Back to Journals » The Application of Clinical Genetics » Volume 13
Germline Sequencing Identifies Rare Variants in Finnish Subjects with Familial Germ Cell Tumors
Authors Crowgey EL ![]() , Soini T
, Soini T ![]() , Shah N, Pauniaho SL
, Shah N, Pauniaho SL ![]() , Lahdenne P, Wilson DB
, Lahdenne P, Wilson DB ![]() , Heikinheimo M, Druley TE
, Heikinheimo M, Druley TE ![]()
Received 7 January 2020
Accepted for publication 19 April 2020
Published 30 June 2020 Volume 2020:13 Pages 127—137
DOI https://doi.org/10.2147/TACG.S245093
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Martin Maurer
Erin L Crowgey,1 Tea Soini,2 Nidhi Shah,1 Satu-Liisa Pauniaho,2,3 Pekka Lahdenne,2 David B Wilson,4 Markku Heikinheimo,2,4 Todd E Druley4
1Nemours Center for Cancer and Blood Disorders, Nemours Alfred I. duPont Hospital for Children, Wilmington, DE, USA; 2Pediatric Research Center, Children’s Hospital, University of Helsinki and Helsinki University Hospital, Helsinki, Finland; 3Tampere Center for Child Health Research, University of Tampere, Tampere University Hospital, Tampere, Finland; 4Department of Pediatrics, Division of Hematology and Oncology, Washington University School of Medicine, St. Louis, MO, USA
Correspondence: Todd E Druley
Department of Pediatrics, Division of Hematology and Oncology, Washington University School of Medicine, 4515 McKinley Avenue, Campus Box 8510, St. Louis, MO 63108, USA
Email [email protected]
Purpose: Pediatric germ cell tumors are rare, representing about 3% of childhood malignancies in children less than 15 years of age, presenting in neonates or adolescents with a greater incidence noted in older adolescents. Aberrations in primordial germ cell proliferation/differentiation can lead to a variety of neoplasms, including teratomas, embryonal carcinoma, choriocarcinoma, and yolk sac tumors.
Patients and Methods: Three Finnish families with varying familial germ cell tumors were identified, and whole-genome sequencing was performed using an Illumina sequencing platform. In total, 22 unique subjects across the three families were sequenced. Family 1 proband (female) was affected by malignant ovarian teratoma, Family 2 proband (female) was affected by sacrococcygeal teratoma with yolk sac tumor in the setting of Cornelia de Lange syndrome, and Family 3 proband (male) was affected by malignant testicular teratoma. Rare variants were identified using an autosomal recessive or de novo model of inheritance.
Results: For family 1 proband (female), an autosomal recessive or de novo model of inheritance identified variants of interest in the following genes: CD109, IKBKB, and CTNNA3, SUPT6H, MUC5AC, and FRG1. Family 2 proband (female) analysis identified gene variants of interest in the following genes: LONRF2, ANO7, HS6ST1, PRB2, and DNM2. Family 3 proband (male) analysis identified the following potential genes: CRIPAK, KRTAP5-7, and CACNA1B.
Conclusion: Leveraging deep pedigrees and next-generation sequencing, rare germline variants were identified that were enriched in three families from Finland with a history of familial germ cell tumors. The data presented support the importance of germline mutations when analyzing complex cancers with a low somatic mutation landscape.
Keywords: genomics, familial germ cell tumors, next generation sequencing, germline analysis
Introduction
Pediatric germ cell tumors (GCT) represent about 3% of all childhood malignancies for children less than 15 years of age; this incidence increases to about 15% in adolescence and young adulthood.1 These tumors may arise from the gonads or extra-gonadal tissue. There are five subtypes of pediatric GCT: teratoma, germinoma, yolk sac tumor (also known as endodermal sinus tumor), embryonal carcinoma, and choriocarcinoma, as well as mixed malignant subtypes.1–3 Although infantile and pediatric GCT are biologically distinct from GCT of older adolescents and adults, treatment for all GCT is essentially the same and generally grounded in large doses of platinum-based chemotherapy combined with surgery. Thus, younger patients suffer more severe side effects from treatment.2,4
Predisposition to developing ovarian and testicular teratomas has been reported in families with multiple affected members.3 While the exact mechanisms underlying GCT development remain unclear, abnormalities in the KIT-ligand (KITLG) and KRAS pathway are thought to play a role.5,6 Amplifications in chromosome 12p and X chromosome have been implicated in nonseminomatous GCT. Yolk sac tumors also show gains in chromosomes 1q, 11q, 20q, as well as gains in chromosome 22; losses in chromosome 1p, 6q, and 16q have also been reported.7
There is evidence to support that methylation plays a role in GCT biology, and that methylation patterns identified in GCT can distinguish the five sub-types.8 Based on prevailing literature and our experience with families enriched with germ cell malignancies, germline variant that predisposes a progenitor germ cell to malignant transformation or makes the microenvironment more permissive to tumor growth is likely to be a strong candidate for unraveling the complex biology of these cancers.
Next-generation sequencing (NGS) has significantly advanced our understanding of the genomic landscape of familial cancers.2 Combining deep phenotyping with deep sequencing has the potential to identify key oncogenes involved with rare and aggressive cancers. The goal of this study was to apply these techniques to 3 independent families from Finland with an extensive history of neoplasia, but no known genomic lesions. Of interest, the people of Finland have a unique ancestry marked by a population bottleneck followed by geographic isolation on a peninsula.9 Consequently, the Finnish population has an enrichment in rare genetic variants. Our study leverages the distinctive genetics of this population.
