Back to Journals » Clinical Ophthalmology » Volume 9
Geographic atrophy in patients with advanced dry age-related macular degeneration: current challenges and future prospects
Authors Danis RP, Lavine J, Domalpally A
Received 14 July 2015
Accepted for publication 7 October 2015
Published 20 November 2015 Volume 2015:9 Pages 2159—2174
DOI https://doi.org/10.2147/OPTH.S92359
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Scott Fraser
Ronald P Danis, Jeremy A Lavine, Amitha Domalpally
Department of Ophthalmology and Visual Sciences, University of Wisconsin-Madison, Madison, WI, USA
Abstract: Geographic atrophy (GA) of the retinal pigment epithelium (RPE) is a devastating complication of age-related macular degeneration (AMD). GA may be classified as drusen-related (drusen-associated GA) or neovascularization-related (neovascular-associated GA). Drusen-related GA remains a large public health concern due to the burden of blindness it produces, but pathophysiology of the condition is obscure and there are no proven treatment options. Genotyping, cell biology, and clinical imaging point to upregulation of parainflammatory pathways, oxidative stress, and choroidal sclerosis as contributors, among other factors. Onset and monitoring of progression is accomplished through clinical imaging instrumentation such as optical coherence tomography, photography, and autofluorescence, which are the tools most helpful in determining end points for clinical trials at present. A number of treatment approaches with diverse targets are in development at this time, some of which are in human clinical trials. Neovascular-associated GA is a consequence of RPE loss after development of neovascular AMD. The neovascular process leads to a plethora of cellular stresses such as ischemia, inflammation, and dramatic changes in cell environment that further taxes RPE cells already dysfunctional from drusen-associated changes. GA may therefore develop secondary to the neovascular process de novo or preexisting drusen-associated GA may continue to worsen with the development of neovascular AMD. Neovascular-associated GA is a prominent cause of continued vision loss in patients with otherwise successfully treated neovascular AMD. Clearly, treatment with vascular endothelial growth factor (VEGF) inhibitors early in the course of the neovascular disease is of great clinical benefit. However, there is a rationale and some suggestive evidence that anti-VEGF agents themselves could be toxic to RPE and enhance neovascular-associated GA. The increasing prevalence of legal blindness from this condition due to the aging of the general population lends urgency to the search for a therapy to ameliorate GA.
Keywords: age-related macular degeneration, geographic atrophy, fundus autofluorescence, optical coherence tomography, clinical trials
Background
The global societal burden of age-related macular degeneration (AMD) is huge due to the disabling vision loss that may ensue. A meta-analysis of the global prevalence of any stage of AMD among 129,664 individuals was 8.69% among those aged 45–85 years.1 Projections of the global prevalence of AMD in 2020 are as high as 196 million. AMD may be classified as early, intermediate, or late based upon features identified by examination or color photographs of the macula.2,3 Late AMD, consisting of geographic atrophy (GA) and neovascular AMD forms, had a prevalence of 0.37% rising to 0.5% among European populations. In the Beaver Dam Eye Study, 42% of subjects with GA were legally blind (visual acuity [VA] ≤20/200 in the worst eye).4 Patients with good acuity frequently have severe visual dysfunction from development of scotomas (blind spots) that interfere with visual tasks out of proportion to performance on standard VA testing.5 Among populations in the US6 and UK,7,8 the proportion of individuals with GA and neovascular AMD is about equal among late AMD patients. While the incidence of neovascular AMD is higher, GA occurs more commonly in individuals over 85 years of age. There is no exclusivity between GA and neovascular AMD, and both forms of the disease may develop in the same eye. The risk factors for development of one form or the other are not readily distinguishable, and the precise mechanisms driving the course of an individual eye toward GA versus neovascular AMD remain to be elucidated. Current thinking is that both late manifestations are initiated by common pathways that culminate in stress and damage to the retinal pigment epithelium (RPE), Bruch’s membrane, and choroid, and can result in either RPE degeneration or choroidal neovascularization with subsequent RPE atrophy. With aging, gradual and cumulative damage to the retina occurs through various factors, including oxidative stress, parainflammatory dysregulation, and vascular sclerosis. Impairment of normal physiologic function of the RPE cells, including the constant degradation of lipid-rich outer photoreceptor discs, can lead to formation of intracellular (lipofuscin) and extracellular (drusen) deposition of toxic debris. Interestingly, while use of nutritional supplements significantly slowed the overall development to late AMD among participants in the Age-Related Eye Disease Study (AREDS), this appeared to be primarily observed due to prevention of development of neovascular AMD with no significant effect on incidence of GA.9,10
GA refers to the clinical appearance of nearly complete loss of the RPE cells under the retina, often with a sharp demarcation between relatively healthy RPE and the region denuded of these cells (hence the term “geographic”) as observed on a color photograph. The term was originally applied by Gass11 to the late manifestation of AMD where one or more circumscribed areas of atrophy develop, slowly enlarge and coalesce over time with eventual involvement of the fovea. Atrophy of the RPE may also be subcategorized as drusen-associated and neovascularization-associated.12,13 Drusen-associated GA arises from the AMD process in which RPE cell dropout develops but neovascularization does not occur. In contrast, in neovascularization-associated atrophy, the RPE loss is at least in part related to cellular damage related to the fibrovascular proliferation that occurs in this type of AMD. Neovascular-associated RPE atrophy has also been called GA in the past because in many cases of neovascular atrophy the clinical appearance can be identical to drusen-associated GA. However, herein we use the terminology neovascular-associated RPE atrophy to avoid confusion. In the past, neovascular-associated atrophy has not been a prominent topic of clinical interest, but has become more so as development of agents that abort the neovascularization process (anti-vascular endothelial growth factor [VEGF] agents) result in eyes that no longer have ongoing bleeding and fluid leakage but suffer from a gradual loss of RPE cells resulting in further vision loss.
Drusen-associated GA
Drusen-associated GA can result in severe irreversible central vision loss due to the formation of dense scotomas resulting in deficits in reading and many other visual tasks common in daily life.14,15 The decline in vision may be minor in early stages if the fovea is spared, but later becomes severe with central involvement. Often, the GA will begin with one or more regions of RPE dropout that are near but not in the fovea (Figure 1). Patients with GA, even in late stages, may maintain good central vision over some time, but visual dysfunction is underestimated until the disease progression involves the central fovea and there is acuity loss. The peripheral spread is typically faster than toward the center for unknown reasons.16 There is loss of measurable retinal sensitivity in regions of GA,17 and patients may notice paracentral scotomas development.15,18,19 Expansion of the atrophic regions inevitably occurs, sometimes with multiple regions merging together and eventually invading the fovea (Figure 2). VA as measured on eye charts does not correlate well with GA lesions or progression, prompting the development of alternative end points for clinical research to test therapeutic agents.
 | Figure 1 Right eye of patient with drusen-associated GA imaged by color photograph (A), fundus autofluorescence (B), infrared reflectance (C), and OCT (D). |
 | Figure 2 The same eye as shown in Figure 1 after 1 year. |
Genetic risk factors
AMD is a complex and multifaceted disease involving contributions from both genetic and environmental influences. AMD may be classified as early, intermediate, or late based upon features identified by examination or color photographs of the macula.2 The late AMD phenotypes are time-dependent variables because they develop from intermediate stages. Therefore, age-matched control populations must be carefully selected for genotypic comparison. Previous work exploring the genetic contributions of AMD has implicated numerous genomic regions and a variety of candidate genes as modulators of AMD susceptibility. Common variants explain approximately 50%–70% of the heritability of AMD. Single nucleotide polymorphisms of genes involved in the complement activation cascade have consistently been implicated in AMD pathogenesis with CFH, C3, CFB, and C2 the most prominent.20–22 There is less association for the involvement of pathways related to extracellular matrix remodeling and lipid metabolism. For most AMD-associated risk loci, the true causative risk variants as well as the affected genes are unknown.23 Mechanisms other than those related to common single nucleotide polymorphisms found with genomic studies, such as rare variants, copy number variations, epigenetics, and mitochondrial inheritance may contribute to the missing heritability. These factors are relatively unexplored and difficult to study.
Some groups have sought genetic polymorphisms that particularly predispose individuals to drusen-associated GA versus neovascular AMD. The common susceptibility loci found in many studies are general risk factors for both subtypes. Using AREDS data, the homozygosity of allelic polymorphism LOC387715/ARMS2/HTRA1 genotype had a nominal association with GA progression.21 A subsequent study pooling participants in the AREDS with the Fundus Autofluorescence in Age-related Macular Degeneration Study, a multicenter study conducted in Germany, confirmed this association with what is now known as the ARMS2_rs10490924 risk allele and also indicated an independent association with C3_rs2230199.24 A small case-control study suggested that some CFH alleles appeared to be associated with GA and that CFB-32Gln might be associated with a more aggressive GA phenotype.25 Overall, there has been little discriminatory value thus far for genotypic risk factors specific to GA manifestations.
