Back to Journals » Clinical, Cosmetic and Investigational Dermatology » Volume 19
Genetically Proxied Biological Aging and Risk of Hypertrophic Scar/Keloid-Coded Phenotypes: An Exploratory Two-Sample Mendelian Randomization Study
Authors Chen Z, Li B, Gao R, Li Y, Liang L, Gao Q, Chen Y, Zhang L
Received 10 April 2026
Accepted for publication 1 July 2026
Published 14 July 2026 Volume 2026:19 615985
DOI https://doi.org/10.2147/CCID.S615985
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Monica K. Li
Zequn Chen,1,* Bingmin Li,2,* Rui Gao,3,* Yanqi Li,4 Liming Liang,4 Quanwen Gao,4 Youbai Chen,4 Lixia Zhang4
1The Third Ward of Department of Burn, Senior Department of Burns and Plastic Surgery, The Fourth Medical Center of Chinese PLA General Hospital, Beijing, People’s Republic of China; 2Department of Dermatology, Fourth Medical Center of the Chinese PLA General Hospital, Beijing, People’s Republic of China; 3Burn and Plastic Surgery, No. 940 Hospital of Joint Logistics Support Force of People’s Liberation Army, Lanzhou, Gansu, People’s Republic of China; 4Department of Plastic and Reconstructive Surgery, Senior Department of Burns and Plastic Surgery, The Fourth Medical Center of Chinese PLA General Hospital, Beijing, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Lixia Zhang; Youbai Chen, Email [email protected]; [email protected]
Purpose: Hypertrophic scars (HS), common fibroproliferative disorders, predominantly affect younger individuals. To investigate potential causal associations between multiple genetically proxied biological aging indicators and the risk of HS using a two-sample Mendelian randomization (MR) approach.
Patients and Methods: We used genome-wide association studies (GWAS) data to examine causal links between biological aging indicators (epigenetic clocks, telomere length, macroscopic aging) and HS. Primary analysis employed inverse-variance weighted MR, with sensitivity analyses (MR-Egger, weighted median, weighted mode) and multivariable MR adjusting for fibrogenic mediators. Results were validated in independent cohorts. Robustness was assessed via MR-PRESSO, Cochran’s Q, MR-Steiger, and leave-one-out analyses to detect statistical evidence of directional and horizontal pleiotropy.
Results: Genetically predicted higher IEAA (Odds Ratio [OR]=0.926, 95% Confidence Interval [CI]: 0.878– 0.976, P=0.004) and higher PhenoAge (OR=0.911, 95% CI: 0.848– 0.979, P=0.011) were nominally associated with a decreased risk of HS. The PhenoAge association was consistently replicated in the independent validation cohort (OR=0.91, P=0.0059). However, these associations did not remain significant after False Discovery Rate correction. Negative control analysis showed no association with general skin fibrosis, suggesting specificity for hyper-proliferative scarring. Multivariable MR indicated that the protective effect of PhenoAge was independent of classical fibrogenic pathways. No significant causal associations were found for other aging indicators. No statistical evidence of directional or horizontal pleiotropy was detected in sensitivity analyses.
Conclusion: This MR study provides exploratory evidence suggesting that genetically proxied faster epigenetic aging may be nominally associated with a reduced risk of HS. These findings offer novel hypotheses regarding the complex interplay between aging processes and scar formation.
Keywords: mendelian randomization, hypertrophic scar, keloid, biological aging, epigenetic age acceleration, PhenoAge
Introduction
Hypertrophic scars (HS) are raised, erythematous, and often pruritic or painful fibroproliferative dermal lesions that typically develop within the boundaries of an original wound following trauma, surgery, or burns.1 They represent a significant clinical challenge, causing cosmetic disfigurement, functional impairment, and substantial psychological distress, thereby impacting patients’ quality of life.2 HS are common fibroproliferative disorders with a variable incidence depending on the type of injury. For instance, HS have been reported to occur in approximately 35% of linear surgical wounds and can affect up to 72% of burn wounds.3 Current treatments, including pressure therapy, silicone gel sheeting, intralesional corticosteroids, and laser therapy, offer limited efficacy and are associated with high recurrence rates.4 A peculiar epidemiological feature of HS is its higher incidence and severity in younger individuals, particularly adolescents and young adults, with a decreased prevalence observed in the elderly.5 This age-related disparity suggests that mechanisms intrinsic to younger physiological states might predispose to HS,6,7 while certain aspects of aging could be protective.
Biological aging, as distinct from chronological aging, reflects an individual’s physiological status and susceptibility to age-related diseases. It can be assessed through various molecular and phenotypic markers.8 Epigenetic clocks, such as Intrinsic Epigenetic Age Acceleration (IEAA), HannumAge, PhenoAge, and GrimAge, are derived from DNA methylation patterns and serve as robust estimators of biological age, with accelerations linked to various adverse health outcomes.9 Epigenetic clocks represent distinct biological domains based on their training models. First-generation clocks, such as HannumAge and IEAA, primarily predict chronological age and relate to hematopoietic stem cell properties. Second-generation clocks, like PhenoAge and GrimAge, incorporate clinical measures of morbidity and mortality, offering a more robust estimation of health span and systemic physiological decline.10 Associated biomarkers like granulocyte proportions and plasminogen activator inhibitor-1 (PAI1) levels are also integrated into some of these clocks. Telomere length (TL), the protective cap at chromosome ends, shortens with cell division and is another widely studied marker of cellular aging. Macroscopic indicators like the frailty index and perceived facial aging capture organismal decline and overall health status.11
The relationship between biological aging processes and HS formation is complex and not fully understood. Observational studies have implicated epigenetic modifications, such as altered DNA methylation and histone modifications, in the pathogenesis of keloids and HS.12 For instance, specific DNA methylation patterns in fibroblasts from keloid scars have been identified,13 and changes in DNA hydroxymethylation have been linked to scar formation.14 Research has also shown telomere shortening and reduced hTERT activity in pathological scar formation.15 Interestingly, cellular senescence, a hallmark of aging, can induce a senescence-associated secretory phenotype (SASP) in fibroblasts that may possess anti-fibrotic properties, including the upregulation of matrix metalloproteinases (MMPs) that degrade collagen and downregulation of collagen synthesis itself.16 This suggests that certain aging-related cellular states might paradoxically counteract excessive fibrosis. However, traditional observational studies are susceptible to confounding factors and reverse causation, making it difficult to establish causal relationships. Establishing a causal relationship between biological aging and HS is of considerable clinical importance. Aging-related biomarkers may improve risk stratification for pathological scarring, and a better understanding of aging-associated fibrotic mechanisms could identify novel therapeutic targets for HS prevention and treatment.
Mendelian randomization (MR) is an epidemiological method that utilizes genetic variants as instrumental variables (IVs) to infer causal relationships between an exposure and an outcome, minimizing biases from confounding and reverse causation inherent in observational studies.17 By leveraging randomly allocated genetic variants associated with biological aging indicators, MR can provide more robust evidence for or against a causal link with HS.18
This study aims to employ a two-sample MR approach to investigate the potential causal associations between a comprehensive panel of genetically proxied biological aging indicators (including epigenetic clocks, related biomarkers, telomere length, and macroscopic aging measures) and the risk of HS. Elucidating these relationships could provide novel insights into HS pathophysiology, identify potential biomarkers for risk stratification, and suggest new avenues for prevention or treatment.
Materials and Methods
Study Design
This study employed a two-sample MR design to assess the causal effects of various biological aging indicators on the risk of HS (Figure 1). The MR design relies on three core assumptions: (1) the IVs are robustly associated with the exposure; (2) the IVs are not associated with any confounding factors that could bias the exposure-outcome association; and (3) the IVs affect the outcome only through their effect on the exposure (ie, no horizontal pleiotropy).14 This study was reported following the Strengthening the Reporting of Observational Studies in Epidemiology using Mendelian Randomization (STROBE-MR) guidelines19 (Table S1).
