UV-visible spectroscopy
Introduction
Graphene is a single-atom-thick, two-dimensional sheet of hexagonally arranged carbon atoms isolated from its three-dimensional parent material – graphite (Gt).1 Graphene exhibits attractive electronic, catalytic, mechanical, optical, and magnetic properties. These properties provide graphene with great potential in various applications ranging from energy storage to biomedical materials.2 Due to its remarkable physical, chemical, and mechanical properties and with a view to important applications, graphene has been emerging as one of the areas of research attracting great interest in academia and industry.3–5 Recently, graphene has attracted much attention in the scientific community for its numerous potential applications in biotechnology, including biosensing,6,7 disease diagnostics,8 antibacterial9–11 and antiviral materials,12 cancer targeting,13 photothermal therapy,14 drug delivery,15 and tissue engineering.16,17
Regarding biomedical applications of graphene materials, Lee et al18 suggested that graphene and graphene oxide (GO) sheets can be used as biocompatible, transferable, and implantable platforms for stem cell culture. Further, Lee et al18 reported that the noncovalent binding capability of graphene allows it to act as a preconcentration platform for osteogenic inducers, which accelerate mesenchymal stem cells growing on it toward the osteogenic lineage. Nayak et al19 reported that graphene-treated human mesenchymal stem cells show enhanced proliferation and differentiation. Chen et al20 reported that graphene and GO can support induced pluripotent stem cell culture and allow for spontaneous differentiation, and also suggested that the different surface properties of graphene and GO influence the performance of induced pluripotent stem cell culture and applications. The different physical and chemical properties could influence the biological responses of cells. Bacillus marisflavi-reduced GO exhibited toxicity to MCF-7 cells in a dose-dependent manner.21 Graphene-family nanomaterials can be either benevolent or toxic to cells, and biological response depends on layer number, lateral size, stiffness, hydrophobicity, surface functionalization, and dose.22
The success of graphene synthesis depends on safe and nontoxic methods, and large-scale production at low cost.23 In this regard, the approach that first led to the isolation of graphene using micromechanical cleavage method was a low-yield, low-throughput process, and therefore not likely to be industrially scalable.1 Simultaneously, several other research groups have developed various approaches for the synthesis of graphene and its derivatives, including exfoliation of Gt,24,25 flash reduction,26 hydrothermal dehydration,27 mechanical exfoliation,28 epitaxial growth,29 photocatalysis,30 and photodegradation.31 Graphene material has been produced physically, chemically, and mechanically for a long time, but recent developments have shown the critical role of microorganisms and biological reducing agents for the production of graphene. The use of biological systems in this area is rapidly developing due to their ease of handling, availability, and nontoxicity. Moreover, biological synthesis of graphene is an environmentally friendly method without the use of harsh, toxic, and expensive chemicals.
Generally, the most common synthesis of graphene has utilized chemical reducing agents, such as hydrazine, sodium citrate, and sodium borohydride to reduce GO,24,25 which can lead to the absorption of harsh chemicals on the surfaces of nanoparticles, raising the toxicity issue. Currently, the green synthesis of graphene is being investigated to decrease the use of toxic chemicals and increase biocompatibility in biotechnology and biomedical applications. For the development of green chemistry, several biological materials, such as wild carrot root,32 yeast,33 Escherichia coli,34,35 E. fergusoni,36 Pseudomonas aeruginosa,37 and the humanin peptide,38 have been reported as serving as both reducing and stabilizing agents for green synthesis of graphene. The organisms used in graphene synthesis vary from simple prokaryotic systems to complex eukaryotes. The biological synthesis of nanomaterials using plants has received more attention as a suitable alternative to chemical procedures and physical methods.39 Extracts from plants may act both as reducing and capping agents in nanoparticle synthesis.40,41 In general, plants contain many biomolecules, such as proteins, amino acids, polysaccharides, alkaloids, alcoholic compounds, polyphenols, enzymes, chelating agents, and vitamins. These molecules play an important role in the bioreduction, formation, and stabilization of metal nanoparticles.40,41 Therefore, plant extracts could be used as an alternative reducing and stabilizing agent for the synthesis of graphene. In general, extracts of Ginkgo biloba leaves, which contain flavonoid, glycosides, and terpenoids, have been used pharmaceutically in the People’s Republic of China, South Korea, and Japan, due to their nootropic properties. Here, we explored the possibility of using G. biloba leaf extract as an alternative reducing agent for the synthesis of graphene, and also evaluated the biocompatibility effect of as-prepared graphene in human breast cancer cells.
Materials and methods
Gt powder was purchased from Sigma-Aldrich (St Louis, MO, USA). NaOH, KMnO4, NaNO3, anhydrous ethanol, 98% H2SO4, 36% HCl, and 30% hydrogen peroxide (H2O2) aqueous solution were purchased from Sigma-Aldrich and used directly without further purification. All aqueous solutions were prepared with deionized water. All other chemicals were purchased from Sigma-Aldrich unless stated otherwise.
Synthesis of graphene oxide
The synthesis of GO was performed as described previously.37,38,42 Briefly, Gt powder was oxidized to Gt oxide (GtO) using KMnO4/H2SO4, and then the GtO was exfoliated into GO sheets by ultrasonication in water. In a typical synthesis process, natural Gt powder (2 g) was added to cooled (0°C) H2SO4 (350 mL v/v), and then KMnO4 (8 g w/v) and NaNO3 (1 g w/v) were added gradually while stirring. The mixture was transferred to a 40°C water bath and stirred for 60 minutes. Deionized water (250 mL) was slowly added, and the temperature was increased to 98°C. The mixture was maintained at 98°C for a further 30 minutes, and the reaction was terminated by the addition of deionized water (500 mL) and 40 mL of 30% H2O2 solution (v/v). The color of the mixture changed to brilliant yellow, indicating the oxidation of pristine Gt to GtO. The mixture was then filtered and washed with diluted HCl to remove metal ions. Finally, the product was washed repeatedly with distilled water until pH 7.0 was achieved. The sample of GtO was obtained after drying. To prepare GO, the synthesized GtO was redispersed in distilled water to create a yellow-brown dispersion, and the exfoliation of GtO to generate GO sheets was achieved by ultrasonication for 30 minutes. The resulting aqueous dispersion of the brown GO sheet was stable.
Preparation of leaf extract
Fresh G. biloba leaves were collected from Konkuk University campus and stored at 4°C until needed. Twenty grams of G. biloba leaves were washed thoroughly with double-distilled water, and were then sliced with a sharp stainless steel knife into fine pieces, about 1–5 cm2. The finely cut G. biloba leaves were mixed in 100 mL of sterile distilled water and then boiled for 5 minutes. After boiling, the mixture was filtered through Whatman grade 1 filter paper. The extract was used for synthesis of graphene. The extract was stored at 4°C until further use.
Reduction of graphene oxide by GbE
The reduction of GO was performed as described previously.38 Briefly, reduced GO was obtained from the reaction of a plant extract with GO. In the typical reduction experiment, 10 mL of G. biloba extract (GbE) was added to 90 mL of 0.5 mg/mL aqueous GO solution, and then the mixture was kept in a tightly sealed glass bottle and stirred at 30°C for 12 and 24 hours. Then, by using a magneto-stirrer heater, reduced GO suspension was stirred at 400 rpm at a temperature of 30°C for 30 minutes. A homogeneous GbE-reduced GO (Gb-rGO) suspension was obtained without aggregation. Then, functionalized Gb-rGO was filtered and washed with deionized water. Finally, a black Gb-rGO dispersion was obtained.
Characterization
Ultraviolet-visible (UV-vis) spectra were obtained using a WPA Biowave II (Biochrom, Cambridge, UK). The aqueous suspension of GO and Gb-rGO was used as the UV-vis samples, and the deionized water was used as the reference. The particle size of dispersions was measured by a Zetasizer Nano ZS90 (Malvern Instruments, Malvern, UK). X-ray diffraction (XRD) analyses were carried out on an X-ray diffractometer (D8 Discover; Bruker AXS, Madison, WI, USA). The high-resolution XRD patterns were measured at 3 Kw with Cu target using a scintillation counter; λ=1.5406 Å at 40 kV and 40 mA was recorded in the range of 2 θ=5°–80°. The dried powder was diluted with potassium bromide in a ratio of 1:100, and Fourier-transform infrared spectroscopy (FTIR) and spectrum GX spectrometry within the range of 500–4,000 cm−1 were recorded. A JSM-6700F (JEOL, Tokyo, Japan) semi-in-lens field-emission scanning electron microscope (SEM) operating at 10 kV was used to acquire SEM images. The solid samples were transferred to a carbon tape held in an SEM sample holder for analyses. The analyses of the samples were carried out at an average working distance of 6 mm. Raman spectra of GO and reduced GO were measured by an Alpha300 (Witec, Ulm, Germany) with a 532 nm laser. The calibration was initially made using an internal silicon reference at 500 cm−1, and gave a peak position resolution of less than 1 cm−1. The spectra were measured from 500 to 4,500 cm−1. All samples were deposited on glass slides in powder form without using any solvent. Surface images were measured using tapping-mode atomic force microscopy (AFM) (SPA 400; Seiko Instruments, Chiba, Japan) operating at room temperature. Height and phase images were recorded simultaneously using nanoprobe cantilevers (SI-DF20; Seiko Instruments).