Pathophysiology
Most known risk alleles are related to factors that affect regulation of the alternative complement pathway. The exact mechanism by which the alternative complement pathway causes AMD is still unknown. Histopathological studies of AMD patients identify complement inhibitors, component C3, and component C5 within drusen.26,27 Factor H is anchored to Bruch’s membrane by heparan sulfate glycosaminoglycans, which decline by ~50% with aging.28 A model has been proposed where aging leads to decreased anchoring of complement inhibitors to Bruch’s membrane, causing localized complement activation. Complement activation leads to drusen formation, release of proinflammatory anaphylatoxins such as C3a and C5a, and membrane attack complex-driven tissue destruction. Based upon histopathological studies, a model has been advanced where localized inflammation and death of the RPE causes GA, while loss of choriocapillaris stimulates RPE ischemia, VEGF production, and choroidal neovascularization.29 GA has been associated with activation of cells endogenous to the retina, such as microglia cells, Muller cells, and RPE cells. In the choroid, a wide spectrum of leukocytes are resident along with pericapillary macrophages, giant cells, and mast cells.30 Drusen, which largely consist of lipoprotein particles and RPE cell remnants, also consist of a number of inflammatory proteins, including apolipoprotein E, coagulation proteins, acute phase proteins, immunoglobulin G, complement components, and complement activators all of which promote a toxic milieu.31,32 Studies have also examined the role of DICER1 deficiency and its role in development of GA.22 In patients with GA, DICER1 levels are greatly reduced, and this leads to accumulation of a toxic double-stranded RNA called Alu in the RPE. With regards to models of exudative AMD, inhibition of complement components C3, C5, or Factor B reduces choroidal neovascularization in the laser-induced mouse model.33–35 However, there are no good animal models to test the role of complement in GA formation.
Ocular risk factors
The pathology of AMD is characterized by thinning of the photoreceptor layer of the retina, variable atrophy and other changes of the RPE, thickening of Bruch’s membrane, drusen formation, and decreased density of the choriocapillaris layer. There is considerable phenotypic variability of the disease, related no doubt to the diverse genotypic contributions, stage of the disease, and environmental influences. In most cases of drusen-associated GA, there is a gradual fading or collapse of large drusen with overlying loss of pigmentation of the RPE as well as pigment migration into the neuroretina.12,13,29 This intraretinal pigment is likely from RPE cells dissociated from Bruch’s membrane.36,37 There is gradual thinning of the photoreceptor layers, manifesting first as shortening of the outer segments, followed eventually by cell death and subsequent thinning of the outer nuclear layer of the retina.13,38 Finally, loss of the RPE occurs which, congruent with photoreceptor layer atrophy, may lead to the appearance of the outer plexiform layer against Bruch’s membrane. Detailed pathological examination shows that the choriocapillary vessels become thinner and the layer becomes less dense along with atrophy of the neuroretinal tissue and RPE. The photoreceptors, RPE, Bruch’s membrane, and choriocapillaris have together been described as a functionally integrated complex with a “mutualistic symbiotic relationship”.29 These layers are so interdependent anatomically and functionally that there can be no damage to one layer without corresponding dysfunction and disruption of the other layers. Consequently, there may be no primary insult leading to atrophy but rather a concerted deterioration affecting the entire complex. There is discussion as to the relative contributions of choriocapillary and choroidal sclerosis, Bruch’s membrane thickening by basal linear deposits, drusen formation/basal laminar deposits, subretinal drusenoid material, and dysfunction of the photoreceptor outer segments.
Clinically and by color fundus photography, the development of GA mirrors the appearance of the lesions pathologically. The photographic definition of GA is at least two out of three of the following characteristics: sharp demarcation, round or ovoid shape, visibility of the choroidal vessels (Figure 1). Typically, large confluent drusen eventually develop overlying pigment clumping with fading of the drusen, accompanying loss of RPE pigment to eventually produce a region of pigment loss with relatively sharp margins, revealing the underlying choroidal vessels.39–41 In the AREDS, the time from drusen confluence to GA averaged 5.9 years, and the time from development of hyperpigmentation was 2.5 years.39 The prediction of development of GA is much greater based upon local ocular features, such as drusen area, hypopigmentation of the RPE, and pigment clumps rather than genetic and demographic risk factors.20 Quantification of AMD fundus photographic features has been proposed by the AREDS group based upon risk of progression to late AMD outcome.42 This scale is based upon reading center grading of fundus photographs as a research tool and is far too detailed for clinical use, but does appear to have predictive value in other research populations.43 The risk of progression to central GA over 5 years ranges from 12% to 20% among eyes with drusen area more than half disc area (1.27 mm2) and depigmentation more than questionably present.42 Progression from noncentral to central GA occurs in 45% of eyes over 5 years. An alternate, highly simplified version for use by clinicians has also been developed by the AREDS group44 and appears to have good utility and demonstrates predictive value in other study populations.43 A feature not taken into account by the AREDS scales is the presence of what has been variably called reticular pseudodrusen and subretinal drusenoid deposits. These have the appearance of drusen-like dots and ribbons that may coalesce into a continuous sheet. There is evidence that subretinal drusenoid deposits, which differ from typical drusen in that their location is between the RPE and photoreceptor layers, may have a strong association with development of late AMD, particularly with GA.45–47 A spatiotemporal relationship between these deposit and GA has been suggested, which also suggests that these deposits may typify at least one pathway that leads to GA. A two-compartment model whereby these deposits are related to disturbed outer segment lipid homeostasis has been proposed.36 Subretinal drusenoid deposits have also been suggested to be a marker for choroidal and choriocapillary atrophy and may be related to the vascular component of degenerative changes associated with GA.45
Origin of the autofluorescence signal
An important observation associated with development of GA is the variation in the autofluorescence associated with the accumulation of lipofuscin with age.48 The natural autofluorescence of the fundus that is excited by 488–535 nm light has spectral features and an age-relationship that indicates a principle origin from the fluorescent pigments that accumulate in RPE cells as lipofuscin.49,50 Lipofuscin is produced in the membranes of photoreceptor outer segments from nonenzymatic reactions of vitamin A aldehyde which is transferred to RPE cells within phagocytosed outer segment disks and becomes deposited in the lysosomal compartment of the cells as compound A2E and related compounds. Fundus autofluorescence (FAF) naturally increases with age, although subjects vary widely in terms of intensities. Lipofuscin is an amalgam of lipoprotein compounds that are related to the accumulation of unreduced retinoids, primarily A2E, which produce reactive oxygen species through photoactivation that can then react with and inactivate cellular constituents. These reactions lead to the formation of advanced glycation end products that in turn incite inflammatory processes. Because advanced glycation end products are detected in drusen, they reflect a link between lipofuscin and the formation of drusen. Photooxidation of A2E stimulates complement activation, which fits with the genotypic risk factors of genes coding for proteins that regulate the complement cascade.49
On clinical imaging, it can be observed that eyes with GA usually have alterations of autofluorescence around them, particularly with blue light excitation (Figure 1). It has also been noted that certain patterns of autofluorescence abnormality have some predictive value for the development of GA.51 This variation in the autofluorescence signal is probably related to disorganization of the RPE with degenerative processes.52
Clinical imaging assessment of GA
Unlike in neovascular AMD where vision loss tends to be rapid, drusen-associated GA more often presents with gradual decline in visual function. Patients may harbor enlarging GA lesions for years before VA is affected. Usually, severe VA loss does not occur until development of central GA.13 GA progression has long been noted to be highly variable between individuals and to have a poor association with VA. In many patients, the GA tends to skirt around the fovea with some preferential sparing of central acuity until late in the disease. Fixation often becomes unsteady as atrophy approaches the center such that there may be an island of functional RPE bordered completely by atrophy, thus leaving the patients with a ring-shaped scotoma. Morphologic changes occur relatively slowly, and use of VA as an end point may require years for changes to manifest (Figures 1 and 2). Other visual function tests may be early markers for risk of GA or GA progression. Dark adaptation, microperimetry, and visual function questionnaires have been advanced as potential end points for clinical prognostication and clinical trials, but these tests rely on subjective patient responses, engender considerable data variability and may be difficult to standardize. Therefore, alternative end points have been considered for clinical trials. These include decreasing the rate of progression of GA area enlargement, which would seem to be a clinically meaningful therapeutic response that has the potential to delay or prevent severe VA loss.53 Other potential anatomic end points might be prevention of worsening of intermediate AMD pre-GA and decrease in volume and area of macular drusen.54 The US Food and Drug Administration has recently accepted the anatomic end point of GA enlargement rate as a main outcome parameter in clinical trials for new therapies.55
Detection and measurement of GA area can be achieved through a number of imaging modalities, including color fundus photography, fluorescein angiography, scanning laser ophthalmoscopy (SLO)-based infrared fundus imaging, SLO- or camera-based autofluorescence, and optical coherence tomography (OCT). There are advantages and disadvantages of each modality, but the most accurate and reproducible reading center grading likely results from using more than one imaging type.
Color fundus photography
Evolution from fundoscopic clinical examination to fundus imaging using film-based stereoscopic color images set the stage to develop standardized methods to measure the atrophic lesion.56,57 Fundus photography facilitated multiple studies to understand the prevalence of GA and the phenotypic risk factors for development and progression of GA.3,14,58–60 While photographic documentation was a major leap from clinical documentation, measurement of the atrophic patch in film-based images was still labor intensive, using grids and transparency based projection methods.