Patients and Methods
Subject Enrollment
Three families from Finland were enrolled for this study. The study was approved by the ethical committee of Helsinki University Hospital, Helsinki, Finland. Written informed consent was obtained from all subjects and/or their parents, as well as the relatives from whom the samples were obtained for this study. A total of 22 unique subjects were sequenced across the 3 families, n=4, n=11, and n=7, respectively (Supplemental Table 1). Family 1 proband had a malignant ovarian teratoma, diagnosed at 8 years of age, with a family history of ovarian teratoma (Supplemental Table 1). Family 2 proband was diagnosed with a neonatal sacrococcygeal teratoma and a subsequent yolk sac tumor at 1.5 years of age, with a family history of malignancies (NOS) (Supplemental Table 1). Family 3 had a proband with a malignant testicular teratoma, diagnosed at 1 year of age, with a family history of malignancies (NOS) (Supplemental Table 1). Peripheral blood was collected from each proband and family members.
De-identified analysis of genomic data from the consented patients was conducted at Nemours (Dr. Crowgey) with IRB research determination (IRB# 1327922–1).
Whole-Genome Sequencing (WGS) and Genome Alignment
Whole-genome sequencing was conducted at Washington University School of Medicine by the McDonnell Genome Institute. Whole-genome sequencing was conducted using the NovaSeq 6000 high-throughput sequencing platform (Illumina, San Francisco, CA) at a depth of coverage of 30X. Samples were prepared using the TruSeq whole genome library preparation (Illumina). Fastq files were processed via fastqc for data quality (Babraham Institute) and trimmed based on adaptor sequences and quality (cutadapt). Trimmed reads were mapped to the human genome hg19 via bwa mem.10 Genome analysis toolkit (GATK) best practices, base quality score recalibration, indel realignment, and duplicate read removal were followed to generate a combined variant call file (VCF) using Haplotyper for all samples analyzed.11–13
Variant Annotation and Data Analysis
VCFs were annotated using VarSeq version 2.2.0 (Golden Helix®). Specifically, variants were annotated using: ExAC Variant Frequencies 0.3, gnomAD exomes and genome variant frequencies 2.1.1,14 dbSNP 151 (http://www.ncbi.nlm.nih.gov/SNP/), Ensemble Genes 87, and functional predictions were determined via sift,15 polyphen2 HVAR,16 mutation taster,17 mutationAssessor,18 FATHMM,19 and FATHMM MKL coding. At least three of the functional predictions needed to predict the variant as damaging. Variants were filtered based on the following inheritance patterns when appropriate autosomal recessive, and de novo, using the following criteria: read depth >10, genotype qualities >20, MAF ExAC <0.01, MAF gnomAD <0.01, and effect of mutation (loss of function or missense for coding variants). The same logic was applied for non-coding variants located in introns and non-exonic (intergenic) locations.
