")
Back to Journals » Clinical Interventions in Aging » Volume 13
Genome-editing applications of CRISPR–Cas9 to promote in vitro studies of Alzheimer’s disease
Authors Giau VV , Lee H, Shim KH, Bagyinszky E, An SSA
Received 25 October 2017
Accepted for publication 15 December 2017
Published 7 February 2018 Volume 2018:13 Pages 221—233
DOI https://doi.org/10.2147/CIA.S155145
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Richard Walker
Vo Van Giau,1,* Hyon Lee,2,* Kyu Hwan Shim,1 Eva Bagyinszky,1 Seong Soo A An1
1Department of Bionano Technology, Gachon University, Seongnam, South Korea; 2Department of Neurology, Gachon University Gil Medical Center, Incheon, South Korea
*These authors contributed equally to this work
Abstract: Genetic variations play an important role in the clinical presentation and progression of Alzheimer’s disease (AD), especially early-onset Alzheimer’s disease. Hundreds of mutations have been reported with the majority resulting from alterations in β-amyloid precursor protein (APP), presenilin 1 (PSEN1), or presenilin 2 (PSEN2) genes. The roles of these mutations in the pathogenesis of AD have been classically confirmed or refuted through functional studies, where the mutations are cloned, inserted into cell lines, and monitored for changes in various properties including cell survival, amyloid production, or Aβ42/40 ratio. However, these verification studies tend to be expensive, time consuming, and inconsistent. Recently, the clustered regularly interspaced short palindromic repeats-CRISPR-associated protein 9 (CRISPR–Cas9) system was developed, which improves sequence-specific gene editing in cell lines, organs, and animals. CRISPR–Cas9 is a promising tool for the generation of models of human genetic diseases and could facilitate the establishment of new animal AD models and the observation of dynamic bioprocesses in AD. Here, we recapitulated the history of CRISPR technology, recent progress, and, especially, its potential applications in AD-related genetic, animal modeling, and functional studies.
Keywords: Alzheimer’s disease, CRISPR–Cas9, mutation, Aβ42/40 ratio
Genetic and current functional studies on Alzheimer’s disease
Alzheimer’s disease (AD) is defined pathologically by extensive neuronal loss and the accumulation of intracellular neurofibrillary tangles and extracellular amyloid plaques in the brain. It comprises 50%–75% of all dementia cases and affects 23–35 million people worldwide (http://www.alz.co.uk/). Age is the most prominent biologic risk factor known,1 and it is used to classify AD patients into early-onset (EOAD, ≤65 years) and late-onset (LOAD, >65 years) groups. Large-scale genome-wide association studies (GWAS) have identified more than 20 genetic loci, including apolipoprotein E (APOE), associated with increased susceptibility to LOAD through pathways involved in Aβ production and clearance.2 On the other hand, the majority of EOAD cases are caused by dominantly inherited mutations in β-amyloid precursor protein (APP), presenilin 1 (PSEN1), and presenilin 2 (PSEN2). APP is a transmembrane neuronal protein that is sequentially cleaved by β-secretase and γ-secretase to produce amyloid beta (Aβ), while PSEN1 and PSEN2 are critical components of the γ-secretase complex. To date, more than 300 mutations have been discovered in APP, PSEN1, and PSEN2 genes worldwide (http://www.alzforum.org/mutations). Most of these mutations impair amyloid metabolism, resulting in an elevated Aβ42/40 ratio, elevated Aβ42 levels, and/or reduced production of Aβ40. Except a few cases, these mutations followed autosomal-dominant inheritance pattern, and associated with positive family history of disease. In addition, several patients were described with de novo case of mutations, where no affected family members were observed. However, autosomal-dominant EOAD is quite rare, because it appeared in <5% of all AD cases. Pathogenicity of sporadic LOAD could be complex, and the main cause remained unclear. Several genes, nongenetic factors, and gene–environment interactions may contribute to its pathogenesis. In LOAD, no exact genetic factor was identified, but several genes were discovered, which could increase the risk for disease onset. The strongest-risk allele for LOAD is the apolipoprotein E4, in both homo- and heterozygous stages. However, only APOE itself could not be the only genetic risk for AD, because several AD patients were negative for E4 allele. GWAS and next generation sequencing (NGS) approaches identified several possible genes, which could contribute to disease progression, such as cluseterin,3 complement component (3b/4b) receptor 1 (CR1), sortilin-related receptor-1 (SORL1), ATP-binding cassette subfamily A member 7 (ABCA7), CD33, or triggering receptor expressed on myeloid cells 2 (TREM2).4 Nongenetic factors, such as oxidative stress, inflammation, and lipid metabolism, could also contribute to diseases. Inflammation was suggested to amplify to be involved in neural damage. Over- and underexpression of pro- and anti-inflammatory molecules, respectively, may result in neuroinflammation and, thus, disease initiation and progression. In addition, levels of several inflammatory factors were reported to be altered in the brain or bodily fluids of patients with AD, reflecting their neuropathologic changes.5 Enhanced microglial activation and overexpression of inflammatory cytokines could be significantly associated with oxidative stress and cognitive impairment, especially in the elderlies. Aging could also be related with impaired brain- and lipid metabolism, which also could contribute to neurodegeneration.6
Probably, pathogenic mutations usually result in elevated Aβ42 levels and Aβ42/40 ratio. The detection of altered Aβ metabolism is used as evidence to suggest the pathogenicity and support the role of these mutations in disease progression.3 A knock-in study of a familial PSEN1 mutation showed increase in Aβ deposition that resulted from a high Aβ42/40 ratio caused by a reduction in Aβ40 production.7 In addition, studies performed to measure APP metabolism by site-directed mutagenesis in different cell lines found that most pathogenic mutations were associated with elevated levels of Aβ42 in the plasma, showing that in vitro studies of these mutations usually correlated with in vivo findings.8 Different cell models have been used for APP, PSEN1, and PSEN2 gene mutation studies, including human embryonic kidney cells 293 (HEK293), green monkey kidney cells (COS-1), Chinese hamster ovary cells (CHO), and neuroblastoma (N2a) cell lines. The mutations were typically transfected to cell lines after polymerase chain reaction (PCR) generated mutagenesis and analyzed for alterations in amyloid metabolism by enzyme-linked immunosorbent assay (ELISA). Many techniques have been employed to determine whether these mutations result in disease phenotype through mechanisms such as altering γ-secretase activity. Several cell-based studies have been performed to determine the roles of APP, PSEN1, and PSEN2 mutations in AD. The most well-modeled mutations of the APP gene are KM670/671NL (“Swedish APP”) and V717I (“London APP”); however, the more stable APP695 isoform is usually used in functional studies of APP mutations. The “Swedish APP” mutation was first analyzed by Citron et al in HEK293 cells. Normal and mutant APP-expressing cells were labeled with S35-methionine and monitored for amyloid metabolism, Aβ expression, and total Aβ levels. They found significantly higher levels of amyloid expression (six- to sevenfold greater) in the mutant cells compared to control cells.9 Cai et al transfected the “Swedish APP” mutation into human neuroblastoma cells (M17) and obtained similar results with 6 times more amyloid being found in the media of mutant cultures.10 Johnston et al screened the changes in amyloid production in skin fibroblast cells from patients with the “Swedish AAP” mutation as well as from unaffected family members and revealed a threefold elevation in total amyloid levels in the affected group.11 Because of the significant increase in amyloid production due to Swedish APP, it could also be used for studying presenilin mutations. APP V717I is one of the most well-known mutations associated with EOAD and has been transfected into several different cell lines. Studies on M17 cells revealed an increased Aβ42/total Aβ ratio as well as elevated Aβ42 expression.12 De Jonghe et al transfected the wild-type and mutant APP695 into different cell lines, including CHO-K1, human neuroglioma cells, and HEK293 cells.13 These approaches detected a significant increase in Aβ42 levels and decrease in Aβ40. Murayama et al used COS-1 cells as a model and studied 28 PSEN1 mutations. They performed co-transfection with “Swedish APP” and measured the Aβ42/total Aβ ratio.14 Several PSEN1 and PSEN2 mutations (PSEN1 A79V, I143T, A231V, L262F, C263F, L282V, G384A, and d9, and PSEN2 N141I) were analyzed by Kumar-Singh et al. They generated stable double mutant cell lines (HEK293), which contained the “Swedish APP” mutation and the selected PSEN mutation. All these mutations resulted in an elevated Aβ42/40 ratio. However, several mutations did not result in elevated Aβ42 levels, suggesting that these mutations may be loss-of-function mutations. Reduced levels of Aβ40 suggest that these mutations could be protective and may enhance Aβ42 clearance.15 Functional analyses of such mutations as demonstrated allow to determine the putative pathogenic nature of some of these mutations in the patients.