High-resolution digital fundus imaging in early 2000 resulted in validation studies to transition to the new technology.61,62 Digital images were easier to acquire and share and were amenable to a number of image analysis tools including calibrated measurement tools and planimetry.63,64 Registration of images has been employed to better understand the natural history of events leading to the development of GA.39 The pathway of drusen regression, pigment clumping, and hypopigmentation has been shown to lead to precede GA. Although digital fundus photography is an efficient way to document clinical interpretation, there are a few difficulties in measurements based on color photographs. Image quality is dependent on having clear media which can be challenging in elderly patients with lens opacities. In addition, lightly pigmented fundus, prominent choroidal vessels, and overlying drusen can affect measurements. Foveal involvement is also difficult to ascertain. Despite this, the intergrader agreement for measurement of GA from color photographs has been high.63
FAF
FAF is an imaging method based on topographic mapping of lipofuscin at the level of the RPE.65 Low-intensity autofluorescence in areas of GA led to keen interest in developing this modality for imaging atrophic lesions.65–67 The measurements and enlargement rates of GA have been comparable between color photographs and FAF images. The confocal imaging systems used for acquiring autofluorescence images reduce the autofluorescence emitted from structures anterior to the retina and focus on the single RPE plane. This provides high-contrast monochromatic images that can be adapted for automated image analysis.68–70 Autofluorescence images provide additional insight into phenotypic markers for progression. As noted, increased autofluorescence surrounding the area of GA has been suggested to be a risk factor for progression.71–73 The increased autofluorescence, sometimes known as “halo”, has been attributed to accumulation of lipofuscin and may precede the development of outer retinal atrophy. The distinct patterns of autofluorescence seen surrounding the atrophic area have also raised interest in potential biomarkers.74,75 There has been significant difference in progression rates of GA between eyes with no pattern or focal pattern as compared with eyes with a “diffuse” or “banded pattern”. The luteal pigment at the macula absorbs short wavelength light used in SLO systems resulting in a dark fovea. Foveal involvement is difficult to assess and requires complementary images such as near infrared reflectance, color photographs, or OCT for confirmation (Figures 1 and 2).
In contrast to SLO systems, the fundus camera-based autofluorescence systems detect signal from all tissue levels. These systems also use green light, which allows better visualization of lesion delineation from the fovea.76 Modifications in the systems using filters have improved the image quality for evaluating retinal changes.77,78 Quantitative assessment of GA has been similar between the fundus camera and SLO based systems.78,79
Spectral domain optical coherence tomography
Spectral domain optical coherence tomography (SDOCT) imaging has become the standard imaging modality for various retinal diseases over the last decade. Its role in imaging of GA has evolved over the last few years to understand the cellular level changes occurring in eyes with GA. The early findings preceding development of GA on SDOCT have been described from longitudinal analysis of eyes with intermediate AMD.80 Hypopigmentation is manifested by increased transmission of the reflected OCT signal beneath the RPE layer. Overlying pigment clumping may be demonstrated by hyperreflective clumps that have migrated from the RPE layer into the outer retina. Typically, there is thinning of the outer nuclear layer coincident with loss of the ellipsoid portion of photoreceptor inner segment and the external limiting membrane. An outward bowing of the outer plexiform layer and the inner nuclear layer occurs. These details may or may not correlate with distinct GA on color photographs, and have been labeled “nascent GA”.80 In fully developed GA, there may be absence of the outer retinal layers and RPE such that the outer plexiform layer appears to be in contact with Bruch’s membrane. SDOCT has also been widely employed to evaluate the cellular level changes occurring at the junction zone of GA, such as outer plexiform layer, external limiting membrane, and the ellipsoid zone thinning.81 Outer retinal disruption has been shown to precede development of atrophy defined as loss of RPE and increased choroidal hyperreflectivity.82 Other changes in the outer retina such as splitting of RPE and Bruch’s membrane complex correlate with autofluorescence patterns and have been associated with faster growth rates.83,84 Additional interesting changes such as outer retinal tubules and hyporeflective wedges in eyes with GA have unknown significance at this time.85,86,87 SDOCT imaging in GA is most useful for accurately and reproducibly identifying foveal involvement (Figure 2).88 Improvement in the resolution of SDOCT will help understand the perilesional cellular level dynamic changes that occur in eyes with GA.
Infrared imaging is another modality that is available as a complementary image to SDOCT and autofluorescence images. GA measurements from SDOCT are comparable to autofluorescence images and are useful for identifying foveal sparing. Slab analysis of SDOCT volume scans, whereby the reflectivity signal within a specific layer or stratum of the retina is imaged, allows en face imaging of custom retinal thickness has led to development of automated measurement of GA lesion size from SDOCT images.89
Autofluorescence imaging has become the gold standard for measurement of GA. Quantitative autofluorescence is an emerging field where index of retinal autofluorescence can be measured over time as a prognostic factor.90 Nascent GA is a novel terminology to consolidate phenotypic features from autofluorescence and SDOCT imaging that will help identify eyes at high risk of developing GA.91 Research is underway to develop modalities that will improve visualization and further our understanding of the growth dynamics of GA, such as polarization-sensitive OCT and adaptive optics.92–94 Multimodal imaging brings together the advantages of various modalities available and is the pathway to quantitative and qualitative assessment of GA.
Enlargement of GA area with time
Most patients with drusen-associated GA develop small parafoveal patches as the earliest manifestation of the condition. At these loci, the lesions are preceded by extensive confluent drusen or a drusenoid pigment epithelial detachment with pigmentary abnormalities and/or refractile drusen. As noted, the presence of subretinal drusenoid deposits (reticular pseudodrusen) and abnormal autofluorescence signals are additional risk factors. Approximately 15%–20% of cases begin with subfoveal involvement,40,60 and some patients develop GA without notable precursor lesions. Of those without central involvement at first detection, the median time to foveal atrophy is approximately 2 years.60 Risk factors for continued GA expansion include hyperautofluorescence of the junctional zone surrounding regions of GA,72,73,95,96 the number of foci of GA and shape configuration of the GA regions,97 and the total perimeter of RPE at risk surrounding areas of GA.98 There appears to be no additional risk conferred by smoking or body mass index.60
Longitudinal studies show wide variability in the growth rates of GA lesions between individuals, although the growth rates of lesions in patients with bilateral involvement tend to be similar between eyes of the same individual.53 In the Beaver Dam Eye Study, the mean enlargement of area of GA was 6.4 mm2 over 5 years. In the AREDS, patients with GA had growth of 1.78 mm2 per year over 5 years,99 but growth was greater in larger lesions compared to smaller ones, as has been noted in other studies.55,100 Growth rates of 1.52 mm2/yr,75 2.2 mm2/yr,97 and 1.61 mm2/yr100 have been reported from other cohort studies. By using the en face OCT projection image, estimated GA growth rate was 1.2 mm2 in a relatively small cohort.101
The AREDS Research Group supported a linear model of GA growth over a quadratic model, in agreement with a prior study.102 However, adjustment for the differential growth rate by baseline area could be an additional source of variability in clinical trial analysis. A square root transformation of baseline GA area appears to ameliorate the effect of baseline area on GA progression, which could simplify clinical trial enrollment and analysis.103 Additionally, a classification based on the extent to which GA lesions in an eye depart from circularity based upon measurement of area and total perimeter of the junctional zone shows association with growth rates by providing a measure of the relative border of RPE at risk for further dropout at the margins of GA.98
Anti-VEGF therapy and neovascular-associated RPE atrophy
The neovascular AMD process may also lead to well-defined regions of RPE loss that meet the clinical and photographic criteria of GA, but are noted in this manuscript as neovascular-associated RPE atrophy for clarity (Figure 3).13 This phenomenon appears over years of treatment or with natural involution of neovascularization. Contributions to loss of RPE cells in this setting include acceleration of the processes seen in drusen-associated GA with choroidal vascular compromise, photoreceptor atrophy, and proinflammatory activation. Separation of the RPE from the choriocapillaris by fibrovascular material is likely to be a cell stressor, but the presence of a fibrovascular pigment epithelial detachment does not necessarily indicate incipient atrophy. Neovascular-associated atrophy is likely to be a result of nonspecific insult from the exudative process that accumulates over years of the disease and is perhaps additive to the nonexudative AMD disease process itself. Anti-VEGF treatment itself has been proposed to accelerate RPE atrophy due to off-target effects through inhibiting the natural constitutive maintenance effects of VEGF on vascular and neuronal health.104–106
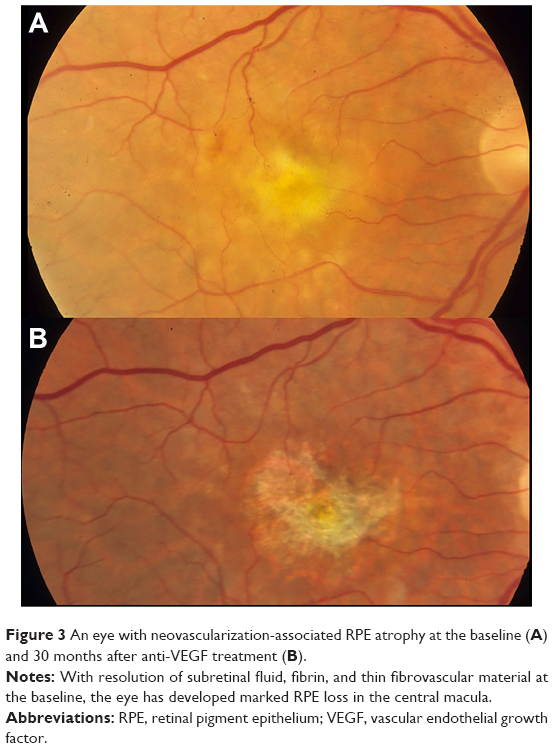 | Figure 3 An eye with neovascularization-associated RPE atrophy at the baseline (A) and 30 months after anti-VEGF treatment (B). |
The development of neovascular-associated atrophy is clinically important because it is associated with poorer visual outcome, as noted in 2-year follow-up of subjects enrolled in the pivotal AMD trials of ranibizumab.107 In a subsequent cross-sectional follow-up study of a cohort drawn from patients enrolled in ranibizumab treatment trials beginning 7 or 8 years previously, atrophy was nearly ubiquitous (98% of subjects).108 A multivariate regression analysis that included retina thickness, presence of fluid, and presence of fibrosis yielded only area of RPE atrophy as the significant determinant of visual outcome. Between 2 and 7 years of follow-up, every patient had enlargement of neovascular-associated atrophy lesions over time.