A variant was considered as autosomal recessive for proband 1 and 2 if both the mother and father were carriers (0/1), unaffected siblings were either carriers or wildtype (0/1 or 0/0), and the proband was homozygous alternative (1/1). Additionally, for family 2 proband, the unaffected extended family members in the pedigree were used to further filter out variants if they were a homozygous alternative (1/1) genotype. Variants were considered as de novo for proband 1 and 2 if both the mother and father were homozygous wildtype (0/0), and the unaffected siblings were also homozygous wildtype (0/0). Additionally, for family 2 proband, the unaffected extended family members in the pedigree were used to further filter out variants if they were a carrier (0/1). For proband 3, unfortunately the mother’s sample was not available for sequencing. The same logic as above for probands 1 and 2 was applied, except that the mother’s parents’ genotypes were used in replacement.
Results
Family 1
Four subjects were sequenced in Family 1 pedigree: mother, father, proband (ovarian teratoma), and unaffected sibling (Figure 1). A history of ovarian teratoma was noted by the primary care physician (Supplemental Table 1); however, germline samples from these extended family members were not available for sequencing. Using an autosomal recessive analysis, three variants of interest were identified in the following genes: CD109 (rs7741152), IKBKB (rs140485496), and CTNNA3 (rs192093851) (Table 1). All of the variants identified were rare within the other Finnish families analyzed with CD109 and IKBKB alleles only identified in Family 1, and 1 allele count outside of Family 1 for rs192093851 (CTNNA3). It was noted that all three variants had a <0.01 minor allele frequency (MAF) within the gnomAD database; however, they were >0.01 within the Finnish-specific genomes within gnomAD (Table 1). Inhibitor of NF-κB kinase subunit beta (IKBKB) is associated with immunodeficiency disorders (IMD15B and 15A) and is a serine kinase that plays a key role in the NF-κB signaling pathway. Gene variants and expression data have linked IKBKB with prostate and ovarian cancers.20 Of interest, IKBKB has been associated with KRAS, which is a known oncogene linked to GCTs.21
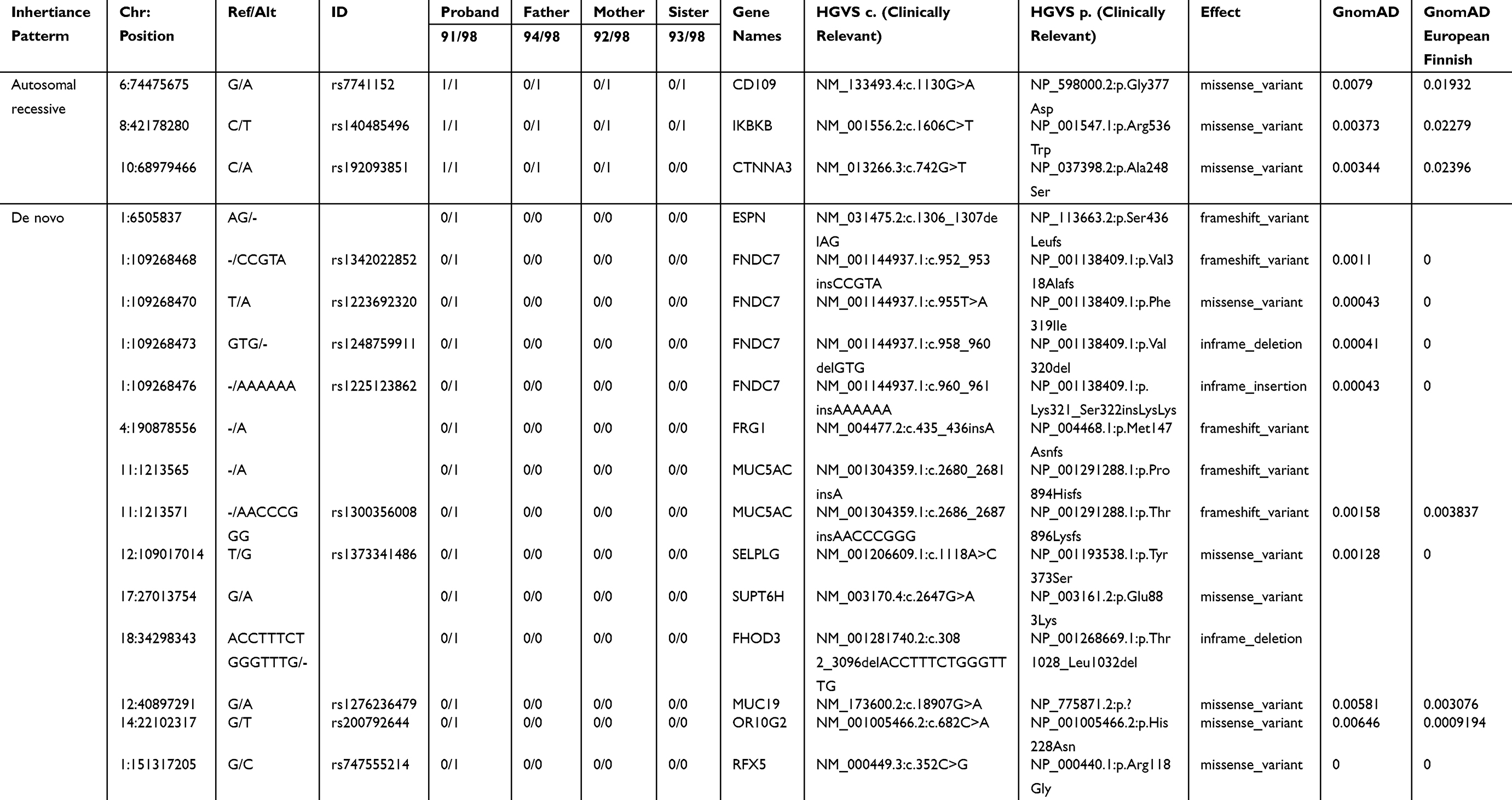 |
Table 1 Rare Autosomal Recessive and De Novo Variants Identified in Family 1 Proband |
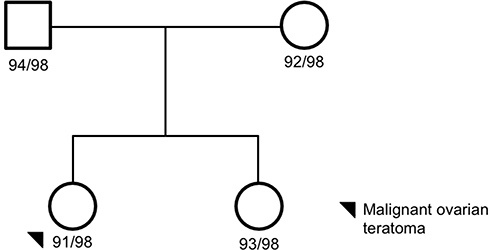 |
Figure 1 Family 1 pedigree. Four family members were sequenced via whole-genome sequencing: proband, mom, dada, and unaffected sister. The proband, black circle, was a malignant ovarian teratoma. Females are represented as circles; males are represented as squares. The number next to the pedigree represents the de-identified subject ID. |
The de novo analysis identified 14 rare variants within proband one across 10 unique genes (Table 1). Nine of the variants had known RefSeq (rs#) identification numbers in the dbSNP database and MAF of >0.01 in the gnomAD genome database as well as within the Finnish-specific genomes within gnomAD (Table 1). Of interest, SUPT6H, a histone chaperone, has previously been shown to control estrogen-related transcription and linked to cancer.22 Additionally, MUC5AC has been associated with ovarian tumors23 and FRG1 expression levels have been shown to be aberrant in several cancers.
