Back to Journals » The Application of Clinical Genetics » Volume 7
Genetics of hypertrophic cardiomyopathy: advances and pitfalls in molecular diagnosis and therapy
Authors Roma-Rodrigues C ![]() , Fernandes AR
, Fernandes AR ![]()
Received 12 June 2014
Accepted for publication 27 July 2014
Published 3 October 2014 Volume 2014:7 Pages 195—208
DOI https://doi.org/10.2147/TACG.S49126
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Prof. Dr. Martin Maurer
Catarina Roma-Rodrigues,1 Alexandra R Fernandes1,2
1UCIBIO, Departamento de Ciências da Vida, Faculdade de Ciências e Tecnologia da Universidade Nova de Lisboa, Campus de Caparica, Caparica, Portugal; 2Centro de Química Estrutural, Instituto Superior Técnico, Universidade de Lisboa, Lisboa, Portugal
Abstract: Hypertrophic cardiomyopathy (HCM) is a primary disease of the cardiac muscle that occurs mainly due to mutations (>1,400 variants) in genes encoding for the cardiac sarcomere. HCM, the most common familial form of cardiomyopathy, affecting one in every 500 people in the general population, is typically inherited in an autosomal dominant pattern, and presents variable expressivity and age-related penetrance. Due to the morphological and pathological heterogeneity of the disease, the appearance and progression of symptoms is not straightforward. Most HCM patients are asymptomatic, but up to 25% develop significant symptoms, including chest pain and sudden cardiac death. Sudden cardiac death is a dramatic event, since it occurs without warning and mainly in younger people, including trained athletes. Molecular diagnosis of HCM is of the outmost importance, since it may allow detection of subjects carrying mutations on HCM-associated genes before development of clinical symptoms of HCM. However, due to the genetic heterogeneity of HCM, molecular diagnosis is difficult. Currently, there are mainly four techniques used for molecular diagnosis of HCM, including Sanger sequencing, high resolution melting, mutation detection using DNA arrays, and next-generation sequencing techniques. Application of these methods has proven successful for identification of mutations on HCM-related genes. This review summarizes the features of these technologies, highlighting their strengths and weaknesses. Furthermore, current therapeutics for HCM patients are correlated with clinically observed phenotypes and are based on the alleviation of symptoms. This is mainly due to insufficient knowledge on the mechanisms involved in the onset of HCM. Tissue engineering alongside regenerative medicine coupled with nanotherapeutics may allow fulfillment of those gaps, together with screening of novel therapeutic drugs and target delivery systems.
Keywords: hypertrophic cardiomyopathy, molecular diagnosis, sarcomere, next-generation sequencing, sudden cardiac death
Introduction
Cardiomyopathies are a heterogeneous group of diseases affecting the heart muscle and frequently lead to progressive heart failure-related disability or cardiovascular death.1,2 According to the morphological and functional phenotype, cardiomyopathies can be divided in hypertrophic cardiomyopathy (HCM), dilated cardiomyopathy, restrictive left ventricular cardiomyopathy, arrhythmogenic cardiomyopathy, and left ventricular non-compaction.1,2 A detailed definition and description of the subtypes of cardiomyopathies is outside the scope of this review and may be found elsewhere.1–3 Many cardiomyopathies are monogenic disorders due to single gene mutations in germinal cells and thereby are inherited.3 HCM, a primary disease of the cardiac muscle that occurs mainly due to mutations in genes encoding for the cardiac contractile apparatus, is the most common familial form of cardiomyopathy, as well as the most phenotypically variable cardiac disease.4–6 Indeed, the pathophysiology of HCM is complex, being characterized by a wide range of disease processes, with varying genetics, clinical manifestations, heart morphology, and prognosis.6 Due to the phenotypic heterogeneity of the disease, the diagnosis of HCM at late adulthood is frequent, when the morphology and functional debility of the heart caused by the disease have progressed.7 This is of special importance for asymptomatic or mildly symptomatic young patients with HCM who may experience ventricular arrhythmias and sudden cardiac death.8 The molecular diagnosis of HCM allows the detection of subjects carrying a mutation on genes that cause the disease, even before the development of symptoms.9 This review outlines the current knowledge about HCM, describing the biochemistry and physiology of HCM, highlighting the genetic and allelic heterogeneity underlying HCM, and pointing out the methods used for molecular diagnosis of HCM, along with their advantages and current limitations. It also emphasizes the current and new directions in HCM therapy.
Hypertrophic cardiomyopathy
Physiology and biochemistry of the normal heart – what goes wrong!
The mechanical events that allow a cardiac cycle to occur require an appropriate venous return, regulation of outflow resistance, a normal myocardial contractile state, heart rate control, and an adequate supply of oxygenated blood.10 Alterations in one of these aspects may lead to cardiac deficiency.10 In HCM, the myocardial contractile state is compromised by cardiomyocyte hypertrophy, disarray, and increased myocardial fibrosis.6 In fact, the myocardium of a heart affected by HCM is composed of myocytes with distorted nuclei and disorganized myofibrils.11 This morphology have a negative impact on cardiac relaxation and lead to arrhythmias and progression of heart failure.11
Despite the limited knowledge of the mechanisms involved in modification of cardiomyocytes in HCM, it is well established that they are related to mutations at the level of the sarcomere proteins and other proteins involved in muscle contraction and force transduction.6 Sarcomeres, which are responsible for muscle contraction, are formed by contractile myofibrils composed of thick filaments (myosin) and thin filaments (actin and associated regulatory proteins, ie, tropomyosin and troponins C, I, and T) that slide past each other (Figure 1).12,13 Other proteins involved in the maintenance and regulation of sarcomere structure and function include myosin binding protein (MyBP)-c, titin, and tropomyosin.13 Several mitochondria, which are responsible for generation of adenosine triphosphate for contraction, are scattered between myofibrils.12 Electrical excitation of the myocyte results in a high increase in the intracellular calcium concentration, an event called excitation-contraction coupling, that initiates further release of calcium from the sarcoplasmic reticulum.12,13 Contractions of the sarcomere are then initiated by calcium binding to troponin C, enabling projections of the myosin molecules to interact with actin molecules, through an adenosine triphosphate-dependent process.12,13 Relaxation occurs by lowering the calcium concentration in the myocyte and consequent detachment from troponin C.12 In HCM, mutations occurring at the level of the sarcomere will ultimately result in inefficient or excessive use of adenosine triphosphate and therefore in an energy deficiency.7,14 Among other consequences, these energy requirements compromise calcium homeostasis and excitation-contraction coupling, in turn compromising the relaxation capacity of the cardiomyocytes and consequently the force of contractibility.7,15 Moreover, mutations in sarcomere proteins seem to trigger an early increase in the synthesis of collagen, which will result in myocardial fibrosis and consequently in overt HCM.16 Myocardial fibrosis, together with an increase in the size of cardiomyocytes, results in myocardial left ventricular hypertrophy (LVH; Figure 2).11