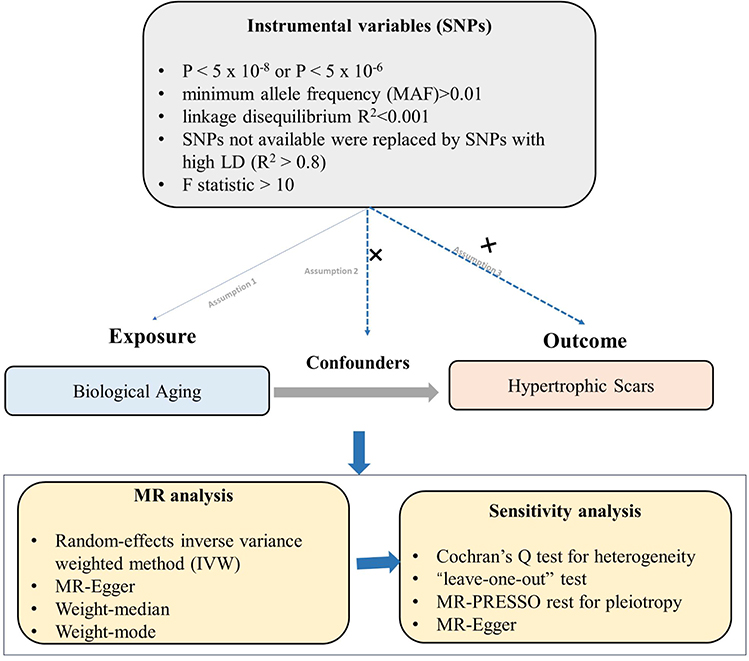 |
Figure 1 Schematic diagram illustrating the two-sample Mendelian randomization approach used to assess the causal relationship between genetically proxied biological aging indicators (exposures) and the risk of hypertrophic scars (outcome). |
To further explore the robustness and potential mechanisms underlying the primary MR findings, we performed several additional analyses: (1) Steiger directionality tests were used to confirm the direction of causality between aging indicators and scar outcomes; (2) an independent validation analysis was conducted in a European ancestry cohort defined as “keloid scar” (with case definition directly mapping to ICD-10 L91.0, consistent with our primary outcome), as well as a negative control analysis using a phenotype representing general scar conditions and non-proliferative skin fibrosis (FinnGen R12, L12_SCARCONDITIONS), which captures non-pathological fibrosis rather than excessive or hyper-proliferative scarring; and (3) for key positive findings, multivariable MR (MVMR) analyses were performed to evaluate whether the observed associations were mediated by major fibrosis-related factors (such as MMPs, tissue inhibitors of MMPs [TIMPs], TGF-β, SMADs, and CTGF), based on their established roles in scar/fibrosis pathogenesis.20
Data Sources for Biological Aging Indicators (Exposures)
To enhance clarity and facilitate verification, detailed information on all exposures and outcomes used in this study, including trait, GWAS source, cohort, ancestry, GWAS ID, sample size (cases/controls), and number of single nucleotide polymorphisms (SNPs), is summarized in Table S2.
A total of 16 biological aging indicators were selected as exposures, based on prior MR studies.21–23 All GWAS summary statistics were derived from participants of European ancestry to minimize population stratification bias; therefore, all analyses were restricted to European-ancestry datasets to ensure validity.
Four epigenetic clocks (PhenoAge, GrimAge, HannumAge, and Intrinsic Epigenetic Age Acceleration (IEAA, based on HorvathAge)) and two epigenetic biomarkers (granulocyte proportions and plasminogen activator inhibitor-1 (PAI1) levels) were obtained from the GWAS meta-analysis by McCartney et al,10 with sample sizes up to 34,710 (GCST90014287–GCST90014292). Overall leukocyte TL GWAS data were sourced from Codd et al (2021) (UK Biobank, 472,174 participants, GWAS ID: ieu-b-4879, and Figshare: https://figshare.com/s/caa99dc0f76d62990195)24 Cell-type specific TL GWAS (B-cell, granulocyte, lymphocyte, memory T-cell, naive T-cell, NK-cell) were from Stoma et al (2022), based on 902 Dutch individuals (ebi-a-GCST90101887–ebi-a-GCST90101892).25 The frailty index was obtained from Atkins et al (2021) (UK Biobank and TwinGene, n=175,226, ebi-a-GCST90020053); facial aging GWAS was based on self-reported perceived age in 423,999 UK Biobank participants (ukb-b-2148).22
Data Source for Hypertrophic Scars (Outcome)
Summary statistics for HS were obtained from the FinnGen consortium (https://r12.finngen.fi/, GWAS ID: L12_HYPETROPHICSCAR), comprising 2068 cases and 465,673 controls of European (Finnish) ancestry. In this cohort, cases were defined based by the International Classification of Diseases (ICD) codes L91.0 (ICD-10), 7014B (ICD-9), and 70130 (ICD-8) from hospital discharge and cause of death registries. To validate the robustness and specificity of our findings, two additional datasets were employed. First, we used summary statistics from the VA Million Veteran Program (MVP) (GCST90476200, N = 2079 cases, 444,929 controls) as an independent replication cohort.26 Although labeled as “Keloid scar” (PheCode 701.4), its case definition maps directly to ICD-10 L91.0, aligning with our primary HS definition. Second, we utilized Scar conditions and fibrosis of skin (FinnGen R12, L12_SCARCONDITIONS, N = 1495 cases, 465,673 controls) as a negative control. This phenotype primarily encompasses non-proliferative fibrosis, defined by ICD-10 code L90.5, allowing us to distinguish between general wound healing and pathological hyper-proliferation.
To minimize potential bias from sample overlap, we utilized datasets from geographically and administratively distinct cohorts: the UK Biobank (United Kingdom), FinnGen (Finland), and the MVP (United States). This diversity in recruitment sources significantly reduces the likelihood of substantial sample overlap between the exposure and outcome datasets.
As this study relied solely on publicly available, de-identified summary statistics, it was exempt from additional institutional ethical review under national legislative guidelines, and no further informed consent was required.
Instrumental Variable Selection
For each exposure, IVs were selected based on the following criteria: SNPs reached genome-wide significance (P < 5 × 10−8). For granulocyte proportions and cell-specific telomere lengths, where fewer SNPs reached this threshold, a more lenient threshold of P < 5 × 10−6 was applied. This was consistent with their original GWAS reports or common practice for exposures with limited IVs, necessitated by the lower number of genome-wide significant SNPs available for these traits. Minor allele frequency (MAF) > 0.01.27 Linkage disequilibrium (LD) clumping was performed (R2 < 0.001, window size = 10,000 kb) using the 1000 Genomes Project European reference panel to ensure independence of IVs.28 When an IV was not available in the outcome GWAS, proxy SNPs in high LD (R2 > 0.8) with the original SNP were identified using the 1000 Genomes European panel, if available. The strength of each IV was assessed using the F-statistic, calculated as F = R2 × (N−2) / (1−R2), where R2 is the proportion of variance in the exposure explained by the SNP, and N is the sample size of the exposure GWAS. IVs with F-statistics <10 were considered weak and excluded to minimize weak instrument bias. The R2 was calculated using the formula: R2 = 2 × MAF × (1−MAF) × β2, where β is the per-allele effect size of the SNP on the quantitative exposure. Finally, during data harmonization steps, SNPs that were palindromic with ambiguous allele frequencies or had incompatible alleles between exposure and outcome datasets were removed.29
Mendelian Randomization Analysis
The primary MR analysis was conducted using the random-effects inverse-variance weighted (IVW) method, which combines the Wald ratios of individual SNPs to provide an overall causal estimate. The results are presented as odds ratios (ORs) with 95% confidence intervals (CIs) representing the risk of HS per one standard deviation (SD) in the genetically proxied exposure. Specifically, for epigenetic age acceleration metrics (IEAA, PhenoAge, GrimAge, and HannumAge), the ORs are expressed per one-year increase in genetically predicted biological age acceleration. This provide an estimate independent of actual chronological age, thereby minimizing age-related confounding. Several additional analyses were performed to assess the robustness of the IVW results and to detect potential violations of MR assumptions: MR-Egger Regression provide a causal estimate adjusted for directional pleiotropy if its intercept term differs significantly from zero (P < 0.05). Weighted Median Estimator provides a consistent estimate if at least 50% of the weight in the analysis comes from valid IVs. Weighted Mode Estimator: This method groups SNPs into clusters based on their causal estimates and provides an estimate based on the largest cluster, robust to outliers.19,27
Multivariable Mendelian Randomization Analysis
For exposures with nominally significant associations in both primary and replication analyses, we conducted MVMR analyses to test whether the observed effect was mediated by major fibrogenic factors, including MMP2, MMP9, TIMP1, TIMP2, TGFB1, TGFBR1, TGFBR2, SMAD2, SMAD3, and CTGF. The selection of these mediators was based on their established roles in skin fibrosis pathogenesis.30 These data were retrieved from published studies31–33 with sample sizes ranging from 5329 to 47,745, all involving European populations. Detailed GWAS source information for these mediators was listed in Table S2. MVMR estimates were interpreted under the standard assumptions of instrument relevance, independence from confounders, and exclusion restriction conditional on included exposures.