Such functional study may also be crucial to fill part of the existing gap in AD genetic research. Hence, in vitro models of AD may provide more extensive information about disease-associated pathways, which could be important for genetic, diagnostic, or drug discovery studies. However, these studies are expensive and require special equipment and settings to be conducted. In addition, technical errors may occur during mutagenesis and transfection.5,14,16–18 Fortunately, recent developments in genome editing with new technologies now make it possible to establish animal models to investigate neurodegenerative diseases as well as AD. Newer gene editing techniques that are widely employed to enhance the mutagenesis in molecular biology may facilitate in vitro AD studies. Since the first reports of the successful use of clustered regularly interspaced short palindromic repeats (CRISPR)-associated protein 9 (CRISPR–Cas9) in mammalian cells in early 2013, genome editing has captured the attention of the lay public to an unusual degree for a scientific advancement.19 How much of the CRISPR–Cas9 story is due to the hype that is commonly witnessed with the introduction of any new and promising technology, and what are the realistic prospects of genome editing, a promising tool for the generation of models of human genetic disease-causing gene mutation in human in the near future? In this review, we outline the basic principles underlying the use of CRISPR–Cas9 for genome editing, potential applications for a new avenue for biomedical and AD research through the generation of new in vitro models, and the substantial obstacles that will need to be overcome if CRISPR–Cas9 is to be used in understanding the pathogenic mutations associated with AD.
History and principles of CRISPR–Cas9
CRISPR was discovered in 1987 by Ishino et al in K-12 strains of Escherichia coli. While sequencing an isozyme of the alkaline phosphatase gene, they uncovered a unique sequence nearby with a set of repeat sequences and spacer regions.20,21 Later, homologous DNA sequences were identified in several bacterial species, including different Mycobacteria, E. coli, Streptococcus, and Campylobacter species. The CRISPR structure in bacteria contained viral (bacteriophage) sequences and could acquire additional repeats inside the sequence. The Yersinia species were found to have 29–132 spacers, with most of them being inactive prophages or conjugative plasmids. The homology between the sequences of the phages/plasmids and the spacers led researchers to believe that CRISPR might be involved in “bacterial immunity” against foreign DNA.22–24 The CRISPR–Cas9 system is an adaptive bacterial and archaeal defense mechanism that recognizes and disables invading bacteriophages or other foreign nucleic acids (Figure 1).25 This system presumably functions via the high nuclease activity of CRISPR–Cas9 that induces highly efficient, targeted double-stranded breaks (DSBs).19,26,27 This can be repaired by nonhomologous end-joining (NHEJ) resulting in nonspecific insertions, deletions, or other mutations (indels).28 DSBs may also be repaired through homology-directed repair (HDR) using a DNA repair template, through which single-stranded oligo DNA nucleotides can be introduced into the target gene, thus allowing knock-ins of specific mutations.19
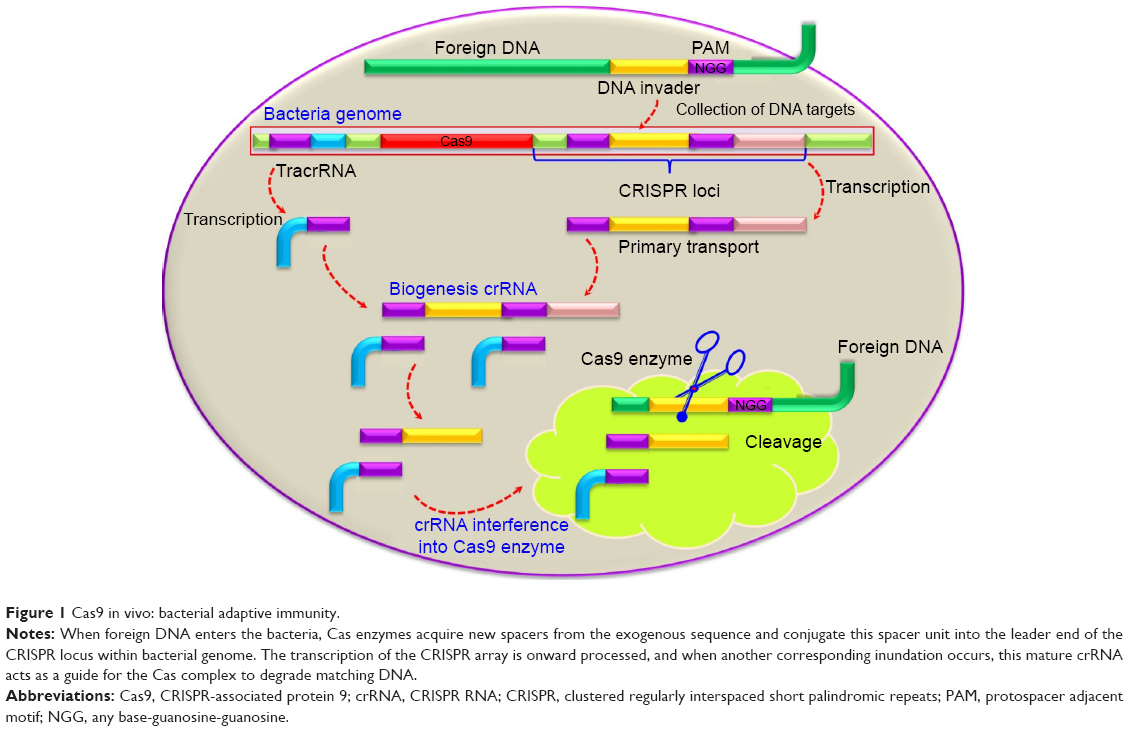 | Figure 1 Cas9 in vivo: bacterial adaptive immunity. |
In 2007, this theory was confirmed after studies found correlations between the presence of spacers and phage sensitivity and resistance. The presence of several additional spacers in bacterial strains was found to impart more resistance to foreign DNA, and thus adding and removing different phage sequences is thought to alter bacterial defense.29 CRISPR also prevents conjugation and foreign DNA transformation, as well as limits horizontal gene transfer and antibiotic resistance in bacteria. In 2010, it was discovered that the CRISPR–Cas (in Streptococcus) system could cleave bacteriophage and plasmid DNA inside the spacer region, at spacer-specific and orientation-dependent sites.31 Small guide RNAs or trans-activating crRNA (tracrRNAs), discovered the following year, were suggested to be involved in the maturation of CRISPR RNAs (crRNAs), which are important in RNAse-III and CRISPR activity against foreign RNAs. In the same year, it was suggested that the Cas9 gene may play a significant role in crRNA maturation and DNA cleavage.32 In 2012, Gasiunas et al showed that the CRISPR/Cas9 RNA-guided DNA endonuclease could cleave double-stranded