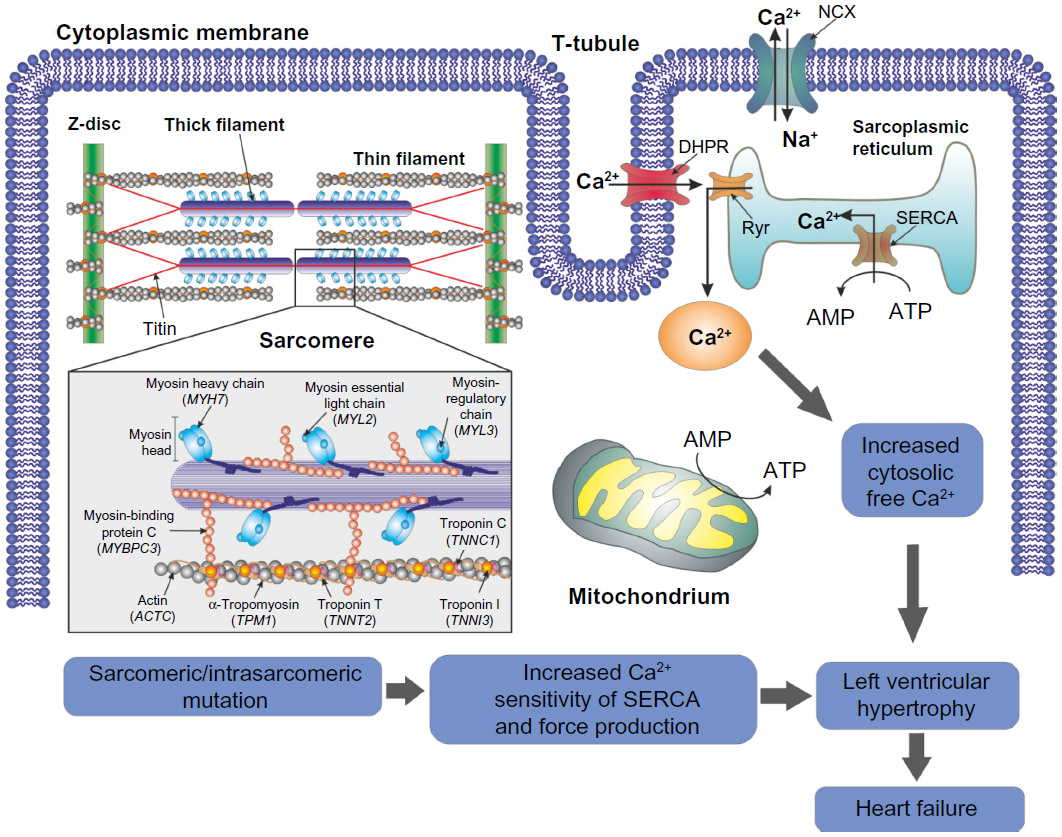 | Figure 1 Schematic representation of heart failure due to sarcomeric/intrasarcomeric mutations. Mutations in the proteins responsible for the maintenance of sarcomere structure and function result in inefficient or excessive use of ATP and a consequent energy deficiency. The energy requirements then compromise calcium homeostasis in the cell, resulting in, among other events, an increase in calcium sensitivity of the ATPase SERCA and increased cytosolic free calcium. The contractibility of the myocyte is then compromised, ultimately resulting in cell death and consequent myocardial fibrosis. This myocardial fibrosis and further ischemia may result in left ventricular hypertrophy, increasing the risk of heart failure. |
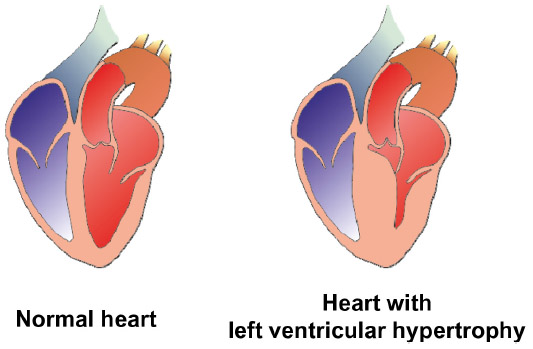 | Figure 2 Morphological features of a normal heart and a heart with left ventricular hypertrophy. |
Physiological and morphological characteristics
Myocardial fibrosis in HCM patients seems to be related to the myocyte death caused by stresses imposed by sarcomere mutations.16 Due to the limited division of cardiomyocytes, maintenance of the structural integrity of the heart is accomplished by accumulation of fibroblasts.17 However, because fibroblasts do not have a contractile capacity, a supplementary effort is required from the surviving cardiomyocytes and a consequent downward spiral that includes neurohormonal activation, further cardiomyocyte death, and increased fibrosis.17,18 Myocardial fibrosis and further ischemia may result in LVH and predispose patients to ventricular arrhythmias and ultimately to sudden cardiac death.16 This clinical outcome occurs unexpectedly, mainly in young people, including trained athletes.8,19 LVH is highly age-dependent, mainly occurring during or soon after the periods of rapid somatic growth that take place during puberty.7,20
HCM is generally characterized at the morphological level by a thickening of the left ventricular wall (Figure 2), usually ≥15 mm, and a consequent decrease in left ventricular chamber size.1 Although asymmetric septal hypertrophy is most common, there is significant heterogeneity in the degree and pattern of LVH.8,18 Due to the morphological and pathological heterogeneity of the disease, the appearance and progression of symptoms is not straightforward.1,5,6 Most HCM patients are asymptomatic, but up to 25% will develop significant symptoms, including chest pain and/or heart failure up to heart transplant.8 Recent work has suggested a correlation between mortality and age, with an inverse relationship being observed between age and sudden death, and a direct relationship between age and stroke-related death.21 Arrhythmias occur in approximately 20% of HCM patients, leading to a poorer prognosis, as these patients have a higher probability of suffering stroke and heart failure.8,22 Furthermore, the clinical outcomes that may develop in HCM patients include systolic anterior motion of the mitral valve with associated obstruction of left ventricular outflow and mitral regurgitation, mid ventricular obstruction as a result of systolic cavity obliteration, and an intracavitary subvalvular dynamic gradient.6
HCM patients must be monitored lifelong by a cardiologist. Clinical diagnosis of HCM requires a hypertrophied, non-dilated left ventricle without evidence of any other cardiac or systemic disease (eg, systemic hypertension or aortic valve stenosis) that could produce the extent of hypertrophy evident.6,9,23 Indications of HCM can be seen on an electrocardiogram, although the diagnosis is based on an echocardiogram and/or magnetic resonance imaging.1,9,23 However, identification of LVH on an echocardiogram or magnetic resonance imaging cannot be uniquely associated with HCM, also due to the existence of HCM phenocopies.5,6 Indeed, a differential diagnosis of HCM is required to discard a LVH associated with metabolic and multiorgan genetic syndromes (eg, amyloidosis, cardiac-specific glycogen storage disease, Noonan syndrome, Danon disease, Anderson-Fabry disease, or Friedrich’s ataxia).1,2,24–26
The treatment (medication, surgical or percutaneous septum reduction, and/or internal cardioverter defibrillator implantation) is determined based on the symptoms and the stratification risk for sudden cardiac death.8 Indeed, the management of HCM patients is directed at control of heart failure symptoms, prevention of sudden cardiac death, treatment of atrial fibrillation, and screening of family members.8 Around 70% of HCM patients have dynamic left ventricular outflow obstruction at rest or provoked by exercise, and 10% of these patients will progress and require therapeutic intervention.6
Genetic causes
From the genetic point of view, more than 90% of HCM is inherited as an autosomal dominant disease with variable expressivity and age-related penetrance, affecting one in every 500 people in the general population.22,27 Hence, the offspring of an affected individual have a 50% probability of inheriting a mutation and risk for HCM. Furthermore, a recent study identified a mutation in the gene encoding the four-and-a-half LIM domain 1 (FHL1) responsible for HCM, suggesting an X-linked inheritance.28 Alternatively, sporadic HCM cases may be due to de novo mutations in the proband (absent in the parents).6 Sixty percent of the mutations occurring in sarcomere genes were described as familial HCM.24 Moreover, 5% of patients with familial HCM are heterozygous carriers, carrying up to three distinct mutations that are associated with a poorer prognosis.5,19,29 In addition, the phenotypic heterogeneity of HCM within families suggests an environmental role in onset of the disease; for example, obesity seems to increase the thickness of the myocardium and to have an impact on the development of LVH and pathology.30,31
Interestingly, a pattern of variability can be distinguished in the types of mutations causing HCM among worldwide populations. While in north European and Southeast Asia Indian populations, HCM is mainly due to founding mutations and hence familial HCM,32–36 in the rest of the world a marked heterogeneity in HCM-associated mutations is observed.37–40
Genes and mutations
To date, around 1,400 mutations have been identified as being responsible for HCM pathology (Table 1).5,22 About 70% of these mutations are in the sarcomere genes encoding cardiac β-myosin heavy chain (MYH7) and cardiac myosin binding protein C (MYBPC3), as shown in Table 1.5,6,19,41 Other sarcomeric genes, eg, regulatory myosin light chain (MYL2), cardiac troponin T (TNNT2), cardiac troponin I (TNNI3), and actin (ACTC), and non-sarcomeric genes, such as genes encoding plasma membrane and mitochondrial proteins, as well as sarcomere adjacent Z-disc encoding genes, account for the other cases (Table 1).6,19,42