IVs for MVMR were selected through a rigorous process. Initially, SNPs associated with PhenoAge or mediators at P<5×10−8 were identified. Due to limited IVs, the threshold for MMP2, TIMP1, TGF-β, and SMADs was relaxed to P<5×10−6 (MMP1 was excluded due to insufficient IVs). After filtering for MAF > 0.01, LD clumping (R2<0.001, window = 10,000kb) was performed for each trait independently. The union of these SNPs was pooled and re-clumped (R2<0.001) to ensure overall independence. During harmonization, palindromic SNPs were removed, and only SNPs present across all relevant datasets were retained as final instruments.
Sensitivity and Pleiotropy Analysis
Cochran’s Q statistic was used to assess heterogeneity among the causal estimates from individual SNPs in the IVW and MR-Egger analyses. A P-value < 0.05 suggests significant heterogeneity.34 The MR-Egger intercept was examined; a P-value > 0.05 for the intercept suggests no significant directional pleiotropy.35 MR-Pleiotropy Residual Sum and Outlier (MR-PRESSO) test detects and corrects for horizontal pleiotropy by identifying outlier SNPs.36 A global P-value < 0.05 from MR-PRESSO indicates significant pleiotropy. If outliers are detected, MR-PRESSO provides an outlier-corrected estimate. Leave-One-Out (LOO) analysis was performed by systematically removing one SNP at a time and recalculating the IVW estimate to assess if the overall causal estimate was driven by any single influential SNP.37 Steiger directionality tests were used to confirm the direction of causality for all positive MR findings.
All MR analyses were performed using the “TwoSampleMR” and “MRPRESSO” packages in R (version 4.0.5). A P-value < 0.05 was considered nominally statistically significant. To account for multiple testing across the 16 exposures, the False Discovery Rate (FDR) method (Benjamini-Hochberg procedure) was applied to the P-values from the primary IVW analyses, with P-FDR < 0.05 considered statistically significant.
Results
Instrumental Variable Selection
After applying selection criteria and harmonization, SNPs used as IVs for each biological aging indicator ranged from 3 (Naive T-cell telomere length) to 159 (leukocyte telomere length). Detailed information for each SNP—including rsID, alleles, EAF, F-statistics, R2, and proxy status—was provided in Table S3a and S3b. The minimum, maximum, range, mean, and median F-statistics for each exposure were calculated for instrument robustness assess (Table S4). The mean and median F-statistics for all exposures exceeded 20, indicating generally adequate instrument strength; the lowest individual F-statistic (19.84) corresponded to granulocyte proportion. A total of 41 SNPs were not present in the outcome dataset, for which 13 suitable proxies were identified and used.