DNA, thus playing an important role in foreign DNA interference.33 The authors suggested that the CRISPR/Cas9 system could be manipulated for use in DNA engineering. Cong et al further revealed that Cas9 nucleases can induce precise cleavage of human DNA and could induce HDR. In addition, they also could simultaneously edit different sites in human or mouse genomes.19 This system was revealed to be useful for in vitro studies of different mutations involved in human diseases. CRISPR–Cas9 is currently being widely used in a variety of biologic applications for genome engineering, which makes the creation of mutant mouse models or mutant cell lines easier and more reliable. Recent advances in genome engineering technologies based on the CRISPR–Cas9 are enabling the systematic interrogation of mammalian genome function. Since these initial studies, Cas9 has been used by many laboratories for genome editing applications in a variety of experimental model systems.34 Genome editing tools play an important role in genetic engineering, and that is the reason why the CRISPR–Cas9 system has been operated and applied in various formats, such as medicine and biology, pharmacology, and biotechnology engineering based on the knock-out, knock-down, and activation screens approaching. The in vivo and in vitro delivery system of CRISPR–Cas9 has been well developed for several important purposes, such as studying the gene functions and designing new therapeutic strategies.35 As illustrated in Figure 2, a large number of publications based on the CRISPR–Cas9 technology have appeared in such a short time (2010–2017).35
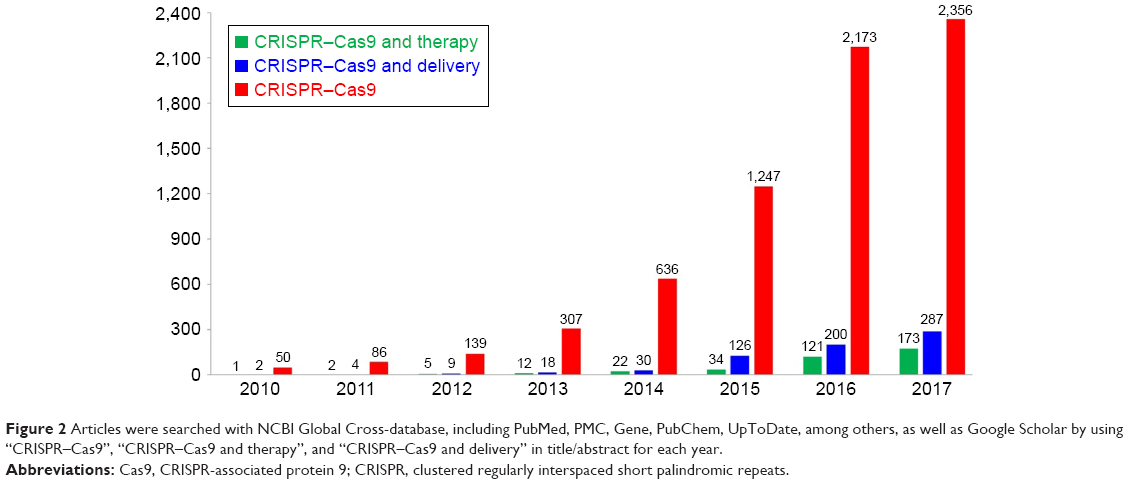 | Figure 2 Articles were searched with NCBI Global Cross-database, including PubMed, PMC, Gene, PubChem, UpToDate, among others, as well as Google Scholar by using “CRISPR–Cas9”, “CRISPR–Cas9 and therapy”, and “CRISPR–Cas9 and delivery” in title/abstract for each year. |
CRISPR–Cas9 sequence-specific gene editing strategies
Genome editing, the process of precisely modifying the nucleotide sequence of the genome, has been performed with custom nucleases designed for specific loci, such as zinc finger nucleases or transcription activator-like effector nucleases. These systems are limited because the molecules are cumbersome, expensive, and time consuming to produce because each target site needs a specific design. Subsequently, several studies adapted the CRISPR–Cas mechanism to create sequence-specific genomic mutations or knock-outs in a variety of eukaryotic cell types and tissues by optimizing the codons in a particular Cas protein, Cas9.19,35–37 Off-target mutagenic effects have also been characterized.28,38,39 To edit the eukaryotic genome, cells are transfected with a plasmid carrying the Cas9 gene and the engineered sequences, which produce specifically targeted RNA guides. Upon cleavage by Cas9, cellular DNA repair mechanisms are relied upon to complete the editing process. There are two general repair pathways: NHEJ and HDR. HDR allows an exogenous “donor” sequence to be provided to the cell and swapped into the genome, causing specific changes in the sequence. NHEJ is a repair mechanism that is useful when disruption of any sort at the DSB site will give the desired effect. As expected, the cell is now a stable genomic knock-out.
They have been widely studied, both in vitro and in vivo. Several variations in the CRISPR–Cas system have been created, such as the single-guide RNA (sgRNA) design,40–42 Cas9 nickases and dCas9-FokI fusion proteins,41–43 CRISPR/Cas9 knock-in mice,26,44 and mutation of genes in a living animal45,46 to determine the most efficient means of introducing CRISPR–Cas9 components into cells. Currently, the sgRNA system, in which chimeric molecules of a specifically designed crRNA template are ligated to the scaffold-like tracrRNA and form a complex with Cas9, has been widely adopted.42 CRISPR/Cas9 allows the genome to be efficiently edited without residual sequences (loxP sites) or the need to breed transgenic animal strains. It also offers the ability to multiplex mutations in order to observe combined effects, and to work directly with cell lines without designing specific enzymes for each cleavage site. Another advantage lies in the way the cleavage sites are determined.