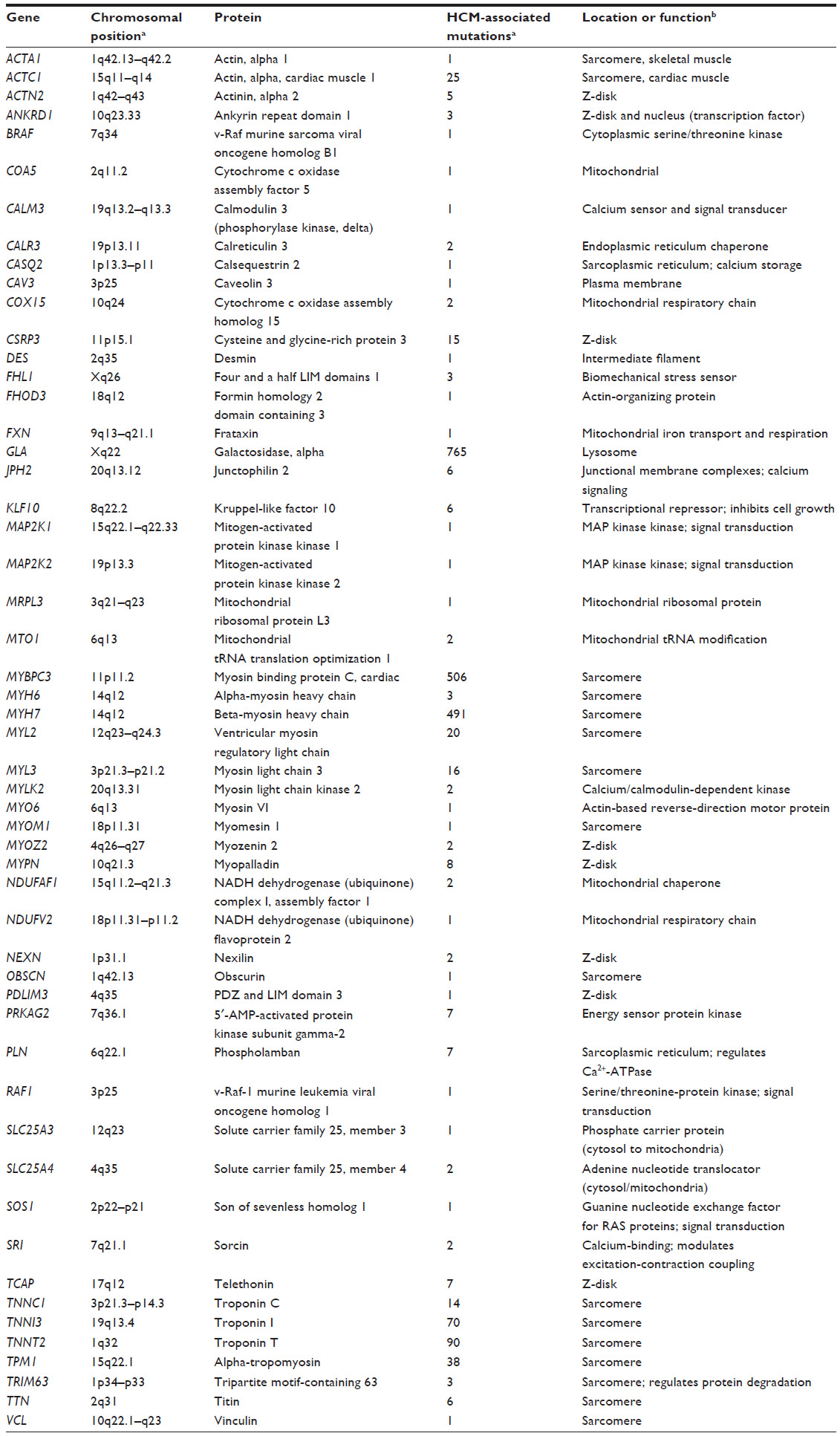 | Table 1 Hypertrophic cardiomyopathy-associated genes, proteins, and number of mutations described for each gene (protein location or function is also described) |
Ultimately, the effect of a mutation on the contraction of the cardiac muscle will depend on the mutated gene, the location of the mutation within the gene, and consequently on the protein and the abundance of the mutated protein within the myocyte.24 The mutations occurring in most part of sarcomere encoding genes are missense with a dominant negative effect resulting in the formation of poison polypeptides incorporated into the sarcomere, with consequences for its function.24,29 However, most MYBPC3 mutations are predicted to result in frame shift and often in protein truncation.24,29,34 Incorporation of the truncated forms in the sarcomere results in disruption of the normal sarcomeric structure and in partial haploinsufficiency.29,34
Due to heterogeneity, age dependence, and incomplete penetrance, the concept of genotype-phenotype has not been established for HCM.9,24 Nevertheless, some TNNT2 mutations have been related to minor LVH and an appreciable risk of arrhythmia.43 Additionally, some mutations in the MYH7 gene result in significant LVH after the second decade of life and seem to be associated with sudden cardiac death;44 on the other hand, it has been suggested that variants of MYBPC3 are associated with later onset of HCM.45
A mutation can be considered pathogenic on the basis of the following criteria: cosegregation with the HCM phenotype (eg, LVH) in family members; previously reported or identified as a cause of HCM; absent from unrelated and ethnic-matched normal controls; protein structure and function is importantly altered (eg, frame shift with truncation); and amino acid sequence change in a region of the protein otherwise highly conserved through evolution (no variation observed) among species.6,24,46 MYH7-R453C, G716R, TNNT2-R92W, and MYBPC3-Δ25p mutations can be considered highly pathogenic, as they result in a poorer prognosis with early manifestation of LVH and a higher risk of sudden cardiac death.29,37,47,48
Many substitutions in DNA sequence do not cause disease and are regarded as benign polymorphisms (ie, variants not expected to be deleterious).9,24 Nevertheless, the relevance for causing disease attached to a significant minority of such identified variants remains unclear, even after applying all criteria for pathogenicity.24 As a result, these mutations are designated in genetic test reports into an ambiguous category, ie, variants of uncertain significance (VUS), with virtually no clinical utility for family screening.24
Interfacing a heterogeneous disease such as HCM with the vast genetic variability of the human genome and the high frequency of novel mutations has created unforeseen difficulties in the molecular diagnosis of HCM and its translation to the clinic.24
Molecular diagnosis
The molecular diagnosis of HCM, which generally consists of screening for mutations in the sarcomere or other HCM-related genes is usually offered to individuals with the HCM phenotype (in the absence of another disease capable of producing similar cardiac wall thickening) and may identify patients with metabolic storage disorders in whom the clinical presentation and pattern of LVH is similar to HCM, eg, Fabry disease (mutations in GLA) and Danon disease (mutations in PRKAG2 and LAMP2).1,2,24–26
In the event of identification of an HCM-related mutation in a proband, it is recommended to offer genetic testing for first-degree relatives who do not present LVH but may be at risk of developing the disease.24,49 Due to the reduced penetrance of HCM, it is recommended to study at least three generations to help define a case of familial HCM.50 Also, if a relative expresses an HCM phenotype and the pathogenic mutation is identified, the genetic status of other members of the family can be clarified, eliminating anxiety and removing the need for future screening.19 Pretest and post-test genetic counseling is highly recommended for discussion of the risks and options available and future management of the individual.5,6,24,49 Individuals found to carry the family-specific mutation despite an absence of clinical manifestations require longitudinal clinical follow-up that depends on age, phenotype, and genotype.8,24,50 In The Netherlands, a pathogenic mutation can be found in 65% of HCM patients.32,33 This means that systematic predictive testing by means of DNA analysis, so-called cascade testing, is technically possible in these patients’ families. Since it is impossible to know in advance which individuals or families will have the greatest advantage from early diagnosis, all index patients who carry an identified mutation should be offered family cascade testing.51
Genetic testing for HCM was initially confined to a few research laboratories that focused on understanding the genetic basis of this disease. In 2003, molecular genetic testing entered the mainstream of the health care system, with automated DNA Sanger sequencing providing more rapid, reliable, and comprehensive molecular diagnosis on a fee-for-service basis.24 Due to its well defined chemistry and sequencing precision, Sanger (dideoxy) sequencing has been considered the “gold standard” for clinical genetic testing.52–54 Most institutional and commercial laboratories screened 5–10 more frequently mutated sarcomere genes (MYH7, MYBPC3, TNNI3, TNNT2, MYL2, TPM1, TNNI3, MYL2, MYL3, ACTC, CSRP3, and TCAP).6,51,55 The order of the genes to be screened was MYBPC3 and MYH7, and if no mutations were found, TNNT2, TPM1, TNNI3, MYL2, MYL3, ACTC, CSRP3, and TCAP were sequenced in this order.51,55 Recently, based on the need to interpret the pathogenic significance of a positive genetic test, Kapplinger et al used Sanger sequencing to annotate genetic variations in the exonic region of nine HCM-associated genes (MYH7, MYLH2, MYLH3, ACTC1, TNNC1, TNNI3, TNNT2, and TPM1) in a cohort of HCM patients and another of healthy individuals.52 With this methodology, it was possible to identify rare non-synonymous variants over-represented in the HCM patient cohort.52
The necessity for electrophoretic separation of DNA fragments is the major obstacle with in Sanger sequencing, increasing experimental time and limiting the number of reactions that can be run in parallel, thus making the technology relatively expensive.56,57 For these reasons, this method is used routinely only for sequencing small amounts of DNA fragments.58 Sanger sequencing is nevertheless important for cosegregation studies of the supposed causative mutation within HCM families and also as a gold standard validation technique in parallel with novel approaches.6,46,56,59–61 The wide genetic heterogeneity of HCM renders Sanger sequencing tedious and too expensive for routine clinical practice,62 prompting DNA microarrays as an alternative technique.63–65
These high-throughput techniques allowed simultaneously testing of more samples and inclusion of established HCM-causative genes, phenocopy associated genes, and other genes with lesser evidence of pathogenicity.6,24
The advantages and disadvantages of the methods currently used for molecular screening and diagnosis in HCM are shown in Table 2. The following sections focus on a description of the most recent methods used in HCM screening and diagnosis.