Cell culture and exposure to GO and Gb-rGO
MDA-MB-231 human breast cancer cells were kindly provided by Professor Kyung Jin Lee, Department of Medicine, University of Ulsan College of Medicine, Asan Medical Center, Seoul. MDA-MB-231 breast cancer cell lines were grown adherently and maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) containing 10% fetal bovine serum (FBS), 50 U/mL penicillin, and 50 mg/ml streptomycin in a humidified atmosphere containing 5% CO2 at 37°C. All experiments were performed in six-well plates, unless stated otherwise. Cells were seeded onto the plates at a density of 1 × 104 cells per well and incubated for 24 hours prior to the experiments. The cells were washed with phosphate-buffered saline (PBS; pH 7.4) and incubated in fresh medium containing different concentrations of GO and Gb-rGO dissolved in water. GO or Gb-rGO suspensions were freshly prepared before the cells were exposed, and diluted to appropriate concentrations from 10 to 100 μg/mL−1 with the culture medium, then immediately applied to the cells. Cells not treated with GO or Gb-rGO served as controls in each experiment.
Cell-viability assay
A water-soluble tetrazolium (WST)-8 assay was performed as described previously in Liao et al.43 Typically, 1 × 104 cells were seeded in a 96-well plate and cultured in DMEM supplemented with 10% FBS at 37°C under 5% CO2. After 24 hours, the cells were washed twice with 100 μL of serum-free DMEM and further incubated with 100 μL of different concentrations of GO or Gb-rGO suspensions in serum-free DMEM. After 24 hours’ exposure, the cells were washed twice with serum-free DMEM, and 15 μL of WST-8 solution was added to each well containing 100 μL of serum-free DMEM. After 1 hour’s incubation at 37°C under 5% CO2, 80 μL of the mixture was transferred to another 96-well plate, because residual GO or Gb-rGO can affect the absorbance values at 450 nm. The absorbance of the mixture solutions was measured at 450 nm using a microplate reader. The cell-free control experiments were performed to see if the GO and Gb-rGO reacted directly with the WST-8 reagents. Typically, 100 μL of GO or Gb-rGO suspensions with different concentrations (10–100 μg/mL) were added to a 96-well plate and 10 μL of WST-8 reagent solution was added to each well; the mixture solution was incubated at 37°C under 5% CO2 for 1 hour. After incubation, the GO or Gb-rGO was centrifuged, and 50 μL of supernatant was transferred to another 96-well plate. Optical density was measured at 450 nm.
TUNEL assay
Cell morphology and apoptosis were evaluated by fluorescence microscopy following 4′,6-diamidino-2-phenylindole (DAPI) staining. MDA-MB-231 human breast cancer cells were grown in six-well plates and treated with 100 μg/mL of GO or Gb-rGO for 24 hours, then the cells were harvested with trypsin, washed with PBS, and fixed with 4% paraformaldehyde in PBS for 10 minutes at room temperature. Fixed cells were washed with PBS and stained with DAPI final concentration 0.5 μg/mL for 10 minutes at room temperature. The cells were washed three times with PBS, and images were captured using a fluorescence microscope. Apoptotic cells were identified by features characteristic of apoptosis. The tests were performed in triplicate.
Apoptotic deoxyribonucleic acid (DNA) fragmentation was also detected by using a DNA-fragmentation imaging kit (Roche, Basel, Switzerland) following the manufacturer’s instructions. Based on the terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick-end labeling (TUNEL) reaction, apoptosis assay was performed, fluorescence detection of cells with apoptotic DNA strand breaks was performed. To examine total cell numbers, nuclei were labeled simultaneously with DAPI. Merged images of both channels were shown using a fluorescence microscope (Olympus, Tokyo, Japan) at 100× magnification.
Alkaline phosphatase activity
MDA-MB-231 human breast cancer cells were cultured in 48-well culture dishes at a density of 5 × 103 cells per well and cultured for 5 days. Then, medium was replaced with treatment medium, which was DMEM containing 5% serum plus GO or Gb-rGO. After 5 days, the alkaline phosphatase (ALP) activity was measured according to the method described by the manufacturer’s instructions (QuantiChrom™ DALP-250 alkaline phosphatase assay kit; Gentaur, Aachen, Germany). The amount of released p-nitrophenol was measured at 405 nm in a 96-well microplate reader. Enzyme activity was evaluated as the amount of nitrophenol released through the enzymatic reaction, and absorbance was recorded using a microplate reader (model 680; Bio-Rad Laboratories, Hercules, CA, USA) at 405 nm. For normalization, the total protein content was measured using a bicinchoninic acid protein assay kit (Thermo Fisher Scientific, Waltham, MA, USA). Thus, the ALP activity was expressed and normalized by the total protein content (U/mg).
Results and discussion
Characterization of GO and Gb-rGO
Figure 1 shows the reduction of GO before and after treatment with the leaf extract of G. biloba. Briefly, the Gt powder was first oxidized into GtO using KMnO4/H2SO4, and then the GtO was exfoliated into GO sheets by ultrasonication in water.37,38,42 In a typical reduction experiment, 10 mL of GbE was added to the 90 mL of GO dispersion (0.5 mg/mL), and the mixture was stirred at 37°C for 24 hours. As shown in Figure 1, after reduction, a homogeneous yellow-brown GO dispersion was converted to black, which indicates the transition of GO to graphene. The color of GO and Gb-rGO appear different because of their distinct structural and physicochemical properties.44 The reduction of GO is also indicated from the color change of the solution before and after reaction (from yellow brown to dark), which could serve as a piece of evidence for the conversion of GO to graphene and also is due to the large number of hydrophilic functional groups, such as carboxyl, hydroxyl, and epoxy groups, on GO nanosheets.44 Further, the reduction of GO was investigated by UV-vis spectroscopy, as shown in Figure 2. GO and Gb-rGO samples were analyzed. GO exhibited peaks at 230 nm attributed to π–π* transitions of aromatic C=C bonds and a shoulder at about 300 nm (ascribed to π–π* transition of C=O bonds). Upon GO incubation with GbE, after stirring for 12 hours at 37°C, the absorption of C=C bonds at 230 nm was red-shifted to 260 nm. With increased time to 24 hours, the absorption of C=C bonds slightly shifted to 270 nm, and also the absorption value increased when compared to 12-hour reduction (Figure 2), indicating that the reduction of GO had been completed in 24 hours. We also observed that the reduction was completed within 24 hours, because the peak shifted no more when the reaction time was extended. The disappearance of the peak at 230 nm and the appearance of a new peak around 270 nm emerged in the UV spectra of the Gb-rGO, suggesting that sp2 carbon was restored and atoms were possibly rearranged within Gb-rGO.39,45
 | Figure 1 Digital photographs of aqueous dispersions of Ginkgo biloba (Gb) extracts (0.5 mg/mL) (A) of graphene oxide (GO) before (B) and after 12 hours (C) and 24 hours (D) reduction of GO using Gb extract. |
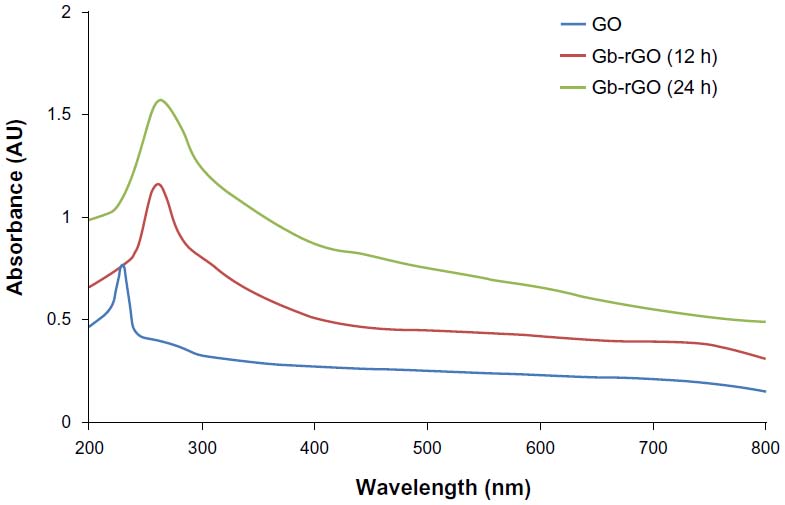 | Figure 2 Ultraviolet-visible absorption spectra of graphene oxide (GO) and Ginkgo biloba extract-reduced GO (Gb-rGO) suspension in water. |
Fernández-Merino et al23 suggested that the maximum red-shift value can be used as a yardstick to estimate the performance of the reducing agent. In this regard, Gb-rGO showed a similar final absorption peak position to the hydrazine,46 tea polyphenol-reduced graphene,45 and phenylhydrazine-reduced graphene.47 The results implied that GO could be reduced and the aromatic structure could be restored using GbE as a reducing agents. The data obtained from our studies consistent with previous reports used various reducing agents to reduce GO, such as vitamin C,23 thiourea,48 and leaf extracts of Colocasia esculenta and Mesua ferrea.39 Therefore, the reduction of GO with plant extract was not only a simple but also safe and environmentally friendly approach. It is a potent green manufacturing technology to produce graphene in large quantities.
