Back to Journals » Drug Design, Development and Therapy » Volume 8
Ginger components as new leads for the design and development of novel multi-targeted anti-Alzheimer’s drugs: a computational investigation
Authors Azam F ![]() , Amer A, Abulifa A
, Amer A, Abulifa A ![]() , Elzwawi M
, Elzwawi M
Received 15 May 2014
Accepted for publication 20 June 2014
Published 23 October 2014 Volume 2014:8 Pages 2045—2059
DOI https://doi.org/10.2147/DDDT.S67778
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Faizul Azam,1,2 Abdualrahman M Amer,1 Abdullah R Abulifa,1 Mustafa M Elzwawi1
1Faculty of Pharmacy, Misurata University, Misurata, Libya; 2Department of Pharmaceutical Chemistry, Nims Institute of Pharmacy, Nims University, Jaipur, Rajasthan, India
Abstract: Ginger (Zingiber officinale), despite being a common dietary adjunct that contributes to the taste and flavor of foods, is well known to contain a number of potentially bioactive phytochemicals having valuable medicinal properties. Although recent studies have emphasized their benefits in Alzheimer’s disease, limited information is available on the possible mechanism by which it renders anti-Alzheimer activity. Therefore, the present study seeks to employ molecular docking studies to investigate the binding interactions between active ginger components and various anti-Alzheimer drug targets. Lamarckian genetic algorithm methodology was employed for docking of 12 ligands with 13 different target proteins using AutoDock 4.2 program. Docking protocol was validated by re-docking of all native co-crystallized ligands into their original binding cavities exhibiting a strong correlation coefficient value (r2=0.931) between experimentally reported and docking predicted activities. This value suggests that the approach could be a promising computational tool to aid optimization of lead compounds obtained from ginger. Analysis of binding energy, predicted inhibition constant, and hydrophobic/hydrophilic interactions of ligands with target receptors revealed acetylcholinesterase as most promising, while c-Jun N-terminal kinase was recognized as the least favorable anti-Alzheimer’s drug target. Common structural requirements include hydrogen bond donor/acceptor area, hydrophobic domain, carbon spacer, and distal hydrophobic domain flanked by hydrogen bond donor/acceptor moieties. In addition, drug-likeness score and molecular properties responsible for a good pharmacokinetic profile were calculated by Osiris property explorer and Molinspiration online toolkit, respectively. None of the compounds violated Lipinski’s rule of five, making them potentially promising drug candidates for the treatment of Alzheimer’s disease.
Keywords: Alzheimer’s disease, ginger, molecular docking, structure–activity relationship, toxicity prediction
Introduction
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder of the central nervous system that accounts for 60%–70% of dementia cases in persons over 65 years of age worldwide.1 Neurodegeneration in AD, and other neurodegenerative diseases appear to be multifactorial, in that a complex set of deleterious reactions including glutamatergic neurotoxicity, increases in iron and nitric oxide, reduced expression of trophic factors, dysfunction of the ubiquitin–proteasome system, depletion of endogenous antioxidants, expression of proapoptotic proteins, and inflammation leads to the decease of neurons.2,3 With human life span increasing and with decreasing cognitive functions in elderly individuals with AD-related dementia, AD has become a major health problem in society.4
As of today, there are only few US Food and Drug Administration (FDA)-approved drugs in the market for treating AD patients. These include acetylcholinesterase (AChE) inhibitors (tacrine, donepezil, rivastigmine) and a noncompetitive inhibitor of N-methyl-D-aspartate (NMDA) receptors, memantine. All of these drugs improved the cognitive functions of AD patients symptomatically and have thus improved the quality of life for these patients; however, these drugs do not modify the disease progression in the long run.5 Additionally, they show limited clinical effects over the shorter term for some patients, mild to moderate cholinergic adverse effects in a minority of patients, and potentially distressing toxicity over the longer term.6
Furthermore, a great number of drugs with a variety of targets and clusters of mechanisms are currently in various stages of basic and clinical investigation. However, the development of therapies for this devastating disorder has been perturbing for physicians, researchers, and the pharmaceutical industry, with many drug candidates showing promise at one stage of clinical research only to fall at the next hurdle.7 Consequently, no ensuing experimental drug in development has been successful thus far; there has not been a new drug marketed for AD in a decade.6 The paucity of currently available drugs for treating AD, and their limited targets in AD pathology, as well as their proven side effects demand the development of a new generation of drugs that not only affect cholinergic functions associated with AD but also target other cellular pathways in AD pathogenesis.
Owing to the complexity of AD pathogenesis, the classic “one molecule, one target” solution may not be effective enough.8 Recently, the novel multi-target-directed strategy has received avid attention of researchers, since single molecules simultaneously interact with multiple targets in complex neurotoxic cascades, may achieve better efficacy in a complementary manner. Meanwhile, multi-targeted drugs would have a larger therapeutic window than those hitting a single target and would thus prove to be safer drugs.9
Since ancient times, the beneficial effects of some natural compounds have been appreciated in preventing various age-related pathologic conditions, including brain aging as well as neurodegeneration, and have been invigorated by a plethora of experimental and epidemiological studies.2 Ginger (Zingiber officinale [Z. officinale]) is a common dietary adjunct that contributes to the taste and flavor of foods. In addition to its flavor, ginger is known to contain a number of potentially bioactive phytochemicals, mainly gingerols and their related dehydrating products, the shogaols (Figure 1) as well as volatile oils.10
 | Figure 1 Chemical structure of potential bioactive phytochemicals from Zingiber officinale. |
Gingerols are pungent principles in the rhizome of ginger and possess the labile β-hydroxy keto functional group, which makes it susceptible to transformation to less-pungent compounds such as shogaols and zingerone by elevated temperature. Gingerols and shogaols have been reported to exhibit many interesting pharmacological and physiological functions, for example, antipyretic, cardiotonic, chemopreventive, anti-inflammatory, and antioxidant properties.11–14 It has been reported that ginger extract inhibits the production of nitric oxide (NO) and proinflammatory cytokines in lipopolysaccharide (LPS)-stimulated BV-2 microglial cells via the NF-kB (nuclear factor kappa-light-chain-enhancer of activated B cells) pathway.15 An inhibitory effect by 6-gingerol was shown on the production of proinflammatory cytokines in murine peritoneal macrophages.16 Likewise, 6-shogaol has been shown to inhibit LPS-induced inducible nitric oxide synthase (iNOS) and cyclooxygenase (COX) gene expression in macrophages.14 Moreover, 6-shogaol showed significant neuroprotective effects in vivo in transient global ischemia via the inhibition of microglia. It suppressed the microglial activation induced by LPS both in primary cortical neuron-glia culture and in an in vivo neuroinflammatory model.17 In vitro data have shown that ginger’s active principles protect nerve cells and may have potential in the treatment of AD.18
Anti-Alzheimer potential of gingerol has been disclosed more precisely when it was reported that it attenuates β-amyloid-induced oxidative cell death via fortifying cellular antioxidant defense system. In the said study, [6]-gingerol pretreatment protected against Aβ25–35-induced cytotoxicity and apoptotic cell death such as DNA fragmentation, disruption of mitochondrial membrane potential, elevated Bax/Bcl-2 ratio, and activation of caspase-3.19 In addition, the potential of traditional Chinese medicinal ginger root extract has been assessed for its ability to prevent behavioral dysfunction in the Aβ-induced AD model in rats very recently.20 Ginger’s potential in AD treatment has been further ascertained when its extract inhibited AChE activity as well as lipid peroxidation in the brain in a dose-dependent manner.21 Various mechanisms by which ginger compounds elicit interesting pharmacological activities have been presented in Table 1.
