Back to Journals » International Journal of General Medicine » Volume 15
Genotype–Phenotype Correlation Analysis and Identification of a Novel SRD5A2 Mutation in Four Unrelated Chinese Patients with 5α-Reductase Deficiency
Authors Gui T, Yao F, Yang X, Wang X, Nie M, Wu X, Tian Q
Received 14 June 2022
Accepted for publication 8 August 2022
Published 18 August 2022 Volume 2022:15 Pages 6633—6643
DOI https://doi.org/10.2147/IJGM.S377675
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Ting Gui,1,* Fengxia Yao,2,* Xinzhuang Yang,2 Xi Wang,3 Min Nie,3 Xueyan Wu,3 Qinjie Tian1
1Department of Obstetrics and Gynecology, National Clinical Research Center for Obstetric and Gynecologic Diseases, State Key Laboratory of Complex Severe and Rare Diseases, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, People’s Republic of China; 2Department of Medical Research Center, State Key Laboratory of Complex Severe and Rare Diseases, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, People’s Republic of China; 3Department of Endocrinology, State Key Laboratory of Complex Severe and Rare Diseases, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Qinjie Tian, Department of obstetrics and gynecology, Peking Union Medical College Hospital, No. 1, Shuaifuyuan Road, Dongcheng District, Beijing, 100730, People’s Republic of China, Tel +86 010-69158335, Email [email protected]
Objective: The 5α-reductase type 2 deficiency is mainly caused by mutations in the SRD5A2 gene. Our study aims to investigate the SRD5A2 gene mutations and their corresponding manifestations.
Methods: Four unrelated Chinese patients with 46, XY ambiguous genitalia were studied. Molecular genetic alterations and clinical presentations were analyzed.
Results: Five variants in the SRD5A2 gene were identified, all highly conserved in vertebrate orthologs. The p.P251A was a novel variant, predicted to “Affect protein function” and to be “probably damaging”. Combining patients’ gene mutations with their external genitalia and male sexual characteristics, we found that three variants, p.Q6X, p.N193S, and p.H90Y, were associated with severe undervirilization of external genitalia, and the other two, p.G203S and p.P251A, probably retained part of the enzyme activity.
Conclusion: Mutation analysis of SRD5A2 gene is crucial for differential diagnosis in patients with 5α-reductase type 2 deficiency. Patients’ variable manifestations depend on the mutation type and residual enzyme activity. The novel variant p.P251A enlarges the spectrum of SRD5A2 mutations.
Keywords: 5α-reductase type 2 deficiency, SRD5A2 gene mutation, androgen receptor insensitivity, disorders of sex development, differential diagnosis
Introduction
The 5α-reductase type 2 deficiency, first described in the 1970s, is characterized by genital ambiguity attributable to undervirilization in genetic males.1,2 The affected individuals usually have normal internal male genitalia but ambiguous external genitalia ranging from a male phenotype with hypospadias or micropenis to a nearly female structure with clitoromegaly.3 This disease has been reported to account for about 7–25% of 46, XY disorders of sex development cases.4 In a previous review published in 2020, 434 cases of 5α-reductase type 2 deficiency were identified in the literature from 44 different countries, indicating that this disease has a worldwide distribution.5
The gene SRD5A2 mutation is considered to be a crucial pathogenic factor for 5α-reductase type 2 deficiency. It is located on chromosome 2p23 containing 5 exons and 4 introns, and encodes a 254 amino acid protein named the steroid 5α-reductase type 2 enzyme which is expressed in external genital tissues and prostate early in gestation.6 This enzyme catalyzes the conversion of testosterone (T) to the more active dihydrotestosterone (DHT). T and DHT interact with the same androgen receptor, but produce distinct biological responses. T plays an important role in transforming the Wolffian ducts into male internal genitalia,3 while DHT is crucial for differentiation and development of the external genitalia into male type.7,8 A defect of the SRD5A2 gene could impair the activity of 5α-reductase type 2 enzyme, which in turn reduces the DHT level and affects the differentiation of external genitalia into male type. Up to now, a total of 185 SRD5A2 gene mutations have been identified (Human Gene Mutation Database, www.hgmd.cf.ac.uk), including 133 (71.89%) missense or nonsense mutations, 12 (6.49%) splicing site changes, 1 (0.54%) regulatory sequence, 19 (0.27%) small deletions, 9 (4.86%) small insertions, 5 (2.7%) small indels, and 5 (3.24%) gross deletions. The homozygous mutations account for about 60–70%, while the proportion of heterozygous mutations is about 30–40%.9
The 5α-reductase type 2 deficiency syndrome is a rare condition but should be considered in front of all 46, XY patients with ambiguous genitalia. Its highly variable manifestations of external genitalia make it difficult to distinguish this condition from other disorders of sex development (DSD), for example, partial androgen insensitivity syndrome (PAIS). Traditionally, the diagnosis depended on measurement of the serum T/DHT ratio. However, normal serum DHT concentration and normal T/DHT ratio could also be observed in some patients.10 The ratio value might be difficult to interpret in prepubertal children and may lead to misdiagnosis. Therefore, molecular genetic analysis quickly becomes the first-line approach for the differential diagnosis of 5α-reductase type 2 deficiency, particularly in patients with partial enzyme deficiency and in prepubertal subjects.11
In the present study, we identified five SRD5A2 gene variations in four unrelated Chinese patients, and evaluated the correlation between phenotype and genotype. One variant, p.P251A, was novel, enlarging the spectrum of SRD5A2 mutations. We hope our study could provide useful information for clinicians on the recognition and management of this rare disease.