XRD analysis of GO and Gb-rGO
To estimate the distance between two layers is an important parameter for the structural information of the graphene.49 The crystalline structure of the GO and Gb-rGO was further studied using XRD. The XRD pattern of GO exhibits a characteristic peak at 11.7° (Figure 3A), whereas, Gb-rGO shows a peak at 26.4° which is close to that of pure rGO.50 As shown in Figure 3A, the XRD pattern of exfoliated GO shows a peak at 2 θ=11.7°, corresponding to an interlayer with a d-spacing of 7.6 Å, indicating the presence of oxygen functional groups, which facilitate hydration and exfoliation of graphene sheets in the aqueous media. Upon oxidation of pristine Gt, the (002) peak shifts to the lower angle at 2 θ=11.7°. This indicates that the distance between the graphene sheets has increased due to the presence of oxygen-containing functional groups attached on both sides of the graphene sheet and the atomic-scale roughness arising from structural defects (sp3 bonding) generated on the originally atomically flat graphene sheet,37,51 the d-spacing of the GO is about 0.76 nm (2 θ ≈ 11.7°), which is significantly larger than the d(002) value of Gt d of around 0.34 nm (2 θ ≈ 26.2°)-thickness single-layer pristine graphene.37,49 In contrast to GO, Gb-rGO has a broad peak centered at 2 θ=26.4°, corresponding to d-spacing of 0.34 nm, which may be due to restacking of graphene layers (Figure 3B). The close d-spacing of Gb-rGO to pristine Gt and disappearance of the peak at 2 θ=11.7° indicates that the oxygen-containing group of GO has been efficiently removed. This shift in the interlayer spacing can be attributed to the reduction of the GO, where the reduction leads rGO to pack more tightly than the GO.37,52 Though there is a decrease in the interlayer spacing compared with GO, the basal spacing of rGO is higher than that of well-ordered Gt. The higher basal spacing may be due to the presence of residual oxygen functional groups, indicating incomplete reduction of GO.49 The large interlayer distance has been attributed to the formation of hydroxyl, epoxy, and carboxyl groups. Due to the removal of various functional groups in the process of significant reduction of GO using GbE as a reducing agent, the interlayer distance was diminished, the peak of the large interlayer distance disappeared completely, and a broad peak near 3.4 Å became visible (Figure 3B). This suggested that most of the functional groups were removed, and the reduction was significant.
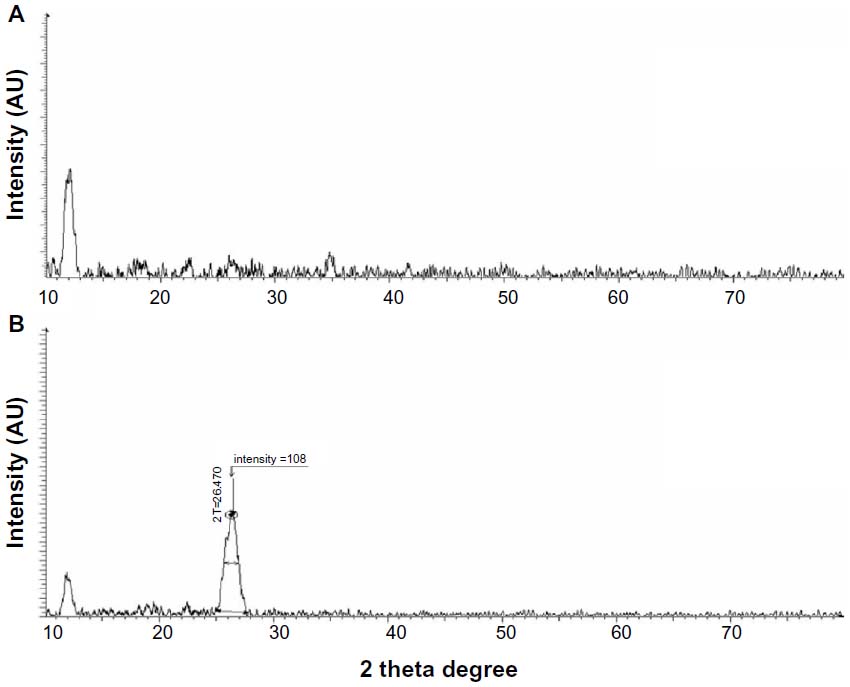 | Figure 3 X-ray diffraction pattern of graphene oxide (A) and Ginkgo biloba extract-reduced graphene oxide (B). |
FTIR analysis of GO and Gb-rGO
The FTIR spectrum of GO in Figure 4 (upper) shows the presence of O–H stretching at 3,440 cm−1, carboxyl C=O and C–O stretching at 1,740 cm−1 and 1,390 cm−1, respectively, aromatic C=C (1,620 cm−1), and alkoxy (1,076 cm−1) indicating the existence of oxygen containing functional groups on the GO.38,53 The substitution of hydroxyl groups on the GO surface by carboxyl groups was confirmed by the CH2-stretching vibration at 2,920 cm−1 (Figure 4, lower spectrum). As for the Gb-rGO (lower spectrum), the new band at 1,536 and 1,636 cm−1 corresponds to amide I and amide II, whereas the carboxyl C=O stretching of GO at 1,740 cm−1 disappeared, indicating that GbE samples could bind to the surface of GO. These observations confirmed that most oxygen functionalities in the GO were removed.54 The FTIR spectrum of the Gb-rGO indicates a significant reduction of the intensity of all oxygen-containing moieties, suggesting efficient conversion of GO to graphene by GbE.