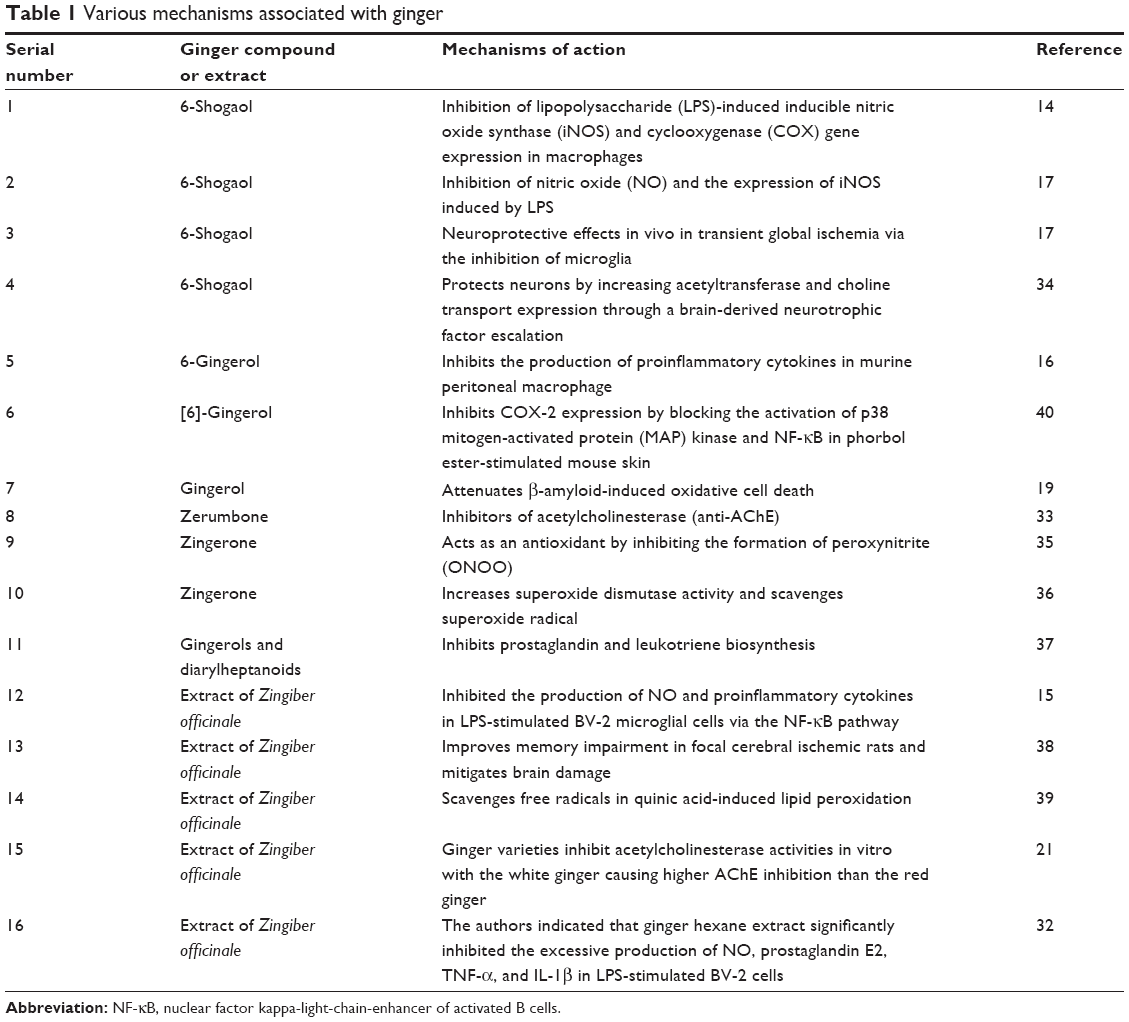 | Table 1 Various mechanisms associated with ginger |
Molecular docking is a computational method for finding out binding modes of ligands to their receptors rapidly, and is being applied consistently to drug design and discovery projects.22–25 Therefore, the present study seeks to employ this technique to investigate the binding interactions between active ginger components and various anti-Alzheimer drug targets. This will not only help in disclosing the interactions of ginger components with multi-targets but will also play an important role in revealing the anti-Alzheimer’s mechanisms as well as assist in lead optimization. In addition, drug-likeness score and molecular properties responsible for good pharmacokinetic profile were calculated by Osiris property explorer (www.organicchemistry.org/prog/peo/) and Molinspiration online toolkit (http://www.molinspiration.com/cgi-bin/properties) respectively.
Materials and methods
Molecular docking
Preparation of the protein receptor
The crystal structures of the protein–ligand complexes for the 13 AD-associated targets were used for the docking calculations (Table 2). They were downloaded from the Research Collaboratory for Structural Bioinformatics (RCSB) Protein Data Bank (PDB; http://www.rcsb.org/pdb/home/home.do). For each crystal structure, the crystallographic water molecules were removed, the missing hydrogen atoms were added, and the inhibitor from the crystal structure was used to define the active site.
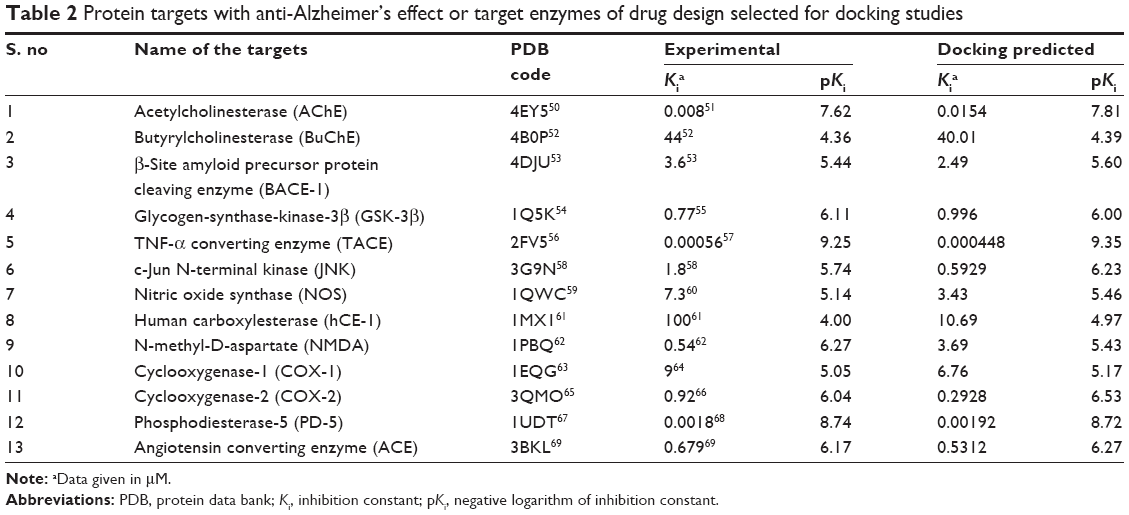 | Table 2 Protein targets with anti-Alzheimer’s effect or target enzymes of drug design selected for docking studies |
Preparation of ligands
Structures of the 12 ginger compounds with proven in vitro and/or in vivo activity against various central nervous system insults were retrieved from the literature.26 The structures of the ligands were drawn in ChemDraw 8.0 (PerkinElmer Informatics, Waltham, MA, USA) and converted to their three-dimensional structures in Chem3D 8.0 (PerkinElmer Informatics). Geometry optimization was done using PM3 method by MOPAC program (http://OpenMOPAC.net). Finally, all the compounds were saved in pdb format for further docking studies.
Docking simulation
Lamarckian genetic algorithm methodology was employed for docking simulations implemented in AutoDock 4.2 (The Scripps Research Institute, La Jolla, CA, USA).27 The standard docking procedure was used for a rigid protein and a flexible ligand whose torsion angles were identified (for ten independent runs per ligand). A grid of 60, 60, and 60 points in x, y, and z directions was built with a grid spacing of 0.375 Å and a distance-dependent function of the dielectric constant was used for the calculation of the energetic map. The default settings were used for all other parameters.
Analysis and visualization of docking simulation results
At the end of docking, the best poses were analyzed for hydrogen bonding or π interactions and root mean square deviation (RMSD) calculations using Discovery Studio Visualizer 2.5 (Accelrys Software Inc., San Diego, CA, USA) and PyMol version 1.3 (The PyMOL Molecular Graphics System; Schrödinger, LLC, New York, NY, USA) programs. From the estimated free energy of ligand binding (ΔGbinding, kcal/mol), the inhibition constant (Ki) for each ligand was calculated (Table 3).