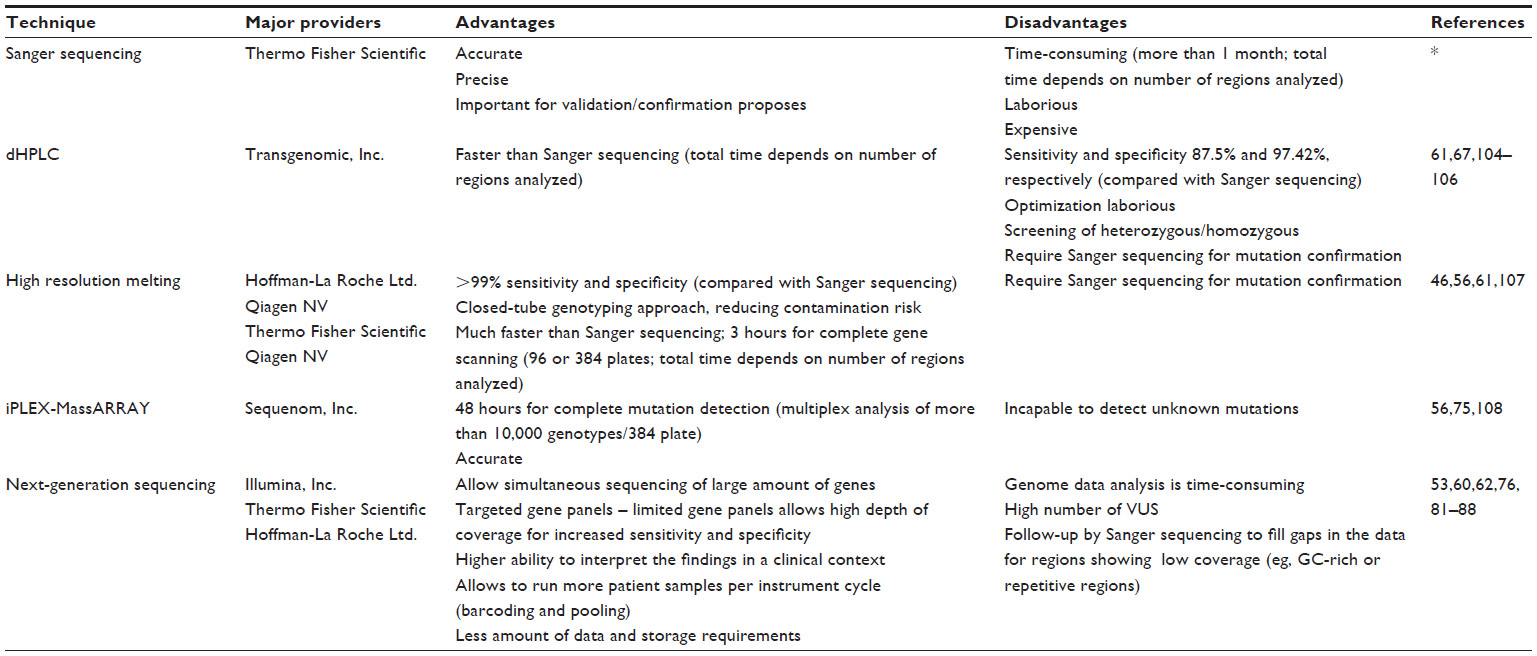 | Table 2 Techniques currently used in diagnosis of hypertrophic cardiomyopathy: major providers, advantages, and disadvantages |
Screening techniques
Although current clinical guidelines recommend genetic testing in patients with HCM and their relatives, its use in everyday clinical practice has been limited by the cost and complexity of conventional sequencing technologies.49,66 Hence, the use of screening techniques that allow analysis of a high number of genes in a shorter period of time, and identifying heteroduplex regions possibly harboring mutations responsible for HCM, is more frequent.56,61 After identification of heteroduplex regions, the region of interest must be sequenced by the Sanger technique in order to identify the mutation.56,61 Several techniques have been used for HCM diagnostics, consisting firstly in the amplification of the selected gene region by polymerase chain reaction (PCR) followed by differential separation of homoduplexes and heteroduplexes. Using denaturing gradient gel electrophoresis and single-strand conformation polymorphism, the amplicons are separated according to the differing electrophoretic mobility between homoduplexes and heteroduplexes,34,67,68 while in denaturing high-performance liquid chromatography, amplicons are separated through the different retention of the denatured amplicons.34,67,68 Screening for mutations using these techniques has proved to be invaluable for identification of mutations in MYBPC3, MYH7, and TNNT2 genes.68–73
High resolution melting has also been used successfully as a screening technique.74 High resolution melting is a high-throughput gene variation scanning technique that relies on the differential melting properties of sequences that vary in at least one nucleotide.74 For this reason, it is used to identify novel mutations in samples.46,56,61 Indeed, this technology was proven effective in screening of mutations in HCM-related genes, providing a closed tube system that reduces the risk of contamination, decreasing analysis time, and requiring no post-PCR sample handling.46,56 Moreover, in addition to its effectiveness in screening of novel mutations, this technique may allow identification of mutations on a specific gene under study if the appropriate controls for all the mutations are simultaneous analyzed.46,56 Recent results reported by our group46,56 and others61 have already confirmed the effectiveness of this technique for identification of alterations in HCM patients. Compared with high resolution melting, the large-scale detection of mutations by other screening techniques is more expensive and technically time-consuming.61,67
Mutation detection
Mutation detection using the iPLEX MassARRAY matrix-assisted laser desorption/ionization time-of-flight system (Sequenom Inc., San Diego, CA, USA) has been used recently for diagnosis of HCM.56,60,62,65,75 In contrast with automated Sanger sequencing, high-throughput techniques such as iPLEX® MassARRAY allow rapid and cost-effective testing for a large number of mutations simultaneously. iPLEX MassARRAY involves multiplex primary PCR using outer primers that flank HCM mutation sites followed by a homogeneous mass extend reaction with multiple, single, inner primers that together generate fragments of different mass specific for each genotype (iPLEX).56 An advantage of iPLEX MassARRAY in comparison with other DNA microarray techniques is its capacity for detecting indels.63,64 iPLEX MassARRAY has been successfully used to identify pathogenic mutations, including indels, in more than 30 genes implicated in HCM.56 Although this technique only detects known mutations, it has been demonstrated to be a high-throughput platform suitable for routine genetic diagnostics, since it allows rapid and relatively inexpensive screening of the most common HCM-related mutations and simultaneous screening of HCM phenocopies.56,65 Using an iPLEX MassArray system, Brion et al were able to routinely analyze 550 mutations of 16 genes.65,75
Next-generation sequencing
Nucleotide-by-nucleotide reading of sequence content by the different next-generation sequencing (NGS) platforms overcomes the limitations of electrophoretic separation and the detection requirements of Sanger sequencing.54 NGS technologies are now becoming more and more adopted in clinical settings with three main levels of analysis and increasing degrees of complexity, ie, disease-targeted gene panels, exome sequencing, and genome sequencing (covering both coding and noncoding regions).76 NGS is composed of three major tasks, ie, sample preparation, sequencing, and data analysis.76 The workflow begins with extraction of genomic DNA from patient samples with targeted panels and exome sequencing requiring enrichment strategies to focus on a subset of genomic targets (usually short DNA fragments [100–500 base pairs] flanked by platform-specific adapters).76 However, GC-rich and repetitive regions within the human genome are still not amenable to be covered by NGS due to sequence complexity and require follow-up by Sanger sequencing to fill the gaps in the data.76 The multiple commercial sequencing platforms developed (Table 2) are able to sequence millions of DNA fragments in parallel, and the different sequencing chemistries result in differences in total sequence capacity, sequence read length, sequence run time, and the final quality and accuracy of the data.76,77 Chemistries include sequencing by synthesis or sequencing by ligation with reversible terminators, bead capture, and ion sensing.76,77
In addition to sequence identification, NGS provides information on variation in the copy number of alleles within the sequenced transcripts.78,79 However, the huge amount of information generated is simultaneously the greatest advantage and greatest disadvantage of NGS.54 A single sequencing experiment generates millions of sequence datasets that need to be analyzed.54 For this reason, clinical application of NGS in HCM has increasingly involved targeted resequencing of genomic subregions, often called “subgenomes”, as a strategy to reduce the amount of data generated by sequencing and facilitate the data analysis.53,60,80 With this methodology, regions of interest are selected by amplification-based methods, such as targeted PCR enrichment or oligonucleotide capture-based methods using either solid surface arrays or in-solution DNA fragment capture.53,60,80 Compared with exome sequencing and genome sequencing, targeted gene panels with a limited gene number allow: a high depth of coverage, with increasing sensitivity and specificity; a high capacity to interpret the findings in a clinical context; running of more patient samples per cycle (barcoding and pooling); less amount of data generated; and less storage requirements.76 The coverage and cost of exome sequencing will be between those of targeted gene panels and genome sequencing; nevertheless, the analytical sensitivity of exome sequencing may be lower than the sensitivity of most targeted gene panels, since a high number of exons in known disease-associated genes may lack sufficient coverage to make a sequence call, and in these cases, follow-up by Sanger sequencing to fill gaps in the data is impractical and expensive.76 In the last 5 years, several of these NGS enrichment methods for genotyping HCM-related mutations have been tested, and were generally observed to provide faster, accurate, and efficient clinical genetic analysis.53,60,62,81–88