Family 2
Eleven subjects were sequenced in Family 2 pedigree: proband (sacrococcygeal teratoma – yolk sac tumor), unaffected brother with a benign sacrococcygeal cyst, unaffected sister, mother, father, two paternal aunts, paternal grandmother, 2 paternal great aunts, and a paternal cousin (Supplemental Table 1; Figure 2). Of interest, the unaffected brother and nephew (unaffected sister) have a history of benign dermoid cysts (sacrococcygeal and ophthalmic area). Furthermore, the proband’s great grandfather (maternal side) had prostate cancer, and a great grandmother with stomach cancer, who has a daughter with leukemia and a granddaughter with cancer. An autosomal recessive analysis identified two rare variants in the following genes LONRF2 (rs116702638) and ANO7 (rs148609049) (Table 2). It was noted that all three variants had a <0.01 MAF within the gnomAD database; however, they were >0.01 within the Finnish-specific genomes within gnomAD (Table 1). Variant rs148609049 has previously been published as a germline variant that associates with significantly shorter survival rates in prostate cancer subjects.24 Aberrant methylation patterns for LON peptidase N-terminal domain and RING finger protein 2 (LONRF2) have been noted in rectal adenocarcinoma. Of interest, ANO7 is associated with aggressive prostate cancer.24
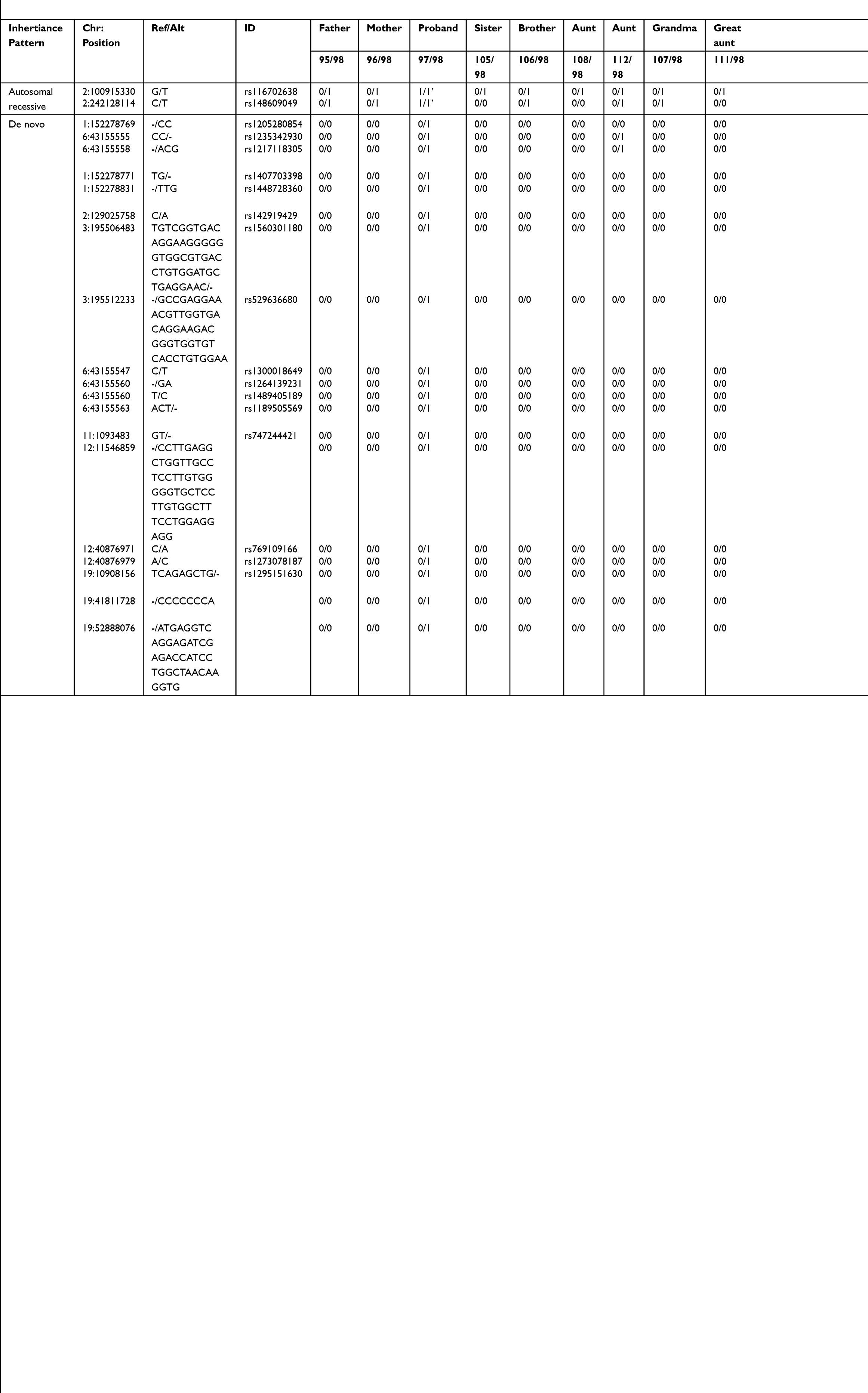 |
Table 2 Rare Autosomal Recessive and De Novo Variants Identified in Family 2 Proband |
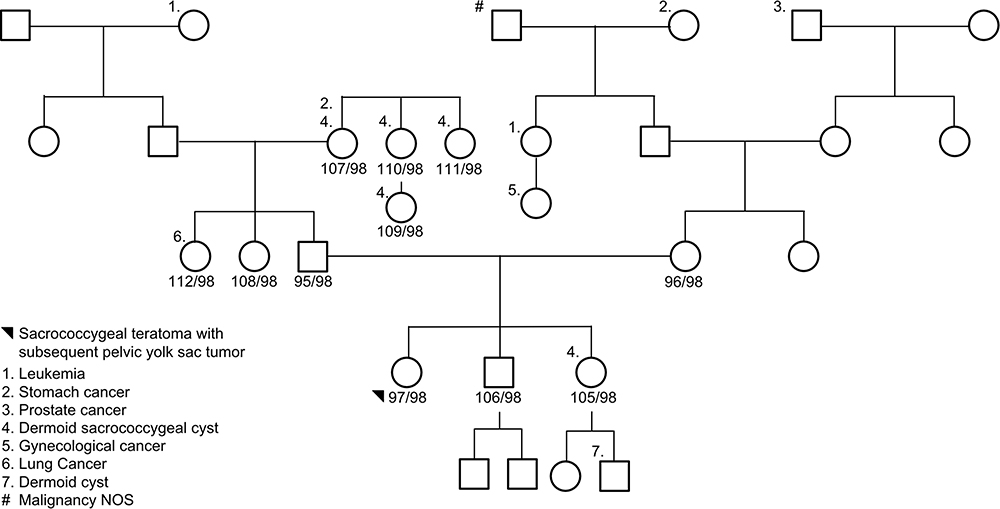 |
Figure 2 Family 2 pedigree. Eleven subjects were sequenced via whole-genome sequencing: proband, unaffected brother, unaffected sister, mother, father, two paternal aunts, paternal grandmother, 2 paternal great aunts, and a paternal cousin. The proband, black circle, was a sacrococcygeal teratoma – yolk sac tumor. Females are represented as circles; males are represented as squares. The number next to the pedigree represents the de-identified subject ID. |