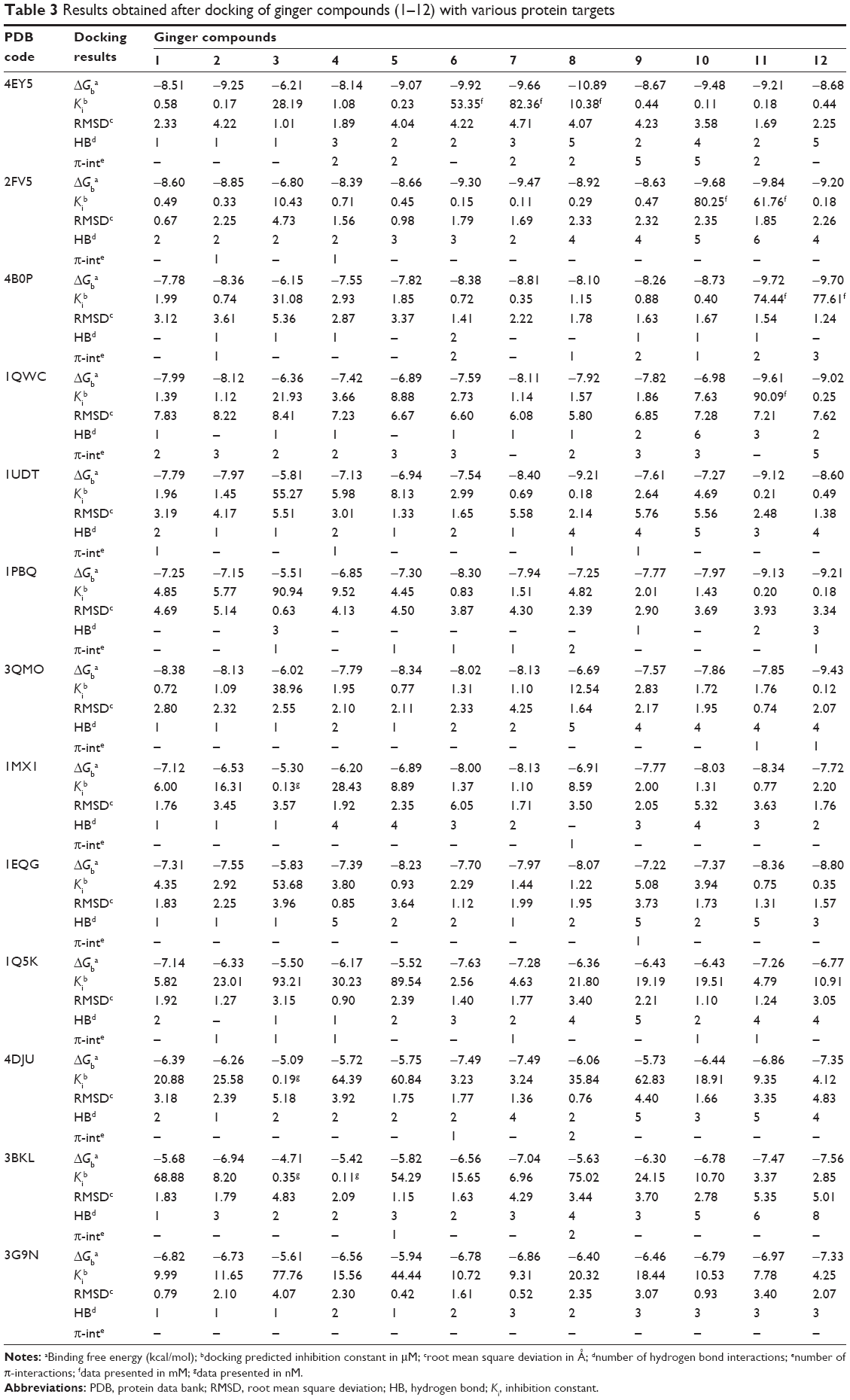 | Table 3 Results obtained after docking of ginger compounds (1–12) with various protein targets |
Calculation of pharmacokinetic parameters
Molinspiration online property calculation toolkit and Osiris property explorer were used to check the pharmaceutical fidelity of the drug candidates. Molecular descriptors, such as miLogP, the number of hydrogen bond donors, the number of hydrogen bond acceptors, the molecular mass of the compounds, topological polar surface area (TPSA), number of rotatable bonds, and violations of Lipinski’s rule of five28 were calculated using Molinspiration online property calculation toolkit. Percentage of absorption (%ABS) was calculated by: %ABS =109−[0.345× TPSA] according to the method of Zhao et al.29
Calculation of toxicity potential
Osiris Property Explorer was used to analyze various attributes of the drugs, such as toxicity, drug-likeness, and drug score.
Results and discussion
Validation of docking protocol
Initially, all the 13 native co-crystallized ligands were extracted from the receptor and re-docked within the inhibitor binding cavity of respective receptors in order to validate the docking calculations, reliability, and reproducibility of the docking parameters for the study. It was evident that the docked conformation of the ligands was almost superimposed with that of the respective co-crystallized ligands (Figure 2). As a general rule, if the best-docked conformation of a ligand resembles the bound native ligand in the experimental crystal structure, the used scoring function is said to be successful. According to the method of validation cited in the literature,30 the successful scoring function is the one in which the RMSD of the best-docked conformation is ≤2.0 Å from the experimental one. In this study, RMSD values of all docked targets were within 2.0 Å (Figure 2), indicating that our docking protocol is valid for the given structures and AutoDock 4.2, therefore deemed reliable for docking ginger components into the inhibitor binding cavity of multiple targets implicated in the pathogenesis of AD.
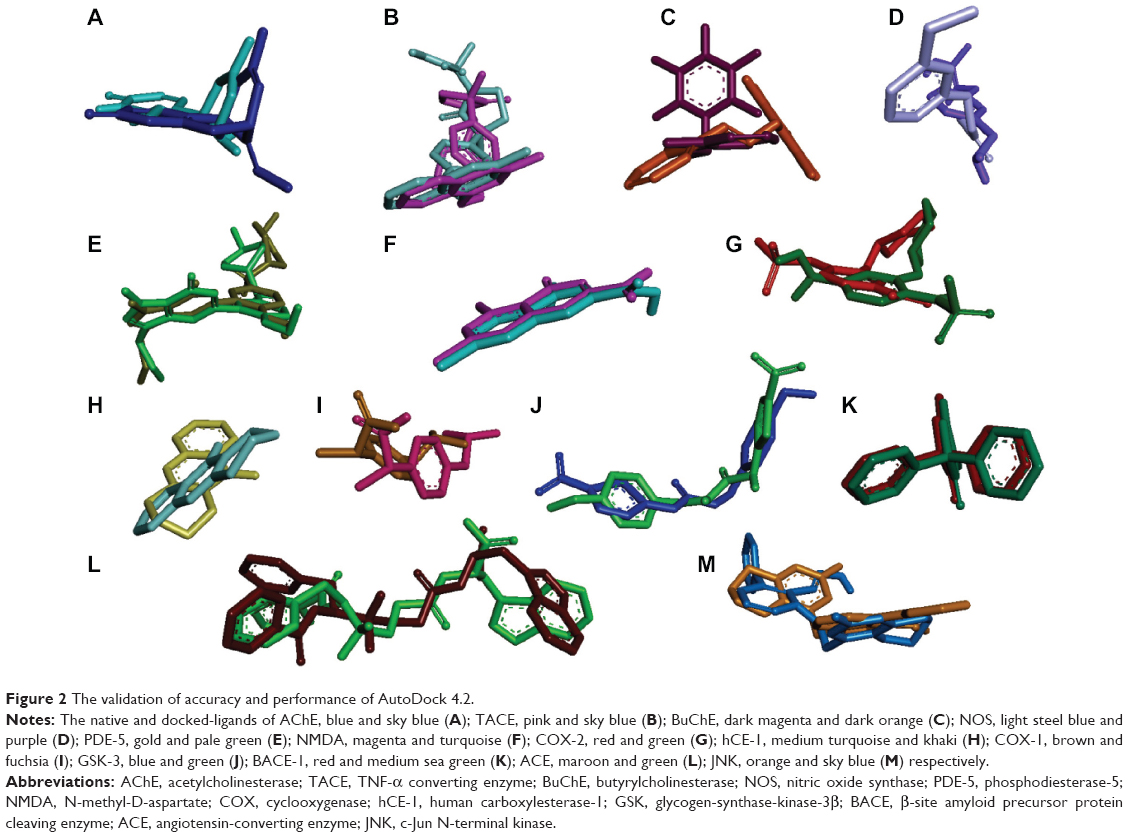 | Figure 2 The validation of accuracy and performance of AutoDock 4.2. |
In addition, all of the experimentally reported and docking predicted binding affinity data were converted to their logarithmic scale and plotted to obtain a correlation coefficient r2 of 0.931 (Figure 3). It means that the docking methodology employed in the present study could be a promising computational tool to aid optimization of lead compounds obtained from ginger. In addition, the docked conformation of each ligand was compared with the respective crystal structure conformation by calculating RMSD values and satisfactory results were achieved (Figure 2).
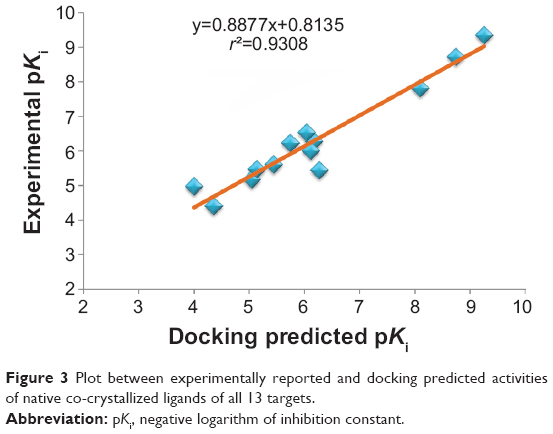 | Figure 3 Plot between experimentally reported and docking predicted activities of native co-crystallized ligands of all 13 targets. |
Interaction of the ginger compounds with potential protein targets
Thirteen potential protein receptors, which are either conventional clinical targets with anti-Alzheimer’s effects or target enzymes of drug design, were selected for docking studies. Docking predicted as well as experimentally reported Ki values for native co-crystallized ligands, PDB codes and related citation is presented in Table 2. For every target protein, a total of ten poses were visualized for each of the 12 ginger compounds to identify the model with minimum binding energy and estimated Ki values as well as best ligand–receptor interaction. The results in terms of binding free energy, predicted Ki, RMSD, number of hydrophobic as well as hydrophilic interaction are presented in Table 3. Since the values of docking predicted Ki, belong to different scales, they were converted to pKi (negative logarithm of Ki) values in order to have uniform data for a comparative study. A comparison of docking predicted activities of ligands against respective targets demonstrated in Figure 4 implicates that all of the docked ginger compounds possess the properties of promiscuous drugs.