NGS data have emerged in HCM, suggesting that double (or triple) or compound pathogenic mutations can be associated with more severe disease expression and an adverse prognosis (eg, heart failure or sudden death, even in the absence of conventional risk markers), with several implications for genetic counselling.6,24 While it is possible that multiple mutations will prove to be prognostic markers or arbitrators of ambiguous risk profiles, the current evidence is preliminary and prospective long-term studies in large populations are required.24
Nevertheless, a major dilemma concerning NGS data for interpreting test results in HCM lies in distinguishing pathogenic mutations from variants of uncertain clinical significance (VUS) or rare nonpathogenic variants.89 This issue has become particularly challenging because the reduced cost of NGS technology now allows comprehensive DNA sequencing of the exome and even the whole genome.84,90,91 Although providing increased scientific insight, NGS substantially increases the recognition of VUS, creating further ambiguity in test reports.24,92,93 Targeted resequencing allows a reduction in the number of VUS identified while increasing the depth of coverage when compared with exome sequencing and genome sequencing.76 Lopes et al, using their targeted NGS strategy, identified a large number of rare non-synonymous sequence variants in non-sarcomeric genes (such as RYR2, ANK2, CAV3, and SCN5A) that may be potential phenotype modifiers in HCM and could explain the genetic heterogeneity of the disease.82 Understanding the clinical significance of these novel variants remains a challenge, reinforcing the need for generally accepted guidelines for interpreting VUS.6,24,82,92,93 The American College of Medical Genetics and Genomics has developed laboratory standards and guidelines for NGS to ensure quality results and more consistent interpretation and reporting of variants found using these technologies.76
Therefore, application of NGS to HCM stands at a crossroads. If genetic testing is to evolve and have a more substantial role in the management of patients with HCM, future efforts should focus on clarifying precisely the pathogenicity of the substantial number of novel variants that are presently recognized and associated with the clinical heterogeneity of HCM and those that will inevitably be identified by new molecular techniques.
Therapeutics for hypertrophic cardiomyopathy
Current treatments
Current therapeutics for HCM patients depend on clinically observed phenotypes and are based on alleviation of symptoms.5,94 The pharmacotherapy applied in HCM is based on β-blockers, calcium channel blockers, and disopyramide. β-blockers improve the myocardial oxygen supply by inactivation of myocardial contractile proteins, and thereby alleviate left ventricular outflow.5,8,94,95 Calcium channel blockers used in HCM include verapamil and diltiazem.8,95 Verapamil acts to decrease the resting outflow gradients, thereby improving diastolic function;8,95 however, the secondary effects of this agent include vasodilation, hypotension, and pulmonary edema, limiting its use to second-line therapy.8,95 Disopyramide is an antiarrhythmic agent that exerts a negative inotropic effect on myocytes by altering sodium–calcium exchange.8,95 However, disopyramide is a third-line therapy and should be given in conjunction with β-blockers and calcium channel blockers.8,95
Only 5% of HCM patients require invasive therapies for symptom control.8,96 Surgical myectomy and alcohol ablation are generally used to relieve left ventricular obstruction in severely symptomatic patients.4,5,8 On the other hand, implantation of an internal cardioverter defibrillator is generally considered for termination of life-threatening arrhythmias, avoiding the possibility of sudden cardiac death and hence extending the lifespan of the patient.5 HCM patients with atrial fibrillation are generally treated with anticoagulation agents to avoid thromboembolism, and can also be considered for pulmonary vein isolation, pacemaker implantation, or a surgical Maze procedure.8
Despite the advantages of early detection of HCM, there is currently no evidence that this will change the course of the disease.95 Indeed, regardless of improvement in quality of life for HCM patients, current pharmacological therapy is not able to overcome the effects of HCM-induced myocardial disorders and cannot improve the long-term prognosis.95 However, given that myocardial dysfunction occurs before development of LVH, and does not necessarily result in symptomatic features, early administration of β-blockers and calcium channel blockers in asymptomatic patients may help to prevent the onset of symptoms and/or reduce the risk of sudden cardiac death.9,20
Tissue engineering and regenerative medicine
The major limitations for the design of new drugs for HCM relate to the lack of in vitro models of human cardiac disorders that accurately reflect disease phenotypes, as well as the genomic differences between humans and the mouse, the most common genetic animal model used for the study of human cardiovascular disorders.97,98 Use of scaffolds that mimic the composition, structure, and biomechanics of the native human heart, together with differentiation of pluripotent stem cells into cardiomyocytes in HCM patients, may aid a deeper understanding of the mechanisms that lead to onset of the disease, and possibly a better understanding of the genotype-phenotype correlation.98,99 Moreover, these systems can be used to screen accurate therapeutic drugs for the pathology induced by particular HCM-related mutations, allowing insights into drug efficiency, safety, and mode of action.99,100
One of the most interesting possibilities in cardiac regenerative medicine is the reprogramming of cardiac fibroblasts to become beating cardiomyocytes.101 This has special importance for HCM patients, since there is a possibility that early reprogramming of fibroblasts into myocytes would reduce the extent of myocardial fibrosis and consequently decrease the risks of development of other pathological HCM-related conditions and sudden cardiac death. Several studies performed in the last decade suggest that such reprogramming is possible, but that it is technically challenging and relatively inefficient, so further studies are needed to achieve the possibility of reprogramming in vivo.101 After optimization of this procedure, nanotherapeutics could be used to target cardiac fibroblasts in the patient using nanoparticles containing miRNAs and the transcription factors that need to be reprogrammed for generation of cardiomyocytes.101