The de novo analysis identified 19 rare variants within proband two across 10 unique genes (Table 2). Sixteen of the variants were found in dbSNP with MAF of >0.01 in the gnomAD genome database and within the Finnish-specific genomes within gnomAD (Table 2). Of interest HS6ST1, PRB2, and DNM2 have been previously linked to cancers.25,26
Family 3
Seven subjects were sequenced in Family 3 pedigree: proband (malignant testicular teratoma), un-affected siblings, dad, uncle (maternal), grandparents (maternal) (Supplemental Table 1; Figure 3). Two InDels were identified in this family using an autosomal recessive pattern in the following genes (Table 3): CRIPAK and KRTAP5-7. CRIPAK encodes for a cysteine-rich PAK1 inhibitor protein that negatively regulates PAK1 expression. PAKs are a family of proteins that are involved in cytoskeletal dynamics, cell survival and proliferation and have been well associated with cancers.27 The de novo analysis identified 22 rare variants in 17 unique genes. Aberrant expression levels of CACNA1B have previously been associated with cancers.28 Of interest, TXNDC2 is solely expressed in testis and is predicted to regulate disulfide bonds, and TFE3 is a transcription factor linked to cancers and is ubiquitous in fetal and adult tissues.
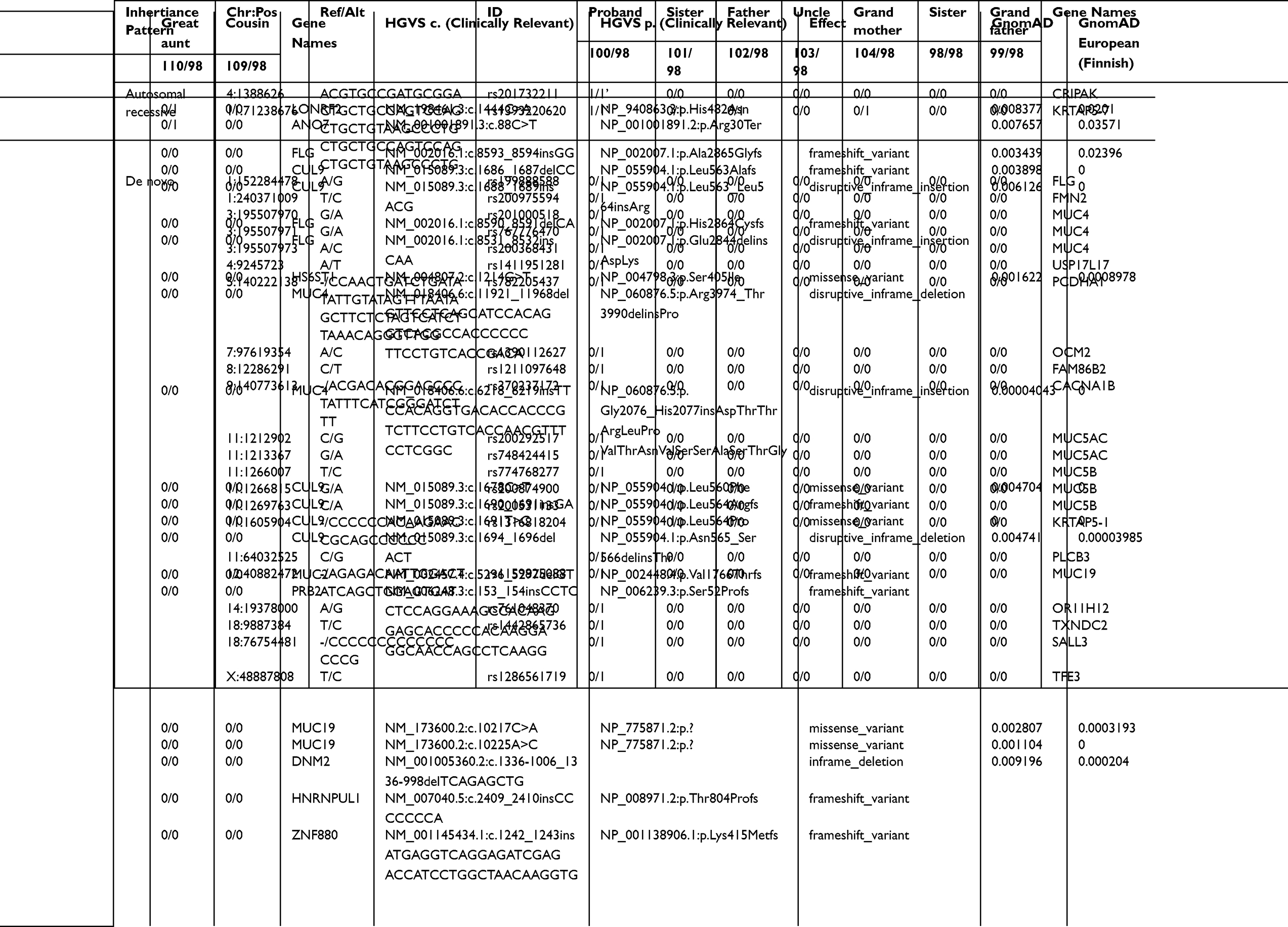 |
Table 3 Autosomal Recessive and De Novo Variants Identified in Family 3 Proband |
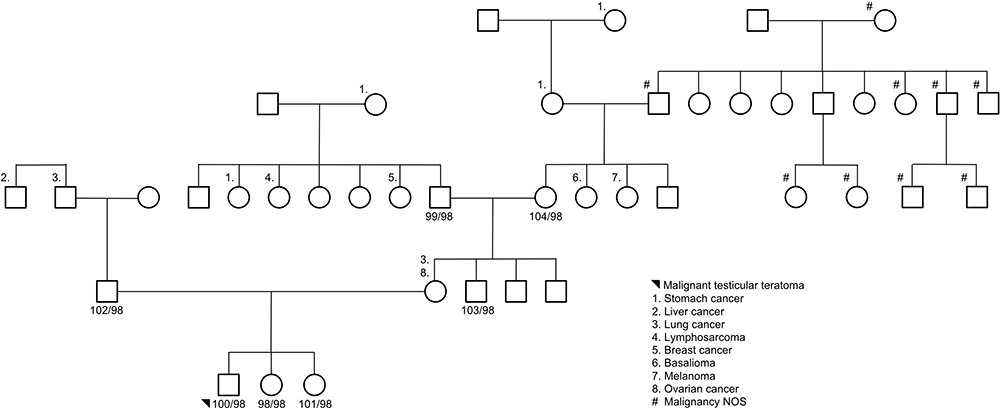 |
Figure 3 Family 3 pedigree. Seven subjects were sequenced via whole-genome sequencing: proband, unaffected siblings, data, maternal uncle, and maternal grandparents. Females are represented as circles; males are represented as squares. The number next to the pedigree represents the de-identified subject ID. |