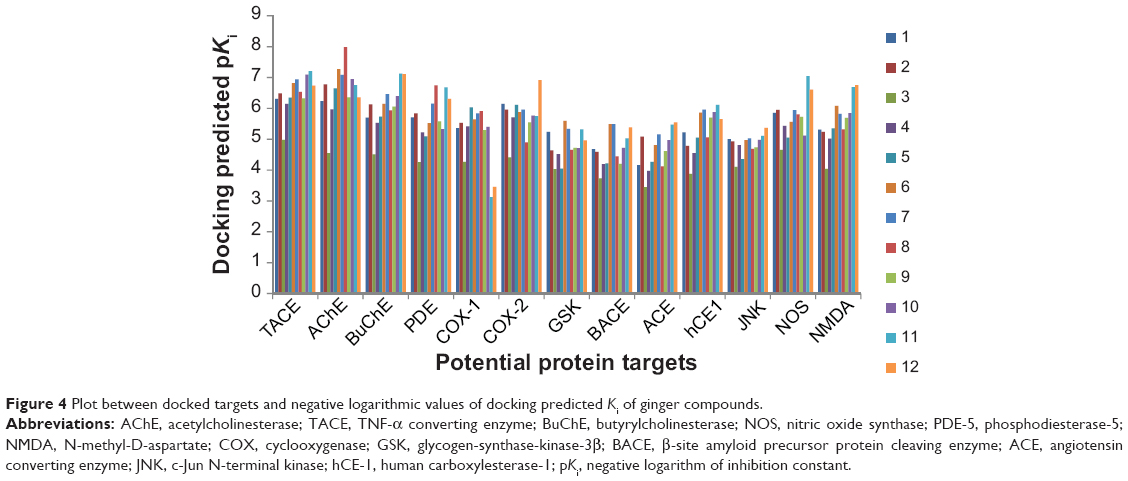 | Figure 4 Plot between docked targets and negative logarithmic values of docking predicted Ki of ginger compounds. |
Natural products are a rich source of lead compounds. Many of today’s medicines are either obtained directly from a natural source or were developed from a lead compound originally obtained from a natural source. In initial stage, the level of activity associated with lead compound may not be prodigious and there may be undesirable side effects, but the lead compound provides a start for the drug design and development process. Drugs interacting with multiple targets might have a better chance of affecting the complex equilibrium of whole cellular networks than drugs that act on a single target. Ginger compounds were capable of interacting with all docked targets showing variable affinities, which indicated that these ligands have broad-spectrum structural features that make them proficient for recognizing numerous significant target proteins. Figure 5 reveals common structural features essential for activity.
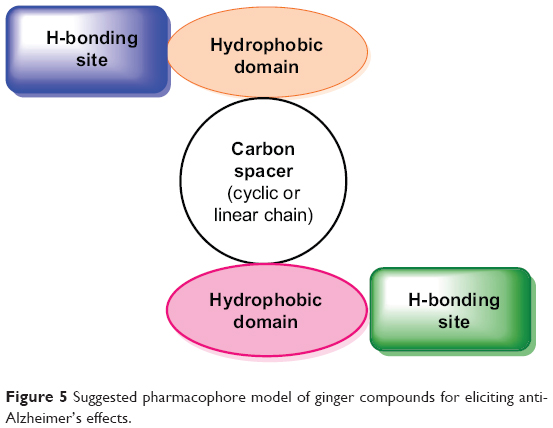 | Figure 5 Suggested pharmacophore model of ginger compounds for eliciting anti-Alzheimer’s effects. |
It is evident from the plot presented in Figure 4 that AChE is the most promising and JNK (c-Jun N-terminal kinase) is the least favorable anti-Alzheimer’s drug target. In addition, butyrylcholinesterase (BuChE), TNF-α converting enzyme, COX-2, NOS, and NMDA are proposed as best putative targets for ginger’s bioactive phytochemicals. Docked ligand–protein complexes of most promising compounds in various potential targets are presented in Figure 6.
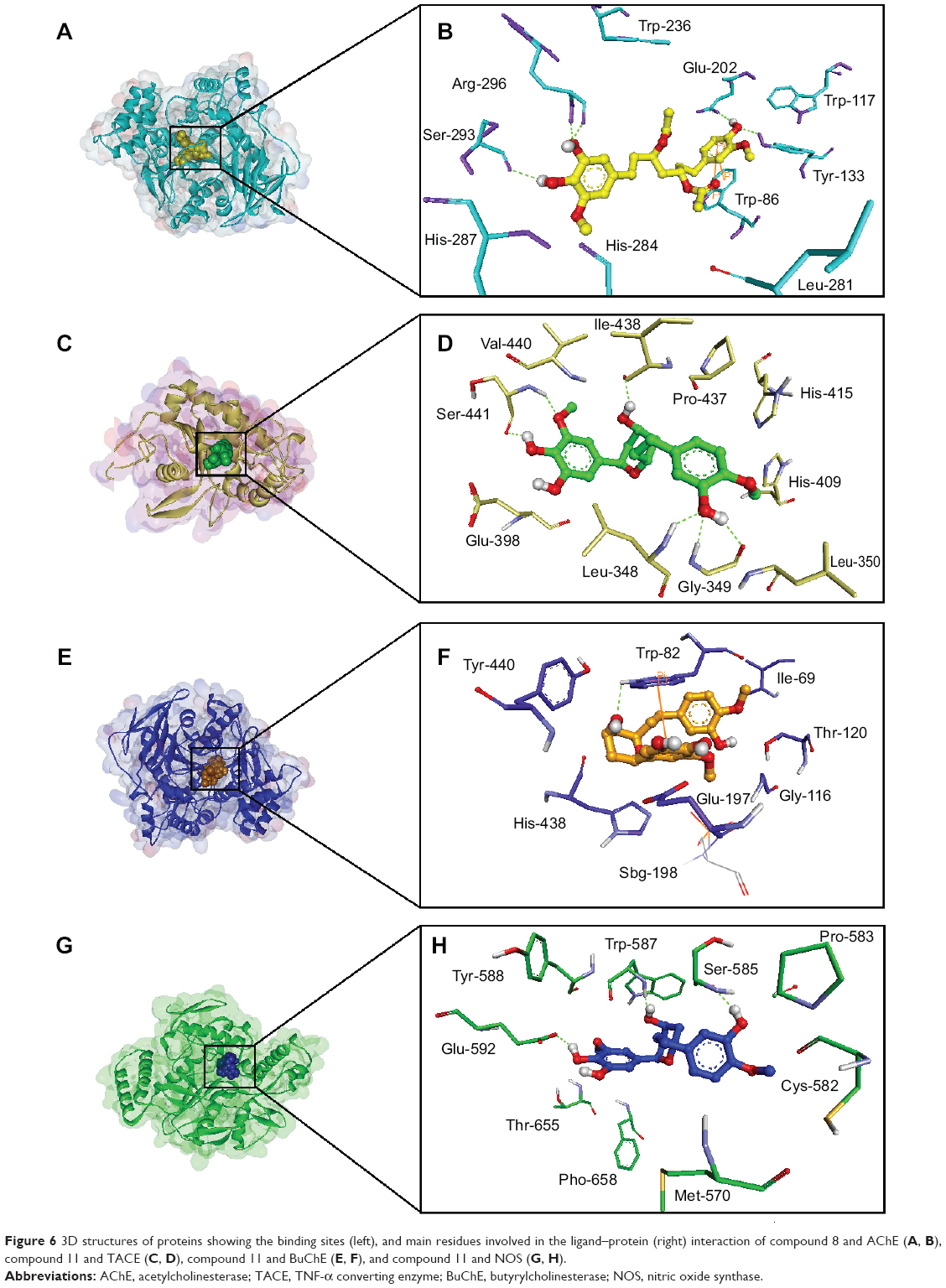 | Figure 6 3D structures of proteins showing the binding sites (left), and main residues involved in the ligand–protein (right) interaction of compound 8 and AChE (A, B), compound 11 and TACE (C, D), compound 11 and BuChE (E, F), and compound 11 and NOS (G, H). |
Various experimental reports have evidenced that ginger extract is capable of inhibiting targets such as AChE, BuChE,31 COX-1, COX-2, JNK, and NOS.32 Table 1 demonstrates the biological activities of ginger compounds or extracts that have been reported in the literature.14–17,19,21,32–40 Due to the molecular complexity of AD, multi-targeted therapies are becoming increasingly important as, in the long-term, they maximize the therapeutic effect and overcome the adverse effects associated with combination therapy.9 Thus, the potential of multi-targeted therapies that have been identified in ginger compounds may be a key explanation for why ginger extract is effective as an anti-Alzheimer’s treatment.14–17,19,21,32–40 In addition, it has been recognized that ginger in folklore medicinal practice possesses various pharmacological properties due to the different components attacking various targets or different steps in the pathologic process of AD.21 Therefore, the components of ginger, which have different mechanisms of anti-Alzheimer action, interact primarily in an additive or synergistic manner. A single drug should be more economical and lead to fewer adverse effects than a combination with each drug targeting a different protein.