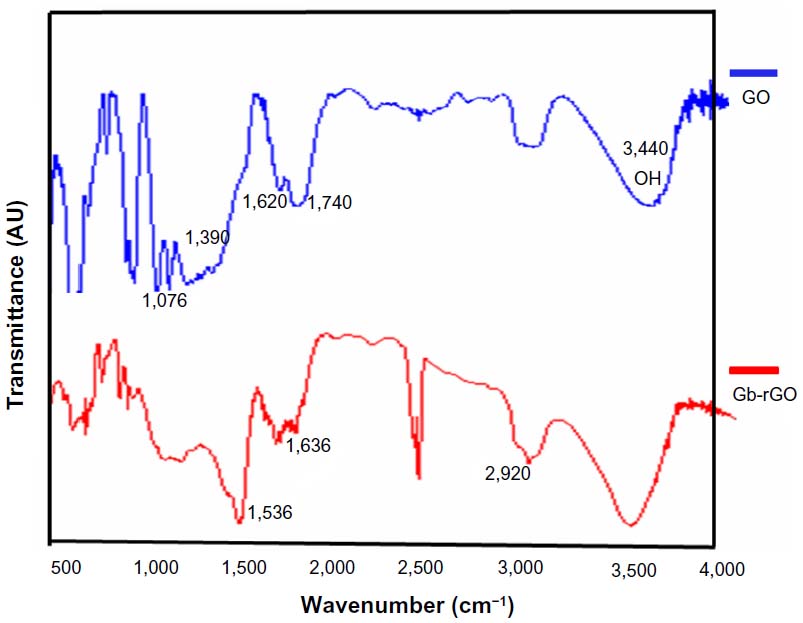 | Figure 4 Fourier-transform infrared spectra of graphene oxide (GO) and Ginkgo biloba extract-reduced GO (Gb-rGO). |
Size-distribution analysis of GO and Gb-rGO
Characterization of the dissolved materials is necessary for particular applications using valuable methods, such as dynamic light scattering (DLS).55 DLS theory is a well-established technique for measuring round-particle size over a size range from a few nanometers to a few microns.9 DLS was employed to measure the approximate size of GO and Gb-rGO samples. As shown in Figure 5A and B, it is clear that the particle sizes of the GO and Gb-rGO were 900±20 nm and 1400±40 nm, respectively. It can be concluded that the particle size of Gb-rGO was increased after reduction. Several studies suggested that the various sizes of reduced GO depend on the type of reducing agent used in the reduction process. Recently, Lammel et al56 observed that the average hydrodynamic diameters of GO and carboxyl graphene nanoplatelets were 385 and 1,110 nm, respectively. Stankovich et al25 reported that functionalized graphene nanoplates treated with isocyanate produce a DLS peak of 560±60 nm. Liu et al44 characterized aqueous dispersions of Gt, GtO, GO, and rGO using DLS, and the nominal effective diameters of the particles in Gt, GtO, GO, and rGO dispersions were 5,250, 4,420, 560, and 2,930 nm, respectively. Pseudomonas aeruginosa- and Bacillus marisflavi-reduced GO show 2,600 nm and 3,800 μm, respectively.37,38 It is reasonable to conclude that the reducing agent from the leaf extract plays an important role in increasing the size of the as-prepared Gb-rGO.
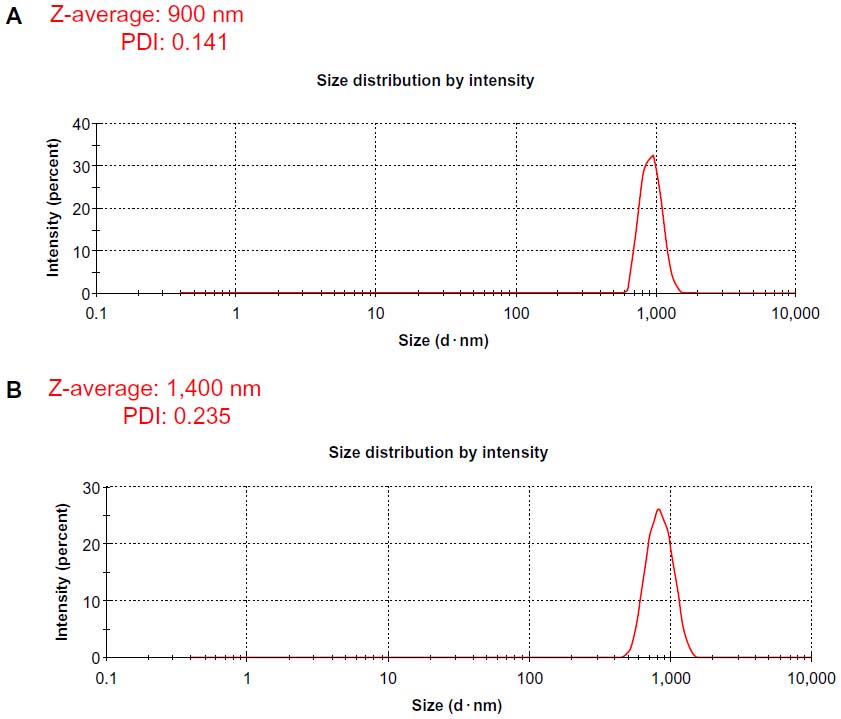 | Figure 5 Hydrodynamic size distribution of graphene oxide (A) and Ginkgo biloba extract-reduced graphene oxide (B). |
Raman spectra of GO and Gb-rGO
Raman spectroscopy is a powerful nondestructive tool to characterize carbonaceous materials, particularly to see the difference between ordered and disordered crystal structures of carbon.57 In our study, Raman spectra were used to analyze significant structural changes occurring during the reduction processing from GO to rGO. In Raman spectra, the typical features of carbon showed that the G line was around 1,582 cm−1 and the D line around 1,350 cm−1. The G line is usually assigned to the E2g phonon of C sp2 atoms, while the D line is a breathing mode of κ-point phonons of A1g symmetry.58,59 In the Raman spectrum of GO, the G band is broadened and shifted to 1,599 cm_1, whereas the D-band peak shows at 1,343 cm_1 (Figure 6A), which is prominent, indicating the reduction in size of the in-plane sp2 domains, possibly due to the extensive oxidation. The Raman spectrum of the Gb-rGO also contains both G and D bands at 1,603 and 1,355 cm_1, respectively (Figure 6B). Remarkably, the Raman spectrum of Gb-rGO shows a higher intensity ratio (2.0) than GO (1.6). Stankovich et al25 suggested a decrease in the average size of the sp2 domains upon reduction of the exfoliated GO. Variations in the relative intensities of the G peak and D peak in the Raman spectra of GO during reduction usually indicate a change in the electronic conjugation state. This change suggests an increase in the number of sp2 domains following reduction of GO.25 The results of the Raman spectroscopy analyses described here agree with those of previous studies that used the sulfur-containing amino acid l-cysteine to reduce GO to graphene,49 or those that developed a green and facile approach for the synthesis of graphene nanosheets using reducing sugars.14,28 Our findings are in significant agreement with earlier observations using various biological molecules such as wild carrot root,32 Baker’s yeast,33 and bacterial biomass.35,37 In comparison to other biological reducing agents, GbE can be obtained from easily available waste plant products.
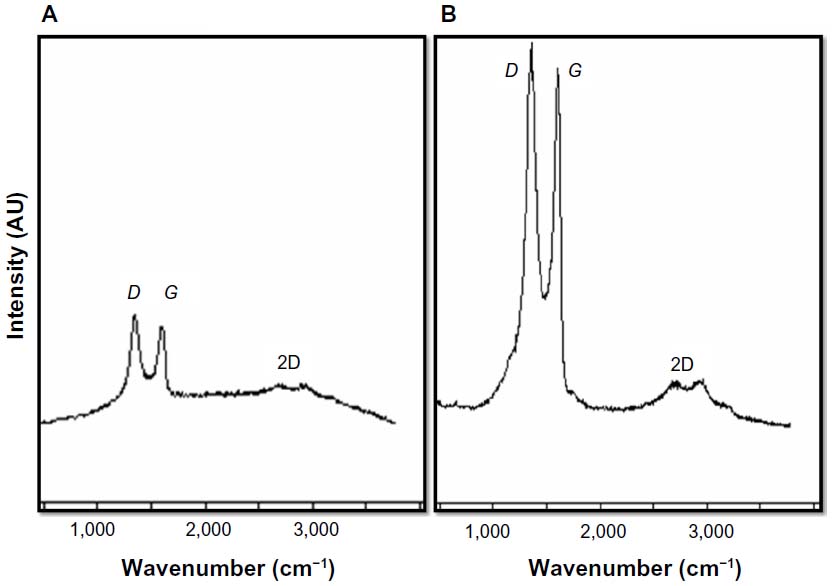 | Figure 6 Raman spectra of graphene oxide (A) and Ginkgo biloba extract-reduced graphene oxide (B). |
Surface morphology of GO and Gb-rGO
GO sheets were prepared from natural Gt flakes using the methods of Hummers and Offeman.60 These sheets had good solubility in water because of their numerous oxygen-containing functional groups.61 In general, Gt appears to be piled up in thick cakes, while GO is exfoliated into thin large flakes with wavy wrinkles.61 The SEM images revealed that the GO material consisted of individual sheets closely associated with each other, with a silky and leaf-like structure (Figure 7A), whereas Gb-rGO sheets exhibited thin layers of nanosheets and were mainly comprised of larger, wavy forms (Figure 7B). Functionalized graphene nanosheets are mainly wrinkled flakes that are similar to GO, but for graphene nanosheets functionalized with long chains and polymers, the surfaces are coarse and hairy, and the edges of the flakes are blurry.61 At higher concentrations, the surfaces of GO sheets have a soft-carpet-like morphology, which may be due to the presence of residual H2O molecules and hydroxyl or carboxyl groups attached to GO.62 Furthermore, individual GO sheets can be stacked layer by layer. The SEM images of Gb-rGO samples resembled transparent and rippled silk waves.