Materials and Methods
Ethics Approval
Our study was approved by the Ethics Committee of Peking Union Medical College Hospital (PUMCH) (IRB Number: JS-2510). Written informed consents were obtained from the parents or legal guardians of all four patients. Our study complied with the Declaration of Helsinki. The data used in this study was confirmed to be anonymized or maintained with confidentiality.
Four unrelated patients with 5α-reductase type 2 deficiency were retrospectively reviewed in our study (named as patient 1, patient 2, patient 3, and patient 4). All data in this study were collected from the hospital’s archived database. This study did not influence the diagnosis or treatment of the patients.
Evaluation of Patients’ Clinical Information
Clinical information, including the chief complaint, male secondary sexual characteristics (such as deep voice, laryngeal prominence, labial palps, dense sex pilus, et al), female secondary sexual characteristics (breast development and menstruation), and physical examination (external genitalia type at birth, palpable masses in major labia or inguinal canals, clitoris, urinary and vaginal orifices, and vaginal length), as well as ultrasound findings, was collected and analyzed. All physical examinations of the external genitalia were performed by the same gynecologic endocrinologist. Regarding the serum hormone measurement, the automated Elecsys Immunoanalyzer (Beckmann, USK) was used to detect serum levels of luteinizing hormone (LH), follicle-stimulating hormone (FSH), estradiol (E2), progesterone (P), prolactin (PRL), testosterone (T), dihydrotestosterone (DHT), 17-OH progesterone (17α-OHP), dehydroepiandrosterone sulfate (DHEAS), adrenocorticotrophic hormone (ACTH), and cortisol. The hCG stimulation test was performed with a daily injection of hCG 2000 U for 3 days in one patient.
Molecular Genetic Analyses
Molecular genetic analyses were performed as follows: 1) Targeted Sanger sequencing for androgen receptor (AR) gene was performed to exclude the diagnosis of AIS, and AR gene in all four patients were intact. 2) Then whole-exome sequencing (WES) and single-nucleotide polymorphism (SNP)-array were performed, and SRD5A2 gene mutations were found. 3) Finally, candidate variants were further confirmed by Sanger sequencing. All data interpretation was based on the GRCh37/hg38 human genome assembly, and the reference sequence for SRD5A2 was NM-000348.3 obtained from GenBank.12 It is worth mentioning that, patient 4 in our study received Sanger sequencing directly for SRD5A2 gene rather than WES in PUMCH, as we were more inclined to consider the diagnosis of 5α-reductase type 2 deficiency and the time and money spent on sequencing was much lesser.
Genomic DNA was extracted from peripheral blood leucocytes (DNA QIAamp DNA blood Mini Kit; Quiagen, Courtaboeuf, France), according to the manufacturer’s protocol. For targeted Sanger sequencing, PCR amplification of exons was performed in a 25-µl reaction volumes containing 20–100 ng genomic DNA, 10 uM of each primer, 250 µM of each dNTP, 1X PCR buffer (containing 1.5 mM Mg2+), and 1 U Taq DNA polymerase. Initial denaturation was 5 min at 95°C, followed by amplification for 30 cycles with denaturation at 95°C for 30 sec, annealing at 60°C for 30 sec, and extension at 72°C for 45 sec. Specific primers are listed in Table 1. The PCR products were sequenced bi-directionally using a dye terminator cycle sequencing ready reaction kit (ABI PRISM 3500, Applied Biosystems).