Structure–activity relationship study presented in Figure 7 identifies the importance of various functionalities for ligand-receptor interactions: 1) replacement of distal hydrophobic domain (phenyl ring) with methyl group (compound 3) is detrimental for activity in all targets; 2) in hydrogen-bonding area, p-OH and m-OCH3 groups are important for all targets; additional m-OH is beneficial for most targets except glycogen-synthase-kinase-3β and β-site amyloid precursor protein cleaving enzyme; 3) double bond between C1 and C2 in compound 2 appreciates the activity at COX-1, COX-2, NOS, and angiotensin converting enzyme (ACE); however, double bond between C4 and C5 in compound 1 improves the activity at COX-2, glycogen-synthase-kinase-3β and JNK; 4) C3 and/or C5 must be substituted with C=O, OH, and OAc groups for optimum activity at all targets; 5) carbons C1 and C5 may be cyclized to form tetrahydropyran, which is essential for the activity of compounds 11 and 12 at NMDA, BuChE, ACE, JNK, and NOS; 6) for maximum activity, R should be aromatic ring substituted with H-bond donor/acceptor group at para and/or meta positions for all targets; 7) compounds having linear chain as carbon spacer are moderate or poor in activity; however, compounds 1 and 5 have exhibited appreciable activity at COX.
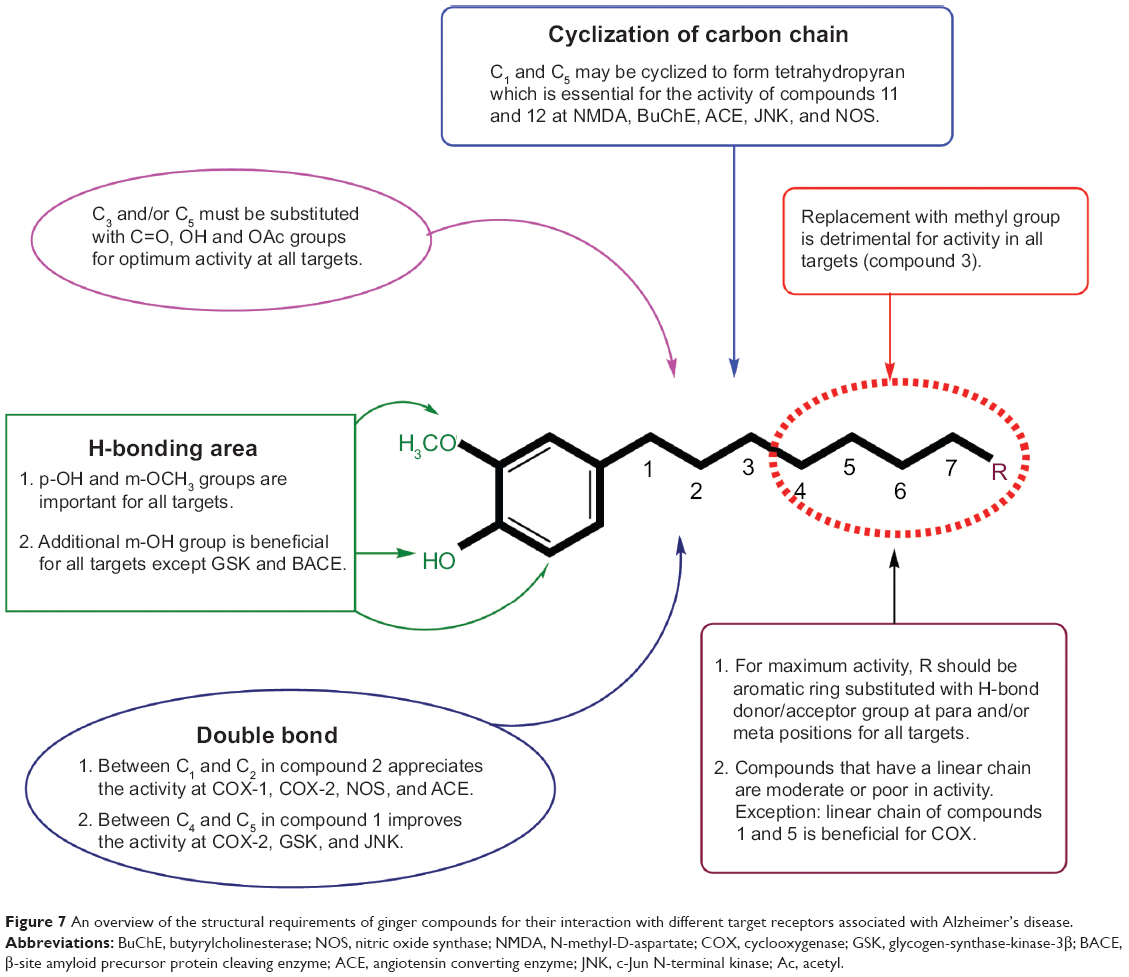 | Figure 7 An overview of the structural requirements of ginger compounds for their interaction with different target receptors associated with Alzheimer’s disease. |
Binding interactions between ginger components and most promising target, AChE
Comparative analysis of the docking results revealed that AChE is the most favorable target for interaction of ginger components (Figure 4). The neuropathology of AD is characterized by early loss of basal forebrain cholinergic neurons, leading to decreased cholinergic transmission, which can be improved with AChE inhibitors or by modulation of muscarinic and nicotinic acetylcholine receptors. In folklore medicine, ginger has been reportedly used for the treatment of AD as ginger extract, ginger tea, or as inclusion in food formulations and preparation.21 Inhibition of AChE activity by extract of Z. officinale has been documented in an in vitro study where white ginger caused higher AChE inhibition than the red ginger.21
Due to differences in substrate specificity and susceptibility to various kinds of inhibitors, cholinesterases were divided into two types: AChE, which hydrolyzes acetylcholine, and BuChE, which is able to hydrolyze larger molecules, such as butyrylcholine.41,42 AChE is the main enzyme metabolizing acetylcholine. It is also responsible for cerebral blood flow modulation, β-amyloid aggregation, activation and expression of APP95 protein, τ protein phosphorylation and has an influence on inflammatory processes. It interacts with β-amyloid, leading to creation of stable complexes and formation of senile plaques.43,44 The other enzyme, BuChE, is also important, because of its ability to hydrolyze acetylcholine and other choline esters. It was observed that the BuChE level increases in AD patients. Its role is not fully understood, but some studies suggested that it could promote amyloid plaque formation, and therefore, the search for inhibitors of both enzymes has been undertaken for the treatment of AD.45,46
Native co-crystallized ligand, Huperzine A is surrounded by residues Trp-86, Tyr-119, Gly-120, Gly-121, Gly-122, Tyr-124, Ser-125, Gly-126, Tyr-133, Glu-202, Ser-203, Tyr-337, and His 447, constituting active site of AChE enzyme (Figure 8). All of the docked compounds utilized the same amino acids of inhibitor binding pocket for polar as well as nonpolar interactions. The active site of AChE is composed of a catalytic triad (Ser-203, Glu-334, and His-447) that sits at the bottom of a narrow ~20 Å deep gorge. Just at the mouth of the gorge is the peripheral anionic binding site, which is composed of Tyr-72, Asp-74, Tyr-124, Glu-285, Trp-286, and Tyr-341 (Figure 8). Compounds 4, 9–10, and 12 utilized Tyr-72 and Tyr-124 for hydrophilic interaction while hydrophobic π–π interaction was noted with Trp-286 and Tyr-341 in the peripheral anionic-binding site. None of the compounds interacted with the residues of the catalytic triad except compound 10, which used phenyl ring for sharing π–π interaction with His-447.