The Comparison of Age-Related Macular Degeneration Treatment Trial Research Group reanalyzed their data at 2 years looking at both the risk of new RPE atrophic lesions in patients with no atrophy at baseline109 and the growth of atrophy lesions in patients with preexisting GA.110 In patients with no GA at baseline, ranibizumab treatment was associated with fewer new neovascular-associated atrophy lesions than bevacizumab. Additionally, the monthly dosing group correlated with more new atrophic lesions than the PRN group that received fewer total injections over time109 further suggesting a dose–response effect. These data led to the hypotheses that ranibizumab may cause more neovascular-associated atrophic lesions than bevacizumab due to its greater effects on resolving fluid, which in some way led to more atrophy. Other possibilities include its smaller antibody size than bevacizumab, penetrating deeper into the outer retina, RPE, and choriocapillaris, and causing relatively increased toxicity. However, in the analysis of atrophy growth rate in patients with preexisting GA lesions, neither the dosing regimen nor the presence of intraretinal or subretinal fluid correlates with GA growth rate.110 Ranibizumab does continue to be associated with greater GA growth rates than bevacizumab treatment.110
Another study retrospectively assessing development of neovascular-associated atrophy in patients with no GA at baseline who were treated with anti-VEGF treatment and followed for a mean of 5 years showed development of RPE loss exclusively within the region previously occupied by the neovascular lesions.111 Eyes with GA at baseline had increase in their total GA area, as would be expected, with the areas developing at the site of the neovascular lesion merging with preexisting GA and enlarging over time. Eyes with dense macular hemorrhage may be predisposed to neovascular-associated atrophy. Given that participants in trials of anti-VEGF treatment for retinal venous occlusion and diabetic macular edema are not noted to develop RPE atrophy as an adverse event, anti-VEGF treatment by itself does not appear to be a risk for GA in eyes without AMD. The question as to whether anti-VEGF accelerates neovascular-associated atrophy development requires additional clarification, which may be forthcoming from other clinical trials of anti-VEGF treatments.
Clinical trials and investigational therapies for GA
Opposed to anti-VEGF therapy for exudative AMD, there are currently no approved treatments for GA. Current clinical trials are investigating remedies for GA through multiple modalities, including complement and inflammation, the visual cycle, neuroprotection, and cell replacement therapy.
Treatments targeting complement and inflammation
Lampalizumab is a humanized mouse anti-Factor D antibody, designed to inhibit the rate limiting step in the alternative complement pathway.112 In preclinical trials, Factor D deficient mice demonstrate reduced photoreceptor apoptosis and electroretinogram (ERG) preservation after exposure to constant light.113 In Phase Ia dose escalation studies, no ocular adverse events were reportable to intravitreal lampalizumab injection, and systemic complement was also unaffected.112 The scientific rationale and safety of the Phase Ia trial led to a Phase II trial (MAHALO) to investigate the role of lampalizumab in GA. At the 31st American Society for Retinal Specialists, the unpublished results of the MAHALO study were presented, demonstrating a 20% reduction in GA growth rate at 18 months with monthly intravitreal lampalizumab injections.114 In a subgroup analysis of patients with the complement factor I biomarker, the effect size increased to a 44% reduction in GA growth rate.115 These results sparked dual Phase III multicenter, randomized, double-masked, sham injection controlled studies called Chroma and Spectri. In 2015, lampalizumab is the potential therapy for GA farthest in clinical development.
Eculizumab is a humanized mouse anti-C5 antibody that is already approved for the treatment of paroxysmal nocturnal hemoglobinuria and atypical hemolytic uremic syndrome. Eculizumab prevents C5 convertase from cleaving C5 into C5a and C5b, decreasing anaphylatoxin release, terminal complement activation, and membrane attack complex. The COMPLETE study randomized 30 patients to high-dose intravenous eculizumab versus low dose versus placebo for 6 months.116 Eculizumab has no effect upon GA size or growth rate at 6 or 12 months despite adequate drug dosing and complement inhibition.116 Possible shortcomings of the study include small sample size, inhibition of the terminal rather than the alternative complement pathway, and lack of genetic testing to determine whether patients had susceptibility single nucleotide polymorphisms in complement genes.
Sirolimus (also called rapamycin) is an inhibitor of the mammalian target of rapamycin, a pleiotropic protein kinase involved in cellular growth, proliferation, survival, and metabolism. Sirolimus is used clinically as an antirejection medication in organ transplantation because of its ability to suppress T-cell and B-cell proliferation and antibody production.117 Additionally, in a mouse of model of RPE degeneration and photoreceptor apoptosis caused by inhibition of oxidative metabolism, sirolimus treatment prevented RPE degeneration, photoreceptor loss, and improves ERG findings.118 Based upon its anti-inflammatory properties and its ability to improve RPE function, sirolimus was investigated as a potential treatment for GA. In a small Phase II trial with eleven patients, subconjunctival administration of sirolimus every 3 months for 24 months of therapy shows no effect upon VA, GA size, or GA growth.117 The lack of effect was hypothesized to be due to poor bioavailability of subconjunctival sirolimus to the posterior segment. A second Phase II trial investigated intravitreal injection of sirolimus every 2 months for 24 months in six patients with bilateral GA. Unfortunately, intravitreal sirolimus demonstrated no effect upon VA, GA size, or GA growth using this dosing and delivery.119
The visual cycle
A major component of lipofuscin is the retinoid A2E, a byproduct of the visual cycle, which is formed when all-trans retinoic acid sequentially condenses with phosphatidylethanolamine.120 Accumulation of lipofuscin and A2E has multiple deleterious effects upon RPE function including impaired lysosomal degradation, generation of reactive oxygen species, induction of proapoptotic proteins, and complement activation.121,122 Based upon these findings and the observation that the edges of GA lesions show increased FAF from presumed A2E accumulation,67 it was hypothesized that reducing A2E accumulation could treat GA. There are currently two drugs being investigated to modulate the visual cycle and decrease A2E formation: fenretinide and ACU-4429 (or emixustat hydrochloride).
Fenretinide is a synthetic vitamin A derivative, which binds retinol-binding protein (RBP), causing excretion of the fenretinide-RBP complex in urine.121 In normal physiology, RBP binds vitamin A in the serum, delivering vitamin A to extrahepatic tissues like the eye. Depletion of RBP by fenretinide reduces vitamin A levels in the eye and slows the visual cycle. In preclinical trials, fenretinide treatment of Abca4 null mice, a genetic model of Stargardt’s disease, causes reduced lipofuscin and A2E accumulation.123 Based upon the known safety profile of fenretinide and history of its usage in humans for treatment of rheumatoid arthritis and psoriasis, a Phase II clinical trial tested the role of fenretinide in GA progression. This study was a randomized, placebo-controlled, double-blinded trial that investigated 246 patients over a 2-year follow-up period.121 In the overall comparison of fenretinide versus placebo, fenretinide shows no effect upon VA, GA size, or GA growth. In a subgroup analysis of patients achieving RBP concentrations less than 2 mg/dL, fenretinide treatment is weakly associated (P=0.1848, N=21) toward a decreased growth rate of GA lesions. However, patients in the high-dose fenretinide group had more ocular adverse events related to delayed dark adaptation and were more likely to withdraw from the study. In 2015, no Phase III trials were underway.
ACU-4429 (or emixustat hydrochloride) is an inhibitor of the RPE65 enzyme. RPE65 is an isomerase, specifically expressed in the RPE and regenerates 11-cis-retinol from all-trans-retinol.124 By inhibiting 11-cis-retinol regeneration, ACU-4429 slows the visual cycle and is hypothesized to reduce A2E and lipofuscin accumulation, similar to fenretinide. Two Phase I studies have been conducted and published, showing slowed rod visual cycling on ERG analysis without an effect upon cones.124,125 Adverse effects were related to delayed dark adaptation, similar to fenretinide, and included nyctalopia, dyschromatopsia, blurred vision, and photophobia. Phase IIb/III trials are being conducted and are expected to be completed in July 2016 (NCT01802866).
Neuroprotection
Vision loss from GA occurs due to death of photoreceptors. A logical treatment is the prevention of photoreceptor apoptosis. Current studies are investigating differing mechanisms of neuroprotection including antioxidants, regulation of amyloid signaling, and modulation of prosurvival growth factors.