Non-Coding Variant Analysis
Rare variants (gnomAD MAF <0.01) in non-coding regions of the genome were analyzed, introns or intergenic regions. Heterozygous variants were only considered for a proband if the parents and unaffected siblings were homozygous wildtype (0/0), and homozygous variants were only considered for a proband if the parents and unaffected siblings were homozygous wildtype or heterozygous (0/1). A variant was considered as known if it was listed in dbSNP 151. The range of intronic variants detected per proband was 2526–3439 (left side, Figure 4), with the majority of variants per proband were known heterozygous. The range of intergenic variants detected was 8320–10826 (right side, Figure 4), with the majority of the variants also being known heterozygous.
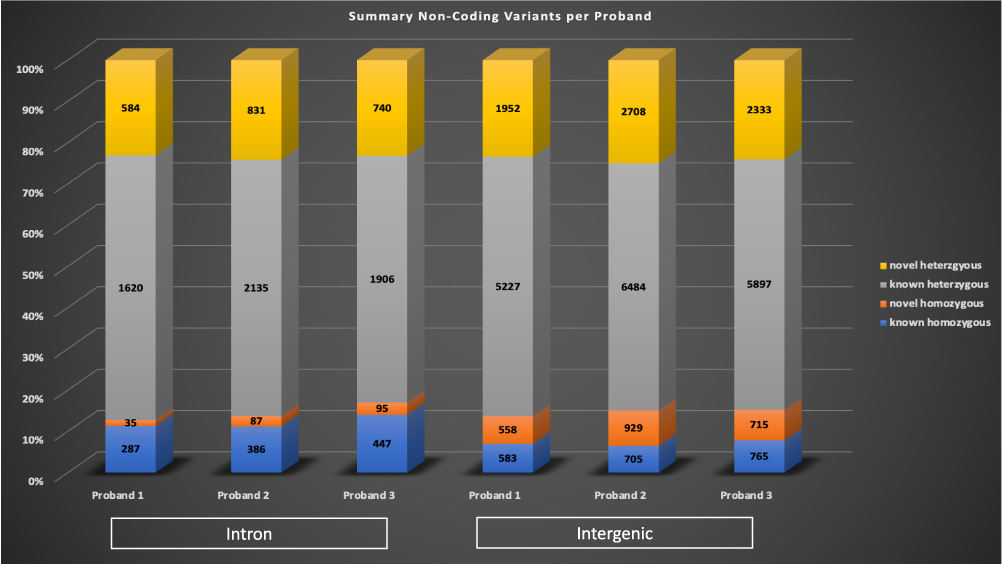 |
Figure 4 Non-coding variants identified in the probands. Rare non-coding variants were identified in introns (left bars) and non-exonic regions (intergenic, right bars) per each proband. A variant was considered to be known if it was listed in dbSNP 151. Known homozygous variants are in blue, novel homozygous variants are in orange, known heterozygous variants are in grey, and novel heterozygous variants are in yellow. |
Discussion
GCT are primarily sporadic cancers in early childhood or adolescence with a variety of subtypes. Thus, the mechanisms involved in GCT are poorly understood. The identification of families with relatively little admixture and multiple GCT cases offers an opportunity to identify novel genes that may drive or modulate tumor onset and subtype. In this study, we performed WGS on three such non-consanguinous families from Finland, where the proband in each family had a different GCT subtype. The NGS analysis revealed different genes with an autosomal recessive or de novo inheritance pattern in each family.