The rapid progress in nanotechnology is changing the foundations for diagnosis, treatment, and prevention of cardiovascular disease. Nanotechnology-derived structures offer “three-in-one functions as imaging systems, target probes, and therapeutic carriers, and are capable of delivering considerable amounts of a drug to cells in a targeted directed manner, so are being developed for use in cardiovascular disease.102 Several ligand-target therapeutic strategies are being developed to overcome the current limitations of conventional drugs and provide additional tools for therapy.102 Nanotechnology offers a wealth of safe tools for the treatment and diagnosis of cardiovascular disease, such as targeted devices with unique therapeutic properties that, because of their small size, can pass biological barriers and deliver multiple therapeutic agents directly to cardiomyocytes.102 Use of molecularly targeted nanovectors affords at least four potential advantages over conventional therapy, and ideally, a delivery system ought to: deliver much greater therapeutic payloads per target; carry multiple, potentially different targeted agents, thereby enhancing selectivity; integrate a means to bypass biological barriers; and enable colocalized delivery of multiple agents, resulting in targeted combination therapy.102
Other “nanotools” are focused on the development of a safe, efficient, specific, and non-pathogenic vehicle for gene delivery.103 Therapeutic and diagnostic agents can be encapsulated, covalently attached, or adsorbed onto such nanocarriers.103 These approaches can easily overcome drug solubility issues, particularly given that large proportions of new drug candidates emerging from high-throughput drug screening initiatives are not water-soluble.103
Conclusion and future directions
It is well established that HCM is a genetically heterogeneous disease, resulting from mutations occurring mainly in sarcomere genes. However, new HCM-related mutations are continuously being revealed, and much more are still to be discovered.6 The dramatic clinical outcomes of HCM, including sudden cardiac death in younger people, including trained athletes, makes early and accurate diagnosis of the pathology essential. Recent advances in the molecular biology field with novel and rapidly evolving strategies for generating genetic data using next-generation technologies opens up the possibility of defining new genes responsible for HCM. Nevertheless, these exome and whole-genome sequencing approaches also reveal a multitude of novel variants and VUS for which clinical relevance is uncertain. The complexity of the information obtained by NGS analysis is being diminished by fractionation of the genome into subgenomes, making this platform one of the most promising for clinical diagnosis of HCM. Nevertheless, future efforts should focus on clarifying more precisely the pathogenicity of the substantial number of novel variants presently known and those that will inevitably be identified by new molecular techniques. It is imperative to create joint collaborations between academia, commercial testing laboratories, and hospitals to promote the exchange of genetic information and to develop standardized mutation classification approaches that allow translation into patient care.
Therapeutics for HCM patients are mainly based on symptom relief, partly due to difficulties in cardiomyocyte cell culture and a lack of suitable animal models. Although in its infancy, cardiac tissue engineering seems to be promising in terms of providing a deeper understanding of the biological mechanisms inherent to HCM and being able to develop new therapeutic approaches for cardiovascular regeneration. Moreover, regenerative medicine together with nanotechnology may improve the therapeutics available for HCM patients by allowing early regeneration of the heart, even in asymptomatic subjects.
Acknowledgment
We thank the Fundação para a Ciência e a Tecnologia/Ministério da Educação e Ciência (FCT/MEC) for financial support via PTDC/CVT-EPI/4651/2012 and PEst-OE/QUI/UI0100/2013.
Disclosure
The authors report no conflicts of interest in this work.
References
Maron BJ, Towbin JA, Thiene G, et al. Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation. 2006;113:1807–1816. | |
Elliott P, Andersson B, Arbustini E, et al. Classification of the cardiomyopathies: a position statement from the European Society of Cardiology Working Group on myocardial and pericardial diseases. Eur Heart J. 2008;29:270–276. | |
Thiene G, Corrado D, Basso C. Cardiomyopathies: is it time for a molecular classification? Eur Heart J. 2004;25:1772–1775. | |
Khouzam RN, Naidu SS. Current status and future perspectives on alcohol septal ablation for hypertrophic obstructive cardiomyopathy. Curr Cardiol Rep. 2014;16:478. | |
Efthimiadis GK, Pagourelias ED, Gossios T, Zegkos T. Hypertrophic cardiomyopathy in 2013: current speculations and future perspectives. World J Cardiol. 2014;6:26–37. | |
Santos S, Freitas AT, Fernandes AR. Overview of HCM genomics and transcriptomics: molecular tools in HCM assessment for application in clinical medicine. Cardiovascular Disease II. iConcept Press. Available from: http://www.iconceptpress.com/books/cardiovascular-disease-ii/. In press 2014. | |
Poggesi C, Ho CY. Muscle dysfunction in hypertrophic cardiomyopathy: what is needed to move to translation? J Muscle Res Cell Motil. 2014;35: 37–45. | |
Enriquez AD, Goldman ME. Management of hypertrophic cardiomyopathy. Ann Glob Health. 2014;80:35–45. | |
Bos JM, Towbin JA, Ackerman MJ. Diagnostic, prognostic, and therapeutic implications of genetic testing for hypertrophic cardiomyopathy. J Am Coll Cardiol. 2009;54:201–211. | |
Wilcken DEL. Physiology of the normal heart. Medicine. 2010;38: 336–339. | |
Teekakirikul P, Kelly MA, Rehm HL, Lakdawala NK, Funke BH. Inherited cardiomyopathies: molecular genetics and clinical genetic testing in the postgenomic era. J Mol Diagn. 2013;15:158–170. | |
Layland J, Shah AM. Biochemistry and physiology of cardiac muscle. Medicine. 2006;34:169–172. | |
Solaro RJ. Sarcomere control mechanisms and the dynamics of the cardiac cycle. J Biomed Biotechnol. 2010;2010:105648. | |
Guclu A, Germans T, Witjas-Paalberends ER, et al. ENerGetIcs in hypertrophic cardiomyopathy: translation between MRI, PET and cardiac myofilament function (ENGINE study). Neth Heart J. 2013;21:567–571. | |
Spudich JA. Hypertrophic and dilated cardiomyopathy: four decades of basic research on muscle lead to potential therapeutic approaches to these devastating genetic diseases. Biophys J. 2014;106: 1236–1249. | |
Ho CY, Lopez B, Coelho-Filho OR, et al. Myocardial fibrosis as an early manifestation of hypertrophic cardiomyopathy. N Engl J Med. 2010;363:552–563. | |
Camelliti P, Borg TK, Kohl P. Structural and functional characterisation of cardiac fibroblasts. Cardiovasc Res. 2005;65:40–51. | |
Jessup M, Brozena S. Heart failure. N Engl J Med. 2003;348: 2007–2018. | |
Maron BJ, Maron MS. Hypertrophic cardiomyopathy. Lancet. 2013; 381:242–255. | |
Ho CY. Hypertrophic cardiomyopathy: preclinical and early phenotype. J Cardiovasc Transl Res. 2009;2:462–470. | |
Spirito P, Autore C, Formisano F, et al. Risk of sudden death and outcome in patients with hypertrophic cardiomyopathy with benign presentation and without risk factors. Am J Cardiol. 2014;113:1550–1555. | |
Nishimura RA, Ommen SR. Hypertrophic cardiomyopathy: the search for obstruction. Circulation. 2006;114:2200–2202. | |
Gersh BJ, Maron BJ, Bonow RO, et al. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy. J Thorac Cardiovasc Surg. 2011;142:e153–e203. | |
Maron BJ, Maron MS, Semsarian C. Genetics of hypertrophic cardiomyopathy after 20 years: clinical perspectives. J Am Coll Cardiol. 2012;60:705–715. | |