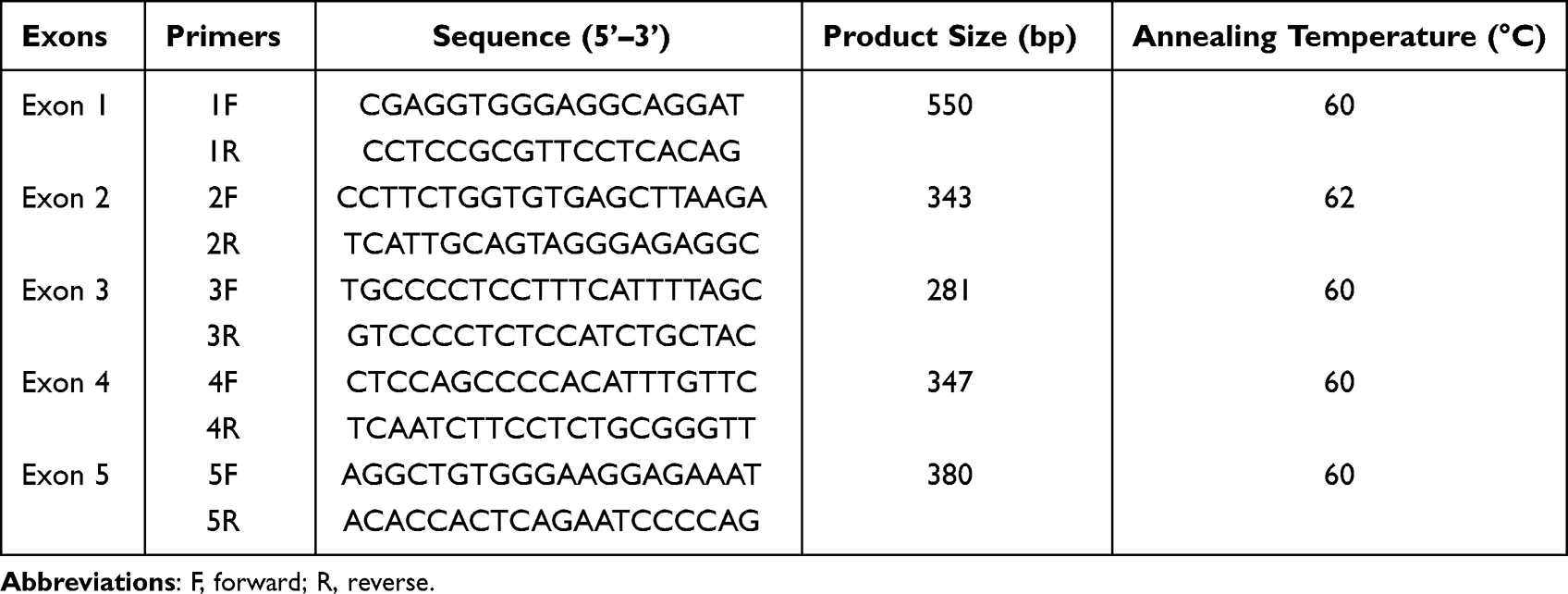 |
Table 1 Primers for SRD5A2 Amplification |
For whole-exome sequencing, the extracted genomic DNA was used for sequencing library preparation using the TruSeq DNA Sample Preparation kit (Illumina, Inc., San Diego, CA, USA) and the exomic regions were enriched using the xGen® Exome Research Panel v1.0 according to manufacturers’ instructions. Paired-end short read sequencing was performed on an Illumina platform with a PE150 read length (Illumina, Inc.). Then identification of pathogenic mutations from exome data was performed. Bioinformatics analyses were performed using an in-house pipeline. Briefly, sequencing reads that passed quality filtering were aligned with the human reference genome hg38, using the Burrows-Wheeler Aligner program. Variants were identified using a combination of the FreeBayes, Genome Analysis Toolkit, CNVnator and in-house software programs.
The human gene mutation database (HGMD), the dbSNP database of NCBI (http://www.ncbi.nlm.nih.gov/SNP), and the 1000 Genomes Project (http://browser.1000genomes.org/) were used to confirm whether the detected mutations had been reported previously. Resulting sequences from patients were compared with the NCBI published human SRD5A2 sequence (NP_000339.2) using the online multiple sequence alignment tool.12
Protein Function Predictions of Novel Mutation
The potential pathogenicity of the novel SRD5A2 mutation was examined by in silico analysis using two softwares: Polyphen-2 (http://genetics.bwh.harvard.edu/pph2) and SIFT (http://sift.bii.a-star.edu.sg/). Polyphen-2 predicts the possible impact of an amino acid substitution on the structure and function of a human protein using straightforward physical and comparative considerations.13 The SIFT predicts the functional importance of amino acid substitutions based on the alignment of orthologous and/or paralogous protein sequences.14 For PolyPhen-2, the outcome “possibly damaging” and “probably damaging” were both considered pathogenic. For SIFT, scores lower than 0.05 suggest a potential pathogenicity. Original sequences of proteins were obtained from the Ensembl and UniProt/Swiss-Prot databases.
Results
Patients’ Clinical Characteristics (Table 2)
Four patients were found from unrelated Chinese families. All four patients were raised as girls with female type of external genitalia at birth. Three patients were directly admitted to the gynecologic endocrine clinic of PUMCH during their puberty with chief complaint of appearance of masculinization or primary amenorrhea and absence of breast development, while the other one was first admitted to the pediatric clinic at 7 months because of palpable masses in the groin areas and then was admitted to the gynecologic endocrine clinic at 28 years old complaining of sexual intercourse failure.
 |
Table 2 Clinical Characteristics of Patients |
Karyotype analysis was then performed to clarify the possible etiology of the absence of female secondary sexual characteristics, with the results indicating 46, XY in all the four patients. These socially female patients were actually genetically male. They were under-masculinity rather than virilization. What caused the contradiction, the gonad (secreting androgen), the receptor of androgen (binding with androgen), or the conversion of T to DHT which is responsible for the differentiation of male external genitalia?