The retina and RPE are highly metabolically active tissues under constant exposure to light. The generation of reactive oxygen species leads to lipid peroxidation and cellular damage, which are hypothesized to cause photoreceptor and RPE apoptosis. In preclinical studies, OT-551 prevents photoreceptor loss and restores ERG amplitudes in a rat model of light-induced retinal degeneration.126 These results led to a Phase II clinical trial that enrolled ten patients with bilateral GA.127 OT-551 was administered topically three times daily for 2 years to one study eye. OT-551 did not influence microperimetry, GA size or growth, or total drusen area in this trial. These results are consistent with AREDS report #35, showing no effect of antioxidant vitamin supplementation on the development of central GA.128
In addition to complement components, histopathological studies of patients with AMD find amyloid β present in drusen.129 Furthermore, amyloid β colocalizes with activated complement components,129 implicating amyloid β as a potential complement activator and driver of local inflammation. In a mouse model of dry AMD, the mouse Apoe4 gene was replaced by the human APOE4 allele linked to AMD. When fed a high fat and high cholesterol diet, these mice demonstrate drusen deposition that contain amyloid β and activated complement, causing diminished ERG responses.130 When these mice are treated with an antibody targeting amyloid β40 and β42 proteins, these phenotypes are abrogated.130 These preclinical trials have led to two completed Phase I trials that are unpublished and a Phase II efficacy study to determine whether intravenous administration of this humanized monoclonal antibody targeting amyloid β can affect GA growth.115
Brimonidine is an α2-adrenergic agonist commonly used in the treatment of ocular hypertension and glaucoma. In addition to its ocular antihypertensive effects, brimonidine diminishes retinal ganglion cell death in models of optic nerve crush, ocular hypertension, retinal ischemia, and phototoxicity.115,131 This neuronal antiapoptotic effect occurs through upregulation of prosurvival factors like fibroblast growth factor, activation of the prosurvival Akt pathway, and inhibition of apoptosis.132,133 These effects led to the hypothesis that an intravitreal sustained release brimonidine implant could protect photoreceptors from apoptosis in patients with GA. A Phase I/II safety and efficacy study is completed (NCT00658619) and results are available on the clinicaltrials.gov website. A trend toward decreased GA size was present and was found to be dose dependent at every time point except month 3.134 In early 2015, these data were unpublished and more sophisticated analysis unavailable.
Ciliary neurotrophic factor (CNTF) is a neuroprotective factor that slows photoreceptor loss in multiple models of retinal degeneration.135 In the form of an encapsulated cell technology (ECT) implant, CNTF can decrease photoreceptor apoptosis in mouse and dog models of retinitis pigmentosa.136 These results led to a Phase I safety trial, demonstrating that the ECT implant delivering CNTF was safe in patients with retinitis pigmentosa.137 In a Phase II efficacy study, 51 patients were randomized to sham, low-dose, and high-dose ECT implant surgery and followed for 12 months.138 The results show no change in GA size or GA growth. Interestingly, analysis of macular volume by OCT demonstrates that CNTF treatment increases outer nuclear layer thickness.138 In terms of a trial to reduce GA, this study was considered a negative result. However, this technology has the potential for the delivery of other prosurvival factors, anti-inflammatory factors, or antiangiogenic agents for patients with AMD.
Cell replacement therapy
Human embryonic stem cells hold great promise as cell replacement therapy for lost photoreceptor and RPE cells in GA lesions. There are many safety concerns, however, in the use of stem cell therapy in the treatment of human disease. These issues include cross-species antigenic contamination from animal proteins, Escherichia coli-derived proteins, and viral vectors that are used in the directed differentiation of human embryonic stem cells.139 Additionally, genetic techniques for directed and efficient differentiation could cause insertional mutagenesis, potentially leading to tumor formation. And finally, dedifferentiation of embryonic stem cells could lead to teratoma formation.
Human embryonic stem cells can differentiate into RPE cells (hES-RPE) spontaneously with 1% efficiency.139 Although inefficient, spontaneous differentiation without specific growth factors or transgenes, abrogates the concern of cross-species antigenicity, insertional mutagenesis, or viral antigen-triggered inflammation. The Royal College of Surgeons (RCS) rat is an animal model of RPE degeneration with loss of outer segment phagocytosis, leading to photoreceptor apoptosis. Subretinal injections of hES-RPE in RCS rats improve ERG responses and decreases photoreceptor loss without causing abnormal proliferation of RPE cells or teratoma formation.140 These cells have been recently transplanted into patients with Stargardt’s macular dystrophy and patients with dry AMD.141 During pars plana vitrectomy, hES-RPE cells were injected into the subretinal space at transition zones between viable RPE and areas of GA. Pigmented cells are visible overlying Bruch’s membrane at the border of the healthy RPE and GA lesions; these areas increase in size and become more dense over time. In patients with dry AMD, VA improved by >10 ETDRS letters in 6/9 patients, with no decline in VA in any patient. On the National Eye Institute Visual Function Questionnaire, patient’s scores improved in areas of mental health, general vision, peripheral vision, near activities, and distance activities. No evidence for cellular rejection, ectopic tissue proliferation, or teratoma formation was found. Complications were related to vitrectomy, including cataract progression and one case of endophthalmitis.142
Conclusion
Atrophy of the RPE with late AMD, whether from the atrophic or neovascular forms, is a highly prevalent cause of severe vision loss worldwide. Precursor ocular characteristics provide some clues as to underlying etiology, and emerging genotypic risk factors and the study of the cell biology of AMD provide others, but in 2015, there are no proven treatments for this condition. A number of pharmacotherapeutic targets are promising, and clinical trials are in various stages of development and execution. Finding new treatments to prevent or delay the development of GA has clear public health import.
Disclosure
The authors report no conflicts of interest in this work.
References
Wong WL, Su X, Li X, et al. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: a systematic review and meta-analysis. Lancet Glob Health. 2014;2(2): e106–e116. | ||
Ferris FL, Wilkinson CP, Bird A, et al. Clinical classification of age-related macular degeneration. Ophthalmology. 2013;120(4):844–851. | ||
The Age-Related Eye Disease Study Research Group. The Age-Related Eye Disease Study system for classifying age-related macular degeneration from stereoscopic color fundus photographs: the Age-Related Eye Disease Study report number 6. Am J Ophthalmol. 2001;132(5):668–681. | ||
Klein R, Klein BE, Moss SE. Age-related eye disease and survival. The Beaver Dam Eye Study. Arch Ophthalmol. 1995;113(3):333–339. | ||
Sunness JS, Rubin GS, Zuckerbrod A, Applegate CA. Foveal-sparing scotomas in advanced dry age-related macular degeneration. J Vis Impair Blind. 2008;102(10):600–610. | ||
Rudnicka AR, Kapetanakis VV, Jarrar Z, et al. Incidence of late-stage age-related macular degeneration in American whites: systematic review and meta-analysis. Am J Ophthalmol. 2015;160(1):85.e83–93.e83. | ||
Owen CG, Fletcher AE, Donoghue M, Rudnicka AR. How big is the burden of visual loss caused by age related macular degeneration in the United Kingdom? Br J Ophthalmol. 2003;87(3):312–317. | ||
Owen CG, Jarrar Z, Wormald R, Cook DG, Fletcher AE, Rudnicka AR. The estimated prevalence and incidence of late stage age related macular degeneration in the UK. Br J Ophthalmol. 2012;96(5):752–756. | ||
Age-related Eye Disease Study Group. A randomized, placebo-controlled, clinical trial of high-dose supplementation with vitamins C and E, beta carotene, and zinc for age-related macular degeneration and vision loss: AREDS report no. 8. Arch Ophthalmol. 2001;119(10):1417–1436. | ||
Chew EY, Clemons TE, Sangiovanni JP, et al. Secondary analyses of the effects of lutein/zeaxanthin on age-related macular degeneration progression: AREDS2 report no. 3. JAMA Ophthalmol. 2014;132(2):142–149. | ||
Gass JD. Drusen and disciform macular detachment and degeneration. Arch Ophthalmol. 1973;90(3):206–217. | ||
Sarks SH. Drusen patterns predisposing to geographic atrophy of the retinal pigment epithelium. Aust J Ophthalmol. 1982;10(2):91–97. | ||