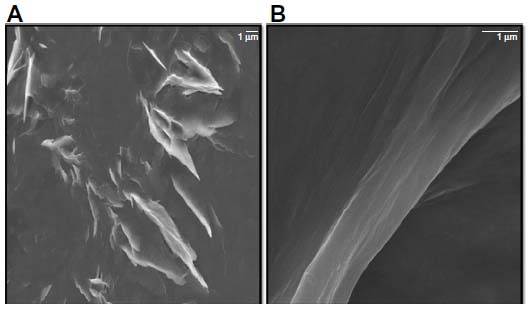 | Figure 7 Scanning electron microscopy images of graphene oxide (A) and Ginkgo biloba extract-reduced graphene oxide (B). |
Surface and height profile of GO and Gb-rGO by AFM
AFM is an important technique for evaluating surface morphology and properties.61 AFM can also be used to measure the surface and height profiles of individual graphene sheets. Therefore, AFM images are regarded as the most important indication of the successful functionalization of graphene.61,63–65 Figure 8A and B shows typical AFM images of GO and Gb-rGO dispersions in water, after their deposition on a freshly cleaned glass surface. The average thickness of as-prepared Gb-rGO, measured from the height profile of the AFM image, is about 57.48 nm. Compared with the well-exfoliated GO sheets with a thickness of about 36.68 nm, Gb-rGO was thicker than GO. Stankovich et al25 reported that GO sheets are “thicker” due to the presence of covalently bound oxygen and the displacement of the sp3 hybridized carbon atoms slightly above and below the original graphene plane. From our results, it is suggested that the reducing agent plays an important role in increasing the thickness of the as prepared Gb-rGO, through removing most of the oxygen-containing functional groups in GO. Su et al66 demonstrated that dispersed molecules with large aromatic structures and extra negative charges are noncovalently immobilized on the basal plane of graphene sheets via strong interactions.
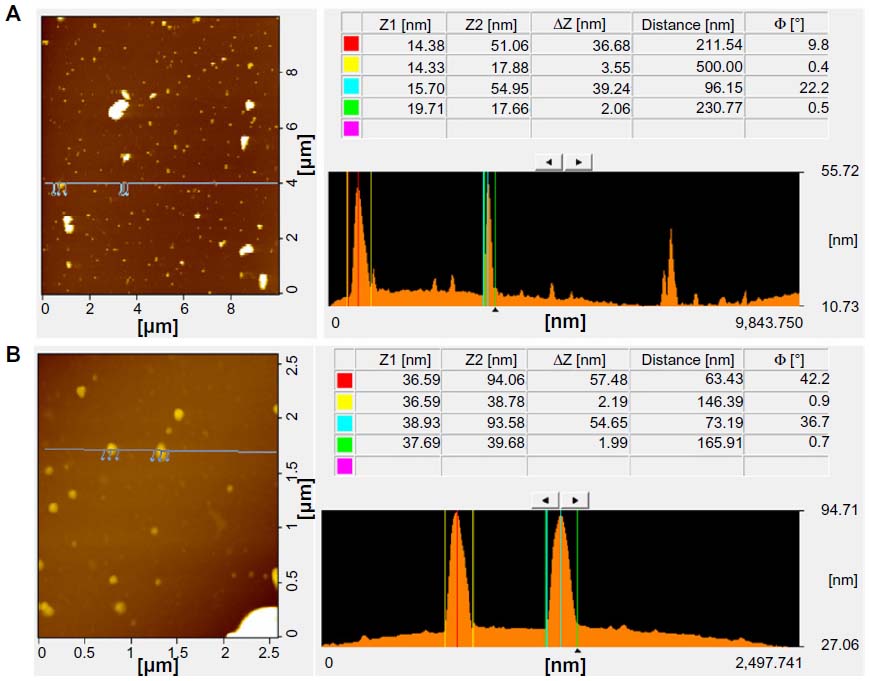 | Figure 8 Atomic force microscopy images of graphene oxide (A) and Ginkgo biloba extract-reduced graphene oxide (B). |
The effect of Gb-rGO on cell viability
Cell viability is one of the most important factors to evaluate the biocompatibility of graphene materials. To estimate the biocompatibility of GO and Gb-rGO, the cells were treated with various concentrations of GO and Gb-rGO (0–100 μg/mL) for 24 hours. At higher GO concentrations (50–100 μg/mL), viability loss was observed and was significantly different from control samples, whereas Gb-rGO-treated cells showed no significant inhibitory effect on cell viability (Figure 9). These results indicated that the viability of cells was not affected by Gb-rGO at any of the working concentrations. Chang et al67 undertook a comprehensive study on the toxicity of GO by examining various parameters, such as morphology, viability, mortality, and membrane integrity of A549 cells, and finally suggested that GO does not enter A549 cells and has no obvious cytotoxicity, but that GO can cause dose-dependent oxidative stress in cells and induce a slight loss of cell viability at higher concentrations. Chen et al68 investigated the biocompatibility of graphene films using a mouse fibroblast cell line (L-929), and the results showed that the cells adhered and proliferated on graphene film well and that graphene film is biocompatible. Agarwal et al16 reported that rGO is more biocompatible than single-wall carbon nanotubes (CNTs), using different cell lines: neuroendocrine PC12 cells, oligodendroglia, and osteoblasts. Cells were cultured in parallel on CNTs and rGO films with the same initial seeding density. The proliferation of PC12 cells on the two types of substrates was monitored over 5 days after seeding. The cells proliferated well on rGO, whereas their proliferation was largely inhibited on NTs. Biris et al69 demonstrated that osteoblast cells (MC3T3-E1) have a high ability to grow on graphene film. Zhang et al70 also reported that few-layer graphene increased intracellular generation of ROS and induced mitochondrial injury in neuronal cells after 4 and 24 hours at a dose of 10 μg/mL. Lu et al71 suggested that surface modification of graphene plays an important role in reducing the toxicity of rGO and carboxylated graphene.
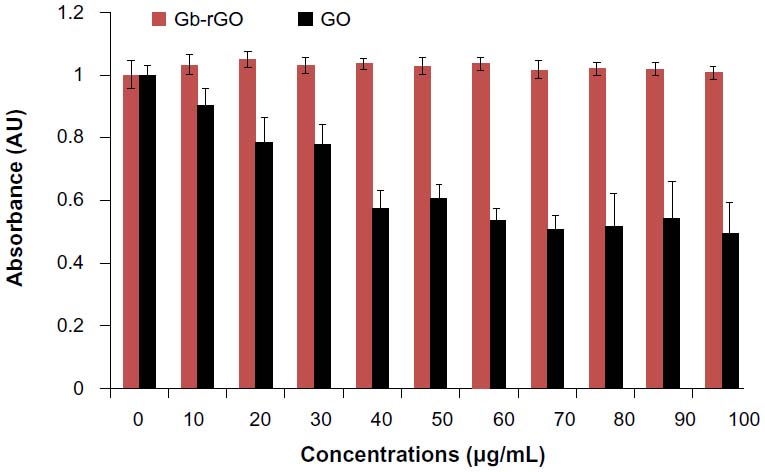 | Figure 9 Effect of graphene oxide (GO) and Ginkgo biloba extract-reduced GO (Gb-rGO) on cell viability of MDA-MB-231 human cancer cells. Cell viability of MDA-MB-231 cells was determined by water-soluble tetrazolium 8 assay after 24-hour exposure to different concentrations of GO or Gb-rGO. The results represent the means of three separate experiments, and error bars represent the standard error of the mean. GO-treated groups showed statistically significant differences from the control group by Student’s t-test (P<0.05). |
Cytocompatibility of Gb-rGO
Cytocompatibility depends on the dispersant used to stabilize the suspension, the type of materials, and the concentration.53 To determine cytocompatibility, the cells were treated with various concentrations of GO and Gb-rGO (100 μg/mL) for 24 hours, and cell death was examined using the TUNEL assay. TUNEL is a rapid method for identification and quantification of the apoptotic cell fraction in cultured cell preparations.72 Cell death is characterized by apoptotic morphology, chromatin condensation, membrane blebbing, internucleosome degradation of DNA, and apoptotic body formation.70,73 Following treatment with GO and Gb-rGO for 24 hours, the morphological features of apoptosis, condensation of chromatin, and fragmentation of the nucleus were examined. Use of the TUNEL assay further confirmed the induction of apoptosis by GO in cancer cells. As shown in Figure 10A, DNA fragmentation and rate of cell death was increased in cells treated with GO compared to control cells. Control and Gb-rGO-treated cells showed round and homogeneous nuclei, and no TUNEL-positive cells were observed, whereas cells treated with GO displayed condensed and fragmented nuclei in TUNEL-positive cells (Figure 10A). Interestingly, in the Gb-rGO-treated dish, all the cells were attached and appeared healthy. In contrast, GO-treated cells were fewer in number, and most were detached from the dish. The rate of apoptosis increased in GO-treated samples compared to Gb-rGO (Figure 10B). Taken together, all these results suggest that Gb-rGO could be a cytocompatible-friendly material. In a similar vein, Wojtoniszak et al53 reported that cells exposed to rGO/polyethylene glycol (PEG) suspension at concentrations between 3.125 μg/mL and 25 μg/mL showed relatively higher cytocompatibility than GO-treated cells. Yang et al13 reported on the pharmacokinetics and biodistribution of graphene functionalized with PEG, examining toxicity in mice, and the results suggested that graphene/PEG does not induce appreciable toxicity at an administered dose of 20 mg/kg for 3 months. Feng et al74 reported that at high concentrations, polyethyleneimine (PEI) graphene complexes showed slightly higher cell viability in human epithelial carcinoma compared to GO, possibly due to the improved stability of GO-PEI in the physiological environment. Sayes et al75 suggested that the toxicity of CNTs depends on the degree of functionalization, which is due to the carboxylation of CNTs making oxygen atoms abundant and decreasing toxicity.