Ultrasonography examination confirmed testicular echoes bilaterally without Mullerian ducts. All the four patients had palpable masses in the inguinal areas or the major labia, and pathological examination of the surgically removed masses confirmed the gonad to be testis. Ectopic testes were capable enough to secret testosterone. After puberty, these patients showed different degrees of masculinity, such as deep voice, laryngeal prominence, labial palps, and dense sex pilus. Evaluation of the external masculinization score (EMS) showed 1.0, 2.0, 2.0, 4.0, for patient 1, patient 2, patient 3, and patient 4, respectively (Table 3). The lower the score, the severer the degree of feminization.15 Consistently, obvious clitoromegaly (5 cm in length) was observed in patient 4, with common orifice for urethra and vagina. These indicated that the function of androgen receptor as well as the conversion of T to DHT in some patients were retained, at least partially.
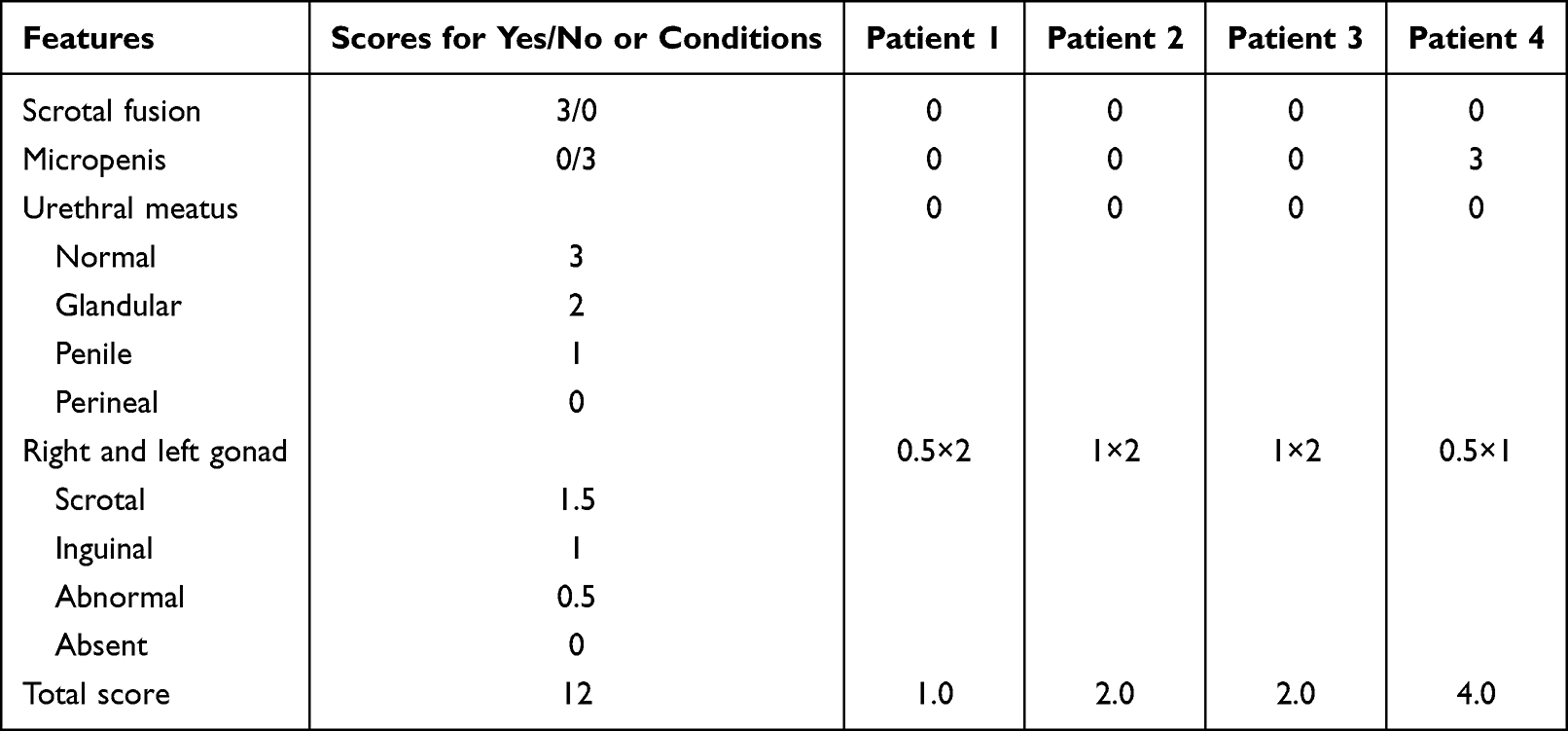 |
Table 3 External Masculinization Score (EMS) of Four Patients with Ambiguous Genitalia |
To clarify the differential diagnosis of partial androgen receptor insufficiency syndrome and 5α-reductase type 2 deficiency, the serum sexual hormones were measured. Three patients had elevated levels of T, except for one patient who had sexual glands removed during the infancy. For certain reasons, only one patient received detection of T/DHT ratio, result indicating apparent elevation after hCG stimulation. For other parameters, gonadotrophins (FSH and LH), showed different degrees of elevation, while E2 presented obvious reduction, in accordance with absence of female gonad ovary. Other parameters for differential diagnosis, such as 17α-OHP, DHEAS, ACTH, cortisol, and 24H-UFC, were found negative.