Sarks JP, Sarks SH, Killingsworth MC. Evolution of geographic atrophy of the retinal pigment epithelium. Eye. 1988;2(Pt 5):552–577. | ||
Sunness JS. The natural history of geographic atrophy, the advanced atrophic form of age-related macular degeneration. Mol Vis. 1999;5:25. | ||
Sunness JS, Gonzalez-Baron J, Applegate CA, et al. Enlargement of atrophy and visual acuity loss in the geographic atrophy form of age-related macular degeneration. Ophthalmology. 1999;106(9):1768–1779. | ||
Schmitz-Valckenberg S, Fleckenstein M, Helb HM, Charbel Issa P, Scholl HP, Holz FG. In-vivo imaging of foveal sparing in geographic atrophy secondary to age-related macular degeneration. Invest Ophthalmol Vis Sci. 2009;50(8):3915–3921. | ||
Meleth AD, Mettu P, Agron E, et al. Changes in retinal sensitivity in geographic atrophy progression as measured by microperimetry. Invest Ophthalmol Vis Sci. 2011;52(2):1119–1126. | ||
Sunness JS, Applegate CA, Haselwood D, Rubin GS. Fixation patterns and reading rates in eyes with central scotomas from advanced atrophic age-related macular degeneration and Stargardt disease. Ophthalmology. 1996;103(9):1458–1466. | ||
Sunness JS, Rubin GS, Applegate CA, et al. Visual function abnormalities and prognosis in eyes with age-related geographic atrophy of the macula and good visual acuity. Ophthalmology. 1997;104(10): 1677–1691. | ||
Seddon JM, Reynolds R, Maller J, Fagerness JA, Daly MJ, Rosner B. Prediction model for prevalence and incidence of advanced age-related macular degeneration based on genetic, demographic, and environmental variables. Invest Ophthalmol Vis Sci. 2009;50(5):2044–2053. | ||
Klein ML, Ferris FL 3rd, Francis PJ, et al. Progression of geographic atrophy and genotype in age-related macular degeneration. Ophthalmology. 2010;117(8):1554–1559. | ||
Grassman F, Fauser S, Weber B. The genetics of age-related macular degeneration (AMD) – novel targets for designing treatment options? Eur J Pharm Biochem. Epub May 16, 2015. | ||
Liu MM, Chan CC, Tuo J. Genetic mechanisms and age-related macular degeneration: common variants, rare variants, copy number variations, epigenetics, and mitochondrial genetics. Hum Genomics. 2012;6(1):13–21. | ||
Grassmann F, Fleckenstein M, Chew EY, et al. Clinical and genetic factors associated with progression of geographic atrophy lesions in age-related macular degeneration. PLoS One. 2015;10(5):e0126636. | ||
Caire J, Recalde S, Velazquez-Villoria A, et al. Growth of geographic atrophy on fundus autofluorescence and polymorphisms of CFH, CFB, C3, FHR1-3, and ARMS2 in age-related macular degeneration. JAMA Ophthalmol. 2014;132(5):528–534. | ||
Johnson LV, Ozaki S, Staples MK, Erickson PA, Anderson DH. A potential role for immune complex pathogenesis in drusen formation. Exp Eye Res. 2000;70(4):441–449. | ||
Johnson LV, Leitner WP, Stapels MK. Complement activation and inflammatory processes in Drusen formation and age related macular degeneration. Exp Eye Res. 2001;73(6):887–896. | ||
Keenan TD, Pickford CE, Holley RJ, et al. Age-dependent changes in heparan sulfate in human Bruch’s membrane: implications for age-related macular degeneration. Invest Ophthalmol Vis Sci. 2014;55(8): 5370–5379. | ||
Bhutto I, Lutty G. Understanding age-related macular degeneration (AMD): relationships between the photoreceptor/retinal pigment epithelium/Bruch’s membrane/choriocapillaris complex. Mol Aspects Med. 2012;33(4):295–317. | ||
Hageman GS, Luthert PJ, Victor Chong NH, Johnson LV, Anderson DH, Mullins RF. An integrated hypothesis that considers drusen as biomarkers of immune-mediated processes at the RPE-Bruch’s membrane interface in aging and age-related macular degeneration. Prog Retin Eye Res. 2001;20(6):705–732. | ||
Anderson DH, Mullins RF, Hageman GS. A role for local inflammation in the formation of drusen in the aging eye. Am J Ophthalmol. 2002;134(3):411–431. | ||
Mullins RF, Skeie JM, Malone EA, Kuehn MH. Macular and peripheral distribution of ICAM-1 in the human choriocapillaris and retina. Mol Vis. 2006;12:224–235. | ||
Bora PS, Sohn JH, Cruz JM, et al. Role of complement and complement membrane attack complex in laser-induced choroidal neovascularization. J Immunol. 2005;174(1):491–497. | ||
Bora NS, Kaliappan S, Jha P, et al. Complement activation via alternative pathway is critical in the development of laser-induced choroidal neovascularization: role of factor B and factor H. J Immunol. 2006;177(3):1872–1878. | ||
Nozaki M, Raisler BJ, Sakurai E, et al. Drusen complement components C3a and C5a promote choroidal neovascularization. Proc Natl Acad Sci U S A. 2006;103(7):2328–2333. | ||
Curcio CA, Messinger JD, Sloan KR, McGwin G, Medeiros NE, Spaide RF. Subretinal drusenoid deposits in non-neovascular age-related macular degeneration: morphology, prevalence, topography, and biogenesis model. Retina. 2013;33(2):265–276. | ||
Zanzottera EC, Messinger JD, Ach T, Smith RT, Curcio CA. Subducted and melanotic cells in advanced age-related macular degeneration are derived from retinal pigment epithelium. Invest Ophthalmol Vis Sci. 2015;56(5):3269–3278. | ||
Curcio CA, Medeiros NE, Millican CL. Photoreceptor loss in age-related macular degeneration. Invest Ophthalmol Vis Sci. 1996;37(7):1236–1249. | ||
Klein ML, Ferris FL, Armstrong J, et al. Retinal precursors and the development of geographic atrophy in age-related macular degeneration. Ophthalmology. 2008;115(6):1026–1031. | ||
Group CoA-rMDPTCR. Risk factors for choroidal neovascularization and geographic atrophy in the complications of age-related macular degeneration prevention trial. Ophthalmology. 2008;115(9):1474–1479, 1479.e1471–1476.e1471. | ||
Ying GS, Maguire MG. Development of a risk score for geographic atrophy in complications of the age-related macular degeneration prevention trial. Ophthalmology. 2011;118(2):332–338. | ||
Davis MD, Gangnon RE, Lee LY, et al. The Age-Related Eye Disease Study severity scale for age-related macular degeneration: AREDS report no. 17. Arch Ophthalmol. 2005;123(11):1484–1498. | ||
Ying GS, Maguire MG, Alexander J, Martin RW, Antoszyk AN. Description of the Age-Related Eye Disease Study 9-step severity scale applied to participants in the Complications of Age-Related Macular Degeneration Prevention trial. Arch Ophthalmol. 2009;127(9):1147–1151. | ||
Ferris FL, Davis MD, Clemons TE, et al. A simplified severity scale for age-related macular degeneration: AREDS report no. 18. Arch Ophthalmol. 2005;123(11):1570–1574. | ||
Marsiglia M, Boddu S, Bearelly S, et al. Association between geographic atrophy progression and reticular pseudodrusen in eyes with dry age-related macular degeneration. Invest Ophthalmol Vis Sci. 2013;54(12):7362–7369. | ||
Holz FG, Strauss EC, Schmitz-Valckenberg S, van Lookeren Campagne M. Geographic atrophy: clinical features and potential therapeutic approaches. Ophthalmology. 2014;121(5):1079–1091. | ||
Klein R, Meuer SM, Knudtson MD, Iyengar SK, Klein BE. The epidemiology of retinal reticular drusen. Am J Ophthalmol. 2008;145(2):317–326. | ||
Holz FG, Steinberg JS, Göbel A, Fleckenstein M, Schmitz-Valckenberg S. Fundus autofluorescence imaging in dry AMD: 2014 Jules Gonin lecture of the Retina Research Foundation. Graefes Arch Clin Exp Ophthalmol. 2015;253(1):7–16. | ||
Sparrow JR, Duncker T. Fundus autofluorescence and RPE lipofuscin in age-related macular degeneration. J Clin Med. 2014;3(4):1302–1321. | ||
Sparrow JR, Yoon K, Wu Y, Yamamoto K. Interpretations of fundus autofluorescence from studies of the bisretinoids of the retina. Invest Ophthalmol Vis Sci. 2010;51(9):4351–4357. | ||
Fleckenstein M, Schmitz-Valckenberg S, Lindner M, et al. The “diffuse-trickling” fundus autofluorescence phenotype in geographic atrophy. Invest Ophthalmol Vis Sci. 2014;55(5):2911–2920. | ||
Rudolf M, Vogt SD, Curcio CA, et al. Histologic basis of variations in retinal pigment epithelium autofluorescence in eyes with geographic atrophy. Ophthalmology. 2013;120(4):821–828. | ||
Sunness JS, Applegate CA, Bressler NM, Hawkins BS. Designing clinical trials for age-related geographic atrophy of the macula: enrollment data from the geographic atrophy natural history study. Retina. 2007;27(2):204–210. | ||
Garcia Filho CA, Yehoshua Z, Gregori G, et al. Change in drusen volume as a novel clinical trial endpoint for the study of complement inhibition in age-related macular degeneration. Ophthalmic Surg Lasers Imaging Retina. 2014;45(1):18–31. | ||
Csaky KG, Richman EA, Ferris FL III. Report from the NEI/FDA Ophthalmic Clinical Trial Design and Endpoints Symposium. Invest Ophthalmol Vis Sci. 2008;49(2):479–489. | ||
Klein R, Davis MD, Magli YL, Segal P, Klein BE, Hubbard L. The Wisconsin age-related maculopathy grading system. Ophthalmology. 1991;98(7):1128–1134. | ||
Bird AC, Bressler NM, Bressler SB, et al. An international classification and grading system for age-related maculopathy and age-related macular degeneration. The International ARM Epidemiological Study Group. Surv Ophthalmol. 1995;39(5):367–374. | ||