Jacoby DL, DePasquale EC, McKenna WJ. Hypertrophic cardiomyopathy: diagnosis, risk stratification and treatment. CMAJ. 2013;185:127–134. | |
Sankaranarayanan R, Fleming EJ, Garratt CJ. Mimics of hypertrophic cardiomyopathy – diagnostic clues to aid early identification of phenocopies. Arrhythmia and Electrophysiology Review. 2013;2:36–40. | |
Semsarian C, Ingles J. Expanding the genetic spectrum of hypertrophic cardiomyopathy: X marks the spot. Circ Cardiovasc Genet. 2013;6:528–530. | |
Hartmannova H, Kubanek M, Sramko M, et al. Isolated X-linked hypertrophic cardiomyopathy caused by a novel mutation of the four-and-a-half LIM domain 1 gene. Circ Cardiovasc Genet. 2013;6:543–551. | |
Kuster DW, Sadayappan S. MYBPC3’s alternate ending: consequences and therapeutic implications of a highly prevalent 25 bp deletion mutation. Pflugers Arch. 2014;466:207–213. | |
Olivotto I, Maron BJ, Tomberli B, et al. Obesity and its association to phenotype and clinical course in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2013;62:449–457. | |
Rowin EJ, Maron MS. The ever expanding spectrum of phenotypic diversity in hypertrophic cardiomyopathy. Am J Cardiol. 2013;112: 463–464. | |
Dhandapany PS, Sadayappan S, Xue Y, et al. A common MYBPC3 (cardiac myosin binding protein C) variant associated with cardiomyopathies in South Asia. Nat Genet. 2009;41:187–191. | |
Alders M, Jongbloed R, Deelen W, et al. The 2373insG mutation in the MYBPC3 gene is a founder mutation, which accounts for nearly one-fourth of the HCM cases in the Netherlands. Eur Heart J. 2003;24:1848–1853. | |
Andersen PS, Havndrup O, Bundgaard H, et al. Genetic and phenotypic characterization of mutations in myosin-binding protein C (MYBPC3) in 81 families with familial hypertrophic cardiomyopathy: total or partial haploinsufficiency. Eur J Hum Genet. 2004;12:673–677. | |
Jääskeläinen P, Miettinen R, Kärkkäinen P, Toivonen L, Laakso M, Kuusisto J. Genetics of hypertrophic cardiomyopathy in eastern Finland: few founder mutations with benign or intermediary phenotypes. Ann Med. 2004;36:23–32. | |
Waldmüller S, Sakthivel S, Saadi AV, et al. Novel deletions in MYH7 and MYBPC3 identified in Indian families with familial hypertrophic cardiomyopathy. J Mol Cell Cardiol. 2003;35:623–636. | |
Brito D, Miltenberger-Miltenyi G, Vale Pereira S, Silva D, Diogo AN, Madeira H. Sarcomeric hypertrophic cardiomyopathy: genetic profile in a Portuguese population. Rev Port Cardiol. 2012;31:577–587. | |
Solomon SD, Jarcho JA, McKenna W, et al. Familial hypertrophic cardiomyopathy is a genetically heterogeneous disease. J Clin Invest. 1990;86:993–999. | |
Ko YL, Lien WP, Chen JJ, Wu CW, Tang TK, Liew CC. No evidence for linkage of familial hypertrophic cardiomyopathy and chromosome 14q1 locus D14S26 in a Chinese family: evidence for genetic heterogeneity. Hum Genet. 1992;89:597–601. | |
Dausse E, Schwartz K. Genetic heterogeneity of familial hypertrophic cardiomyopathy. Neuromuscul Disord. 1993;3:483–486. | |
Gajendrarao P, Krishnamoorthy N, Kassem HS, et al. Molecular modeling of disease causing mutations in domain C1 of cMyBP-C. PLoS One. 2013;8:e59206. | |
Bos JM, Ackerman MJ. Z-disc genes in hypertrophic cardiomyopathy: stretching the cardiomyopathies? J Am Coll Cardiol. 2010;55: 1136–1138. | |
Watkins H, McKenna WJ, Thierfelder L, et al. Mutations in the genes for cardiac troponin T and alpha-tropomyosin in hypertrophic cardiomyopathy. N Engl J Med. 1995;332:1058–1064. | |
Ho CY. Hypertrophic cardiomyopathy. Heart Fail Clin. 2010;6: 141–159. | |
Niimura H, Patton KK, McKenna WJ, et al. Sarcomere protein gene mutations in hypertrophic cardiomyopathy of the elderly. Circulation. 2002;105:446–451. | |
Santos S, Marques V, Pires M, et al. High resolution melting: improvements in the genetic diagnosis of hypertrophic cardiomyopathy in a Portuguese cohort. BMC Med Genet. 2012;13:17. | |
Ackerman MJ, VanDriest SL, Ommen SR, et al. Prevalence and age-dependence of malignant mutations in the beta-myosin heavy chain and troponin T genes in hypertrophic cardiomyopathy: a comprehensive outpatient perspective. J Am Coll Cardiol. 2002;39:2042–2048. | |
Landstrom AP, Ackerman MJ. Mutation type is not clinically useful in predicting prognosis in hypertrophic cardiomyopathy. Circulation. 2010;122:2441–2449. | |
Ho CY. Genetics and clinical destiny: improving care in hypertrophic cardiomyopathy. Circulation. 2010;122:2430–2440. | |
Hershberger RE, Lindenfeld J, Mestroni L, et al. Genetic evaluation of cardiomyopathy – a Heart Failure Society of America practice guideline. J Card Fail. 2009;15:83–97. | |
van Langen IM, Arens Y, Baars H, et al; ICIN Working Group on Hereditary Heart Diseases. Genetic diagnostics and genetic counseling in hypertrophic cardiomyopathy (HCM). Neth Heart J. 2010;18:144–159. | |
Kapplinger JD, Landstrom AP, Bos JM, et al. Distinguishing hypertrophic cardiomyopathy-associated mutations from background genetic noise. J Cardiovasc Transl Res. 2014;7:347–361. | |
Dames S, Durtschi J, Geiersbach K, Stephens J, Voelkerding KV. Comparison of the Illumina genome analyzer and Roche 454 GS FLX for resequencing of hypertrophic cardiomyopathy-associated genes. J Biomol Tech. 2010;21:73–80. | |
Rizzo JM, Buck MJ. Key principles and clinical applications of “next-generation” DNA sequencing. Cancer Prev Res (Phila). 2012;5:887–900. | |
Marian AJ. Hypertrophic cardiomyopathy: from genetics to treatment. Eur J Clin Invest. 2010;40:360–369. | |
Santos S, Lança V, Oliveira H, et al. Genetic diagnosis of hypertrophic cardiomyopathy using mass spectrometry DNA arrays and high resolution melting. Rev Port Cardiol. 2011;30:7–18. | |
Hert DG, Fredlake CP, Barron AE. Advantages and limitations of next-generation sequencing technologies: a comparison of electrophoresis and non-electrophoresis methods. Electrophoresis. 2008;29:4618–4626. | |
Mardis ER. A decade’s perspective on DNA sequencing technology. Nature. 2011;470:198–203. | |
Li Z, Huang J, Zhao J, et al. Rapid molecular genetic diagnosis of hypertrophic cardiomyopathy by semiconductor sequencing. J Transl Med. 2014;12:173. | |
Mook ORF, Haagmans MA, Soucy JF, et al. Targeted sequence capture and GS-FLX titanium sequencing of 23 hypertrophic and dilated cardiomyopathy genes: implementation into diagnostics. J Med Genet. 2013;50:614–626. | |
Millat G, Chanavat V, Crehalet H, Rousson R. Development of a high resolution melting method for the detection of genetic variations in hypertrophic cardiomyopathy. Clin Chim Acta. 2010;411:1983–1991. | |
Fokstuen S, Makrythanasis P, Nikolaev S, et al. Multiplex targeted high-throughput sequencing for Mendelian cardiac disorders. Clin Genet. 2014;85:365–370. | |
Waldmuller S, Freund P, Mauch S, Toder R, Vosberg HP. Low-density DNA microarrays are versatile tools to screen for known mutations in hypertrophic cardiomyopathy. Hum Mutat. 2002;19:560–569. | |
Fokstuen S, Lyle R, Munoz A, et al. DNA resequencing array for pathogenic mutation detection in hypertrophic cardiomyopathy. Hum Mutat. 2008;29:879–885. | |
Brion M, Allegue C, Monserrat L, et al. Large scale analysis of HCM mutations in sudden cardiac death. Forensic Sci Int. 2008;1:549–550. | |
Charron P, Arad M, Arbustini E, et al. Genetic counselling and testing in cardiomyopathies: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2010;31:2715–2726. | |