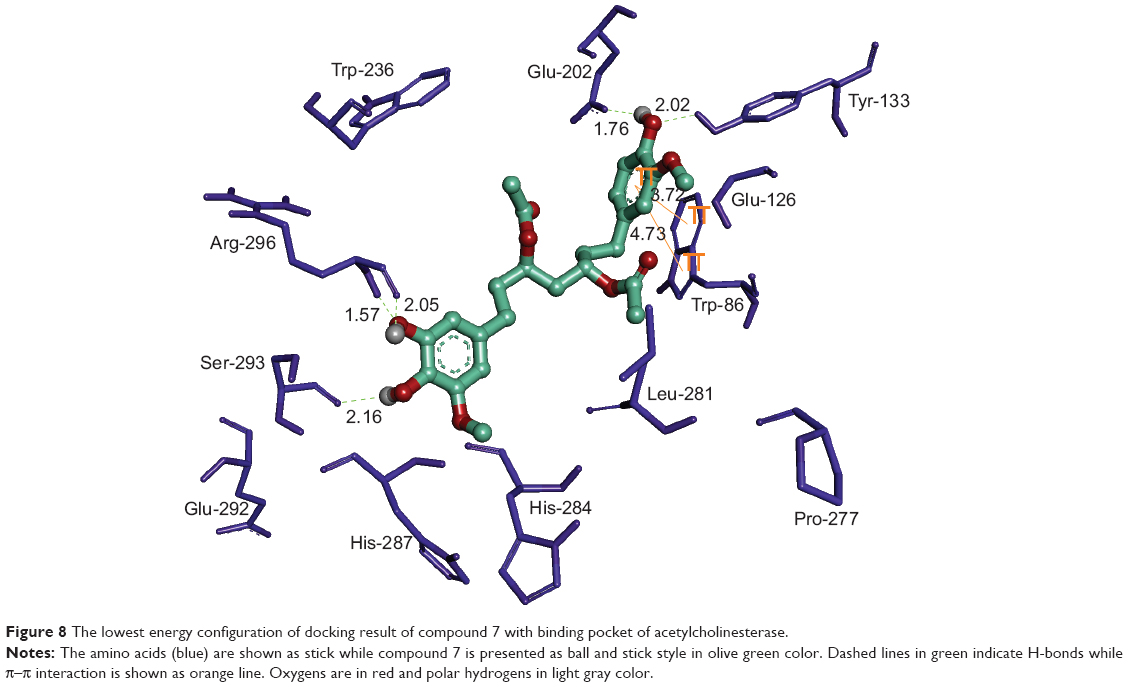 | Figure 8 The lowest energy configuration of docking result of compound 7 with binding pocket of acetylcholinesterase. |
The oxy-anionic hole comprises Gly-120, Gly-121, and Ala-204. Compound 4 and 12 shared H-bond with Gly-120 of oxy-anionic hole. The choline-binding site is defined mostly by Trp-86. Compounds 1–4, and 6 used Trp-86 for hydrophobic interaction with choline binding site. Compound 7 appears to interact with AChE most persuasively (Figure 9), conferring minimum binding energy among the docked compounds. The binding interactions are dominated by polar interactions involving hydroxyl group of Tyr-133, and carboxylic groups of Glu-202, Ser-293, and Arg-296. van der Waals contacts in terms of π–π interactions were noted with Trp-86. This compound has exploited the residues of ligand-binding area (Glu-202 and Trp-86) to demonstrate its maximum potency (Figure 8). In general, the docking energies are lower for all ligands in BuChE in comparison to AChE (Table 3).
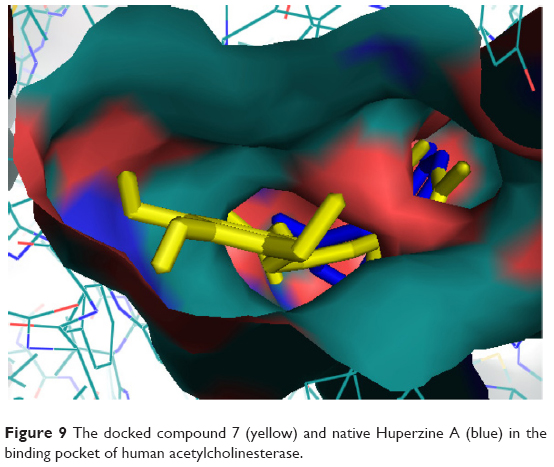 | Figure 9 The docked compound 7 (yellow) and native Huperzine A (blue) in the binding pocket of human acetylcholinesterase. |
Prediction of pharmacokinetic properties
A successful oral drug is one that is promptly and completely absorbed from the gastrointestinal tract, distributed specifically to its site of action in the body, metabolized in a way that does not instantly remove its activity, and eliminated in a suitable manner, without causing any harm to the organs. It is a well-known fact that approximately half of all drugs in development fail to make it to the market because of poor pharmacokinetics (PK).28 The PK properties depend on the chemical descriptors of the molecule. Computational prediction of PK properties such as absorption, distribution, metabolism, excretion, and toxicity (ADMET) have become progressively important in drug selection and promotion process and are promising tools to determine the success of the compound for human therapeutic use.47 Therefore, early prediction of ADMET properties has been done with the objective of increasing the success rate of the ginger compounds in future development processes.
Molinspiration online property calculation toolkit was utilized to screen the ginger compounds as drug candidate based on Lipinski’s rule of five and the results are presented in Table 4. This rule is based on the surveillance that most orally administered drugs have a molecular weight of ≤500, a LogP (logarithm of partition coefficient) ≤5, five or fewer hydrogen bond donor sites, and ten or fewer hydrogen bond acceptor sites. Molecules violating more than one of these rules may have problems with bioavailability. Fortunately, none of the compounds under study has violated these criterions.
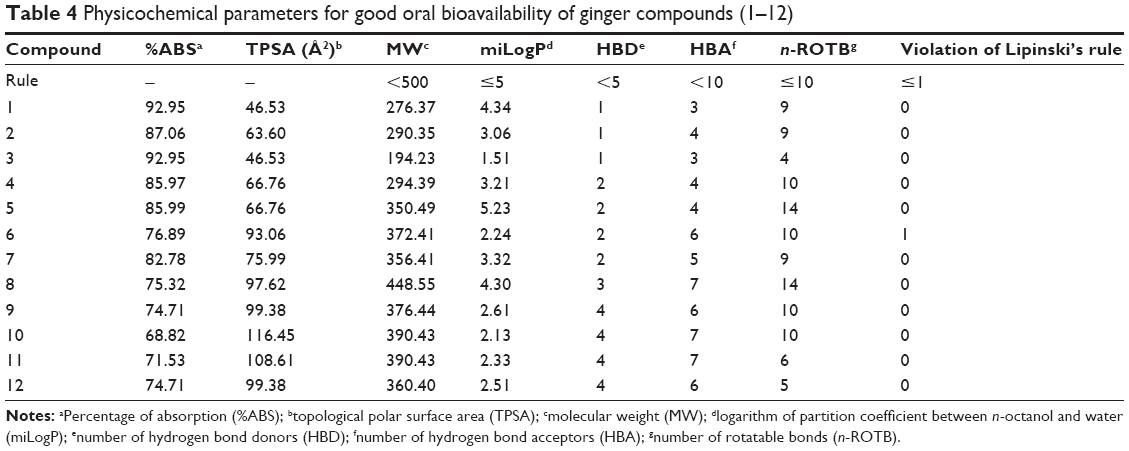 | Table 4 Physicochemical parameters for good oral bioavailability of ginger compounds (1–12) |
In addition, the bioavailability of ginger compounds was judged through TPSA analysis. This descriptor has been reported to correlate with passive molecular transport through membranes and therefore, allows prediction of transport properties of drugs and has been linked to drug bioavailability. As per the Veber’s rule for good oral bioavailability, the number of rotatable bond must be ≤10, and TPSA values ≤140 Å2.48 The number of rotatable bonds has been shown to be a very good descriptor of oral bioavailability of drugs. Rotatable bond is defined as any single non-ring bond, bounded to non-terminal heavy (ie, non-hydrogen) atom. Amide C–N bonds are not considered because of their high rotational energy barrier. With exception of compounds 5 and 8, number of rotatable bonds was found to be appropriate in all ginger compounds.
Percentage of absorption was estimated using the equation: %ABS =109−0.345× TPSA, according to Zhao et al.29 TPSA was also calculated using Molinspiration online property calculation toolkit according to the fragment-based method of Ertl et al.49 Generally, it has been seen that passively absorbed molecules with a TPSA >140 Å2 are thought to have low oral bioavailability. According to the above criterions, calculated percentages of absorption for ginger compounds 1−12, ranged between 68.82% and 92.95%.
Toxicity risks and drug score assessment
Nowadays, it is much more convenient to predict the toxicity risks of compounds through reliable bioinformatics tools. In the present study, Osiris property explorer was used to calculate toxicity risks parameters such as mutagenicity, tumorigenicity, irritation, and reproductive or developmental toxicity of all the ginger compounds, 1−12 (Table 5). The predictions are based on the functional group similarity for the query molecule with the in vitro and in vivo validated compounds present in the database of this online program. The toxicity risk predictor locates fragments within a molecule, which indicate a potential toxicity risk. The results can be visualized using color codes; green color shows low toxic tendency, yellow shows the mediocre, and red color shows high tendency of toxicity. Toxicity screening results presented in Figure 9 showed that none of the compounds (1−12) pose the risk of tumorogenicity and reproductive toxicity; however, compound 1 indicated high risk of mutagenicity. On the other hand, compounds 2 and 3 indicated high risk of irritation.