Klein R, Klein BE, Linton KL. Prevalence of age-related maculopathy. The Beaver Dam Eye Study. Ophthalmology. 1992;99(6):933–943. | ||
Sunness JS, Bressler NM, Tian Y, Alexander J, Applegate CA. Measuring geographic atrophy in advanced age-related macular degeneration. Invest Ophthalmol Vis Sci. 1999;40(8):1761–1769. | ||
Lindblad AS, Lloyd PC, Clemons TE, et al. Change in area of geographic atrophy in the Age-Related Eye Disease Study: AREDS report number 26. Arch Ophthalmol. 2009;127(9):1168–1174. | ||
Scholl HP, Dandekar SS, Peto T, et al. What is lost by digitizing stereoscopic fundus color slides for macular grading in age-related maculopathy and degeneration? Ophthalmology. 2004;111(1):125–132. | ||
van Leeuwen R, Chakravarthy U, Vingerling JR, et al. Grading of age-related maculopathy for epidemiological studies: is digital imaging as good as 35 mm film? Ophthalmology. 2003;110:1540–1544. | ||
Danis RP, Domalpally A, Chew EY, et al. Methods and reproducibility of grading optimized digital color fundus photographs in the Age-Related Eye Disease Study 2 (AREDS2 report number 2). Invest Ophthalmol Vis Sci. 2013;54(7):4548–4554. | ||
Hubbard LD, Danis RP, Neider MW, et al. Brightness, contrast, and color balance of digital versus film retinal images in the Age-Related Eye Disease Study 2. Invest Ophthalmol Vis Sci. 2008;49(8):3269–3282. | ||
von Ruckmann A, Fitzke FW, Bird AC. Distribution of fundus autofluorescence with a scanning laser ophthalmoscope. Br J Ophthalmol. 1995;79(5):407–412. | ||
von Ruckmann A, Fitzke FW, Bird AC. Fundus autofluorescence in age-related macular disease imaged with a laser scanning ophthalmoscope. Invest Ophthalmol Vis Sci. 1997;38(2):478–486. | ||
Holz FG, Bellman C, Staudt S, Schutt F, Volcker HE. Fundus autofluorescence and development of geographic atrophy in age-related macular degeneration. Invest Ophthalmol Vis Sci. 2001;42(5):1051–1056. | ||
Schmitz-Valckenberg S, Jorzik J, Unnebrink K, Holz FG. Analysis of digital scanning laser ophthalmoscopy fundus autofluorescence images of geographic atrophy in advanced age-related macular degeneration. Graefes Arch Clin Exp Ophthalmol. 2002;240(2):73–78. | ||
Ramsey DJ, Sunness JS, Malviya P, Applegate C, Hager GD, Handa JT. Automated image alignment and segmentation to follow progression of geographic atrophy in age-related macular degeneration. Retina. 2014;34(7):1296–1307. | ||
Schmitz-Valckenberg S, Brinkmann CK, Alten F, et al. Semiautomated image processing method for identification and quantification of geographic atrophy in age-related macular degeneration. Invest Ophthalmol Vis Sci. 2011;52(10):7640–7646. | ||
Holz FG, Bellmann C, Margaritidis M, Schutt F, Otto TP, Volcker HE. Patterns of increased in vivo fundus autofluorescence in the junctional zone of geographic atrophy of the retinal pigment epithelium associated with age-related macular degeneration. Graefes Arch Clin Exp Ophthalmol. 1999;237(2):145–152. | ||
Schmitz-Valckenberg S, Bindewald-Wittich A, Dolar-Szczasny J, et al. Correlation between the area of increased autofluorescence surrounding geographic atrophy and disease progression in patients with AMD. Invest Ophthalmol Vis Sci. 2006;47(6):2648–2654. | ||
Bearelly S, Khanifar AA, Lederer DE, et al. Use of fundus autofluorescence images to predict geographic atrophy progression. Retina. 2011;31(1):81–86. | ||
Bindewald A, Bird AC, Dandekar SS, et al. Classification of fundus autofluorescence patterns in early age-related macular disease. Invest Ophthalmol Vis Sci. 2005;46(9):3309–3314. | ||
Holz FG, Bindewald-Wittich A, Fleckenstein M, Dreyhaupt J, Scholl HP, Schmitz-Valckenberg S. Progression of geographic atrophy and impact of fundus autofluorescence patterns in age-related macular degeneration. Am J Ophthalmol. 2007;143(3):463–472. | ||
Wolf-Schnurrbusch UE, Wittwer VV, Ghanem R, et al. Blue-light versus green-light autofluorescence: lesion size of areas of geographic atrophy. Invest Ophthalmol Vis Sci. 2011;52(13):9497–9502. | ||
Spaide RF. Fundus autofluorescence and age-related macular degeneration. Ophthalmology. 2003;110(2):392–399. | ||
Schmitz-Valckenberg S, Fleckenstein M, Gobel AP, et al. Evaluation of autofluorescence imaging with the scanning laser ophthalmoscope and the fundus camera in age-related geographic atrophy. Am J Ophthalmol. 2008;146(2):183–192. | ||
Park SP, Siringo FS, Pensec N, et al. Comparison of fundus autofluorescence between fundus camera and confocal scanning laser ophthalmoscope-based systems. Ophthalmic Surg Lasers Imaging Retina. 2013;44(6):536–543. | ||
Wu Z, Luu CD, Ayton LN, et al. Optical coherence tomography-defined changes preceding the development of drusen-associated atrophy in age-related macular degeneration. Ophthalmology. 2014;121(12):2415–2422. | ||
Simader C, Sayegh RG, Montuoro A, et al. A longitudinal comparison of spectral-domain optical coherence tomography and fundus autofluorescence in geographic atrophy. Am J Ophthalmol. 2014; 158(3):557–566.e551. | ||
Nunes RP, Gregori G, Yehoshua Z, et al. Predicting the progression of geographic atrophy in age-related macular degeneration with SD-OCT en face imaging of the outer retina. Ophthalmic Surg Lasers Imaging Retina. 2013;44(4):344–359. | ||
Moussa K, Lee JY, Stinnett SS, Jaffe GJ. Spectral domain optical coherence tomography-determined morphologic predictors of the age-related macular degeneration-associated geographic atrophy progression. Retina. 2013;33(8):1590–1599. | ||
Fleckenstein M, Schmitz-Valckenberg S, Martens C, et al. Fundus autofluorescence and spectral-domain optical coherence tomography characteristics in a rapidly progressing form of geographic atrophy. Invest Ophthalmol Vis Sci. 2011;52(6):3761–3766. | ||
Hariri A, Nittala MG, Sadda SR. Outer retinal tubulation as a predictor of the enlargement amount of geographic atrophy in age-related macular degeneration. Ophthalmology. 2015;122(2):407–413. | ||
Cohen SY, Dubois L, Nghiem-Buffet S, et al. Retinal pseudocysts in age-related geographic atrophy. Am J Ophthalmol. 2010;150(2):211–217.e211. | ||
Mones J, Biarnes M, Trindade F. Hyporeflective wedge-shaped band in geographic atrophy secondary to age-related macular degeneration: an underreported finding. Ophthalmology. 2012;119(7):1412–1419. | ||
Forte R, Querques G, Querques L, Leveziel N, Benhamou N, Souied EH. Multimodal evaluation of foveal sparing in patients with geographic atrophy due to age-related macular degeneration. Retina. 2013;33(3):482–489. | ||
Yehoshua Z, Garcia Filho CA, Penha FM, et al. Comparison of geographic atrophy measurements from the OCT fundus image and the sub-RPE slab image. Ophthalmic Surg Lasers Imaging Retina. 2013;44(2):127–132. | ||
Schachar IH, Zahid S, Comer GM, et al. Quantification of fundus autofluorescence to detect disease severity in nonexudative age-related macular degeneration. JAMA Ophthalmol. 2013;131(8):1009–1015. | ||
Wu Z, Luu CD, Ayton LN, et al. Fundus autofluorescence characteristics of nascent geographic atrophy in age-related macular degeneration. Invest Ophthalmol Vis Sci. 2015;56(3):1546–1552. | ||
Schutze C, Bolz M, Sayegh R, et al. Lesion size detection in geographic atrophy by polarization-sensitive optical coherence tomography and correlation to conventional imaging techniques. Invest Ophthalmol Vis Sci. 2013;54(1):739–745. | ||
Panorgias A, Zawadzki RJ, Capps AG, Hunter AA, Morse LS, Werner JS. Multimodal assessment of microscopic morphology and retinal function in patients with geographic atrophy. Invest Ophthalmol Vis Sci. 2013;54(6):4372–4384. | ||
Gocho K, Sarda V, Falah S, et al. Adaptive optics imaging of geographic atrophy. Invest Ophthalmol Vis Sci. 2013;54(5):3673–3680. | ||
Hwang JC, Chan JW, Chang S, Smith RT. Predictive value of fundus autofluorescence for development of geographic atrophy in age-related macular degeneration. Invest Ophthalmol Vis Sci. 2006;47(6):2655–2661. | ||
Batioglu F, Gedik Oguz Y, Demirel S, Ozmert E. Geographic atrophy progression in eyes with age-related macular degeneration: role of fundus autofluorescence patterns, fellow eye and baseline atrophy area. Ophthalmic Res. 2014;52(2):53–59. | ||
Sunness JS, Margalit E, Srikumaran D, et al. The long-term natural history of geographic atrophy from age-related macular degeneration: enlargement of atrophy and implications for interventional clinical trials. Ophthalmology. 2007;114(2):271–277. | ||
Domalpally A, Danis RP, White J, et al. Circularity index as a risk factor for progression of geographic atrophy. Ophthalmology. 2013; 120(12):2666–2671. | ||
Klein R, Meuer SM, Knudtson MD, Klein BE. The epidemiology of progression of pure geographic atrophy: the Beaver Dam Eye study. Am J Ophthalmol. 2008;146(5):692–699. | ||
Biarnes M, Arias L, Alonso J, et al. Increased fundus autofluorescence and progression of geographic atrophy secondary to age-related macular degeneration: the GAIN study. Am J Ophthalmol. 2015;160(2):345–353.e5. | ||