Final molecular genetic analyses revealed intact androgen receptor and SRD5A2 gene mutations in all four patients, confirming the diagnosis of 5α-reductase type 2 deficiency. Finally, all four patients chose to be reared as females. Three patients received perineoplastic surgery and bilateral gonadectomy; one patient received bilateral gonadectomy during infancy and secondary perineoplastic surgery and vaginal toppling before marriage. Post-operative histopathological examination confirmed the masses were testicular tissues.
Molecular Genetic Analyses
In aggregate, five variants in the SRD5A2 gene were identified (Figure 1, Table 4). The most prevalent variant was p.Q6* (3/4), followed by p.N193S (2/4), and p.G203S, p.H90Y, and p.P251A (1/4), respectively. Among the five variants, p.P251A was novel, while the other four had been previously reported. The p.Q6* resulted in a nonsense mutation; the p.N193S, p.G203S, p.H90Y, and p.P251A resulted in missense mutations. These mutations were clustered in exon 1 and exon 4.
 |
Table 4 Sequencing Analyses of SRD5A2 Gene in Four Unrelated Chinese Patients |
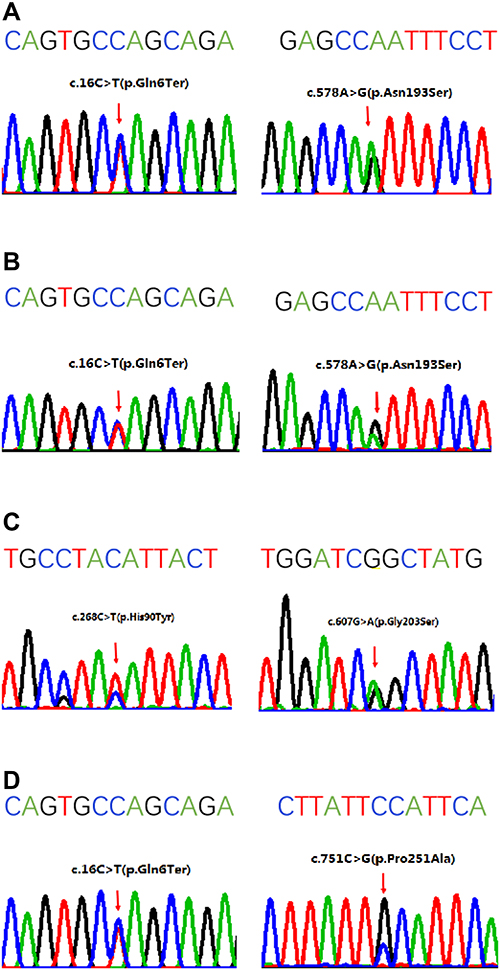 |
Figure 1 Sanger sequencing of the SRD5A2 gene in four patients. The sequence above the four-color chromatogram is the reference sequence. (A) The c.16C>T:p.Q6* at exon 1 and c.578A>G:p.N193S at exon 4 in patient 1. (B) The c.16C>T:p.Q6* at exon 1 and c.578A>G:p.N193S at exon 4 in patient 2. (C) The c.268C>T:p.H90Y at exon 1 and c.607G>A:p.G203S at exon 4 in patient 3. (D) The c.16C>T:p.Q6* at exon 1 and c.751C>G:p.P251A at exon 4 in patient 4. |
In our study, heterozygous mutations were observed in all four patients. Pedigree analyses suggested that all the mutations were inherited from the patients’ father or mother (Table 4). It was worth to mention that the younger sister of patient 4 was also found to have the same SRD5A2 gene mutation.
Multiple sequence alignment analysis was performed to analyze the evolutionary conservancy in different species. Our results showed that the five variants found in our study, p.Q6*p.N193S, p.G203S, p.H90Y, and p.P251A, all affected the strictly conserved domains among vertebrate orthologs, including five species, H. sapiens (human), P. troglodytes (chimpanzee), M. mulatta (monkey), C. lupus (wolf), and B. taurus (cow) (Figure 2).
 |
Figure 2 Multiple sequence alignment analysis of the SRD5A2 gene. Multiple amino acid alignment of SRD5A2 including sequences from species in the following order: H. sapiens (human), P. troglodytes (chimpanzee), M. mulatta (monkey), C. lupus (wolf), and B. Ttaurus (cow). The mutant amino acids of SRD5A2 gene and corresponding residues of aligned sequences are shown in orange box. |
Prediction of the Effects of the Novel Mutations
The novel missense mutation, p.P251A, was evaluated as “probably damaging” with a score of 1.000 (sensitivity: 0.00; specificity: 1.00) by PolyPhen 2. In SIFT analysis, the score obtained for the new substitution was 0.00, which placed the mutation in the “Affect protein function” class. According to the ACMG/AMP guidelines, the novel variant p.P251A was classified as VUS (variant of uncertain significance).16
Discussion
Patients with 5α-reductase type 2 deficiency could present with variable degrees of undervirilization. In a previous study of 190 Chinese patients with this condition, the most common malformation of the external genitalia was hypospadias, followed by microphallus and cryptorchidism.4 However, in our study, the most common manifestation was cryptorchidism, followed by the blocking out of vaginal opening by the elevated perineal body but separated from the urethral orifice. Patients in our study all had complete female type of external genitalia at birth and did not present any male characteristics until puberty. That is why they came to our gynecological endocrine clinic, rather than the pediatric clinic, at a later age in life.