Family 1, proband with malignant ovarian teratoma, analysis yielded three potential autosomal recessive variants of interest in the following genes: CD109, IKBKB, and CTNNA3. Although variants had a MAF <0.01 in the gnomAD genome database, they did have higher MAF in the Finnish-specific genome data. We hypothesize that germline variants might cause a predisposition, but these higher allele frequencies in the Finnish population indicate that a second hit/additional variant in combination would be necessary. CD109 is highly associated with cancers29 and aberrant expression patterns are associated with squamous cell carcinoma.30 IKBKB is a potent activator of the NF-kB pathway, which plays a major regulatory role during developmental transitions and the ability of KRAS to transform tissues with aberrant NF-κB signaling is well established.31 Additionally, dysregulation of microRNAs that target IKBKB has been associated with ovarian tumors.32 The de novo variant analysis identified rare variants within the gnomAD Finnish data, with the most interesting gene candidates being SUPT6H, a histone chaperone, MUC5AC has been associated with ovarian tumors,23 and FRG1.
An autosomal recessive analysis for family 2 yielded two potential variants of interest in the following genes: LONRF2, and ANO7. ANO7 encodes for the protein anoctamin-7, which has calcium-dependent phospholipid scramblase activity. The variant detected is a loss of function variant resulting in a stop gain at amino acid 30 in the translated protein and has previously been published as a germline variant that associates with significantly shorter survival rates in prostate cancer subjects.24 The de novo analysis identified rare variants within the gnomAD Finnish data and included HS6ST1, PRB2, and DNM2, which all have been previously linked to cancers.25,26
Family 3 had variants of interest identified in CRIPAK and KRTAP5-7. CRIPAK is a novel interacting partner of PAK1.33 PAK1, also referred to as p21-activated kinase 1, is a complex gene involved in many cellular signaling pathways including mitosis. Recent studies have described PAK1 has a relevant oncogene in ovarian carcinoma34 and breast carcinoma,35 phenotypes recorded within this family history. However, the pedigree and variant results for Family 3’s proband suggest a potential sporadic case of a neoplasm. A de novo analysis did identify rare variants in TXNDC2, which is solely expressed in testis and is predicted to regulate disulfide bonds, and TFE3, a transcription factor linked to pediatric cancers that is ubiquitously expressed in fetal and adult tissues.
A current limitation of this study is the lack of functional studies, which are required to further validate the biological significance of the variants identified. It is challenging to functionally validate these types of complex variants associated with a complex phenotype, as models are difficult to establish. Unfortunately, tumor testing, which looks for abnormalities in cancer cells, was not conducted at the time of diagnosis for the probands. These data could have provided additional insight for interpreting the germline variants identified. Furthermore, the non-coding variant analysis identified numerous variants that are difficult to interpret without additional functional testing.
Abbreviations
GCT, germ cell tumors; NGS, next-generation sequencing; WGS, whole-genome sequencing; GATK, genome analysis toolkit; VCF, variant call file; MAF, minor allele frequency; SISu, Sequencing Initiative Suomi.
Acknowledgments
The authors would like to thank the families for participating in this research effort. Furthermore, the authors acknowledge support from the McDonnell Genome Institute at Washington University for conducting the next-generation sequencing, and Krishna Kanchi for providing bioinformatics support for the raw data.
Author Contributions
All authors contributed to data analysis, drafting and/or revising the article, gave final approval of the version to be published, and agreed to be accountable for all aspects of the work.
Funding
The authors acknowledge grant support to MH via Sigrid Jusélius Foundation, Leukemia Research Foundation of Delaware and the National Institute of General Medical Sciences of the National Institutes of Health grant number U54-GM104941 (PI: Binder-Macleod) for supporting ELC, and the Kellsie’s Hope Foundation to TED.
Disclosure
Dr Todd E Druley reports ownership, salary from ArcherDX, Inc., outside the submitted work. In addition, Dr Todd E Druley has a patent #62/106,967 pending to Canopy Biosciences. The authors report no other conflicts of interest in this work.
References
1. Olson TA, Murray MJ, Rodriguez-galindo C, et al. Pediatric and adolescent extracranial germ cell tumors: the road to collaboration. J Clin Oncol. 2015;33(27):3018–3028. doi:10.1200/JCO.2014.60.5337
2. Pierce JL, Frazier AL, Amatruda JF. Pediatric germ cell tumors: a developmental perspective. Adv Urol. 2018;2018. doi:10.1155/2018/9059382.
3. Variants in BAK1, SPRY4, and GAB2 are associated with pediatric germ cell tumors: a report from the Children’s Oncology GroupHHS Public Access: Genes Chromosome Cancer. 2018;56(7):612–625. doi:10.1002/gcc.22457.Variants
4. Acharya S, Dewees T, Shinohara ET, Perkins SM. Long-term outcomes and late effects for childhood and young adulthood intracranial germinomas. Neuro-Oncology. 2015;17(5):741–746. doi:10.1093/neuonc/nou311
5. Ferlin A, Pengo M, Pizzol D, Carraro U, Frigo AC, Foresta C. Variants in KITLG predispose to testicular germ cell cancer independently from spermatogenic function. Endocr Relat Cancer. 2012;101–108. doi:10.1530/ERC-11-0340
6. Olie RA, Looijenga LHJ, Boerrigter L, Top B, Rodenhuis S, Langeveld A. N- and KRAS mutations in primary testicular germ cell tumors: incidence and possible biological implications. Genes Chromosomes Cancer. 1995;116:110–116.
7. Mostert M, Rosenberg C, Stoop H, Schuyer M, Timmer A. Comparative genomic and in situ hybridization of germ cell tumors of the infantile testis. Lab Invest. 2000;80(7):1055–1064.