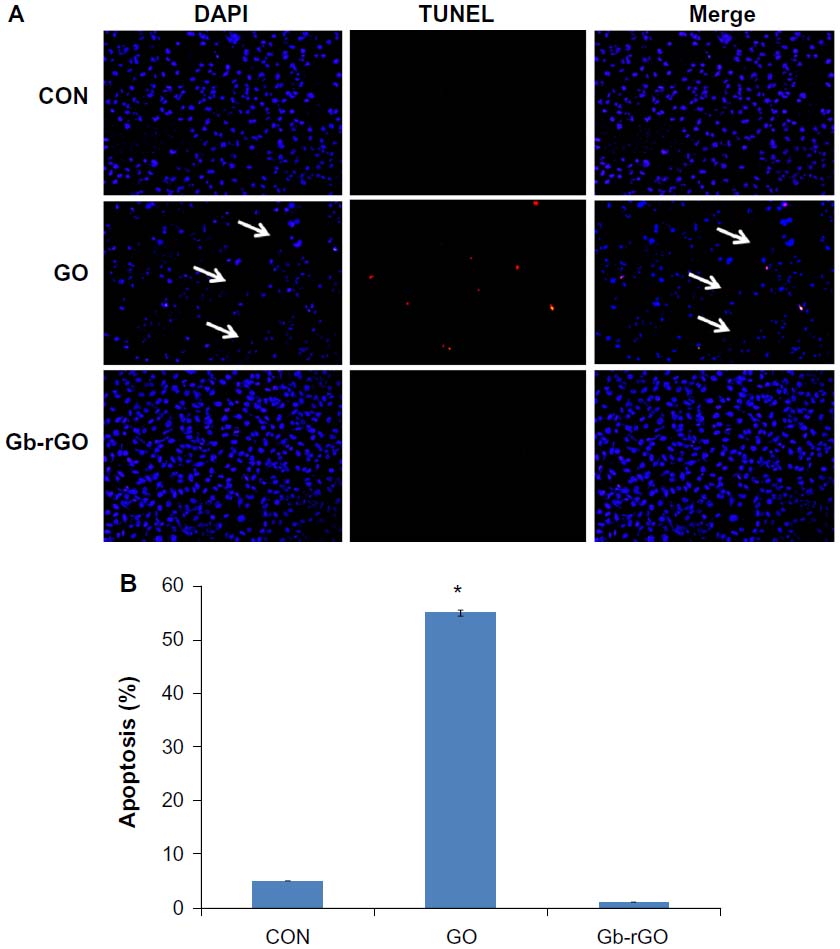 | Figure 10 (A and B) Detection of apoptosis by terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick-end labeling (TUNEL) assay. Fluorescent staining of MDA-MB-231 cells after 24 hours’ treatment with graphene oxide (GO) and Ginkgo biloba extract-reduced GO (Gb-rGO) (100 μg/mL) using TUNEL assay. Representative images are shown for apoptotic deoxyribonucleic acid fragmentation (red staining) and corresponding nuclei (blue staining). The arrows indicate the detachment of cells from the dish (A). MDA-MB-231 cells were treated with GO and Gb-rGO (100 μg/mL) for 24 hours. The percentage of death cells was quantified using the cell-death detection kit. The experiments were performed in triplicate; data shown represent means ± standard deviation of three independent experiments. *P<0.05 compared with untreated cells (B). |
Impact of Gb-rGO on ALP activity
Biocompatibility and nontoxicity are essential parameters for potential applications of any kind of nanomaterial in biomedical applications. ALP is a membrane enzyme that is involved in the mineralization of skeletal tissues, and the activity of this enzyme was used as a marker of osteoblastic differentiation.76 Biological responses of GO and Gb-rGO were preliminarily examined using the cell-viability assay, the results of which showed that Gb-rGO was neither enhanced nor proliferation of MDA-MB-231 human breast cancer cells inhibited. ALP results showed that cells treated with GO showed significantly lower ALP activity than Gb-rGO treated cells and untreated cells (Figure 11), which indicates that Gb-rGO was significantly biocompatible. Liu et al77 investigated the effect of pure hydroxyapatite (HA) and HA–rGO composites on ALP activity, and concluded that HA–rGO-treated cells showed higher ALP than untreated cells. Recently, Liu et al and Kalbacova et al77,78 reported that single-walled CNTs show better adherence and proliferation of human osteoblasts and mesenchymal stromal cells on them than silica. MG63 cells cultured with lower concentrations of nanographene platelets showed higher ALP expression when compared to those without nanographene platelets.79 Li et al80 demonstrated that MG-63 cells cultured with GO–HA and CS-GO–HA composites showed higher ALP activities than the control groups.
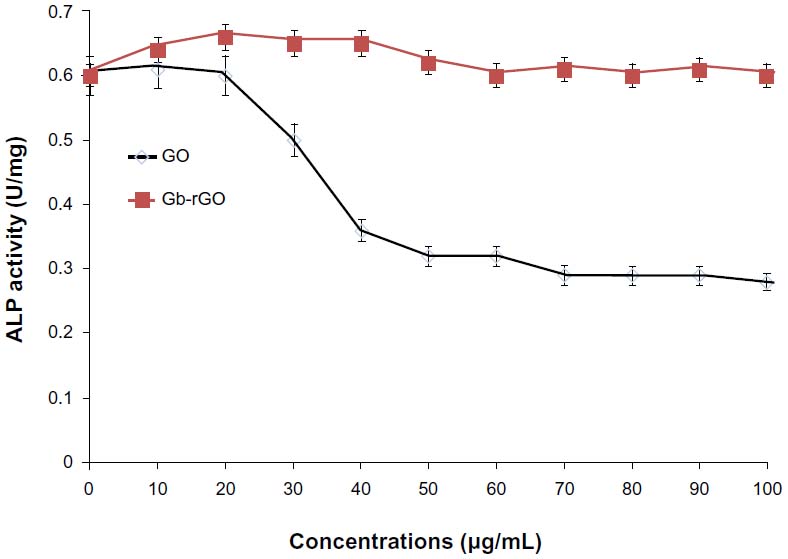 | Figure 11 The effect of graphene oxide (GO) and Ginkgo biloba extract-reduced GO (Gb-rGO) on alkaline phosphatase (ALP) activity. MDA-MB-231 cells were treated with various concentrations of GO and Gb-rGO for 5 days. The results represent the means of three separate experiments, and error bars represent the standard error of the mean. GO-treated groups showed statistically significant differences from the control group by Student’s t-test (P<0.05). |
Conclusion
Graphene nanomaterials are being used in increasingly large quantities for many applications in daily life, due to their novel characteristics, such as thermal, electrical, mechanical, and biological properties. Here, we developed an environmentally friendly approach for the reduction of GO by using GbE as a reducing and stabilizing agent. This is the first report about the use of G. biloba as bioreductant for the preparation of graphene. This biological reduction method is simple, cost-effective, and avoids the use of any toxic reagents, and also it offers another route to the production of graphene. Further, we assessed the cytocompatibility effect of biologically synthesized graphene in human cancer cells using various assays, such as cell viability, TUNEL, and ALP activity in human breast cancer cells. The results showed that the parent-material GO exhibited dose-dependent toxicity in cancer cells, whereas Gb-rGO showed significant cytocompatibility even at a high concentration of 100 μg/mL. Biologically synthesized graphene could be a favorable nanoplatform and the best candidate for biomedical applications, such as tissue engineering, imaging, and drug delivery.