The variability of manifestations is speculated to be related with multiple factors. The SRD5A2 gene mutation is the most important one, determining the residual enzyme activity and the degree of undervirilization. Most severe phenotypes are usually caused by nonsense mutations, which lead to absence of enzymatic activity. Conversely, missense and splicing junction mutations that do not totally inactivate the enzyme produce a wide phenotype variability.
The variant p.Q6* has been identified common in Asian patients and had been reported to be associated with severe phenotype and lower external masculinization score and urethral meatus score17,18 This is probably because the p.Q6* mutation creates a drastically truncated protein and abolishes all enzyme activity, since the transversion of C to T (c.16C>T) led to the transversion of amino acid Glutamine to a premature stop codon at position 6 in exon 1.19 The complete loss of enzymatic activity has been reported to be due to the lack of both the testosterone and the NADPH binding domains.20
The variant p.N193S has been frequently reported in the whole world.21 The asparagine at position 193 was a highly conservative residue in mammals and the variant was classified as damaging and pathogenic.22 Site-directed mutagenesis and in vitro assays confirmed that p.N193S severely impaired enzyme activity by decreasing the affinity of the enzyme for NADPH.23
The variant p.G203S has also been found common in Asian patients.18 Zhang et al reported that in vitro, approximately 60% of enzymatic activity was reduced by the p.G203S mutation.19 Patients with mutations retaining residual enzyme activity usually have a milder and variable phenotype.24
At present, only two studies reported the variant p.H90Y, and no specific discussions were described in the two papers. In a study of prevalence of endocrine and genetic abnormalities in 122 boys for disorders of sex development in 2017, Nixon et al found the mutation of p.H90Y in SRD5A2 gene was involved in the disorder of androgen synthesis.25 In another multi-center study of regional distribution and genotype–phenotype profiling of SRD5A2 gene 190 Chinese patients in 2019, Gui et al found the variant p.H90Y was a heterozygous missense mutation located in exon 1.4
In addition, we found a new variant of SRD5A2 gene, c.751C>G:p.P251A, enlarging the molecular spectrum of SRD5A2 gene abnormalities. This new mutation led to the transversion of Proline to Alanine. Multiple sequence alignment analysis showed that the residue Pro251 was conserved across the species, and in silico analysis of the potential pathogenicity showed that the new variant was predicted to “Affect protein function” and to be “probably damaging”.
The enzyme 5α-reductase utilizes NADPH as a cofactor to reduce the double bonds of a variety of steroid substrates such as testosterone through 3-oxo-delta(4,5) structures.26 The SRD5A2 gene contains the 3-oxo-5-alpha-steroid 4-dehydrogenase domain, which encodes 149 amino acids starting from position 106 to position 254. The variants p.G203S and p.P251A are located within this domain. This indicates that mutation of amino acids at position 203 and 251 could probably damage the affinity of enzyme 5α-reductase type 2 to its cofactor NADPH, which in turn, leads to the reduction of enzyme catalyzing activity of converting T to DHT.
In our study, patients 1 and 2 had the same gene mutation, p.Q6*/p.N193S, but they showed different clinical manifestations. Patient 1 had apparent male sexual secondary characteristics during adolescence, while patient 2 did not present any virilization. This was because patient 2 received gonadectomy at 7 months, resulting in serum T concentration remaining at a very low level even after puberty. Furthermore, the variant p.Q6* and p.N193S both severely damage the enzyme activity of 5α-reductase, so the conversion of T to DHT was almost completely interdicted, leading to the differentiation of external genitalia into female type.
Patient 1 (p.Q6*/p.N193S) and patient 4 (p.Q6*/p.P251A) had a common variant p.Q6* with another different variant. Comparing their clinical manifestations, we found both patients had apparent male sexual secondary characteristics such as deep voice, labial palps, and dense sex pilus, but their external genitalia had different appearance. Patient 1 had an almost complete female type of external genitalia and separate opening for urethra and vagina; patient 4 was observed clitoromegaly (5cm in length like a microphallus) and common opening for urethra and vagina. It was speculated that the variant p.P251A retained partial enzymatic activity, and the retained DHT could stimulate the differentiation of the external genitalia into male type during the embryonic phase, resulting in dysplasia of vagina and common opening with the urethra. This indicated that variant p. N193S impaired the enzyme activity more severely than the variant p.P251A, consistent with previous studies mentioned above.
Patient 3 (p.H90Y/p.G203S) presented milder male sexual characteristics after puberty compared with patient 1 and patient 4, and almost female-type external genitalia similar to that of patient 1 and patient 2. This indicated that the serum T level was mildly elevated, while the serum DHT level was not corresponding increased but still kept at a very low level. This proved that the variant p.H90Y severely damage the conversion of T to DHT, although the variant p.G203S could retain the enzyme activity about 40%.19
Previous research from different countries revealed that mutations have been predominantly confined to exons 1, 4, and 5. Exon 4 is the most frequently involved, followed by exon 1 and exon 5, and mutations located within exon 4 often lead to complete loss of the enzyme activity.4,17,27 In our study, exon 4 and exon 1 accounted for 50%, respectively, consistent with previous researches.