Many researchers have explored ways of managing the off-target effects created when using CRISPR/Cas9. Disrupting gene expression is a common approach to study gene function and understand loss-of-function neurodegenerative disease-associated mutations. CRISPR can enhance sequence-specific gene editing models by correcting or introducing genetic aberrations linked to a particular disorder. Many of these models have been made for multiple neurologic disorders, including Alzheimer’s, Parkinson’s, schizophrenia, and autism as a mapping way affected by disease-causing mutations (Table 1). Cho et al reported that off-target cleavage was nearly undetectable when the cleavage sites were unique with no homologous sequences elsewhere in the genome.56 A more recent study revealed that reducing the target sequence length from 20 nt to 17 or 18 nt decreased the off-target effects without drastic reduction of on-target efficiency.38 When inserting a Cas9/sgRNA plasmid into eukaryotic cells, transcription is often under the control of a U6 promoter, which requires a leading G nucleotide on the transcript. This mechanism was also demonstrated to increase target fidelity via the leading G nucleotide.42 Interestingly, synthetic GG(N)20 sgRNAs produced by T7 enzymes had greater discriminatory abilities compared to plasmid-encoded sgRNAs, though some were then less active at their on-target sites.57 However, it seems difficult to thoroughly eliminate the off-target effects of CRISPR genome editing. One strategy could involve the use of a reporter gene construct that is affected by the on-target mutation, or one could alternatively attempt to distinguish wild types and mutants by phenotypes. The use of various sgRNAs to edit the same target area in separate cell populations has also been suggested by Sander and Joung.34
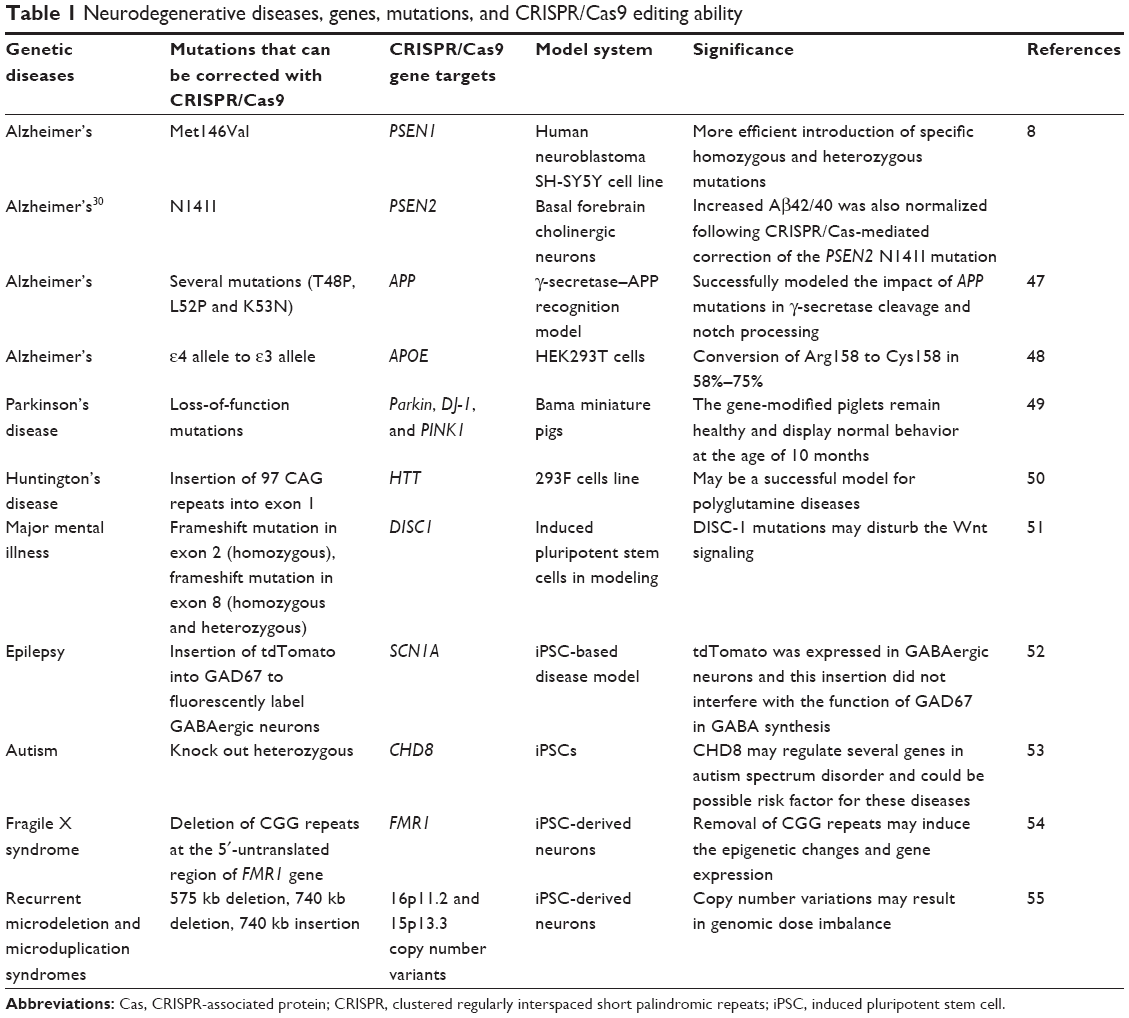 | Table 1 Neurodegenerative diseases, genes, mutations, and CRISPR/Cas9 editing ability |
Recently, some studies have attempted to improve HDR efficiency by biochemically altering the HDR or NHEJ pathways. The treatment of various mammalian cells with Scr7, a DNA ligase IV inhibitor, resulted in up to 19-fold increase in efficiency of the HDR pathway.58 Likewise, Song et al demonstrated that the HDR enhancer RS-1 increased the knock-in efficiency both in vitro and in vivo by two- to fivefold in rabbit embryos.18 Most recently, a study in human cells showed that the use of asymmetric ssDNA donors of optimal length increased the rate of HDR by up to 60% for a single nucleotide substitution.59 Interestingly, Liang et al achieved up to 56% efficiency in precise genome editing in HEK293 cells and 45% in induced pluripotent stem cell (iPSC) lines after normalizing for the cutting efficiency of gRNAs, though the HDR pathway remains a rate-limiting step for seamless genome editing.43 To explore in detail how specific genetic errors can lead to disease, researchers need to perform experiments in cells that carry these exact mutations. Figure 3 shows the general workflow of CRISPR–Cas9-mediated gene knock-ins in cultured cells including CRISPR design (http://crispr.mit.edu/), sgRNA design (http://www.broadinstitute.org/rnai/public/analysis-tools/sgrna-design), cloning, delivery of the genomic alteration, validation of regeneration, screening of transgenic cells for gene editing events, and in vitro measurement of protein levels in CRISPR/Cas9-edited HEK293T cells using techniques such as ELISA and Southern blot.
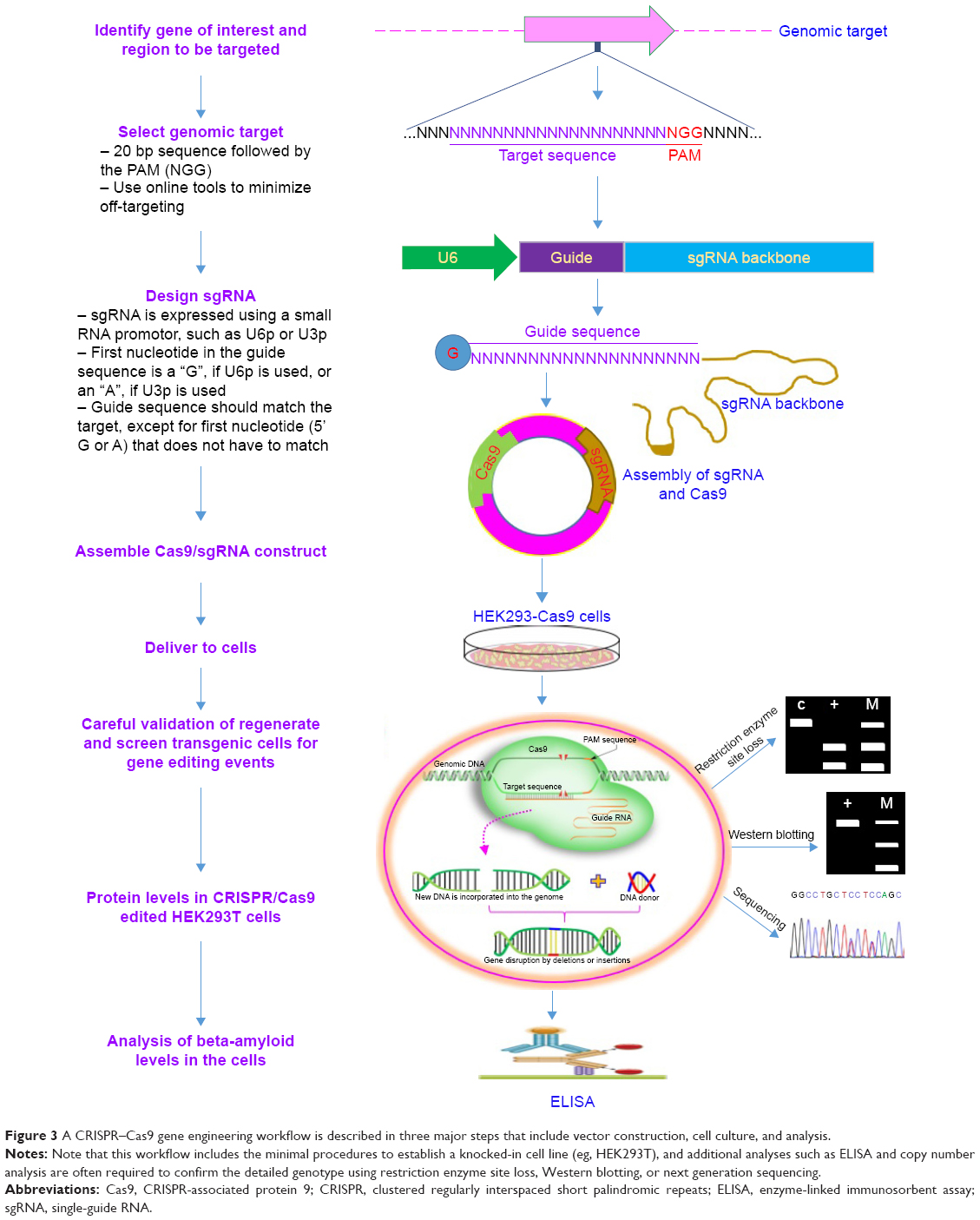 | Figure 3 A CRISPR–Cas9 gene engineering workflow is described in three major steps that include vector construction, cell culture, and analysis. |
Over the past few years, gene sequencing has uncovered numerous genes that are important in brain development and neurologic diseases like AD and Parkinson’s disease. Most diseases are known to be associated with dominant mutations in single or multiple genes. However, new cellular functional models need to be developed to address whether these mutations can affect the phenotype. The CRISPR/Cas9 system has the potential to create better cellular replicas of these mutations with greater ease and precision in inserting the disease-causing genes into the genome, thereby developing models that mimic the conditions of the disease more closely. This creates more opportunities for AD research to explore a wide range of potential mutations that lead to debilitating or fatal neuronal damage. Specific mutations from an EOAD patient that are suspected to be causative of dementia could be introduced into the cell via the CRISPR–Cas9 system and measured for amyloid production and Aβ42/40 ratio, because several of these mutations are associated with an elevated Aβ42/40 ratio. Cell models and many other animal models using CRISPR/Cas9 can also be developed to study AD at a cheaper, faster, and more precise rate.