8. Rijlaarsdam MA, Tax DMJ, Gillis AJM, Dorssers LCJ. Genome wide DNA methylation profiles provide clues to the origin and pathogenesis of germ cell tumors. PLoS One. 2015;1–34. doi:10.1371/journal.pone.0122146
9. Chheda H, Palta P, Pirinen M, et al. Whole-genome view of the consequences of a population bottleneck using 2926 genome sequences from Finland and United Kingdom. Nat Publ Gr. 2017;25(4):477–484. doi:10.1038/ejhg.2016.205
10. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi:10.1093/bioinformatics/btp324
11. Auwera GA, Carneiro MO, Hartl C, et al. From FastQ data to high‐confidence variant calls: the genome analysis toolkit best practices pipeline. Curr Protoc Bioinformatics. 2013;43(1). doi:10.1002/0471250953.bi1110s43
12. DePristo MA, Banks E, Poplin R, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43(5):491–498. doi:10.1038/ng.806
13. McKenna A, Hanna M, Banks E, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–1303. doi:10.1101/gr.107524.110
14. Karczewski KJ, Francioli LC, Tiao G, et al. Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of- function intolerance across human protein-coding genes. BioRxiv. 2019:531210.
15. Sim N-L, Kumar P, Hu J, Henikoff S, Schneider G, Ng PC. SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012;40(W1):W452–W457. doi:10.1093/nar/gks539
16. Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–249. doi:10.1038/nmeth0410-248
17. Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11(4):361–362. doi:10.1038/nmeth.2890
18. Reva B, Antipin Y, Sander C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res. 2011;39(17):e118. doi:10.1093/nar/gkr407
19. Rogers MF, Shihab HA, Mort M, Cooper DN, Gaunt TR, Campbell C. FATHMM-XF: accurate prediction of pathogenic point mutations via extended features. Hancock J, ed. Bioinformatics. 2018;34(3):511–513. doi:10.1093/bioinformatics/btx536
20. Page A, Navarro M. Context-dependent role of IKK β in cancer. Genes. 2017;8(12):376. doi:10.3390/genes8120376
21. Hacioglu BM, Kodaz H, Erdogan B, Cinkaya A, Tastekin E. K-RAS and N-RAS mutations in testicular germ cell tumors. Bosn J Basic Med Sci. 2017;17:159–163.
22. Bedi U, Scheel AH, Hennion M, Begus-Nahrmann Y, Rüschoff J, Johnsen SA. SUPT6H controls estrogen receptor activity and cellular differentiation by multiple epigenomic mechanisms. Oncogene. 2015;34(4):465–473. doi:10.1038/onc.2013.558
23. Hirabayashi K, Yasuda M, Kajiwara H, et al. Alterations in mucin expression in ovarian mucinous tumors: immunohistochemical analysis of MUC2, MUC5AC, MUC6, and CD10 expression. Acta Histochem Cytochem. 2008;41(2):15–21. doi:10.1267/ahc.08008
24. Kaikkonen E, Rantapero T, Zhang Q, et al. ANO7 is associated with aggressive prostate cancer. Int J Cancer. 2018;143(10):2479–2487. doi:10.1002/ijc.31746
25. Backen AC, Cole CL, Lau SC, et al. Heparan sulphate synthetic and editing enzymes in ovarian cancer. Br J Cancer. 2007;96(10):1544–1548. doi:10.1038/sj.bjc.6603747
26. Joshi HP, Subramanian IV, Schnettler EK, et al. Dynamin 2 along with microRNA-199a reciprocally regulate hypoxia-inducible factors and ovarian cancer metastasis. Proc Natl Acad Sci U S A. 2014;111(14):5331–5336. doi:10.1073/pnas.1317242111
27. Dummler, Bettina et al. “Pak protein kinases and their role in cancer.” Cancer metastasis reviews. 2009;28(1-2):51–63. doi:10.1007/s10555-008-9168-1
28. Phan NN, Wang C-Y, Chen C-F, Sun Z, Lai M-D, Lin Y-C. Voltage-gated calcium channels: novel targets for cancer therapy. Oncol Lett. 2017;14(2):2059–2074. doi:10.3892/ol.2017.6457
29. Hashimoto M, Ichihara M, Watanabe T, et al. Expression of CD109 in human cancer. Oncogene. 2004:3716–3720. doi:10.1038/sj.onc.1207418.
30. Qi R, Dong F, Liu Q, Murakumo Y, Liu J. CD109 and squamous cell carcinoma. J Transl Med. 2018;1–8. doi:10.1186/s12967-018-1461-3
31. Kitajima S, Thummalapalli R, Barbie DA. Seminars in cell & developmental biology inflammation as a driver and vulnerability of KRAS mediated oncogenesis. Semin Cell Dev Biol. 2016;58:127–135. doi:10.1016/j.semcdb.2016.06.009
32. Edson MA, Nagaraja AK, Matzuk MM. The mammalian ovary from genesis to revelation. Endocr Rev. 2009;30:624–712. doi:10.1210/er.2009-0012
33. Talukder AH, Meng Q, Kumar R. CRIPak, a novel endogenous Pak1 inhibitor. Oncogene. 2006;1311–1319. doi:10.1038/sj.onc.1209172
34. Schraml P, Schwerdtfeger G, Burkhalter F, et al. Combined array comparative genomic hybridization and tissue microarray analysis suggest PAK1 at 11q13. 5-q14 as a critical oncogene target in ovarian carcinoma. Ame J Pathol. 2003;163(3):985–992.
35. Ong CC, Jubb AM, Haverty PM, et al. apoptosis of tumor cells. Proc Natl Acad Sci. 2011;1(18):2–7. doi:10.1073/pnas.1103350108
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.