Furthermore, it has been reported that approximately two-thirds of patients with 5α-reductase deficiency had homozygous mutations, approximately one-third was heterozygous, and deletion and disruptive mutations were rare, from different families in the whole world.28 However, all the SRD5A2 mutations in our study were heterozygous, which might be ascribable to the vast population, ethnic diversity, and prohibition of consanguineous marriage in China.24 This suggested that the carrier frequency of a single mutant allele was probably underestimated due to the rarity of the disease.
Previously, it was believed that the increase in the ratio of T/DHT could indicate the diagnosis of 5α-reductase deficiency. In our study, only one patient (patient 1) received the detection of T/DHT, and the ratio was indeed dramatically elevated after the administration of HCG, helping in the diagnosis of 5α-reductase deficiency. However, it was recently reported that the plasma T/DHT ratio at baseline or after stimulation was not associated with genital ambiguity grade in the newborn.29 A normal T/DHT ratio could not rule out this disease.10 The origin of the divergent phenotype is not precisely known.
The presentations of 5α-reductase deficiency strongly resemble that of PAIS.30 Regarding the differential diagnosis, molecular analysis is considered to be the most effective approach, as biochemical analysis might not always be unequivocal. In our study, three patients did not receive the detection of serum T/DHT ratio for certain unknown reason. The sequencing analyses showed that AR gene intact but the SRD5A2 gene mutated, thus excluding the diagnosis of PAIS and confirming the diagnosis of 5α-reductase type 2 deficiency.
After confirming the diagnosis of 5α-reductase type 2 deficiency, patients and their family are confronted with the problem of choosing the gender. Gender role selection is dependent upon certain social and cultural factors. All these factors should be considered by the physicians and the psychiatrists, working in concert with the patients and their families. Most patients may admit their maleness but choose not to change to a male gender role, since they are reared as females with adequate femaleness after birth and do not attend clinic until puberty or even adulthood. Social sex during childhood, rather than the biological genetic gender, more strongly influences gender selection. In China, the choice of male gender may be attributable to the traditional patriarchal mentality.
The main shortcoming of this work was that the number of cases recruited in this research was small. In the future, a multicenter study with a larger sample size should be performed.
Conclusions
Five variants in the SRD5A2 gene were identified in four unrelated Chinese patients. The p.P251A, a novel variant, was highly conservative in species, and was predicted to “Affect protein function” and to be “probably damaging”. Comprehensively analyzing patients’ external genitalia and male secondary sexual features, three variants, p.Q6*, p.N193S, and p.H90Y, were speculated to severely damage the enzymatic activity, and the other two, p.G203S and p.P251A, were speculated to retain part of the enzymatic activity. Sequencing analysis of SRD5A2 gene is crucial for differential diagnosis in patients with 5α-reductase type 2 deficiency.
Funding
This study was financially supported by the National Natural Science Foundation of China under Grant No. 81671424.
Disclosure
Ting Gui and Fengxia Yao share first authorship. The authors report there are no competing interests to declare.
References
1. Imperato-McGinley J, Guerrero L, Gautier T, Peterson RE. Steroid 5α-reductase deficiency in man: an inherited form of male pseudohermaphroditism. Science. 1974;186:1213–1216. doi:10.1126/science.186.4170.1213
2. Walsh PC, Madden JD, Harrod MJ, Goldstein JL, MacDonald PC, Wilson JD. Familial incomplete male pseudohermaphroditism, type 2. Decreased dihydrotestosterone formation in pseudovaginal perineoscrotal hypospadias. N Engl J Med. 1974;291:944–949. doi:10.1056/NEJM197410312911806
3. Imperato-McGinley J, Zhu YS. Androgens and male physiology the syndrome of 5 alpha-reductase-2 deficiency. Mol Cell Endocrinol. 2002;198(1–2):51–59. doi:10.1016/S0303-7207(02)00368-4
4. Gui B, Song Y, Su Z, et al. New insights into 5α-reductase type 2 deficiency based on a multi-center study: regional distribution and genotype-phenotype profiling of SRD5A2 in 190 Chinese patients. J Med Genet. 2019;56(10):685–692. doi:10.1136/jmedgenet-2018-105915
5. Bstista RL, Mendonca BB. Integrative and analytical review of the 5-alpha-reductase type 2 deficiency worldwide. Appl Clin Genet. 2020;13:83–96. doi:10.2147/TACG.S198178
6. Chan AOK. Performance of in silico analysis in predicting the effect of non-synonymous variants in inherited steroid metabolic disease. Steroids. 2013;78(7):726–730. doi:10.1016/j.steroids.2013.04.002
7. Banerjee PP, Banerjee S, Brown TR, Zirkin BR. Androgen action in prostate function and disease. Am J Clin Exp Urol. 2018;6(2):62–77.
8. Imperato-McGinley J. 5 alpha-reductase-2 deficiency and complete androgen insensitivity: lessons from nature. Adv Exp Med Biol. 2002;511:121–131.