Millat G, Bouvagnet P, Chevalier P, et al. Prevalence and spectrum of mutations in a cohort of 192 unrelated patients with hypertrophic cardiomyopathy. Eur J Med Genet. 2010;53:261–267. | |
Zeller R, Ivandic BT, Ehlermann P, et al. Large-scale mutation screening in patients with dilated or hypertrophic cardiomyopathy: a pilot study using DGGE. J Mol Med (Berl). 2006;84:682–691. | |
Ehlermann P, Weichenhan D, Zehelein J, et al. Adverse events in families with hypertrophic or dilated cardiomyopathy and mutations in the MYBPC3 gene. BMC Med Genet. 2008;9:95. | |
Rodríguez-García MI, Monserrat L, Ortiz M, et al. Screening mutations in myosin binding protein C3 gene in a cohort of patients with hypertrophic cardiomyopathy. BMC Med Genet. 2010;11:67. | |
Annapurna SD, Reena TR, Nallari P, Calambur N. Genetic variation in exon 5 of troponin-I gene in hypertrophic cardiomyopathy cases. Indian J Hum Genet. 2007;13:50–53. | |
van Driest SL, Ellsworth EG, Ommen SR, Tajik AJ, Gersh BJ, Ackerman MJ. Prevalence and spectrum of thin filament mutations in an outpatient referral population with hypertrophic cardiomyopathy. Circulation. 2003;108:445–451. | |
van Driest SL, Vasile VC, Ommen SR, et al. Myosin binding protein C mutations and compound heterozygosity in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2004;44:1903–1910. | |
Erali M, Wittwer CT. High resolution melting analysis for gene scanning. Methods. 2010;50:250–261. | |
Brion M, Allegue C, Gil R, et al. Identification of a novel MYBPC3 gene variant in a patient with hypertrophic cardiomyopathy. Ann Clin Lab Sci. 2010;40:285–289. | |
Rehm HL, Bale SJ, Bayrak-Toydemir P, et al. ACMG clinical laboratory standards for next-generation sequencing. Genet Med. 2013;15:733–747. | |
Glenn TC. Field guide to next-generation DNA sequencers. Mol Ecol Resour. 2011;11:759–769. | |
Wang H, Nettleton D, Ying K. Copy number variation detection using next generation sequencing read counts. BMC Bioinformatics. 2014;15:109. | |
Boonpeng H, Yusoff K. The utility of copy number variation (CNV) in studies of hypertension-related left ventricular hypertrophy (LVH): rationale, potential and challenges. Mol Cytogenet. 2013;6:8. | |
Voelkerding KV, Dames S, Durtschi JD. Next generation sequencing for clinical diagnostics-principles and application to targeted resequencing for hypertrophic cardiomyopathy: a paper from the 2009 William Beaumont Hospital Symposium on Molecular Pathology. J Mol Diagn. 2010;12:539–551. | |
Ellims AH, IIes LM, Ling LH, et al. A comprehensive evaluation of myocardial fibrosis in hypertrophic cardiomyopathy with cardiac magnetic resonance imaging: linking genotype with fibrotic phenotype. Eur Heart J Cardiovasc Imaging. May 12, 2014. [Epub ahead of print]. | |
Lopes LR, Zekavati A, Syrris P, et al. Genetic complexity in hypertrophic cardiomyopathy revealed by high-throughput sequencing. J Med Genet. 2013;50:228–239. | |
Wang Y, Wang Z, Yang Q, et al. Autosomal recessive transmission of MYBPC3 mutation results in malignant phenotype of hypertrophic cardiomyopathy. PLoS One. 2013;8:e67087. | |
Meder B, Haas J, Keller A, et al. Targeted next-generation sequencing for the molecular genetic diagnostics of cardiomyopathies. Circ Cardiovasc Genet. 2011;4:110–122. | |
Wheeler M, Pavlovic A, DeGoma E, Salisbury H, Brown C, Ashley EA. A new era in clinical genetic testing for hypertrophic cardiomyopathy. J Cardiovasc Transl Res. 2009;2:381–391. | |
Millat G, Chanavat V, Rousson R. Evaluation of a new NGS method based on a custom AmpliSeq library and ion torrent PGM sequencing for the fast detection of genetic variations in cardiomyopathies. Clin Chim Acta. 2014;433C:266–271. | |
Bick AG, Flannick J, Ito K, et al. Burden of rare sarcomere gene variants in the Framingham and Jackson Heart Study cohorts. Am J Hum Genet. 2012;91:513–519. | |
Herman DS, Lam L, Taylor MR, et al. Truncations of titin causing dilated cardiomyopathy. N Engl J Med. 2012;366:619–628. | |
Tester DJ, Ackerman MJ. Genetic testing for potentially lethal, highly treatable inherited cardiomyopathies/channelopathies in clinical practice. Circulation. 2011;123:1021–1037. | |
Mestroni L, Taylor MR. Hearing the noise the challenges of human genome variation in genetic testing. J Am Coll Cardiol. 2011;57: 2328–2329. | |
The 1000 Genomes Project Consortium. A map of human genome variation from populations – scale sequencing. Nature. 2010;467:1061–1073. | |
Watkins H. Assigning a causal role to genetic variants in hypertrophic cardiomyopathy. Circ Cardiovasc Genet. 2013;6:2–4. | |
Das K J, Ingles J, Bagnall RD, Semsarian C. Determining pathogenicity of genetic variants in hypertrophic cardiomyopathy: importance of periodic reassessment. Genet Med. 2014;16:286–293. | |
McKenna WJ, Behr ER. Hypertrophic cardiomyopathy: management, risk stratification, and prevention of sudden death. Heart. 2002;87: 169–176. | |
Hamada M, Ikeda S, Shigematsu Y. Advances in medical treatment of hypertrophic cardiomyopathy. J Cardiol. 2014;64:1–10. | |
Efthimiadis GK, Pitsis A, Pagourelias ED, et al. Surgical septal myectomy for hypertrophic cardiomyopathy in Greece: a single-center initial experience. Hellenic J Cardiol. 2014;55:132–138. | |
Moretti A, Laugwitz KL, Dorn T, Sinnecker D, Mummery C. Pluripotent stem cell models of human heart disease. Cold Spring Harb Perspect Med. 2013;3:a014027. | |
Garbern JC, Mummery CL, Lee RT. Model systems for cardiovascular regenerative biology. Cold Spring Harb Perspect Med. 2013;3:a014019. | |
Vunjak Novakovic G, Eschenhagen T, Mummery C. Myocardial tissue engineering: in vitro models. Cold Spring Harb Perspect Med. 2014;4:a014076. | |
Liang P, Du J. Human induced pluripotent stem cell for modeling cardiovascular diseases. Regenerative Medicine Research. 2014;2:4. | |
Yi BA, Mummery CL, Chien KR. Direct cardiomyocyte reprogramming: a new direction for cardiovascular regenerative medicine. Cold Spring Harb Perspect Med. 2013;3:a014050. | |
Kumar N, Kumar R. Nanomedicine for treatment of cardiovascular diseases. In: Nanotechnology and Nanomaterials in the Treatment of Life-threatening Diseases. Waltham, MA, USA: Elsevier; 2014. | |
Wagner V, Dullaart A, Bock AK, Zweck A. The emerging nanomedicine landscape. Nat Biotechnol. 2006;24:1211–1217. | |
Bahrudin U, Mizuta E, Ariani MD, Nindita Y, Hisatome I. Sensitivity and specificity of denaturing HPLC to detect MYBPC3 gene mutations in hypertrophic cardiomyopathy. Yonago Acta Med. 2009;52: 121–125. | |
Bos JM, Poley RN, Ny M, et al. Genotype-phenotype relationships involving hypertrophic cardiomyopathy-associated mutations in titin, muscle LIM protein, and telethonin. Mol Genet Metab. 2006;88:78–85. | |
van Driest SL, Jaeger MA, Ommen SR, et al. Comprehensive analysis of the beta-myosin heavy chain gene in 389 unrelated patients with hypertrophic cardiomyopathy. J Am Coll Cardiol. 2004;44:602–610. | |
Chen D, Wang YY, Chuai ZR, et al. High-resolution melting analysis for accurate detection of BRAF mutations: a systematic review and meta-analysis. Sci Rep. 2014;4:4168. | |
Allegue C, Gil R, Blanco-Verea A, et al. Prevalence of HCM and long QT syndrome mutations in young sudden cardiac death-related cases. Int J Legal Med. 2011;125:565–572. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.