Yehoshua Z, de Amorim Garcia Filho CA, Nunes RP, et al. Comparison of geographic atrophy growth rates using different imaging modalities in the COMPLETE study. Ophthalmic Surg Lasers Imaging Retina. 2015;46(4):413–422. | ||
Dreyhaupt J, Mansmann U, Pritsch M, Dolar-Szczasny J, Bindewald A, Holz FG. Modelling the natural history of geographic atrophy in patients with age-related macular degeneration. Ophthalmic Epidemiol. 2005;12(6):353–362. | ||
Feuer WJ, Yehoshua Z, Gregori G, et al. Square root transformation of geographic atrophy area measurements to eliminate dependence of growth rates on baseline lesion measurements: a reanalysis of Age-Related Eye Disease Study report no. 26. JAMA Ophthalmol. 2013;131(1):110–111. | ||
Lee S, Chen TT, Barber CL, et al. Autocrine VEGF signaling is required for vascular homeostasis. Cell. 2007;130(4):691–703. | ||
Saint-Geniez M, Kurihara T, Sekiyama E, Maldonado AE, D’Amore PA. An essential role for RPE-derived soluble VEGF in the maintenance of the choriocapillaris. Proc Natl Acad Sci U S A. 2009;106(44):18751–18756. | ||
Nishijima K, Ng YS, Zhong L, et al. Vascular endothelial growth factor-A is a survival factor for retinal neurons and a critical neuroprotectant during the adaptive response to ischemic injury. Am J Pathol. 2007;171(1):53–67. | ||
Rosenfeld PJ, Shapiro H, Tuomi L, et al. Characteristics of patients losing vision after 2 years of monthly dosing in the phase III ranibizumab clinical trials. Ophthalmology. 2011;118(3):523–530. | ||
Bhisitkul RB, Mendes TS, Rofagha S, et al. Macular atrophy progression and 7-year vision outcomes in subjects from the ANCHOR, MARINA, and HORIZON studies: the SEVEN-UP study. Am J Ophthalmol. 2015;159(5):915–924.e912. | ||
Grunwald JE, Daniel E, Huang J, et al. Risk of geographic atrophy in the comparison of age-related macular degeneration treatments trials. Ophthalmology. 2014;121(1):150–161. | ||
Grunwald JE, Pistilli M, Ying GS, et al. Growth of geographic atrophy in the comparison of age-related macular degeneration treatments trials. Ophthalmology. 2015;122(4):809–816. | ||
Tanaka E, Chaikitmongkol V, Bressler SB, Bressler NM. Vision-threatening lesions developing with longer-term follow-up after treatment of neovascular age-related macular degeneration. Ophthalmology. 2015;122(1):153–161. | ||
Do DV, Pieramici DJ, van Lookeren Campagne M, et al. A phase Ia dose-escalation study of the anti-factor D monoclonal antibody fragment FCFD4514S in patients with geographic atrophy. Retina. 2014;34(2):313–320. | ||
Rohrer B, Guo Y, Kunchithapautham K, Gilkeson GS. Eliminating complement factor D reduces photoreceptor susceptibility to light-induced damage. Invest Ophthalmol Vis Sci. 2007;48(11): 5282–5289. | ||
Williams DF. MAHALO Phase II study: Safety, tolerability and activity of lampalizumab (anti-factor D) in patients with geographic atrophy. Paper presented at: American Society of Retinal Specialists, August 27, 2013, Toronto, Ontario, Canada. | ||
Patel HR, Eichenbaum D. Geographic atrophy: clinical impact and emerging treatments. Ophthalmic Surg Lasers Imaging Retina. 2015; 46(1):8–13. | ||
Yehoshua Z, de Amorim Garcia Filho CA, Nunes RP, et al. Systemic complement inhibition with eculizumab for geographic atrophy in age-related macular degeneration: the COMPLETE study. Ophthalmology. 2014;121(3):693–701. | ||
Wong WT, Dresner S, Forooghian F, et al. Treatment of geographic atrophy with subconjunctival sirolimus: results of a phase I/II clinical trial. Invest Ophthalmol Vis Sci. 2013;54(4):2941–2950. | ||
Zhou H, Zhao X, Johnson EJ, et al. Serum carotenoids and risk of age-related macular degeneration in a Chinese population sample. Invest Ophthalmol Vis Sci. 2011;52(7):4338–4344. | ||
Petrou PA, Cunningham D, Shimel K, et al. Intravitreal sirolimus for the treatment of geographic atrophy: results of a phase I/II clinical trial. Invest Ophthalmol Vis Sci. 2015;56(1):330–338. | ||
Radu RA, Mata NL, Nusinowitz S, Liu X, Sieving PA, Travis GH. Treatment with isotretinoin inhibits lipofuscin accumulation in a mouse model of recessive Stargardt’s macular degeneration. Proc Natl Acad Sci U S A. 2003;100(8):4742–4747. | ||
Mata NL, Lichter JB, Vogel R, Han Y, Bui TV, Singerman LJ. Investigation of oral fenretinide for treatment of geographic atrophy in age-related macular degeneration. Retina. 2013;33(3):498–507. | ||
Zhou J, Jang YP, Kim SR, Sparrow JR. Complement activation by photooxidation products of A2E, a lipofuscin constituent of the retinal pigment epithelium. Proc Natl Acad Sci U S A. 2006; 103(44):16182–16187. | ||
Radu RA, Han Y, Bui TV, et al. Reductions in serum vitamin A arrest accumulation of toxic retinal fluorophores: a potential therapy for treatment of lipofuscin-based retinal diseases. Invest Ophthalmol Vis Sci. 2005;46(12):4393–4401. | ||
Kubota R, Al-Fayoumi S, Mallikaarjun S, Patil S, Bavik C, Chandler JW. Phase 1, dose-ranging study of emixustat hydrochloride (ACU-4429), a novel visual cycle modulator, in healthy volunteers. Retina. 2014;34(3):603–609. | ||
Kubota R, Boman NL, David R, Mallikaarjun S, Patil S, Birch D. Safety and effect on rod function of ACU-4429, a novel small-molecule visual cycle modulator. Retina. 2012;32(1):183–188. | ||
Tanito M, Li F, Elliott MH, Dittmar M, Anderson RE. Protective effect of TEMPOL derivatives against light-induced retinal damage in rats. Invest Ophthalmol Vis Sci. 2007;48(4):1900–1905. | ||
Wong WT, Kam W, Cunningham D, et al. Treatment of geographic atrophy by the topical administration of OT-551: results of a phase II clinical trial. Invest Ophthalmol Vis Sci. 2010;51(12):6131–6139. | ||
Chew EY, Clemons TE, Agrón E, et al. Long-term effects of vitamins C and E, β-carotene, and zinc on age-related macular degeneration: AREDS report no. 35. Ophthalmology. 2013;120(8):1604–1611.e1604. | ||
Johnson LV, Leitner WP, Rivest AJ, Staples MK, Radeke MJ, Anderson DH. The Alzheimer’s A beta-peptide is deposited at sites of complement activation in pathologic deposits associated with aging and age-related macular degeneration. Proc Natl Acad Sci U S A. 2002;99(18):11830–11835. | ||
Ding JD, Johnson LV, Herrmann R, et al. Anti-amyloid therapy protects against retinal pigmented epithelium damage and vision loss in a model of age-related macular degeneration. Proc Natl Acad Sci U S A. 2011;108(28):E279–E287. | ||
Damico FM, Gasparin F, Scolari MR, Pedral LS, Takahashi BS. New approaches and potential treatments for dry age-related macular degeneration. Arq Bras Oftalmol. 2012;75(1):71–76. | ||
Lai RK, Chun T, Hasson D, Lee S, Mehrbod F, Wheeler L. Alpha-2 adrenoceptor agonist protects retinal function after acute retinal ischemic injury in the rat. Vis Neurosci. 2002;19(2):175–185. | ||
Tatton W, Chen D, Chalmers-Redman R, Wheeler L, Nixon R, Tatton N. Hypothesis for a common basis for neuroprotection in glaucoma and Alzheimer’s disease: anti-apoptosis by alpha-2-adrenergic receptor activation. Surv Ophthalmol. 2003;48(Suppl 1):S25–S37. | ||
Allergan. Safety and efficacy of brimonidine intravitreal implant in patients with geographic atrophy due to age-related macular degeneration (AMD) [updated: March 13, 2013]. Available from: https://clinicaltrials.gov/ct2/show/results/NCT00658619?sect=X870156&term=NCT00658619&rank=1#outcome2. NLM identifier: NCT00658619. Accessed October 8, 2015. | ||
LaVail MM, Yasumura D, Matthes MT, et al. Protection of mouse photoreceptors by survival factors in retinal degenerations. Invest Ophthalmol Vis Sci. 1998;39(3):592–602. | ||
Tao W, Wen R, Goddard MB, et al. Encapsulated cell-based delivery of CNTF reduces photoreceptor degeneration in animal models of retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2002;43(10):3292–3298. | ||
Sieving PA, Caruso RC, Tao W, et al. Ciliary neurotrophic factor (CNTF) for human retinal degeneration: phase I trial of CNTF delivered by encapsulated cell intraocular implants. Proc Natl Acad Sci U S A. 2006;103(10):3896–3901. | ||
Zhang K, Hopkins J, Heier JS, et al. Ciliary neurotrophic factor delivered by encapsulated cell intraocular implants for treatment of geographic atrophy in age-related macular degeneration. Proc Natl Acad Sci U S A. 2011;108(15):6241–6245. | ||
Mu Y, Zhao M, Su G. Stem cell-based therapies for age-related macular degeneration: current status and prospects. Int J Clin Exp Med. 2014;7(11):3843–3852. | ||
Lund RD, Wang S, Klimanskaya I, et al. Human embryonic stem cell-derived cells rescue visual function in dystrophic RCS rats. Cloning Stem Cells. 2006;8(3):189–199. | ||
Schwartz SD, Hubschman JP, Heilwell G, et al. Embryonic stem cell trials for macular degeneration: a preliminary report. Lancet. 2012; 379(9817):713–720. | ||
Schwartz SD, Regillo CD, Lam BL, et al. Human embryonic stem cell-derived retinal pigment epithelium in patients with age-related macular degeneration and Stargardt’s macular dystrophy: follow-up of two open-label phase 1/2 studies. Lancet. 2015;385(9967):509–516. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.