Mendelian Randomization Analyses of Biological Aging Indicators and Hypertrophic Scar Risk
Genetically predicted PhenoAge and IEAA showed nominal associations with a reduced risk of HS (PhenoAge: OR = 0.918, 95% CI: 0.847–0.978, P = 0.011, FDR = 0.0877; IEAA: OR = 0.926, 95% CI: 0.878–0.976, P = 0.004, FDR = 0.063), but these associations did not survive FDR correction (Table 1). The protective effect of PhenoAge and IEAA was consistent across multiple MR methods, as shown in the forest plots (Figure 2) and individual SNP-level visualizations (Figure 3A–D). In contrast, while granulocyte proportions initially suggested an association (IVW: OR = 0.015, 95% CI: 0.0003–0.7405, P = 0.0348, FDR = 0.1856), the result was characterized by wide confidence intervals and inconsistent effect directions across models, indicating potential statistical instability (Table 1). Therefore, results for granulocyte proportion should be interpreted with caution. As for the remaining aging indicators, including GrimAge, HannumAge, various telomere lengths, frailty index, and facial aging, no evidence of association with HS risk was observed (all P > 0.05; Table 1).
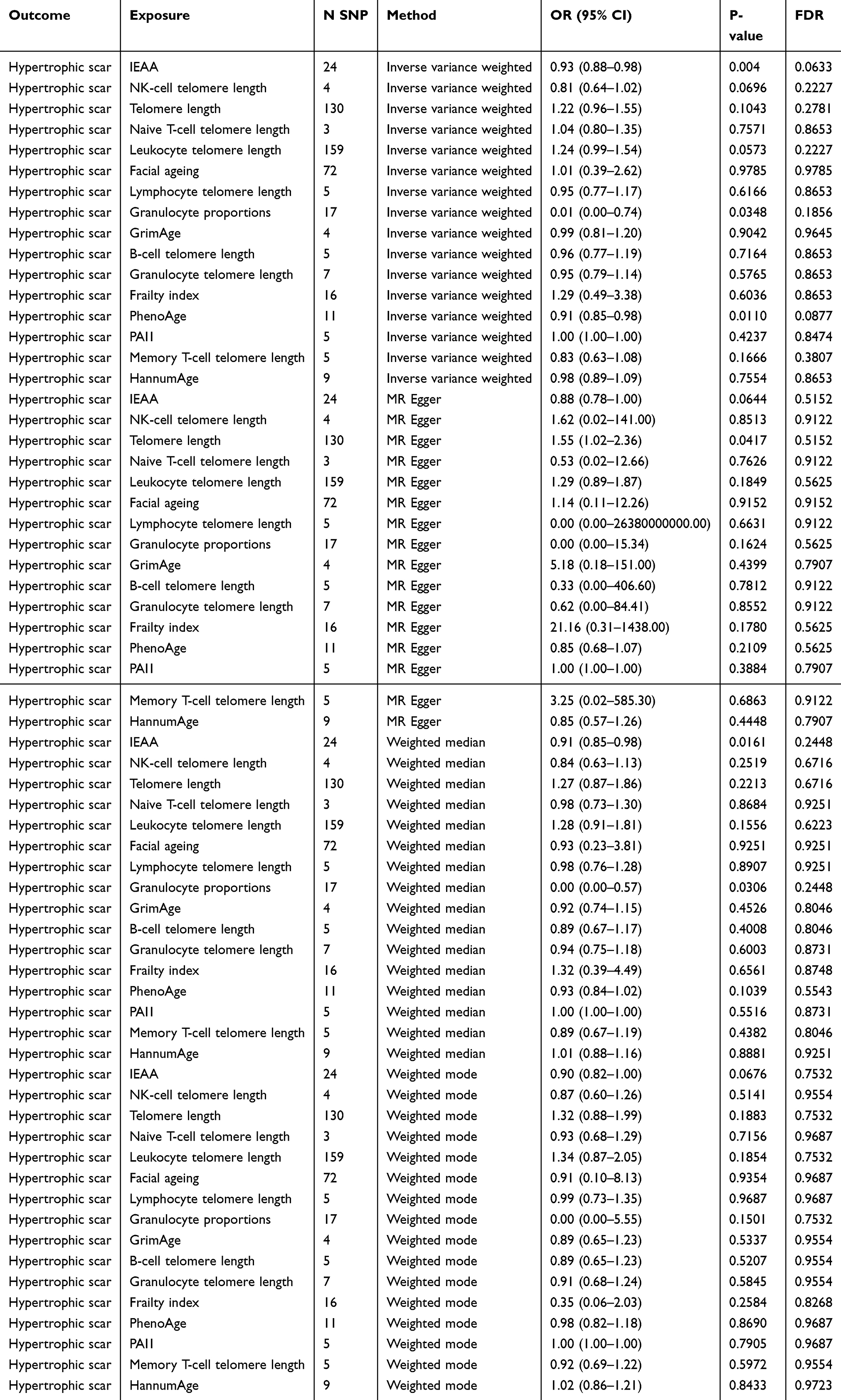 |
Table 1 Mendelian Randomization Estimates for the Causal Association of Biological Aging Indicators with Hypertrophic Scar Risk |
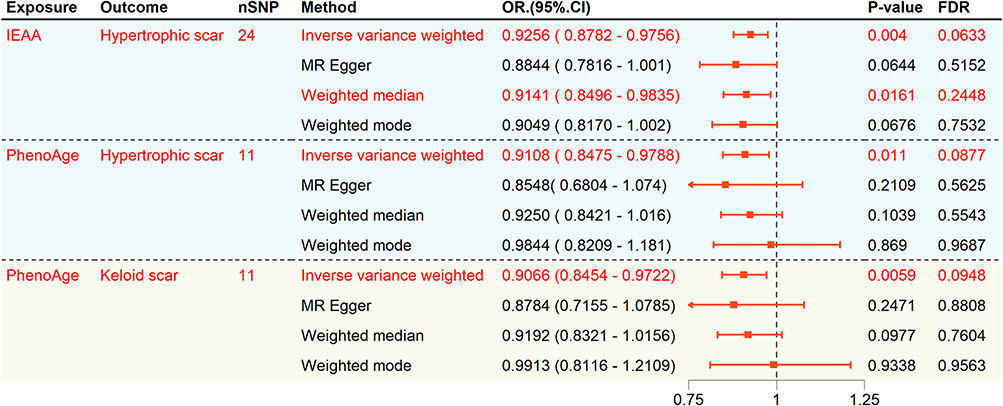 |
Figure 2 Integrated Forest Plot of MR Results. |
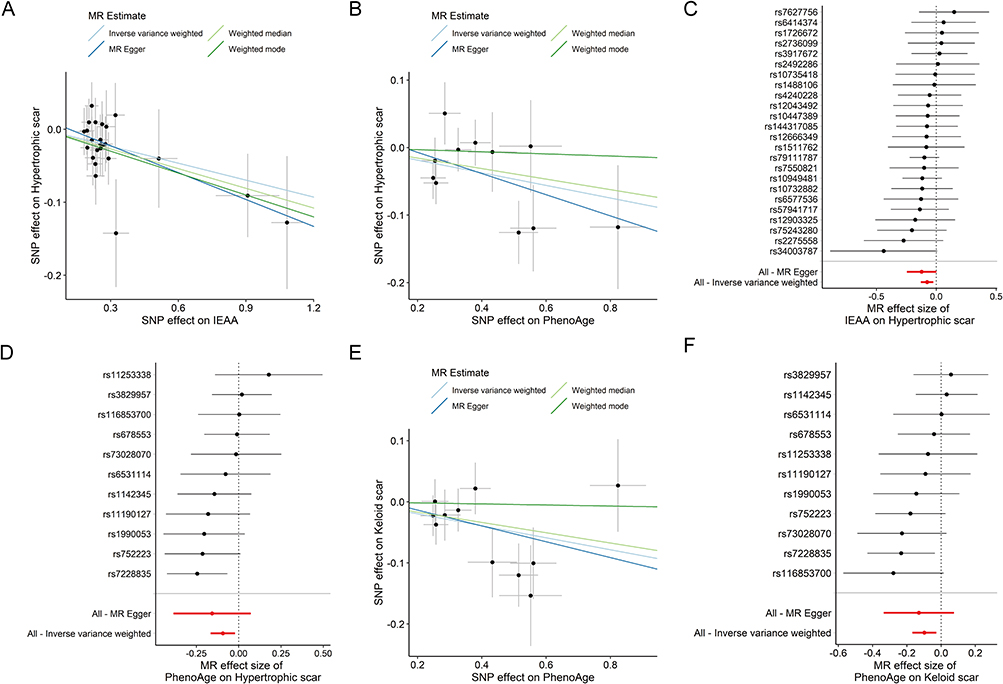 |
Figure 3 Analysis of discovery and replication cohorts. Scatter plots of Mendelian randomization estimates for discovery cohort: (A) IEAA (FinnGen), (B) PhenoAge (FinnGen); paired forest plots of SNP-level effects and overall IVW estimates for discovery: (C) PhenoAge (FinnGen), (D) IEAA (FinnGen). Replication cohort (PhenoAge, Keloid in MVP): scatter plot (E), forest plot (F). In scatter plots, each dot represents one SNP, and line slopes indicate causal estimates from multiple MR methods. In forest plots, outcomes are Odds Ratios (ORs) and 95% Confidence Intervals (CIs) for hypertrophic scars per SD increase in exposure. |
Replication in Independent Cohorts
To evaluate the reproducibility of our findings, we performed MR analyses in an independent cohort from the MVP. PhenoAge consistently demonstrated a nominally significant association with reduced risk (OR = 0.91, 95% CI: 0.85–0.97, P = 0.0059; FDR = 0.0948), mirroring the results from the discovery cohort (Figures 2, 3E and F). To assess whether these findings were specific to hyper-proliferative scarring, we performed a negative control analysis using general skin fibrosis (L90.5). No significant associations were observed for this phenotype (Table S5), suggesting that the identified biological aging markers specifically influence the pathways governing excessive scar proliferation rather than general fibrotic healing processes.