Acknowledgments
This paper was supported by the SMART Research Professor Program of Konkuk University. Dr Sangiliyandi Gurunathan was supported by a Konkuk University SMART full-time professorship. This work was supported by Woo Jang-Choon project (PJ007849) and next generation of Biogreen 21 (PJ009107) from the Rural Development Administration (RDA), Republic of Korea.
Disclosure
The authors report no conflicts of interest in this work.
References
Novoselov KS, Geim AK, Morozov SV, et al. Electric field effect in atomically thin carbon films. Science. 2004;306(5696):666–669. | |
Wang Y, Shi Z, Yin J. Facile synthesis of soluble graphene via a green reduction of graphene oxide in tea solution and its biocomposites. ACS Appl Mater Interfaces. 2011;3(4):1127–1133. | |
Geim KA, Novoselov KS. The rise of graphene. Nat Mater. 2007;6(3):183–191. | |
Rao CNR, Sood AK, Subrahmanyam KS, Govindaraj A. Graphene: the new two-dimensional nanomaterial. Angew Chem Int Ed Engl. 2009;48(42):7752–7777. | |
Allen MJ, Tung VC, Kaner RB. Honeycomb carbon: a review of graphene. Chem Rev. 2010;110:132–145. | |
Shao Y, Wang J, Wu H, Liu J, Aksay IA, Lin Y. Graphene based electrochemical sensors and biosensors. Electroanalysis. 2010;22(10):1027–1036. | |
Akhavan O, Ghaderi E, Rahighi R. Toward single-DNA electrochemical biosensing by graphene nanowalls. ACS Nano. 2012;6(4):2904–2916. | |
Mohanty N, Berry V. Graphene-based single-bacterium resolution bio device and DNA transistor: interfacing graphene derivatives with nanoscale and microscale biocomponents. Nano Lett. 2000;8(12):4469–4476. | |
Hu W, Peng C, Luo W, et al. Graphene-based antibacterial paper. ACS Nano. 2010;4(7):4317–4323. | |
Akhavan O, Ghaderi E. Toxicity of graphene and graphene oxide nanowalls against bacteria. ACS Nano. 2010;4(10):5731–5736. | |
Gurunathan S, Han JW, Eppakayala V, Kim JH. Oxidative stress-mediated antibacterial activity of graphene oxide and reduced graphene oxide in Pseudomonas aeruginosa. Int J Nanomedicine. 2012;7:5901–5914. | |
Akhavan O, Choobtashani M, Ghaderi E. Protein degradation and RNA efflux of viruses photocatalyzed by graphene-tungsten oxide composite under visible light irradiation. J Phys Chem C. 2012;116(17):9653–9659. | |
Yang K, Zhang S, Zhang G, Sun X, Lee ST, Liu Z. Graphene in mice: ultrahigh in vivo tumor uptake and efficient photothermal therapy. Nano Lett. 2010;10(9):3318–3323. | |
Akhavan O, Ghaderi E, Aghayee S, Fereydooni Y, Talebi A. The use of a glucose-reduced graphene oxide suspension for photothermal cancer therapy. J Mater Chem. 2012;22:13773–13781. | |
Zhang X, Yin J, Peng C, et al. Distribution and biocompatibility studies of graphene oxide in mice after intravenous administration. Carbon. 2011;49(3):986–995. | |
Agarwal S, Zhou X, Ye F, et al. Interfacing live cells with nanocarbon substrates. Langmuir. 2010;26(4):2244–2247. | |
Park S, Mohanty N, Suk JW, et al. Biocompatible, robust free-standing paper composed of a TWEEN/graphene composite. Adv Mater. 2010; 22(15):1736–1740. | |
Lee WC, Lim CH, Shi H, et al. Origin of enhanced stem cell growth and differentiation on graphene and graphene oxide. ACS Nano. 2011;5(9):7334–7341. | |
Nayak TR, Andersen H, Makam VS, et al. Graphene for controlled and accelerated osteogenic differentiation of human mesenchymal stem cells. ACS Nano. 2011;5(6):4670–4678. | |
Chen GY, Hwang SM, Su HJ, et al. Defective antiviral responses of induced pluripotent stem cells to baculoviral vector transduction. J Virol. 2012;86(15):8041–8049. | |
Gurunathan S, Han JW, Eppakayala V, Kim JH. Green synthesis of graphene and its cytotoxic effects in human breast cancer cells. Int J Nanomedicine. 2013;8:1015–1027. | |
Sanchez VC, Jachak A, Hurt RH, Kane AB. Biological interactions of graphene-family nanomaterials: an interdisciplinary review. Chem Res Toxicol. 2011;25(1):15–34. | |
Fernández-Merino MJ, Guardia L, Paredes JL, et al. Vitamin C is an ideal substitute for hydrazine in the reduction of graphene oxide suspensions. J Phys Chem C. 2010;114(14):6426–6432. | |
Stankovich S, Piner RD, Chen X, Wu N, Nguyen ST, Ruoff RS. Aqueous dispersions of graphitic nanoplatelets via the reduction of exfoliated graphite oxide in the presence of poly(sodium 4-styrenesulfonate). J Mater Chem. 2006;16:155–158. | |
Stankovich S, Dikin DA, Piner RD, et al. Synthesis of graphene-based nanosheets via chemical reduction of exfoliated graphite oxide. Carbon. 2007;45(7):1558–1565. | |
Cote LJ, Silva RC, Huang J. Flash reduction and patterning of graphite oxide and its polymer composite. J Am Chem Soc. 2009;131(31):11027–11032. | |
Zhou M, Zhai Y, Dong S. Electrochemical sensing and biosensing platform based on chemically reduced graphene oxide. Anal Chem. 2009;81(14):5603–5613. | |
Zhu C, Guo S, Fang Y, Dong S. Reducing sugar: new functional molecules for the green synthesis of graphene nanosheets. ACS Nano. 2010;4(4):2429–2437. | |
Hass J, de Heer WA, Conrad EH. The growth and morphology of epitaxial multilayer graphene. J Phys Condens Matter. 2008;20(32):323202. | |
Akhavan O, Ghaderi EJ. Photocatalytic reduction of graphene oxide nanosheets on TiO2 thin film for photoinactivation of bacteria in solar light irradiation. J Phys Chem C. 2009;113(47):20214–20220. | |
Akhavan O, Abdolahad M, Esfandiar A, Mohatashamifar M. Photodegradation of graphene oxide sheets by TiO2 nanoparticles after a photocatalytic reduction. J Phys Chem C. 2010;114(30):12955–12959. | |
Kuila T, Bose B, Khanra P, Mishra AK, Kim NH, Lee JH. A green approach for the reduction of graphene oxide by wild carrot root. Carbon. 2012;50(3)914–921. | |
Khanra P, Kuila T, Kim NH, Bae SH, Yu DS, Lee JH. Simultaneous bio-functionalization and reduction of graphene oxide by baker’s yeast. Chem Eng J. 2012;183:526–533. | |
Akhavan O, Ghaderi E. Escherichia coli bacteria reduce graphene oxide to bactericidal graphene in a self-limiting manner. Carbon. 2012;50(5):1853–1860. | |
Gurunathan S, Han JW, Eppakayala V, Kim JH. Microbial reduction of graphene oxide by Escherichia coli: a green chemistry approach. Colloids Surf B Biointerfaces. 2013;102:772–777. | |
Gurunathan S, Han JW, Eppakayala V, Kim JH. An environmentally friendly approach to the reduction of graphene oxide by Escherichia fergusoni. J Nanosci Nanotechnol. 2013;13(3):2091–2098. | |
Gurunathan S, Han JW, Dayem AA, Eppakayala V, Kim JH. Biocompatibility of microbially reduced graphene oxide in primary mouse embryonic fibroblast cells. Colloids Surf B Biointerfaces. 2013;105:58–66. | |
Gurunathan S, Han JW, Kim JH. Humanin: a novel functional molecule for the green synthesis of graphene. Colloids Surf B Biointerfaces. 2013;111:376–383. | |
Thakur S, Karak N. Green reduction of graphene oxide by aqueous phytoextracts. Carbon. 2012;50(14):5331–5339. | |