CRISPR–Cas9 in AD
An area where the CRISPR–Cas9 system has provided its advantages was that of genetic recessive disorders. Because of its highly successful genome editing abilities, several studies have used CRISPR/Cas9 to generate genome-edited animals carrying the genetic mutations responsible for a number of human diseases including mouse models of tyrosinemia46 and lung cancer60 and nonhuman primate61 models of muscular dystrophy. The CRISPR–Cas9 system was successfully used in the modification of the genomes of different animals and cell types, including mice, zebrafish, pigs, human pluripotent cells, and human somatic cells. The ability of CRISPR/Cas9 to directly target any gene in the embryo genome opens up a new avenue for us to generate animal models of neurodegenerative diseases. As we mentioned above, CRISPR/Cas9 can cause mutations in one or two alleles, which can mimic heterozygous or homozygous knock-out of a specific gene. This can generate mutations anywhere in the genome resulting in homozygous and heterozygous mutations, which could be useful in exploring neurodegenerative diseases. For example, CRISPR–Cas9 could facilitate the generation of animal models that mimic Parkinson’s disease-associated mutation phenotypes such as loss-of-function mutations observed in PINK1 or Parkin gene mutations. The CRISPR–Cas9 system can also be used for modeling gain-of-function mutations, for example, in Huntington’s disease and PD. CRISPR–Cas9 could also target the nerve cells in adult animals. However, the knock-in rate of genome editing still remains low with this approach, and it may be difficult to model mosaic mutations.62 Swine disease models may reflect the phenotypes of human diseases more closely than mouse models. Holm et al attempted to generate disease-expressing mutations (AD, PD, and amyotrophic lateral sclerosis) in transgenic pigs in 2016 using the CRISPR–Cas9 technology. Although this method may also facilitate the development of larger nonhuman vertebrates, further studies on swine models are needed, especially for complex diseases such as AD or PD.63
iPSCs were established as a useful approach for studying different diseases including AD. They were suggested to reflect certain aspects of the AD phenotype more clearly than rodent models and could be used more effectively to study AD genetics, biomarkers, and the therapeutic candidates against the disease.64 In 2016, Paquet et al used the CRISPR/Cas9 system to model the APP and PSEN1 mutations, demonstrating a more precise genome editing method using gRNA synthesis.65 In addition, the CRISPR-based gene knock-outs in mammalian cells could be created with high efficiency and precise insertion of genetic elements through the cellular HDR pathway.43 The bacterial CRISPR–Cas9 system is capable of sequence-specific gene editing in many organisms and holds promise as a tool to generate models of human diseases, for instance, in human pluripotent stem cells,28,35 as well as AD-causing mutations such as APP (APPSwe) and PSEN1 (PSEN1M146V).65
Modeling AD-like phenotypes via CRISPR–Cas9 may also be useful for analyzing disease progression, phenotypes, and therapies, because the new genome editing technique improves reproducibility among researchers.66 Komor et al managed to transform APOE E4 to APOE E3 in mouse astrocytes by generating a C->T exchange at codon 158 (R->C exchange). The success rate of Arg158 conversion to Cys158 was 58%–78%. This study revealed that gene editing by CRISPR–Cas9 could correct several point mutations involved in human diseases.48 So far, many of the causal genetic mutations in AD have been discovered through high-throughput sequencing technologies (http://www.alzforum.org/mutations). Gene editing with CRISPR/Cas9 can be used to demonstrate whether these identified mutations are indeed directly responsible for the disease phenotypes (Figure 4).
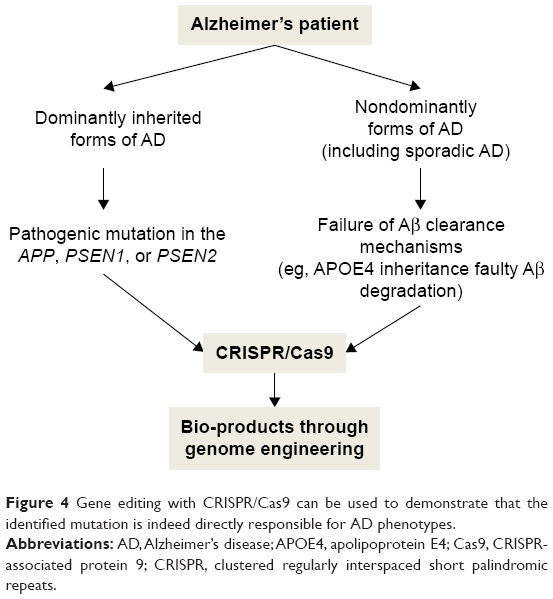 | Figure 4 Gene editing with CRISPR/Cas9 can be used to demonstrate that the identified mutation is indeed directly responsible for AD phenotypes. |
In 2016, the CRISPR–Cas9 system was used for the first time to model AD mutations (co-transfection of APP Swedish mutation and PSEN1 M146V mutation).65 The authors used iPSCs, neural precursor cells, and neurons for their analysis and analyzed the Aβ42 levels and Aβ42/40 ratio in human in APPSwe and M146V knock-in cell lines. M146V was examined in both homozygous and heterozygous stages, and the data were compared with controls.65 Recently, Sun et al modeled 138 mutations in PSEN1 from neuroblastoma (N2a) cell lines and used the CRISPR–Cas9 system to generate mutant cells. This study analyzed the gain- or loss-of-function effects of mutations depending on amyloid production and the Aβ42/40 ratio. Several of these mutations were associated with an elevated Aβ42/40 ratio; however, there was no significant correlation between amyloid levels or ratio and age at the onset.67
AD was known as a complex disease, and several mutations affect through alternative mechanisms, which may be independent from amyloid pathway. Impairment in immune and metabolic pathways was established to contribute in AD progression. Several genes, associated with these mechanisms (ABCA7, SORL1, and CR1), may be genetic risk factors for neurodegeneration. As the mutations in the classic familial AD-associated genes are rare, mutations in genes should be screened to verify their possible involvement in AD. Recently, they were also suggested that they could contribute to EOAD.68 Several novel mutations were discovered in these genes, whose possible effect remained unclear. Genome-specific engineering could be helpful for mapping the impairments in inflammatory mechanisms, in terms of mutations, and the expression of immune molecules in terms of mutations.69,70 CRISPR–Cas9 could facilitate the screening of the possible impairments in inflammatory and metabolic pathways, associated with mutations, in cell models or mouse models. In addition, CRISPR CAS9 could be involved in epigenetic modifications too. These approaches may be beneficial in treatment by regulating the expression of inflammatory molecules.71 For example, microRNA 155, edited by CRISPR–Cas9, was verified to downregulate the proinflammatory cytokines.72
New model systems in vitro approach and CRISPR/Cas9 can be used to generate genetic mutations that can faithfully mimic pathology in AD patients. CRISPR/Cas9 is a new genome modification tool that can efficiently and readily target any gene in the genome in germline cells and somatic cells of different species. It should also be pointed out that analysis of the phenotypes of animal models requires further development of functional assays that can be applied to the AD genetic research. All these obstacles remain to be overcome so that this model can be more widely utilized. The newly developed CRISPR/Ca9 technology will support to develop models of AD, a genetic disease, and enhance our understanding of the pathogenesis of these important diseases.