Sensitivity Analyses
Comprehensive sensitivity analyses were undertaken to assess the robustness of the MR findings. Steiger directionality tests confirmed the hypothesized direction of causality for all exposure-outcome pairs with positive associations (Table S6). Assessment of heterogeneity using Cochran’s Q test revealed no evidence of significant heterogeneity in any of the IVW analyses (all P > 0.05). Furthermore, both MR-Egger intercept tests and MR-PRESSO global tests provided no indication of significant directional or horizontal pleiotropy (all P > 0.05; Table 2 and Table 3). Leave-one-out analyses did not suggest that any single SNP disproportionately influenced the results (Figure 4A–C). Importantly, exclusion of outlier SNPs did not materially change the overall pattern of associations, further supporting the robustness of these findings (Figure 4D–F).
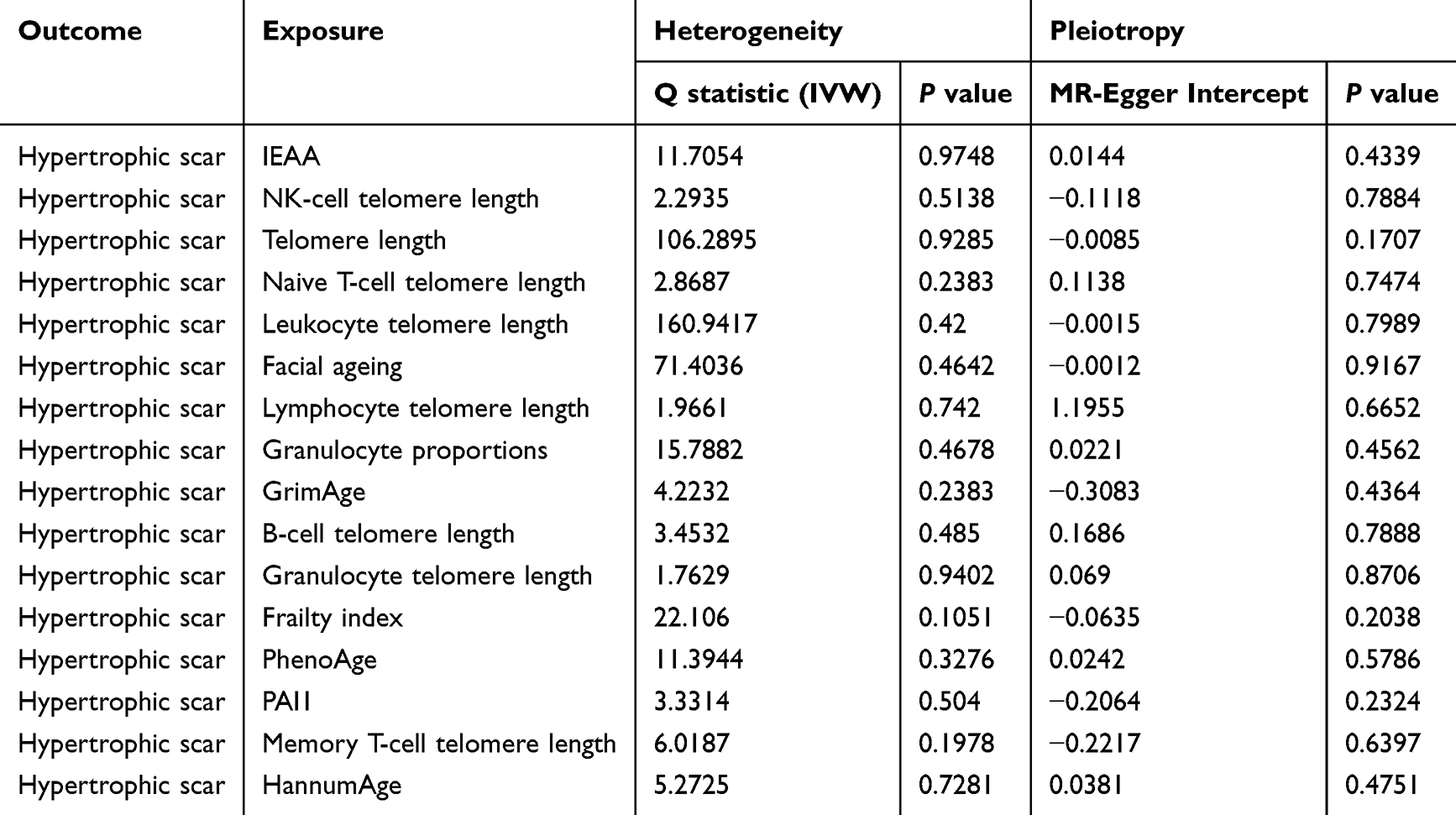 |
Table 2 Heterogeneity and Horizontal Pleiotropy Assessment for Associations with Hypertrophic Scar Risk |
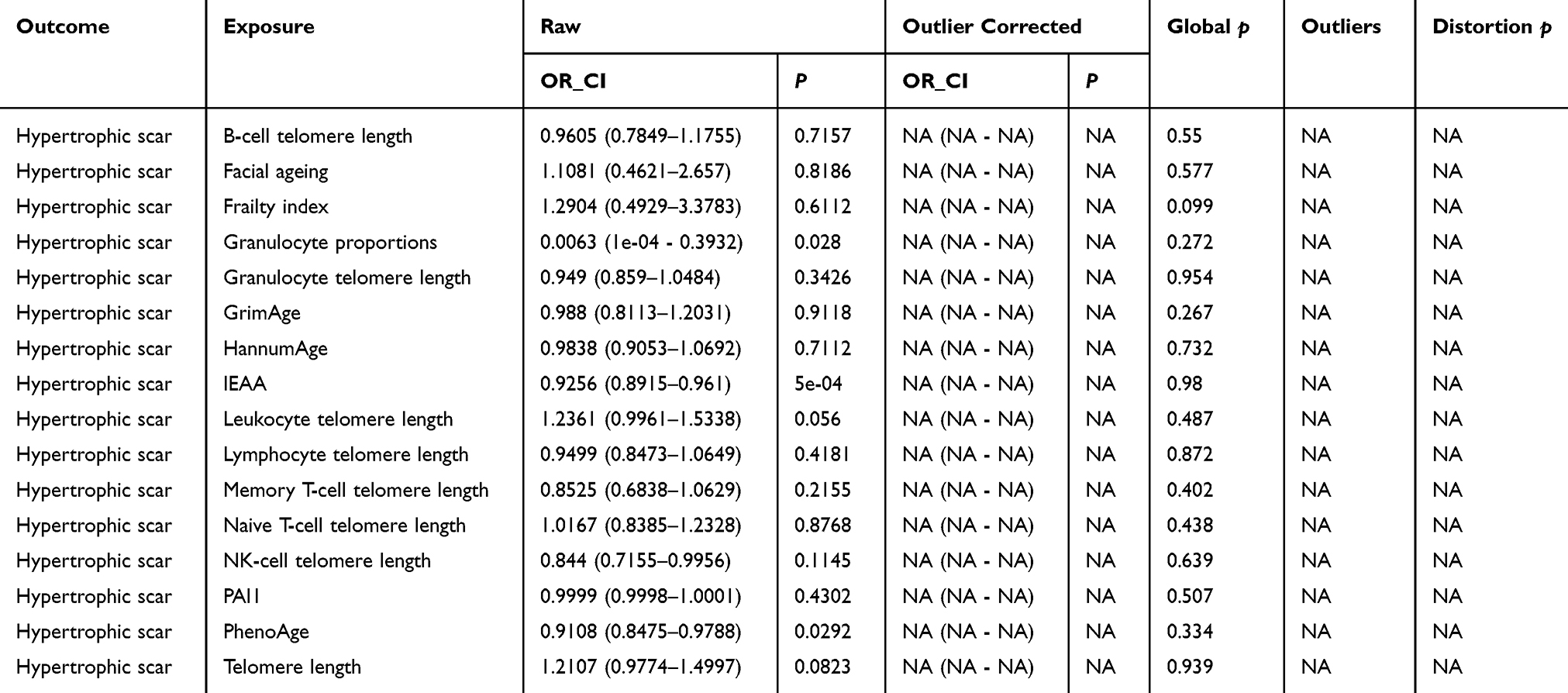 |
Table 3 MR-PRESSO Global Test for Horizontal Pleiotropy |
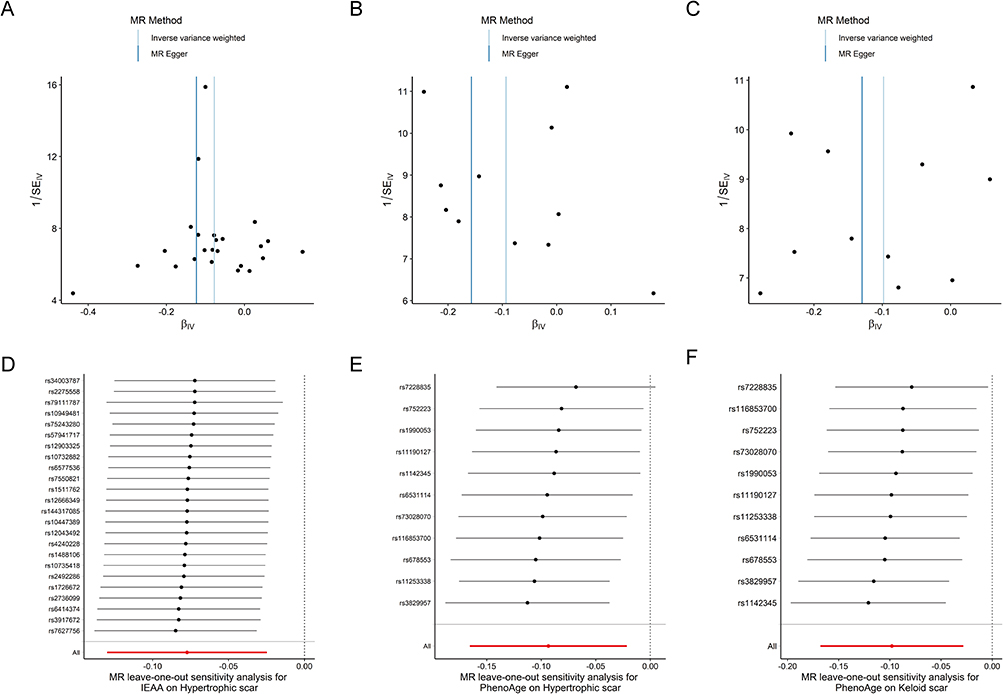 |
Figure 4 Sensitivity analyses for key associations. Funnel plots assessing potential symmetry and pleiotropy for (A) IEAA (FinnGen), (B) PhenoAge (FinnGen), and (C) PhenoAge (Replication/Keloid in MVP). Leave-one-out plots for (D) IEAA (FinnGen), (E) PhenoAge (FinnGen), and (F) PhenoAge (Replication/Keloid in MVP), demonstrating the stability of the IVW estimate upon systematic removal of individual SNPs. |
Multivariable Mendelian Randomization (MVMR) Mediation Analyses
To determine if the observed association between PhenoAge and scar risk is mediated by classical fibrogenic pathways, we performed MVMR adjusting for ten key mediators, including TGF-β1, SMAD proteins, and various MMPs. Detailed information on all SNPs included in each MVMR analysis is provided in Table S7. After instrument selection, LD clumping, and harmonization, no overlapping SNPs were identified between PhenoAge and any mediator, indicating that instrument overlap was unlikely to bias the MVMR estimates.
The protective effect of PhenoAge remained robust and statistically significant (P < 0.05) across all models, with effect sizes remaining largely unchanged after individual adjustment for each mediator (Table 4). For HS, the adjusted ORs for PhenoAge ranged from 0.906 (95% CI: 0.839–0.979) when accounting for MMP2, to 0.934 (95% CI: 0.875–0.996) when accounting for TGFBR2. Similar stability was observed in the validation cohort, with adjusted ORs ranging from 0.902 to 0.914.