9. Zhu H, Liu W, Han B, et al. Phenotypic and molecular characteristics in eleven Chinese patients. Clin Endocrinol. 2014;81(5):711–720.
10. Mazen I, Gad YZ, Hafez M, Sultan C, Lumbroso S. Molecular analysis of 5 alpha-reductase type 2 gene in eight unrelated Egyptian children with suspected 5 alpha-reductase deficiency: prevalence of the G34R mutation. Clin Endocrinol. 2003;58:627–631. doi:10.1046/j.1365-2265.2003.01763.x
11. Chan AO, But BW, Lee CY, et al. Diagnosis of 5α-reductase 2 deficiency: is measurement of dihydrotestosterone essential? Clin Chem. 2013;59:798–806. doi:10.1373/clinchem.2012.196501
12. Lassmann T, Sonnhammer E. Kaling-an accurate and fast multiple sequence alignment algorithm. BMC Bioinform. 2005;6:298. doi:10.1186/1471-2105-6-298
13. Adzhubei LA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi:10.1038/nmeth0410-248
14. Ng PC, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–3814. doi:10.1093/nar/gkg509
15. Ahmed SF, Khwaja O, Hughes IA. The role of a clinical score in the assessment of ambiguous genitalia. BJU Int. 2000;85(1):120–124. doi:10.1046/j.1464-410x.2000.00354.x
16. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. doi:10.1038/gim.2015.30
17. Abaci A, Catli G, Kirbiyik O, et al. Genotype-phenotype correlation, gonadal malignancy risk, gender preference, and testosterone/dihydrotestosterone ratio in steroid 5-alpha-reductase type 2 deficiency: a multicenter study from Turkey. J Endocrinol Invest. 2019;42(4):453–470. doi:10.1007/s40618-018-0940-y
18. Avendano A, Paradisi I, Cammarata-Scalisi F, Callea M. 5-α-reductase type 2 deficiency: is there a genotype-phenotype correlation? A review. Hormones. 2018;17(2):197–204. doi:10.1007/s42000-018-0013-9
19. Zhang MN, Yang J, Zhang HJ, Ning G, Li XY, Sun SY. A novel SRD5A2 mutation with loss of function identified in Chinese patients with hypospadias. Hormone Res Peadiatr. 2011;76(1):44–49. doi:10.1159/000324692
20. Wang Y, Li Q, Xu J, et al. Mutation analysis of five candidate genes in Chinese patients with hypospadias. Eur J Hum Genet. 2004;12(9):706–712. doi:10.1038/sj.ejhg.5201232
21. Avendano A, Gonzalez-Coira M, Paradisi I, et al. 5α-reductase type 2 deficiency in families from an isolated Andean population in Venezuela. Ann Hum Genet. 2020;84(2):151–160. doi:10.1111/ahg.12358
22. Landrum M, Lee J, Riley G, et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acid Res. 2014;42:D980–D985. doi:10.1093/nar/gkt1113
23. Mendonca BB, Batista RL, Domenice S, et al. Steroid 5α-reductase 2 deficiency. J Steroid Biochem Mol Biol. 2016;163:206–211. doi:10.1016/j.jsbmb.2016.05.020
24. Fan L, Song Y, Polak M, et al. Clinical characteristics and genotype-phenotype correlations of 130 Chinese children in a high-homogeneity single-center cohort with 5 α-reductase 2 deficiency. Mol Genet Genom Med. 2020;8. doi:10.1002/mgg3.1431
25. Nixon R, Cerqueira V, Kyriakou A, et al. Prevalence of endocrine and genetic abnormalities in boys evaluated systematically for a disorder of sex development. Hum Reprod. 2017;32(10):2130–2137. doi:10.1093/humrep/dex280
26. Wigley WC, Prihoda JS, Mowszowicz I, et al. Natural mutagenesis study of the human steroid 5 alpha-reductase 2 isozyme. Biochemistry. 1994;33(5):1265–1270. doi:10.1021/bi00171a029
27. Vilchis F, Valdez E, Ramos L, Garcia R, Gomez R, Chavez B. Novel compound heterozygous mutations in the SRD5A2 gene from 46, XY infants with ambiguous external genitalia. J Hum Genet. 2008;53:401–406. doi:10.1007/s10038-008-0274-2
28. Wilson JD, Griffin JE, Russell DW. Steroid 5 alpha-reductase 2 deficiency. Endocr Rev. 1993;14:577–593. doi:10.1210/edrv-14-5-577
29. Bertelloni S, Scaramuzzo RT, Parrini D, Baldinotti F, Tumini S, Ghirri P. Early diagnosis of 5 alpha reductase deficiency in newborns. Sex Dev. 2007;1:147–151. doi:10.1159/000102103
30. Zhang D, Yao F, Tian T, Deng S, Luo M, Tian Q. Clinical characteristics and molecular genetics of complete androgen insensitivity syndrome patients: a series study of 30 cases from a Chinese tertiary medical center. Fertil Steril. 2021;115(5):1270–1279. doi:10.1016/j.fertnstert.2020.12.008
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.