Challenges in CRISPR/Cas9 for generating new model systems in vitro
Our understanding of brain function at the cellular and circuit level has been greatly advanced by functional genomics and the availability of a variety of genetic tools to uncover variants function based on model human brain disorders. For example, many neurologic disorders, such as AD, are associated with genetic risk factors that can be introduced and studied in animal models.73 CRISPR–Cas9 seems to be a promising approach in human genomics as it simplifies genome editing and could facilitate the study of various genes and the discovery of novel gene functions. However, there are several issues associated with the system for which further studies are required. This approach appears to have a high risk of off-target mutations because of the recognition of similar or homologous sequences. The CRISPR–Cas9 system can cleave identical and homologous DNA sites resulting in mutations at undesired sites, which could result in enhanced toxicity or dysfunction in target cells. Target sequences with high GC content could increase the risk of off-target effects. The use of recently developed Cas9 variants (spCas9), more powerful guide RNAs, and the use of Cas9 proteins could improve the precision of genome editing and reduce off-target effects.74 Optimizing the delivery methods for CRISPR–Cas9 is also important for designing more successful genome editing systems. Combinatorial approaches based on iPSC technology and genome editing offer another approach to model human neurologic disorders in vitro. A key advantage of this approach is that genetic modifications can be studied in different human genetic backgrounds because iPSCs retain all of the individual donor’s genetic information. Several neurologic disorders including Parkinson’s,75,76 Alzheimer’s,77 and Huntington’s78 disease, which have been generated through iPSC-based disease models, have been proven to closely mimic cellular and molecular features of human diseases.
Plasmids containing guide RNAs and Streptococcus pyogenes Cas9 (SpCas9) sequences have been also developed, which can target several cell lines. However, these plasmids could also be inserted randomly into the host genome, which could result in detection of problems. Epigenetics-associated immune responses could also be induced in the case of bacterial sequences, which may disturb the genome editing process. Plasmids may also cause toxic effects in host cells. As methods using plasmids pose several challenges, other methods have been developed including the delivery of Cas9 proteins with guide RNAs through electroporation or microinjection. CRISPR/Cas9 has been used in several cell lines such as zebrafish, mice, or pigs. However, this method could induce stress inside the host’s cells. Chemical conjugation of the Cas9 protein and sgRNA may be an alternative approach that could result in less off-target effects. However, it could be limited by an increased risk of chimera formation and insufficient efficiency. Viral vectors for the system are also under development and were suggested to be less toxic and more successful at genome editing. However, viral and eukaryotic genomes are very different and their chromosomal integration may be less effective.79 Among the repair pathways for the DSBs generated by CRISPR–Cas9, HDR seems to be a precise and high-fidelity mechanism for error-fixing, but NHEJ could result in unwanted insertions or deletions. One of the major challenges of genome editing is to induce HDR mechanisms and reduce the degree of NHEJ. However, there is no specific method to measure HDR and NHEJ, but digital PCR-based methods are currently under development for the simultaneous detection of both. Conditions of genome editing also need to be optimized for the cells to favor HDR genome editing.80 An additional issue with CRISPR–Cas9 is that it is uncertain how genetic editing could affect future generations. Germline editing may be problematic, because it could result in a loss of “eugenics” and human diversity.81
Taken together, in addition to generation of new model systems as in vitro models, genome editing in combination with single-cell transcriptomics provides an approach to understanding a specific mutation function within animal cells, which could be also allowing for precise dissection of genetic networks in the brain, and understanding the molecular pathologic changes that occur throughout the AD patients. To realize these advances, however, several open challenges have to be addressed. Currently, methods for delivering Cas proteins and RNA guides to the cells need to be optimized and new methods developed to achieve sufficient levels of specificity and efficiency. In addition, insertion and correction of the gene in target cells need to be more controlled. Nevertheless, precise and efficient gene editing using CRISPR–Cas systems has the potential to advance both basic and translational research in the AD as well as neuroscience.
Conclusions and future perspectives
Similar to other genetic disorders, AD has been studied by deriving iPSCs from patients with EOAD mutations,77,82–85 but this approach requires several months to culture and is limited by the availability of the patients’ cells and variability of genetic backgrounds. Moreover, this disease has mostly been studied in animal models relying on nonphysiologic mutant gene overexpression,86 and only TALEN-mediated gene editing has been used to knock-in an EOAD mutation.87 The rapid progress in the development of Cas9 into a tool for cell and molecular biology research has been remarkable, likely due to the simplicity, high efficiency, and versatility of the system. Since Paquet et al successfully introduced knock-ins of specific homozygous and heterozygous mutations of EOAD mutations (APPSwe and PSEN1M146V) into iPS cells using the CRISPR/Cas9 system,65 this approach now opens up the possibility of developing models for potentially pathogenic mutations from other genes of interest in AD research including both EOAD- and LOAD-associated genes.88
As GWAS and NGS studies introduced several possible risk genes for AD progression, CRISPR–Cas9 could be helpful to screen their AD progression. Several genes could act through different mechanisms than amyloid pathways, like Tau mechanism or inflammation or metabolism. These approaches could facilitate the improvement of therapies against AD. Transcriptomics could be a promising target for gene editing, because RNA editing, or editing the RNA-binding molecules, could be important in regulating the gene expression and in epigenetics.89 In addition, CRISPR–Cas9 could also modify the epigenome, by recruiting enzymes, involved in histone modifications, DNA methylation/demethylation, or RNA interference. It could be important to study the structure–function relationships, associated with epigenetic changes. However, further research may be needed on refining the mechanism of acetylation or chromatin accessibility. Epigenetics could play a role in several diseases, including AD, and studying the epigenetic modifications could be helpful in understanding the disease mechanism.90,91
In addition, the generation of new model systems based on CRISPR/Cas9 including iPSC-derived in vitro models will provide a route to understanding cell type-specific gene function and its networks in the brain with more precision. Furthermore, along with whole/exome genome sequencing and GWAS, in vitro genome editing holds potential for personalized therapeutic applications for patients with AD. Recently, several commercially available products for CRISPR–Cas9 are beginning to provide convenient and effective methods to generate gene knock-outs/ins using programmable nucleases. The newly developed CRISPR–Cas9 technology will not only support the development of genetic models of AD but also enhance our understanding of the pathogenesis of the related disorders including Parkinson’s and Huntington’s diseases, all of which have become more prevalent as the life expectancy of humans has increased.92 Novel genome editing technologies based on the CRISPR–Cas9 system together with powerful readout methods might help us better understand the logic of neuronal circuits and unravel some of the mysteries of AD and dementias in general in the near future.
Acknowledgments
This work was supported by the Internal Seeding grant (GCU-2016–0185) and MD-PhD grant (FRD2016–19) through Gachon University and Gachon University GilYa Lee Hospital, respectively.
Disclosure
The authors report no conflicts of interest in this work.