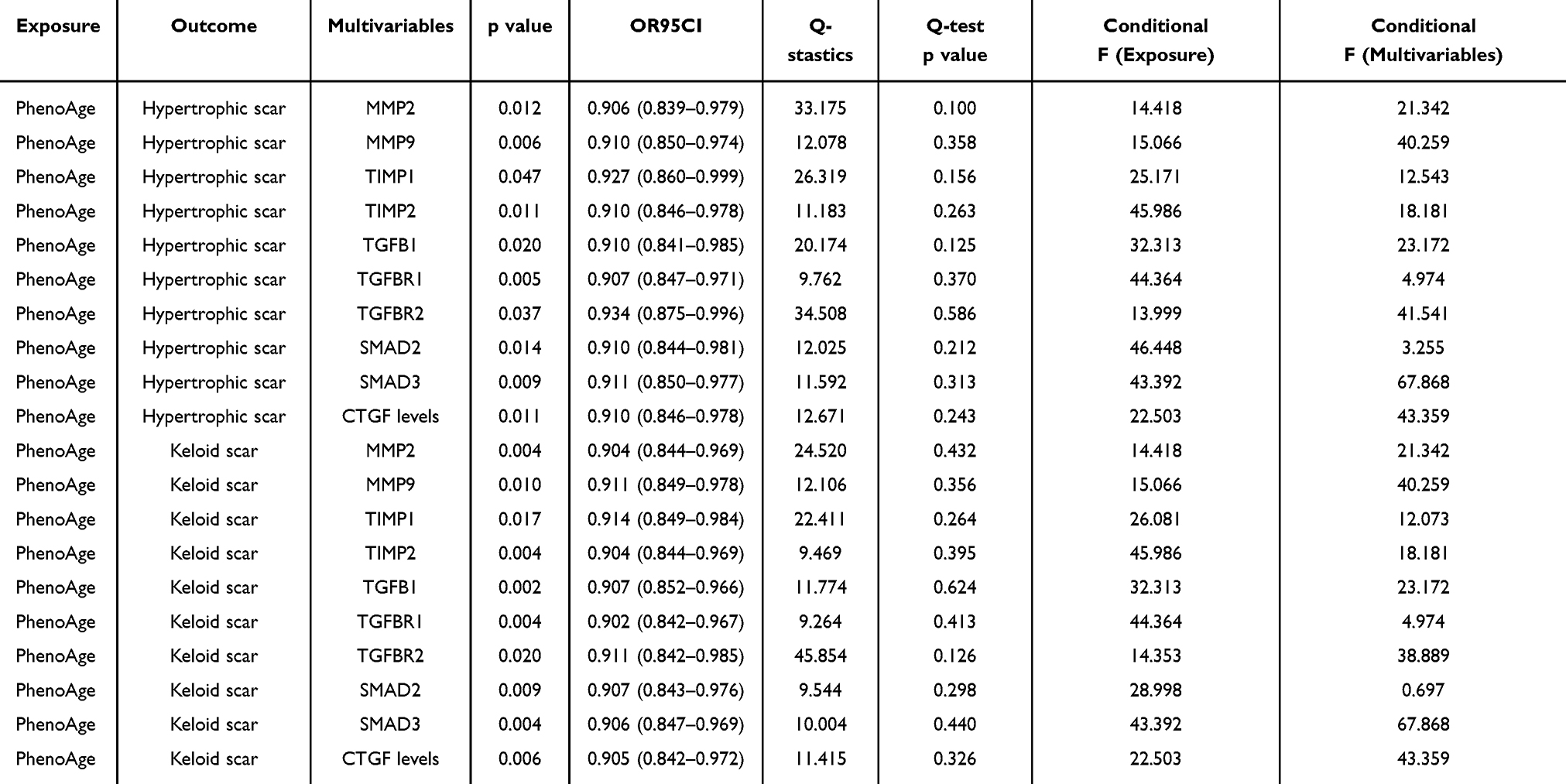 |
Table 4 Multivariable Mendelian Randomization (MVMR) Analyses of the Association Between PhenoAge and Risk of Hypertrophic Scar or Keloid Scar Adjusted for Fibrosis-Related Mediators |
To assess the validity of the MVMR models, conditional F-statistics and heterogeneity statistics were evaluated for each exposure–mediator pair (Table 4). No evidence of significant heterogeneity was observed (all Q-test P values >0.05). Most mediators showed adequate instrument strength (conditional F >10). However, TGFBR1 and SMAD2 exhibited relatively low conditional F-statistics, suggesting potential weak instrument bias due to limited independent genetic variation after accounting for PhenoAge. Therefore, the corresponding MVMR estimates should be interpreted with caution.
Discussion
In this two-sample MR study, we investigated the potential causal relationships between a comprehensive set of 16 genetically proxied biological aging indicators and the risk of HS. Our primary analyses provided nominal evidence suggesting that genetically predicted higher IEAA and higher PhenoAge are associated with a reduced risk of HS. These findings remained directionally consistent across several sensitivity analyses, which also showed no strong evidence of heterogeneity or horizontal pleiotropy. The protective trend for PhenoAge was supported by an independent replication in the MVP cohort. Furthermore, our results potentially suggest a degree of biological specificity for hyper-proliferative scarring, as no significant associations were observed in the negative control analysis for general, non-proliferative skin fibrosis. Multivariable MR analyses further hint that the observed association for PhenoAge might be independent of several canonical fibrogenic mediators. However, it is crucial to emphasize that these associations did not survive FDR correction and should be regarded as hypothesis-generating. Importantly, while our MR findings provide statistical evidence of these associations, they do not directly test biological mechanisms. In the absence of functional validation, the underlying biological pathways remain speculative and require cautious interpretation.
The observation that markers of accelerated epigenetic aging (IEAA, PhenoAge) and higher granulocyte proportions are nominally associated with a decreased risk of HS is intriguing, particularly given that HS are more prevalent and severe in younger individuals.6,7 This finding suggests a complex interplay between specific aging pathways and fibrotic processes. While “aging” is broadly associated with increased disease risk, certain aspects of cellular or systemic aging might confer protection against specific conditions like HS. Indeed, the development of pathological skin fibrosis may be driven by specific aging-related pathways rather than a global decline.38 For example, while our data suggest a protective effect of accelerated DNA methylation-based aging, other epigenetic mechanisms like N6-methyladenosine (m6A) modification have been shown to be pro-fibrotic. A recent study found that the m6A methyltransferase METTL3 promotes HS in rabbit models by enhancing autophagy-driven fibroblast differentiation, directly implicating a specific epigenetic regulator in scar pathogenesis.38 This underscores the need to investigate distinct biological aging pathways, as they may have opposing effects on HS risk.
One biologically plausible hypothesis for the observed protective associations with IEAA and PhenoAge relates to the role of cellular senescence in tissue repair and fibrosis. While chronic senescence can be detrimental, acute or targeted senescence can play beneficial roles in wound healing and limiting fibrosis.39 Senescent fibroblasts can adopt an anti-fibrotic SASP, characterized by increased secretion of MMPs (eg, MMP-1, MMP-3) that degrade excess collagen and decreased collagen synthesis.40 Specifically, an increased MMP/TIMP ratio within the SASP could shift the balance towards matrix degradation, counteracting fibrosis. Although SASP is heterogeneous and can also include pro-fibrotic factors (eg, TGF-β, CTGF), the net effect in the specific context of wound healing and HS formation might lean towards anti-fibrotic if particular SASP profiles are induced.41 Based solely on our MR causal estimates, we cannot directly verify downstream biological mechanisms; we therefore propose the speculative hypothesis that genetic predispositions leading to higher epigenetic age acceleration values might reflect putatively greater potential for, or earlier entry into, such a protective senescent state in response to wounding, thereby limiting excessive matrix deposition characteristic of HS. Furthermore, accelerated epigenetic aging, as reflected by IEAA and PhenoAge, may hypothetically influence HS risk through pathways beyond SASP. For instance, it could alter the behavior of other skin-resident cells like keratinocytes or immune cells, affecting inflammation resolution, angiogenesis, or re-epithelialization in ways that ultimately reduce excessive scarring. Changes in DNA methylation associated with these clocks could also directly modulate genes involved in fibroblast proliferation, differentiation into myofibroblasts, or extracellular matrix production.42 Indeed, epigenetic mechanisms, including DNA methylation (the basis of IEAA and PhenoAge), are deeply intertwined with cellular senescence and fibroblast behavior in scarring.14,43 For instance, global DNA hypomethylation is a feature of cellular senescence, and specific methylation changes regulate fibrotic gene expression.44 Future research is required to explore whether the genetic variants driving IEAA and PhenoAge actually influence pathways that modulate fibroblast senescence and SASP composition in the context of wound healing.
The nominal association between higher granulocyte proportions and lower HS risk also warrants particularly careful consideration. Despite a P-value of 0.0348, the exceptionally wide confidence interval (OR = 0.015, 95% CI: 0.0003–0.741) underscores the extreme imprecision of this effect estimate. Such imprecision may stem from a lower variance explained by these IVs, and it suggests that the result could be highly unstable and potentially influenced by individual IVs or sample fluctuations. Granulocytes, primarily neutrophils, are key players in the early inflammatory phase of wound healing. While excessive or prolonged inflammation can promote fibrosis, an efficient and well-regulated acute inflammatory response is crucial for proper tissue repair.45 The granulocyte proportion measured in the exposure GWAS reflects systemic levels, which are also components of epigenetic clocks like GrimAge.9 The genetic variants influencing these systemic granulocyte proportions might modulate the inflammatory milieu that ultimately curbs excessive fibrotic responses.