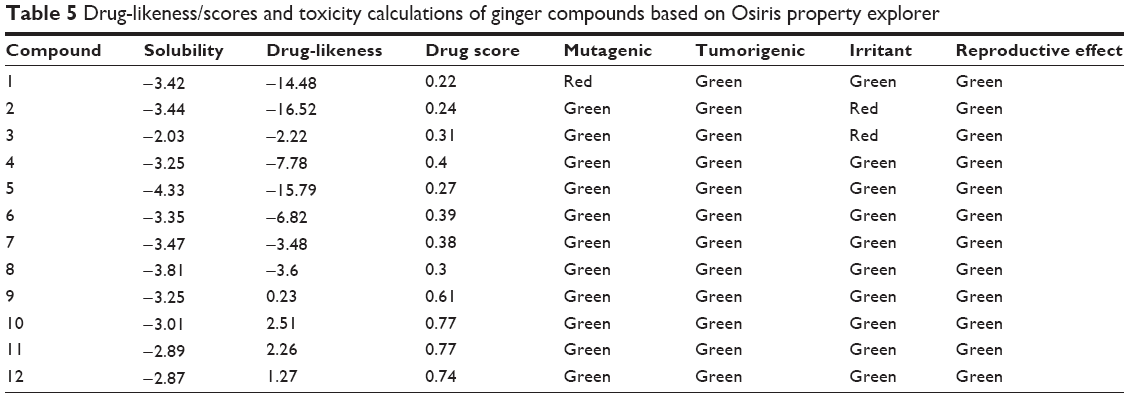 | Table 5 Drug-likeness/scores and toxicity calculations of ginger compounds based on Osiris property explorer |
To assess the ginger compound’s overall potential to qualify for a drug, overall drug score was calculated, which combines drug-likeness, hydrophobicity (LogP), aqueous solubility (LogS), MW, and toxicity risk parameters. The hydrophobicity of drugs could be inferred from LogP value. LogP values are directly proportional to the oral hydrophobicity of the drug. The more hydrophobic the drug, higher is the ability of the drug to circulate longer in our body. It would not be easy to excrete such a drug. In the present investigation, the miLogP values of the drug molecules were observed to be in the range of 1.51–5.23 (Table 4).
Conclusion
In present study, a comparative molecular docking approach using AutoDock was taken to identify the potential anti-Alzheimer receptors for ginger bioactive phytochemicals such as gingerols, shogaols, zingerone, and related compounds. The results show that: 1) several targets such as AChE, BuChE, TNF-α converting enzyme, NOS, COX-2, and NMDA, identified in this study are proposed as best putative targets for ginger phytochemicals; 2) a number of targets identified by docking, such as AChE, BuChE, COX-1, COX-2, JNK, and NOS, have already been verified by experiments for their inhibition by ginger extracts; 3) it is being proposed that 1,3-diacetoxy derivative (compound 7) binds to the AChE active site with certain orientation and conformation so that it may act as an inhibitor of that enzyme. Structure analysis shows that electrostatic interaction and hydrogen bonding play an important role in their binding process. The study provides important information for optimizing lead compounds of ginger for the treatment of AD.
Disclosure
The authors have no conflicts of interest to disclose.
References
Fan LY, Chiu MJ. Combotherapy and current concepts as well as future strategies for the treatment of Alzheimer’s disease. Neuropsychiatr Dis Treat. 2014;10:439–451. | ||
Azam F. Therapeutic potential of free radical scavengers in neurological disorders. In: Kozyrev D, Slutsky V, editors. Handbook of Free radicals: Formation, Types and Effects. New York: Nova Publishers; 2010:57–97. | ||
Mandel S, Youdim MB. Catechin polyphenols: neurodegeneration and neuroprotection in neurodegenerative diseases. Free Radic Biol Med. 2004;37(3):304–317. | ||
Chancellor B, Duncan A, Chatterjee A. Art therapy for Alzheimer’s disease and other dementias. J Alzheimers Dis. 2014;39(1):1–11. | ||
Allain H, Bentue-Ferrer D, Tribut O, Gauthier S, Michel BF, Drieu-La Rochelle C. Alzheimer’s disease: the pharmacological pathway. Fundam Clin Pharmacol. 2003;17(4):419–428. | ||
Schneider LS. Alzheimer disease pharmacologic treatment and treatment research. Continuum (Minneap Minn). 2013;19(2 Dementia):339–357. | ||
Hampel H. Current insights into the pathophysiology of Alzheimer’s disease: selecting targets for early therapeutic intervention. Int Psychogeriatr. 2012;24(Suppl 1):S10–S17. | ||
Azam F, Madi AM, Ali HI. Molecular docking and prediction of pharmacokinetic properties of dual mechanism drugs that block MAO-B and adenosine A2A. J Young Pharm. 2012;4(3):184–192. | ||
Morphy JR, Harris CJ. Designing Multi-Target Drugs. RSC Drug Discovery Series No. 21. Cambridge: Royal Society of Chemistry; 2012. | ||
Silva MI, de Melo CTV, Vasconcelos LF, de Carvalho AMR, Sousa FCF. Bioactivity and potential therapeutic benefits of some medicinal plants from the Caatinga (semi-arid) vegetation of Northeast Brazil: a review of the literature. Brazilian Journal of Pharmacognosy. 2012;22:193–207. | ||
Shukla Y, Singh M. Cancer preventive properties of ginger: a brief review. Food Chem Toxicol. 2007;45(5):683–690. | ||
Surh Y. Molecular mechanisms of chemopreventive effects of selected dietary and medicinal phenolic substances. Mutat Res. 1999;428(1–2): 305–327. | ||
Ali BH, Blunden G, Tanira MO, Nemmar A. Some phytochemical, pharmacological and toxicological properties of ginger (Zingiber officinale Roscoe): a review of recent research. Food Chem Toxicol. 2008;46(2):409–420. | ||
Pan MH, Hsieh MC, Hsu PC, et al. 6-Shogaol suppressed lipopolysaccharide-induced up-expression of iNOS and COX-2 in murine macrophages. Mol Nutr Food Res. 2008;52(12):1467–1477. | ||
Jung HW, Yoon CH, Park KM, Han HS, Park YK. Hexane fraction of Zingiberis Rhizoma Crudus extract inhibits the production of nitric oxide proinflammatory cytokines in LPS-stimulated BV2 microglial cells via the NF kappaB pathway. Food Chem Toxicol. 2009;47(6):1190–1197. | ||
Tripathi S, Maier KG, Bruch D, Kittur DS. Effect of 6-gingerol on proinflammatory cytokine production and costimulatory molecule expression in murine peritoneal macrophages. J Surg Res. 2007;138(2):209–213. | ||
Ha SK, Moon E, Ju MS, et al. 6-Shogaol a ginger product modulates neuroinflammation: a new approach to neuroprotection. Neuropharmacology. 2012;63(2):211–223. | ||
Kim DS, Kim DS, Oppel MN. Shogaols from Zingiber officinale protect human neuroblastoma and normal human umbilical vein endothelial cells from beta-amyloid (25–35) insult. Planta Med. 2002;68(4):375–376. | ||
Lee C, Park GH, Kim CY, Jang JH. [6]-Gingerol attenuates β-amyloid-induced oxidative cell death via fortifying cellular antioxidant defense system. Food Chem Toxicol. 2011;49(6):1261–1269. | ||
Zeng GF, Zhang ZY, Lu L, Xiao DQ, Zong SH, He JM. Protective effects of ginger root extract on Alzheimer disease-induced behavioral dysfunction in rats. Rejuvenation Res. 2013;16(2):124–133. | ||
Oboh G, Ademiluyi AO, Akinyemi AJ. Inhibition of acetylcholinesterase activities and some pro-oxidant induced lipid peroxidation in rat brain by two varieties of ginger (Zingiber officinale). Exp Toxicol Pathol. 2012;64(4):315–319. | ||
Shushni MA, Azam F, Lindequist U. Oxasetin from Lophiostoma sp. of the Baltic Sea: identification, in silico binding mode prediction and antibacterial evaluation against fish pathogenic bacteria. Nat Prod Commun. 2013;8(9):1223–1226. | ||
Azam F, Prasad MV, Thangavel N, Shrivastava AK, Mohan G. Structure-based design, synthesis and molecular modeling studies of 1-(substituted aryl)-3-(thiazol-2-yl)urea derivatives as novel anti-Parkinsonian agents. Med Chem. 2012;8(6):1057–1068. | ||
Azam F, Prasad MV, Thangavel N. Structure-based design, synthesis and molecular modeling studies of 1-(benzo[d]thiazol-2-yl)-3-(substituted aryl)urea derivatives as novel anti-Parkinsonian agents. Med Chem Res. 2012;21(9):2630–2643. | ||
Ahmed MA, Azam F, Rghigh AM, Gbaj A, Zetrini AE. Structure-based design, synthesis, molecular docking and biological activities of 2-(3 benzoylphenyl) propanoic acid derivatives as dual mechanism drugs. J Pharm Bioallied Sci. 2011;4(1):43–50. | ||
Peng F, Tao Q, Wu X, et al. Cytotoxic, cytoprotective and antioxidant effects of isolated phenolic compounds from fresh ginger. Fitoterapia. 2012;83(3):568–585. | ||
Morris GM, Goodsell DS, Halliday RS, et al. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. Journal of Computational Chemistry. 1998;19(14):1639–1662. | ||
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46(1–3):3–26. | ||
Zhao Y, Abraham MH, Le J, et al. Rate-limited steps of human oral absorption and QSAR studies. Pharm Res. 2002;19(10):1446–1457. | ||
Wang R, Lu Y, Wang S. Comparative evaluation of 11 scoring functions for molecular docking. J Med Chem. 2003;46(12):2287–2303. | ||