References
Carr DB, Goate A, Phil D, Morris JC. Current concepts in the pathogenesis of Alzheimer’s disease. Am J Med. 1997;22;103(3A):3S–10S. | ||
Karch CM, Goate AM. Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biol Psychiatry. 2015;77(1):43–51. | ||
Giau VV, An SSA. Optimization of specific multiplex DNA primers to detect variable CLU genomic lesions in patients with Alzheimer’s disease. BioChip J. 2015;9(4):278–284. | ||
Van Cauwenberghe C, Van Broeckhoven C, Sleegers K. The genetic landscape of Alzheimer disease: clinical implications and perspectives. Genet Med. 2016;18(5):421–430. | ||
Bagyinszky E, Giau VV, Shim K, Suk K, An SSA, Kim S. Role of inflammatory molecules in the Alzheimer’s disease progression and diagnosis. J Neurol Sci. 2017;15(376):242–254. | ||
Yin F, Yao J, Brinton RD, Cadenas E. Editorial: the metabolic-inflammatory axis in brain aging and neurodegeneration. Front Aging Neurosci. 2017;9:209. | ||
Xia D, Watanabe H, Wu B, et al. Presenilin-1 knockin mice reveal loss-of-function mechanism for familial Alzheimer’s disease. Neuron. 2015;85(5):967–981. | ||
Fang B, Jia L, Jia J. Chinese Presenilin-1 V97L mutation enhanced Abeta42 levels in SH-SY5Y neuroblastoma cells. Neurosci Lett. 2006;406(1–2):33–37. | ||
Citron M, Oltersdorf T, Haass C, et al. Mutation of the beta-amyloid precursor protein in familial Alzheimer’s disease increases beta-protein production. Nature. 1992;360(6405):672–674. | ||
Cai XD, Golde TE, Younkin SG. Release of excess amyloid beta protein from a mutant amyloid beta protein precursor. Science. 1993;259(5094):514–516. | ||
Johnston JA, Cowburn RF, Norgren S, et al. Increased beta-amyloid release and levels of amyloid precursor protein (APP) in fibroblast cell lines from family members with the Swedish Alzheimer’s disease APP670/671 mutation. FEBS Lett. 1994;354(3):274–278. | ||
Suzuki N, Cheung TT, Cai XD, et al. An increased percentage of long amyloid beta protein secreted by familial amyloid beta protein precursor (beta APP717) mutants. Science. 1994;264(5163):1336–1340. | ||
De Jonghe C, Zehr C, Yager D, et al. Flemish and Dutch mutations in amyloid beta precursor protein have different effects on amyloid beta secretion. Neurobiol Dis. 1998;5(4):281–286. | ||
Murayama O, Tomita T, Nihonmatsu N, et al. Enhancement of amyloid beta 42 secretion by 28 different presenilin 1 mutations of familial Alzheimer’s disease. Neurosci Lett. 1991;265(1):61–63. | ||
Kumar-Singh S, Theuns J, Van Broeck B, et al. Mean age-of-onset of familial Alzheimer disease caused by presenilin mutations correlates with both increased Aβ42 and decreased Aβ40. Hum Mutat. 2006;27(7):686–695. | ||
Herl L, Thomas AV, Lill CM, et al. Mutations in amyloid precursor protein affect its interactions with presenilin/gamma-secretase. Mol Cell Neurosci. 2009;41(2):166–174. | ||
Kaneko H, Kakita A, Kasuga K, et al. Enhanced accumulation of phosphorylated alpha-synuclein and elevated beta-amyloid 42/40 ratio caused by expression of the presenilin-1 deltaT440 mutant associated with familial Lewy body disease and variant Alzheimer’s disease. J Neurosci. 2007;27(48):13092–13097. | ||
Song J, Yang D, Xu J, Zhu T, Chen YE, Zhang J. RS-1 enhances CRISPR/Cas9- and TALEN-mediated knock-in efficiency. Nature Communications. 2016;7:10548. | ||
Cong L, Ran FA, Cox D, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339(6121):819–823. | ||
Ishino Y, Shinagawa H, Makino K, Amemura M, Nakata A. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J Bacteriol. 1987;169(12):5429–5433. | ||
Militello KT, Lazatin JC. Discovery of Escherichia coli CRISPR sequences in an undergraduate laboratory. Biochem Mol Biol Educ. 2017;45(3):262–269. | ||
Bolotin A, Quinquis B, Sorokin A, Ehrlich SD. Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology. 2005;151(8):2551–2561. | ||
Mojica FJ, Diez-Villasenor C, Garcia-Martinez J, Soria E. Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J Mol Evol. 2005;60(2):174–182. | ||
Pourcel C, Salvignol G, Vergnaud G. CRISPR elements in Yersinia pestis acquire new repeats by preferential uptake of bacteriophage DNA, and provide additional tools for evolutionary studies. Microbiology. 2005;151(3):653–663. | ||
Reis A, Hornblower B, Robb B, Tzertzinis G. CRISPR/Cas9 and targeted genome editing: a new era in molecular biology. NEB Expressions. 2014;1:3–6. | ||
Dow LE, Fisher J, O’Rourke KP, et al. Inducible in vivo genome editing with CRISPR-Cas9. Nat Biotechnol. 2015;33(4):390–394. | ||
Yang H, Wang H, Shivalila CS, Cheng AW, Shi L, Jaenisch R. One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell. 2013;154(6):1370–1379. | ||
Hsu PD, Scott DA, Weinstein JA, et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 2013;31(9):827–832. | ||
Barrangou R, Fremaux C, Deveau H, et al. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315(5819):1709–1712. | ||
Ortiz-Virumbrales M, Moreno CL, Kruglikov I, et al. CRISPR/Cas9-Correctable mutation-related molecular and physiological phenotypes in iPSC-derived Alzheimer’s PSEN2N141I neurons. Acta Neuropathol Commun. 2017;5:77. | ||
Garneau JE, Dupuis ME, Villion M, et al. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature. 2010;468(7320):67–71. | ||
Deltcheva E, Chylinski K, Sharma CM, et al. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature. 2011;471(7340):602–607. | ||
Gasiunas G, Barrangou R, Horvath P, Siksnys V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. PNAS. 2012;109(39):E2579–E2586. | ||
Sander JD, Joung JK. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol. 2014;32(4):347–355. | ||
Liu C, Zhang L, Liu H, Cheng K. Delivery strategies of the CRISPR-Cas9 gene-editing system for therapeutic applications. J Control Release. 2017;266:17–26. | ||
Jinek M, East A, Cheng A, Lin S, Ma E, Doudna J. RNA-programmed genome editing in human cells. Elife. 2013;2:e00471. | ||
Kaulich M, Lee YJ, Lonn P, Springer AD, Meade BR, Dowdy SF. Efficient CRISPR-rAAV engineering of endogenous genes to study protein function by allele-specific RNAi. Nucleic Acids Res. 2015;43(7):e45. | ||
Fu Y, Sander JD, Reyon D, Cascio VM, Joung JK. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat Biotechnol. 2014;32(3):279–284. | ||
Kuscu C, Arslan S, Singh R, Thorpe J, Adli M. Genome-wide analysis reveals characteristics of off-target sites bound by the Cas9 endonuclease. Nat Biotechnol. 2014;32(7):677–683. | ||
Murovec J, Pirc Ž, Yang B. New variants of CRISPR RNA-guided genome editing enzymes. Plant Biotechnol J. 2017;15(8):917–926. | ||
Guilinger JP, Thompson DB, Liu DR. Fusion of catalytically inactive Cas9 to FokI nuclease improves the specificity of genome modification. Nat Biotechnol. 2014;32(6):577–582. | ||
Ran FA, Hsu PD, Lin CY, et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 2013;154(6):1380–1389. | ||
Liang X, Potter J, Kumar S, Ravinder N, Chesnut JD. Enhanced CRISPR/Cas9-mediated precise genome editing by improved design and delivery of gRNA, Cas9 nuclease, and donor DNA. J Biotechnol. 2017;241:136–146. | ||
Doench JG, Hartenian E, Graham DB, et al. Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nat Biotechnol. 2014;32(12):1262–1267. | ||
Swiech L, Heidenreich M, Banerjee A, et al. In vivo interrogation of gene function in the mammalian brain using CRISPR-Cas9. Nat Biotechnol. 2015;33(1):102–106. | ||
Yin H, Xue W, Chen S, et al. Genome editing with Cas9 in adult mice corrects a disease mutation and phenotype. Nat Biotechnol. 2014;32(6):551–553. | ||
Xu TH, Yan Y, Kang Y, Jiang Y, Melcher K, Xu HE. Alzheimer’s disease-associated mutations increase amyloid precursor protein resistance to γ-secretase cleavage and the Aβ42/Aβ40 ratio. Cell Discov. 2016;2:16026. | ||
Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016;533(7603):420–424. | ||
Wang X, Cao C, Huang J, et al. One-step generation of triple gene-targeted pigs using CRISPR/Cas9 system. Sci Rep. 2016;6:20620. | ||
An MC, O’Brien RN, Zhang N, Patra BN, De La Cruz M, Ray A, Ellerby LM. Polyglutamine Disease Modeling: Epitope Based Screen for Homologous Recombination using CRISPR/Cas9 System. PLoS Currents Huntington Disease. 2014 Apr 15 . Edition 1. doi: 10.1371/currents.hd.0242d2e7ad72225efa72f6964589369a. | ||
Srikanth P, Han K, Callahan DG, et al. Genomic DISC1 disruption in hiPSCs alters Wnt signaling and neural cell fate. Cell Rep. 2015;12(9):1414–1429. | ||
Liu J, Gao C, Chen W, et al. CRISPR/Cas9 facilitates investigation of neural circuit disease using human iPSCs: mechanism of epilepsy caused by an SCN1A loss-of-function mutation. Transl Psychiatry. 2016;6:e703. | ||
Wang P, Lin M, Pedrosa E, et al. CRISPR/Cas9-mediated heterozygous knockout of the autism gene CHD8 and characterization of its transcriptional networks in neurodevelopment. Mol Autism. 2015;6:55. | ||
Park CY, Halevy T, Lee DR, et al. Reversion of FMR1 methylation and silencing by editing the triplet repeats in fragile X iPSC-derived neurons. Cell Rep. 2015;13(2):234–241. | ||
Tai DJ, Ragavendran A, Manavalan P, et al. Engineering microdeletions and microduplications by targeting segmental duplications with CRISPR. Nat Neurosci. 2016;19(3):517–522. | ||
Cho SW, Kim S, Kim JM, Kim JS. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat Biotechnol. 2013;31(3):230–232. | ||
Jiang W, Cox D, Zhang F, Bikard D, Marraffini LA. RNA-guided editing of bacterial genomes using CRISPR-CAS systems. Nat Biotechnol. 2013;31(3):233–239. | ||
Maruyama T, Dougan SK, Truttmann MC, Bilate AM, Ingram JR, Ploegh HL. Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nat Biotechnol. 2015;33(5):538–542. | ||
Richardson CD, Ray GJ, DeWitt MA, Curie GL, Corn JE. Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nat Biotechnol. 2016;34(3):339–344. | ||
Chen S, Sanjana NE, Zheng K, et al. Genome-wide CRISPR screen in a mouse model of tumor growth and metastasis. Cell. 2015;160(6):1246–1260. | ||
Chen Y, Zheng Y, Kang Y, et al. Functional disruption of the dystrophin gene in rhesus monkey using CRISPR/Cas9. Hum Mol Genet. 2015;24(13):3764–3774. | ||
Tu Z, Yang W, Yan S, Guo X, Li XJ. CRISPR/Cas9: a powerful genetic engineering tool for establishing large animal models of neurodegenerative diseases. Mol Neurodegener. 2015;10:35. | ||
Holm IE, Alstrup AK, Luo Y. Genetically modified pig models for neurodegenerative disorders. J Pathol. 2016;238(2):267–287. | ||
Yang J, Li S, He X-B, Cheng C, Le W. Induced pluripotent stem cells in Alzheimer’s disease: applications for disease modeling and cell-replacement therapy. Mol Neurodegener. 2016;11:39. | ||
Paquet D, Kwart D, Chen A, et al. Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9. Nature. 2016;533(7601):125–129. | ||
Mungenast AE, Siegert S, Tsai LH. Modeling Alzheimer’s disease with human induced pluripotent stem (iPS) cells. Mol Cell Neurosci. 2016;73:13–31. | ||
Sun L, Zhou R, Yang G, Shi Y. Analysis of 138 pathogenic mutations in presenilin-1 on the in vitro production of Aβ42 and Aβ40 peptides by γ-secretase. Proc Natl Acad Sci U S A. 2017;114(4):E476–E485. | ||
Onos KD, Sukoff Rizzo SJ, Howell GR, Sasner M. Toward more predictive genetic mouse models of Alzheimer’s disease. Brain Res Bull. 2016;122:1–11. | ||
Brunger JM, Zutshi A, Willard VP, Gersbach CA, Guilak F. CRISPR/Cas9 editing of murine induced pluripotent stem cells for engineering inflammation-resistant tissues. Arthritis Rheumatol. 2017;69(5):1111–1121. | ||
Kim EJ, Kang KH, Ju JH. CRISPR-Cas9: a promising tool for gene editing on induced pluripotent stem cells. Korean J Intern Med. 2017;32(1):42–61. | ||
Pivniouk VI, Rosenbaum D, Pivniouk O, Vercelli D. Downregulation of human IL4 and IL13 expression by CRISPR/Cas9-mediated deletion of DNase I hypersensitive site HS11/12 from a transgenic human Th2 cytokine locus. J Immunol. 2016;196(1 Suppl):58. | ||
Jing W, Zhang X, Sun W, Hou X, Yao Z, Zhu Y. CRISPR/CAS9-mediated genome editing of miRNA-155 inhibits proinflammatory cytokine production by RAW264.7 cells. Biomed Res Int. 2015;2015:326042. | ||
Oddo S, Caccamo A, Shepherd JD, et al. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39(3):409–421. | ||
Yi L, Li J. CRISPR-Cas9 therapeutics in cancer: promising strategies and present challenges. Biochim Biophys Acta. 2016;1866(2):197–207. | ||
Ryan SD, Dolatabadi N, Chan SF, et al. Isogenic human iPSC Parkinson’s model shows nitrosative stress-induced dysfunction in MEF2-PGC1α transcription. Cell. 2013;155(6):1351–1364. | ||
Soldner F, Laganière J, Cheng AW, et al. Generation of isogenic pluripotent stem cells differing exclusively at two early onset Parkinson point mutations. Cell. 2011;146(2):318–331. | ||
Yagi T, Ito D, Okada Y, et al. Modeling familial Alzheimer’s disease with induced pluripotent stem cells. Hum Mol Genet. 2011;20(23):4530–4539 | ||
Jeon I, Lee N, Li JY, et al. Neuronal properties, in vivo effects, and pathology of a Huntington’s disease patient-derived induced pluripotent stem cells. Stem Cells. 2012;30(9):2054–2062. | ||
Peng R, Lin G, Li J. Potential pitfalls of CRISPR/Cas9-mediated genome editing. FEBS J. 2016;283(7):1218–1231. | ||
Miyaoka Y, Berman JR, Cooper SB, et al. Systematic quantification of HDR and NHEJ reveals effects of locus, nuclease, and cell type on genome-editing. Sci Rep. 2016;31;6:23549. | ||
Yang A. Thinking towards the future of CRISPR/Cas9. Brevia. 2015. | ||
Israel MA, Yuan SH, Bardy C, et al. Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature. 2012;482(7384):216–220. | ||
Kondo T, Asai M, Tsukita K, et al. Modeling Alzheimer’s disease with iPSCs reveals stress phenotypes associated with intracellular Aβ and differential drug responsiveness. Cell Stem Cell. 2013;12(4):487–496. | ||
Muratore CR, Rice HC, Srikanth P, et al. The familial Alzheimer’s disease APPV717I mutation alters APP processing and Tau expression in iPSC-derived neurons. Hum Mol Genet. 2014;23(13):3523–3536. | ||
Sproul AA, Jacob S, Pre D, et al. Characterization and molecular profiling of PSEN1 familial Alzheimer’s disease iPSC-derived neural progenitors. PLoS One. 2014;9:e84547. | ||
Götz J, Ittner LM. Animal models of Alzheimer’s disease and frontotemporal dementia. Nature Rev Neurosci. 2008;9(7):532–544. | ||
Woodruff G, Young JE, Martinez FJ, et al. The presenilin-1 ΔE9 mutation results in reduced γ-secretase activity, but not total loss of PS1 function, in isogenic human stem cells. Cell Rep. 2013;5(4):974–985. | ||
Giau VV, An SSA, Bagyinszky E, Kim SY. Gene panels and primers for next generation sequencing studies on neurodegenerative disorders. Mol Cell Toxicol. 2015;11(2):89–143. | ||
Vorvis C, Hatziapostolou M, Mahurkar-Joshi S, et al. Transcriptomic and CRISPR/Cas9 technologies reveal FOXA2 as a tumor suppressor gene in pancreatic cancer. Am J Physiol Gastrointest Liver Physiol. 2016;310(11):G1124–G1137. | ||
Enríquez P. CRISPR-mediated epigenome editing. Yale J Biol Med. 2016;89(4):471–486. | ||
Sperber H, Mathieu J, Wang Y, et al. The metabolome regulates the epigenetic landscape during naive-to-primed human embryonic stem cell transition. Nat Cell Biol. 2015;17(12):1523–1535. | ||
Yan S, Tu Z, Li S, Li XJ. Use of CRISPR/Cas9 to model brain diseases. Prog Neuropsychopharmacol Biol Psychiatry. 2018;81:488–492. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.