The lack of association for other aging markers, such as various telomere length measures, frailty index, and PAI1 suggests that not all aspects of biological aging are equally relevant to HS pathogenesis. The lack of association for PAI1, despite its inclusion in the GrimAge clock (which also showed no association with HS), might suggest that PAI1’s contribution to HS risk is minimal, or that other components of GrimAge (eg, smoking-related DNAm markers) do not collectively or individually drive HS risk in a detectable way in our study. It is also possible that PAI1’s role in HS is context-dependent and not captured by its systemic genetic proxies. It is also possible that some of these aging indicators (eg, different epigenetic clocks, PAI1, granulocyte proportions) are correlated, although our MR analyses assess each independently. Understanding the interrelations between these aging markers could provide a more holistic view in future studies. Previous observational studies on telomere length and pathological scarring have yielded mixed results,46 and our MR study does not support a causal role for genetically determined telomere length in HS. The smaller sample size for cell-specific telomere length GWAS compared to overall leukocyte telomere length GWAS might also limit power to detect associations for these specific cell types. The lack of association in our negative control analysis (L12_SCARCONDITIONS) provides preliminary evidence for the specificity of biological aging markers in hyper-proliferative scarring processes. However, this interpretation must be balanced against the possibility that the null findings stem from limited statistical power, inherent phenotype heterogeneity within the L90.5 code, or measurement differences across datasets rather than true biological specificity.
This study benefits from the MR design, which minimizes confounding and reverse causation, and utilizes large-scale GWAS data with rigorous sensitivity analyses supporting the primary findings. However, several limitations warrant careful consideration. First and foremost, our nominal associations did not survive FDR correction. This may reflect modest true effects or limited statistical power due to the size of the HS GWAS (n=2068 cases) and the multiple exposures tested, potentially making FDR correction overly stringent. Statistical uncertainty was particularly pronounced for the association with granulocyte proportions, which, though nominally significant, had an extremely wide confidence interval (OR = 0.015, 95% CI: 0.0003–0.741). Such imprecision often results from a small number of IVs and limited statistical power, undermining the robustness of the point estimate. Consequently, while this result may point to a possible association, it must be regarded as highly preliminary and interpreted with caution. A crucial limitation is the potential for undetected pleiotropy. This risk is heightened for exposures like granulocyte proportions and cell-specific telomere length, for which a relaxed IV selection threshold (P < 5×10−6) was necessary due to limited SNP availability. This strategy increases the possibility of weak instrument bias and horizontal pleiotropy, which could influence the causal estimates. Additionally, our findings are derived predominantly from individuals of European ancestry (FinnGen, MVP and UK Biobank). While this design mitigates population stratification bias, susceptibility to pathological scarring differs greatly across ethnicities and skin pigmentation groups, so results cannot be readily generalized to Asian, African or admixed populations. Furthermore, we acknowledge the potential for phenotype ambiguity. The primary outcome was based on ICD-10 code L91.0, which is commonly utilized for both HS and keloids. As these two conditions have distinct clinical behaviors despite shared fibrotic features, our results should be interpreted as applying to the broad spectrum of hyper-proliferative scarring rather than being specific to one subtype. Other inherent considerations include the lifelong nature of genetic predisposition assessed by MR, minor potential sample overlap, and the multiple testing burden. Most importantly, our study lacks direct functional or experimental validation of the identified associations. We were unable to conduct wet-lab experiments, such as CRISPR-Cas9 editing of IEAA/PhenoAge-associated genes in fibroblasts to test effects on collagen deposition, SASP markers, or cell senescence, or the use of mouse HS models to manipulate granulocyte levels and assess scar severity. Additionally, we did not perform transcriptomic or epigenomic analyses, such as DNA methylation or gene expression profiling of HS tissues, to identify overlapping pathways with genetically predicted aging markers. The absence of such functional and omics validation means that the biological mechanisms underlying our MR findings remain unconfirmed, and the causal relationships between biological aging markers (IEAA, PhenoAge, granulocyte proportions) and HS risk should be regarded as speculative.
Future research should prioritize several key directions to address these limitations and advance our understanding. First, functional validation is necessary, including in vitro experiments such as CRISPR-Cas9 editing of relevant genes in fibroblasts to assess effects on fibrogenic processes, SASP, and cellular senescence, as well as in vivo studies manipulating granulocyte levels in mouse models of HS. Second, comprehensive transcriptomic and epigenomic profiling of HS tissues should be performed to directly evaluate the enrichment of aging-related pathways and bridge the gap between genetic risk and tissue-level biology. Third, replication studies in larger and more diverse cohorts are needed to increase statistical power and improve the generalizability of findings. If these preliminary, hypothesis-generating results are validated by future functional and omics studies, they may reveal novel insights into the complex roles of biological aging and immune cell dynamics in skin fibroproliferative disorders, challenge the notion that all aspects of aging are detrimental, and ultimately contribute to more refined approaches for risk prediction and therapeutic intervention in HS.
This two-sample MR study provides preliminary, hypothesis-generating evidence that genetically predicted higher IEAA and PhenoAge may be nominally associated with a reduced risk of HS and keloids. These results expand current knowledge by identifying potential genetic links between specific biological aging markers and HS risk. However, these associations did not remain significant after correction for multiple testing, and no mechanistic conclusions can be drawn in the absence of experimental validation. The observed results may reflect specific biological pathways, but could also be influenced by phenotype heterogeneity or limited statistical power in the control groups. Future studies—including replication in larger, more diverse cohorts and direct functional investigations—are needed to determine whether these associations are robust and to clarify the underlying biological pathways.
Data Sharing Statement
All data generated or analyzed during this study are included in this article and Supplementary Information Files.
Ethical Statements
This study utilized publicly available summary-level data from genome-wide association studies (GWAS). According to Items 1 and 2 of Article 32 of the “Measures for Ethical Review of Life Science and Medical Research Involving Human Subjects” (dated February 18, 2023, China), this research is exempt from institutional ethical review as it involves the use of de-identified, publicly accessible data. All original studies from which the data were derived had obtained necessary ethical approvals and informed consent from participants.
Acknowledgments
The authors would like to express their sincere gratitude to the participants and investigators of the FinnGen project, the UK Biobank (UKB), and the VA Million Veteran Program (MVP) for their dedication and for providing the large-scale genetic summary statistics that made this research possible. We also extend our thanks to the authors and consortia of the original genome-wide association studies (GWAS) for the biological aging indicators and fibrogenic mediators for making their summary-level data publicly accessible.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agreed to be accountable for all aspects of the work.
Funding
There is no funding to report.
Disclosure
The authors declare that they have no competing interests in this work.