Ghayur MN, Gilani AH, Ahmed T, et al. Muscarinic, Ca++ antagonist and specific butyrylcholinesterase inhibitory activity of dried ginger extract might explain its use in dementia. J Pharm Pharmacol. 2008; 60(10):1375–1383. | ||
Choi DK, Koppula S, Suk K. Inhibitors of microglial neurotoxicity: focus on natural products. Molecules. 2011;16(2):1021–1043. | ||
Bustamam A, Ibrahim S, Al-Zubairi AS, Met M, Syam MM. Zerumbone: a natural compound with anti-cholinesterase activity. Am J Pharmacol Toxicol. 2008;3(3):209–211. | ||
Shim S, Kwon J. Effects of [6]-shogaol on cholinergic signaling in HT22 cells following neuronal damage induced by hydrogen peroxide. Food Chem Toxicol. 2012;50(5):1454–1459. | ||
Shin SG, Kim JY, Chung HY, Jeong JC. Zingerone as an antioxidant against peroxynitrite. J Agric Food Chem. 2005;53(19):7617–7622. | ||
Kabuto H, Nishizawa M, Tada M, Higashio C, Shishibori T, Koho M. Zingerone [4-(4-hydroxy-3-methoxyphenyl)-2-butanone] prevents 6-hydroxydopamine-induced dopamine depression in mouse striatum and increases superoxide scavenging activity in serum. Neurochem Res. 2005;30(3):325–332. | ||
Kiuchi F, Iwakami S, Shibuya M, Hanaoka F, Sankawa U. Inhibition of prostaglandin and leukotriene biosynthesis by gingerols and diarylheptanoids. Chem Pharm Bull. 1992;40(2):387–391. | ||
Wattanathorn J, Jittiwat J, Tongun T, Muchimapura S, Ingkaninan K. Zingiber officinale mitigates brain damage and improves memory impairment in focal cerebral ischemic rat. Evid Based Complement Alternat Med. 2011;2011:429–505. | ||
Belle NA, Dalmolin GD, Fonini G, Rubim MA, Rocha JB. Polyamines reduces lipid peroxidation induced by different prooxidants agents. Brain Res. 2004;1008(2):245–251. | ||
Kim SO, Kundu JK, Shin YK, et al. [6]-Gingerol inhibits COX-2 expression by blocking the activation of p38 MAP kinase and NF-kappaB in phorbol ester-stimulated mouse skin. Oncogene. 2005;24(15): 2558–2567. | ||
Ollis DL, Cheah E, Cygler M, et al. The alpha/beta hydrolase fold. Protein Eng. 1992;5(3):197–211. | ||
Cygler M, Schrag JD, Sussman JL, et al. Relationship between sequence conservation and three-dimensional structure in a large family of esterases, lipases, and related proteins. Protein Sci. 1993;2(3):366–382. | ||
Alvarez A, Alarcon R, Opazo C, et al. Stable complexes involving acetylcholinesterase and amyloid-beta peptide change the biochemical properties of the enzyme and increase the neurotoxicity of Alzheimer’s fibrils. J Neurosci. 1998;18(9):3213–3223. | ||
Silman I, Sussman JL. Acetylcholinesterase: “classical” and “non-classical” functions and pharmacology. Curr Opin Pharmacol. 2005;5(3):293–302. | ||
Musial A, Bajda M, Malawska B. Recent developments in cholinesterases inhibitors for Alzheimer’s disease treatment. Curr Med Chem. 2007;14(25):2654–2679. | ||
Darvesh S, Cash MK, Reid GA, Martin E, Mitnitski A, Geula C. Butyrylcholinesterase is associated with β-amyloid plaques in the transgenic APPSWE/PSEN1dE9 mouse model of Alzheimer disease. J Neuropathol Exp Neurol. 2012;71(1):2–14. | ||
Moroy G, Martiny VY, Vayer P, Villoutreix BO, Miteva MA. Toward in silico structure-based ADMET prediction in drug discovery. Drug Discov Today. 2012;17(1–2):44–55. | ||
Veber DF, Johnson SR, Cheng HY, Smith BR, Ward KW, Kopple KD. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem. 2002;45(12):2615–2623. | ||
Ertl P, Rohde B, Selzer P. Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J Med Chem. 2000;43(20):3714–3717. | ||
Cheung J, Rudolph MJ, Burshteyn F, et al. Structures of human acetylcholinesterase in complex with pharmacologically important ligands. J Med Chem. 2012;55(22):10282–10286. | ||
McKinney M, Miller JH, Yamada F, Tuckmantel W, Kozikowski AP. Potencies and stereoselectivities of enantiomers of huperzine A for inhibition of rat cortical acetylcholinesterase. Eur J Pharmacol. 1991; 203(2):303–305. | ||
Wandhammer M, de Koning M, van Grol M, et al. A step toward the reactivation of aged cholinesterases – crystal structure of ligands binding to aged human butyrylcholinesterase. Chem Biol Interact. 2013;203(1):19–23. | ||
Cumming JN, Smith EM, Wang L, et al. Structure based design of iminohydantoin BACE1 inhibitors: identification of an orally available, centrally active BACE1 inhibitor. Bioorg Med Chem Lett. 2012;22(7): 2444–2449. | ||
Bhat R, Xue Y, Berg S, et al. Structural insights and biological effects of glycogen synthase kinase 3 specific inhibitor AR-A014418. J Biol Chem. 2003;278(46):45937–45945. | ||
Hicks JW, Wilson AA, Rubie EA, Woodgett JR, Houle S, Vasdev N. Towards the preparation of radiolabeled 1-aryl-3-benzyl ureas: radiosynthesis of [(11)C- carbonyl] AR- A014418 by [(11)C]CO(2) fixation. Bioorg Med Chem Lett. 2012;22(5):2099–2101. | ||
Niu X, Umland S, Ingram R, et al. IK682, a tight binding inhibitor of TACE. Arch Biochem Biophys. 2006;451(1):43–50. | ||
Duan JJ, Chen L, Wasserman ZR, et al. Discovery of gamma-lactam hydroxamic acids as selective inhibitors of tumor necrosis factor alpha converting enzyme: design, synthesis, and structure-activity relationships. J Med Chem. 2002;45(23):4954–4957. | ||
Cao J, Gao H, Bemis G, et al. Structure-based design and parallel synthesis of n-benzyl isatin oximes as jnk3 map kinase inhibitors. Bioorg Med Chem Lett. 2009;19(10):2891–2895. | ||
Fedorov R, Hartmann E, Ghosh DK, Schlichting I. Structural basis for the specificity of the nitric-oxide synthase inhibitors w1400 and nomega-propyl-l-arg for the inducible and neuronal isoforms. J Biol Chem. 2003;278(46):45818–45825. | ||
Congreve M, Chessari G, Tisi D, Woodhead AJ. Recent developments in fragment-based drug discovery. J Med Chem. 2008;51(13):3661–3680. | ||
Bencharit S, Morton CL, Hyatt JL, et al. Crystal structure of human carboxylesterase 1 complexed with the Alzheimer’s drug tacrine: from binding promiscuity to selective inhibition. Chem Biol. 2003;10(4):341–349. | ||
Furukawa H, Gouaux E. Mechanisms of activation, inhibition and specificity: crystal structures of the NMDA receptor nr1 ligand-binding core. EMBO J. 2003;22(12):2873–2885. | ||
Selinsky BS, Gupta K, Sharkey CT, Loll PJ. Structural analysis of NSAID binding by prostaglandin H2 synthase: time-dependent and time-independent inhibitors elicit identical enzyme conformations. Biochemistry. 2001;40(17):5172–5180. | ||
Viegas A, Manso J, Corvo MC, Marques MM, Cabrita EJ. Binding of ibuprofen, ketorolac, and diclofenac to COX-1 and COX-2 studied by saturation transfer difference NMR. J Med Chem. 2011;54(24): 8555–8562. | ||
Vecchio AJ, Malkowski MG. The structure of ns-398 bound to cyclooxygenase-2. J Struct Biol. 2011;176(2):254–258. | ||
Cryer B, Feldman M. Cyclooxygenase-1 and cyclooxygenase-2 selectivity of widely used nonsteroidal anti-inflammatory drugs. Am J Med. 1998;104(5):413–421. | ||
Sung BJ, Hwang KY, Jeon YH, et al. Structure of the catalytic domain of human phosphodiesterase 5 with bound drug molecules. Nature. 2003;425(6953):98–102. | ||
Jiang W, Guan J, Macielag MJ, et al. Pyrroloquinolone PDE5 inhibitors with improved pharmaceutical profiles for clinical studies on erectile dysfunction. J Med Chem. 2005;48(6):2126–2133. | ||
Watermeyer JM, Kroger WL, O’Neill HG, Sewell BT, Sturrock ED. Probing the basis of domain-dependent inhibition using novel ketone inhibitors of Angiotensin-converting enzyme. Biochemistry. 2008; 47(22):5942–5950. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.