Park Y, Hong YN, Weyers A, Kim YS, Linhardt RJ. Polysaccharides and phytochemicals: a natural reservoir for the green synthesis of gold and silver nanoparticles. IET Nanobiotechnol. 2011;5(3):69–78. | |
Iravani S. Green synthesis of metal nanoparticles using plants. Green Chem. 2011;13(10):2638–2650. | |
Esfandiar A, Akhavan O, Irajizad A. Melatonin as a powerful bio-antioxidant for reduction of graphene oxide. J Mater Chem. 2011;21: 10907–10914. | |
Liao KH, Lin YS, Macosko CW, Haynes CL. Cytotoxicity of graphene oxide and graphene in human erythrocytes and skin fibroblasts. ACS Appl Mater Interfaces. 2011;3(7):2607–2615. | |
Liu S, Zeng TH, Hofmann M, et al. Antibacterial activity of graphite, graphite oxide, graphene oxide, and reduced graphene oxide: membrane and oxidative stress. ACS Nano. 2011;5(9):6971–6980. | |
Wang Y, Shi Z, Yin J. Facile synthesis of soluble graphene via a green reduction of graphene oxide in tea solution and its biocomposites. ACS Appl Mater. Interfaces. 2011;3(4):1127–1133. | |
Li D, Müller MB, Gilje S, Kaner RB, Wallace GG. Processable aqueous dispersions of graphene nanosheets. Nat Nanotechnol. 2008;3(2):101–105. | |
Pham TA, Choi BC, Jeong YT. Facile covalent immobilization of cadmium sulfide quantum dots on graphene oxide nanosheets: preparation, characterization, and optical properties. Nanotechnology. 2010;21(46):465603. | |
Wang J, Zhou T, Deng H, et al. An environmentally friendly and fast approach to prepare reduced graphite oxide with water and organic solvents solubility. Colloids Surf B Biointerfaces. 2013;101: 171–176. | |
Chen D, Li L, Guo L. An environment-friendly preparation of reduced graphene oxide nanosheets via amino acid. Nanotechnology. 2011;22(32):325601. | |
Wu Q, Xu YX, Yao ZY, Liu AR, Shi GQ. Supercapacitors based on flexible graphene/polyaniline nanofiber composite films. ACS Nano. 2010;4(4):1963–1970. | |
Shen J, Hu Y, Shi M, et al. Fast and facile preparation of graphene oxide and reduced graphene oxide nanoplatelets. Chem Mater. 2009;21(15):3514–3520. | |
Park S, An J, Jung I, et al. Colloidal suspensions of highly reduced graphene oxide in a wide variety of organic solvents. Nano Lett. 2009;9(4):1593–1597. | |
Wojtoniszak M, Chen X, Kalenczuk RJ, et al. Synthesis, dispersion, and cytocompatibility of graphene oxide and reduced graphene oxide. Colloids Surf B Biointerfaces. 2012;89:79–85. | |
Gao J, Liu F, Liu Y, Ma N, Wang Z, Zhang X. Environment-friendly method to produce graphene that employs vitamin C and amino acid. Chem Mater. 2010;22(7):2213–2218. | |
Pénicaud A, Drummond C. Deconstructing graphite: graphenide solutions. Acc Chem Res. 2013;46(1):129–137. | |
Lammel T, Boisseaux P, Fernández-Cruz ML, Navas J. Internalization and cytotoxicity of graphene oxide and carboxyl graphene nanoplatelets in the human hepatocellular carcinoma cell line Hep G2. Part Fibre Toxicol. 2013;10(1):27. | |
Wang G, Yang J, Park J, et al. Facile synthesis and characterization of graphene nanosheets. J Phys Chem C. 2008;112(22):8192–8195. | |
Tuinstra F, Koenig JL. Raman spectrum of graphite. J Chem Phys. 1970;53:1126–1130. | |
Ferrari AC, Robertson J. Interpretation of Raman spectra of disordered and amorphous carbon. Phys Rev B Condens Matter Mater Phys. 2000;61(20):14095–14107. | |
Hummers WS, Offeman RE. Preparation of graphitic oxide. J Am Chem Soc. 1958;80(6):1339–1339. | |
He H, Gao C. General approach to individually dispersed, highly soluble, and conductive graphene nanosheets functionalized by nitrene chemistry. Chem Mater. 2010;22(17):5054–5064. | |
Jeong HK, Lee YP, Lahaye RJ, et al. Evidence of graphitic AB stacking order of graphite oxides. J Am Chem Soc. 2008;130(4):1362–1366. | |
Xu Z, Gao C. Aqueous liquid crystals of graphene oxide. ACS Nano. 2011;5(4):2908–2915. | |
Hu X, Xu Z, Gao C. Multifunctional, supramolecular, continuous artificial nacre fibres. Sci Rep. 2012;2:767. | |
Xu Z, Sun H, Zhao X, Gao C. Ultrastrong fibers assembled from giant graphene oxide sheets. Adv Mater. 2013;25(2):188–193. | |
Su CY, Xu Y, Zhang W, et al. Electrical and spectroscopic characterizations of ultra-large reduced graphene oxide monolayers. Chem Mater. 2009;21(23):5674–5680. | |
Chang Y, Yang ST, Liu JH, et al. In vitro toxicity evaluation of graphene oxide on A549 cells. Toxicol Lett. 2011;200(3):201–210. | |
Chen H, Mu¨ller MB, Gilmore KJ, Wallace GG, Li D. Mechanically strong, electrically conductive, and biocompatible graphene paper. Adv Mater. 2008;20(18):3557–3561. | |
Biris AR, Mahmood M, Lazar MD, et al. Novel multicomponent and biocompatible nanocomposite materials based on few-layer graphenes synthesized on a gold/hydroxyapatite catalytic system with applications in bone regeneration. J Phys Chem C. 2011;115(39):18967–18976. | |
Zhang Y, Ali SF, Dervishi E, et al. Cytotoxicity effects of graphene and single-wall carbon nanotubes in neural phaeochromocytoma-derived PC12 cells. ACS Nano. 2010;4(6):3181–3186. | |
Lu CH, Zhu CL, Li J, Liu JJ, Chen X, Yang HH. Using graphene to protect DNA from cleavage during cellular delivery. Chem Commun (Camb). 2010;46(18):3116–3118. | |
Negoescu A, Lorimier P, Labat-Moleur F, et al. In situ apoptotic cell labeling by the TUNEL method: improvement and evaluation on cell preparations. J Histochem Cytochem. 1996;44(9):959–968. | |
Zhang Y, Zhou L, Bao YL, et al. Butyrate induces cell apoptosis through activation of JNK MAP kinase pathway in human colon cancer RKO cells. Chem Biol Interact. 2010;185(3):174–181. | |
Feng L, Zhang S, Liu Z. Graphene based gene transfection. Nanoscale. 2011;3(3):1252–1257. | |
Sayes CM, Liang F, Hudson JL, et al. Functionalization density dependence of single-walled carbon nanotubes cytotoxicity in vitro. Toxicol Lett. 2006;161(2):135–142. | |
Gotoh Y, Hiraiwa K, Nagayama M. In vitro mineralization of osteoblastic cells derived from human bone. Bone Miner. 1990;8(3):239–250. | |
Liu Y, Huang J, Li H. Synthesis of hydroxyapatite-reduced graphite oxide nanocomposites for biomedical applications: oriented nucleation and epitaxial growth of hydroxyapatite. J Mater Chem B. 2013;1(13):1826–1834. | |
Kalbacova M, Broz A, Kromka A, Babchenko O, Kalbac M. Controlled oxygen plasma treatment of single-walled carbon nanotube films improves osteoblastic cells attachment and enhances their proliferation. Carbon. 2011;49(9):2926–2934. | |
Zhang X, Li M, Wang YB, et al. Cell response of nanographene platelets to human osteoblast-like MG63 cells. J Biomed Mater Res Part A. Epub April 16, 2013. | |
Li M, Wang Y, Liu Q, et al. In situ synthesis and biocompatibility of nanohydroxyapatite on pristine and chitosan functionalized graphene oxide. J Mater Chem B. 2013;1(4):475–484. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.