References
1. Frech FS, Hernandez L, Urbonas R, Zaken GA, Dreyfuss I, Nouri K. Hypertrophic scars and keloids: advances in treatment and review of established therapies. Am J Clin Dermatol. 2023;24(2):225–15. doi:10.1007/s40257-022-00744-6
2. Bock O, Schmid-Ott G, Malewski P, Mrowietz U. Quality of life of patients with keloid and hypertrophic scarring. Arch Dermatol Res. 2006;297(10):433–438. doi:10.1007/s00403-006-0651-7
3. Li Y, Sun Q, Hao L, et al. Liposomes loaded with 5-fluorouracil can improve the efficacy in pathological scars. Int J Nanomedicine. 2024;19:7353–7365. doi:10.2147/IJN.S466221
4. Barone N, Safran T, Vorstenbosch J, Davison PG, Cugno S, Murphy AM. Current advances in hypertrophic scar and keloid management. Semin Plast Surg. 2021;35(3):145–152. doi:10.1055/s-0041-1731461
5. Fang X, Wang Y, Chen H, et al. Hypertrophic scarring and keloids: epidemiology, molecular pathogenesis, and therapeutic interventions. MedComm. 2025;6(10):e70381. doi:10.1002/mco2.70381
6. Sugimoto A, Ono S, Usami S, Nitta T, Ogawa R. Older patients and patients with severe arteriosclerosis are less likely to develop keloids and hypertrophic scars after thoracic midline incision: a survey-based analysis of 328 cases. Plast Reconstr Surg. 2022;150(3):659–669. doi:10.1097/PRS.0000000000009451
7. Cho MY, Lee SG, Kim JE, Lee YS, Chang HS, Roh MR. Analysis of risk factors to predict occurrence and prognosis of postsurgical hypertrophic scar development: a review of 4238 cases. Yonsei Med J. 2023;64(11):687–691. doi:10.3349/ymj.2023.0003
8. Ferrucci L, Gonzalez-Freire M, Fabbri E, et al. Measuring biological aging in humans: a quest. Aging Cell. 2020;19(2):e13080. doi:10.1111/acel.13080
9. Gems D, Virk RS, de Magalhães JP. Epigenetic clocks and programmatic aging. Ageing Res Rev. 2024;101:102546. doi:10.1016/j.arr.2024.102546
10. McCartney DL, Min JL, Richmond RC, et al. Genome-wide association studies identify 137 genetic loci for DNA methylation biomarkers of aging. Genome Biol. 2021;22(1):194. doi:10.1186/s13059-021-02398-9
11. Bao H, Cao J, Chen M, et al. Biomarkers of aging. Sci China Life Sci. 2023;66(5):893–1066. doi:10.1007/s11427-023-2305-0
12. Lv W, Ren Y, Hou K, et al. Epigenetic modification mechanisms involved in keloid: current status and prospect. Clin Epigenetics. 2020;12(1):183. doi:10.1186/s13148-020-00981-8
13. Alghamdi MA, Wallace HJ, Melton PE, et al. Identification of differentially methylated CpG sites in fibroblasts from keloid scars. Biomedicines. 2020;8(7):181. doi:10.3390/biomedicines8070181
14. Liu Y, Xu S, Zu T, et al. Reversal of TET-mediated 5-hmC loss in hypoxic fibroblasts by ascorbic acid. Lab Invest. 2019;99(8):1193–1202. doi:10.1038/s41374-019-0235-8
15. Ilieș RF, Halmagyi SR, Cătană A, et al. Role of hTERT rs2736100 in pathological scarring. Exp Ther Med. 2022;23(4):260. doi:10.3892/etm.2022.11186
16. Dyachkova U, Vigovskiy M, Basalova N, Efimenko A, Grigorieva O. M2-macrophage-induced chronic inflammation promotes reversible mesenchymal stromal cell senescence and reduces their anti-fibrotic properties. Int J Mol Sci. 2023;24(23):17089. doi:10.3390/ijms242317089
17. Pan W, Huang Q, Zhou L, et al. Epigenetic age acceleration and risk of aortic valve stenosis: a bidirectional mendelian randomization study. Clin Epigenetics. 2024;16(1):41. doi:10.1186/s13148-024-01647-5
18. Shucheng H, Li J, Liu YL, Chen X, Jiang X. Causal relationship between gut microbiota and pathological scars: a two-sample Mendelian randomization study. Front Med. 2024;11:1405097. doi:10.3389/fmed.2024.1405097
19. Skrivankova VW, Richmond RC, Woolf BAR, et al. Strengthening the reporting of observational studies in epidemiology using mendelian randomization: the STROBE-MR statement. JAMA. 2021;326(16):1614–1621. doi:10.1001/jama.2021.18236
20. Lin Z, Xue H, Pan W. Robust multivariable Mendelian randomization based on constrained maximum likelihood. Am J Hum Gene. 2023;110(4):592–605. doi:10.1016/j.ajhg.2023.02.014
21. Roberts JD, Vittinghoff E, Lu AT, et al. Epigenetic age and the risk of incident atrial fibrillation. Circulation. 2021;144(24):1899–1911. doi:10.1161/CIRCULATIONAHA.121.056456
22. Chen Z, Chen Z, Jin X. Mendelian randomization supports causality between overweight status and accelerated aging. Aging Cell. 2023;22(8):e13899. doi:10.1111/acel.13899
23. Zhao M, He Z, Liu L, et al. Causal and mediating effects of lipid and facial aging: association study integrating GWAS, eQTL, mQTL, and pQTL data. Lipids Health Dis. 2024;23(1):342. doi:10.1186/s12944-024-02328-1
24. Codd V, Wang Q, Allara E, et al. Polygenic basis and biomedical consequences of telomere length variation. Nat Genet. 2021;53(10):1425–1433. doi:10.1038/s41588-021-00944-6
25. Andreu-Sánchez S, Aubert G, Ripoll-Cladellas A, et al. Genetic, parental and lifestyle factors influence telomere length. Commun Biol. 2022;5(1):565. doi:10.1038/s42003-022-03521-7
26. Verma A, Huffman JE, Rodriguez A, et al. Diversity and scale: genetic architecture of 2068 traits in the VA million veteran program. Science. 2024;385(6706):eadj1182. doi:10.1126/science.adj1182
27. Burgess S, Butterworth A, Thompson SG. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet Epidemiol. 2013;37(7):658–665. doi:10.1002/gepi.21758
28. Slatkin M. Linkage disequilibrium--understanding the evolutionary past and mapping the medical future. Nat Rev Genet. 2008;9(6):477–485. doi:10.1038/nrg2361
29. Sanderson E, Spiller W, Bowden J. Testing and correcting for weak and pleiotropic instruments in two-sample multivariable Mendelian randomization. Stat Med. 2021;40(25):5434–5452. doi:10.1002/sim.9133
30. Wang Y, Zheng L, Zhang L, Tai Y, Lin X, Cai Z. Roles of MMP-2 and MMP-9 and their associated molecules in the pathogenesis of keloids: a comprehensive review. Front Pharmacol. 2024;15:1444653.
31. Gudjonsson A, Gudmundsdottir V, Axelsson GT, et al. A genome-wide association study of serum proteins reveals shared loci with common diseases. Nat Commun. 2022;13(1):480. doi:10.1038/s41467-021-27850-z
32. Folkersen L, Gustafsson S, Wang Q, et al. Genomic and drug target evaluation of 90 cardiovascular proteins in 30,931 individuals. Nat Metab. 2020;2(10):1135–1148. doi:10.1038/s42255-020-00287-2
33. Loya H, Kalantzis G, Cooper F, Palamara PF. A scalable variational inference approach for increased mixed-model association power. Nat Genet. 2025;57(2):461–468. doi:10.1038/s41588-024-02044-7
34. Hagag MM, Bayomy NR, El-Horish DBM. Study of vitamin D and calcium levels in patient with rosacea. Egypt J Hosp Med. 2021;84(1):2405–2410. doi:10.21608/ejhm.2021.184656
35. Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol. 2015;44(2):512–525. doi:10.1093/ije/dyv080
36. Bowden J, Del Greco MF, Minelli C, et al. Improving the accuracy of two-sample summary-data mendelian randomization: moving beyond the NOME assumption. Int J Epidemiol. 2019;48(3):728–742. doi:10.1093/ije/dyy258
37. Nolte IM. Metasubtract: an R-package to analytically produce leave-one-out meta-analysis GWAS summary statistics. Bioinformatics. 2020;36(16):4521–4522. doi:10.1093/bioinformatics/btaa570
38. Tu L, Gu S, Xu R, et al. ALKBH3-mediated M(1)A demethylation of METTL3 endows pathological fibrosis: interplay between M(1)A and M(6)A RNA methylation. Adv Sci. 2025;12(19):e2417067. doi:10.1002/advs.202417067
39. O’Reilly S, Markiewicz E, Idowu OC. Aging, senescence, and cutaneous wound healing-a complex relationship. Front Immunol. 2024;15:1429716. doi:10.3389/fimmu.2024.1429716
40. Wang WJ, Chen XM, Cai GY. Cellular senescence and the senescence-associated secretory phenotype: potential therapeutic targets for renal fibrosis. Exp Gerontol. 2021;151:111403. doi:10.1016/j.exger.2021.111403
41. Han X, Lei Q, Xie J, et al. Potential regulators of the senescence-associated secretory phenotype during senescence and aging. J Gerontol. 2022;77(11):2207–2218. doi:10.1093/gerona/glac097
42. Berlanga-Acosta J, Garcia-Ojalvo A, Guillen-Nieto G, Ayala-Avila M. Endogenous biological drivers in diabetic lower limb wounds recurrence: hypothetical reflections. Int J Mol Sci. 2023;24(12):10170. doi:10.3390/ijms241210170
43. Amjadian S, Moradi S, Mohammadi P. The emerging therapeutic targets for scar management: genetic and epigenetic landscapes. Skin Pharmacol Physiol. 2022;35(5):247–265. doi:10.1159/000524990
44. Xue T, Qiu X, Liu H, et al. Epigenetic regulation in fibrosis progress. Pharmacol Res. 2021;173:105910. doi:10.1016/j.phrs.2021.105910
45. Soliman AM, Barreda DR. Acute Inflammation in Tissue Healing. Int J Mol Sci. 2022;24(1). doi:10.3390/ijms24010641
46. De Felice B, Wilson RR, Nacca M. Telomere shortening may be associated with human keloids. BMC Med Genet. 2009;10:110. doi:10.1186/1471-2350-10-110
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Comment on “Multi-Omics Association Analysis of Mitochondrial Genes in Hypertrophic Scars: Application of Mendelian Randomization” [Letter]
Deng X, Zhao Z, Zhang Z
Journal of Inflammation Research 2026, 19:594248
Published Date: 5 February 2026