Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 13
Genetic Testing for BCHE Variants Identifies Patients at Risk of Prolonged Neuromuscular Blockade in Response to Succinylcholine
Authors Zhu G ![]() , Dawson E, Huskey A, Gordon RJ, Del Tredici AL
, Dawson E, Huskey A, Gordon RJ, Del Tredici AL ![]()
Received 30 June 2020
Accepted for publication 5 September 2020
Published 30 September 2020 Volume 2020:13 Pages 405—414
DOI https://doi.org/10.2147/PGPM.S263741
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Guang-dan Zhu,1 Eric Dawson,1 Angela Huskey,1 Ronald J Gordon,2 Andria L Del Tredici1
1Millennium Health, LLC, San Diego, CA, USA; 2Department of Anesthesiology, University of California, UC San Diego School of Medicine, La Jolla, CA, USA
Correspondence:Andria L Del Tredici
Millennium Health, LLC, 16981 Via Tazon, San Diego, CA 92127, USA
Tel +1 858 217 1175
Fax +1 858 451 3636
Email [email protected]
Background: Genetic variants in the BCHE (butyrylcholinesterase) gene are associated with reduced BChE enzyme activity and prolonged post-succinylcholine neuromuscular blockade, which can lead to postanesthetic apnea and respiratory depression. Testing for BChE deficiency is usually performed by biochemical methods and is generally only offered to patients who have a personal or family history of prolonged post-succinylcholine neuromuscular blockade.
Purpose: Using a clinical test, we investigated the frequencies of BCHE genotypes that are associated with increased risk for prolonged post-succinylcholine neuromuscular blockade.
Materials and Methods: Five BCHE variants, including the A (atypical, rs1799807), K (Kalow, rs1803274), F1 (fluoride-1, rs28933389), F2 (fluoride-2, rs28933390), and S1 (silent-1, rs398124632), were genotyped in a large (n = 13,301), multi-ethnic cohort in the United States. Subjects were recipients of pharmacogenetic testing ordered by their physicians as part of routine care.
Results: The minor allele frequencies of A, K, F1, F2, and S1 were 1.60%, 19.93%, 0.08%, 0.47%, and 0.04%, respectively, in this cohort. Based on a review of biochemical and clinical data of these variants, we grouped BCHE genotypes into four phenotypic categories to stratify the risk for prolonged post-succinylcholine neuromuscular blockade. Approximately 0.06% of patients were predicted to have severe BChE deficiency, 8% were predicted to have moderate BChE deficiency, and 29% were predicted to have mild BChE deficiency. Compared to other ethnic groups, Caucasians were predicted to have the highest frequency of BChE deficiency.
Conclusion: While severe BChE deficiency is rare in the United States, approximately 8% of Americans are at moderate risk of prolonged post-succinylcholine neuromuscular blockade, suggesting that a sizable percentage of patients may benefit from preoperative genetic testing of BCHE.
Keywords: succinylcholine, BCHE, genotyping, prolonged neuromuscular blockade, pharmacogenetics
Introduction
Succinylcholine is a neuromuscular blocking agent listed on the World Health Organization’s Model List of Essential Medicines.1 Because of its rapid onset and short duration of action, it is frequently used to facilitate rapid sequence intubation.2 However, succinylcholine is sometimes associated with prolonged neuromuscular blockade, which can lead to postanesthetic apnea and possibly respiratory depression and death. Prolonged post-succinylcholine apnea is estimated to occur in approximately 1 in 1800 anesthesias,3 and as such is more common than malignant hyperthermia.4 Moreover, affected patients may become aware of paralysis during emergence from anesthesia, leading to distress consistent with post-traumatic stress disorder.5,6 Prolonged neuromuscular blockade can also lead to increased post-operative recovery time and costs.7
The BCHE gene encodes butyrylcholinesterase (BChE; also known as plasma cholinesterase or pseudocholinesterase), which metabolizes succinylcholine. At least 75 genetic variants of BCHE have been identified.8 Genetic variants in BCHE have been shown to cause deficiency of BChE enzyme activity, leading to higher-than-expected plasma levels of succinylcholine, and, thus, prolonged neuromuscular blockade. Patients with these genetic variants are usually asymptomatic until they are exposed to succinylcholine as part of a surgical procedure.9 The most well-known BCHE variant is the atypical variant (the “A” variant). The FDA-approved label for succinylcholine recommends that the medication should be used carefully in patients with reduced BChE activity and that it should be used with caution, if at all, in patients homozygous for the atypical variant.10
Genetic testing for BCHE variants, however, is not common practice. Currently, patients who have a personal or family history of prolonged post-succinylcholine neuromuscular blockade may be offered biochemical testing for BChE deficiency.11 Such biochemical tests are used to assign BCHE genotypes indirectly. In one study of 6688 patients, however, biochemical tests were unable to assign a genotype for 16.3% individuals.12
DNA-based methods determine genotypes directly and thus are inherently more accurate than biochemical methods in identifying genetic variants.11 However, studies of BCHE genetic variants have been mostly in small cohorts and data are lacking for their frequencies in ethnic populations in the US. Frequency information of BCHE genetic variation in the general population is an important factor in determining whether BCHE genotyping should be broadly used as a screening tool in patients being scheduled for procedures requiring succinylcholine. In this study, we report the development of a PCR-based 5-variant BCHE genotyping panel and the frequency of these genetic variants in a large, multi-ethnic, US cohort that was unselected for succinylcholine response.
Materials and Methods
A retrospective analysis was conducted utilizing data from 13,301 patients receiving pharmacogenetic testing from Millennium Health, LLC (San Diego, CA). Pharmacogenetic testing was ordered by authorized healthcare providers as part of routine care using a requisition form in which the patient ethnicity could be selected from the following categories: African-American, Asian, Caucasian, Hispanic, or “Other”. Patients who had more than one ethnicity selected were also considered as “Other”. This study was conducted in accordance with the principles expressed in the Declaration of Helsinki and under a research protocol approved by Aspire Independent Review Board (Santee, CA), which includes a waiver of written informed patient consent for use of de-identified data. This was a retrospective data collection study and the manuscript adheres to the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) Statement (http://www.strobe-statement.org).
Sample Preparation and DNA Genotyping
Patient samples were collected with oral swabs (OCD-100, DNA Genotek, Ottawa, Ontario, Canada) and DNA was extracted using Chemagic DNA Saliva Kit (Perkin Elmer, Waltham, MA). BCHE variants (Figure 1) were detected using TaqMan® chemistry-based qPCR using standard primer and probe design techniques. TaqMan® primers and probes (Table 1) were manufactured by Thermo Fisher Scientific (Waltham, Massachusetts, US). The study laboratory is certified under the Clinical Laboratory Improvement Amendments of 1988 (CLIA) and is accredited by the College of American Pathologists.
 |
Table 1 Primers and Probes Used in This Studya |
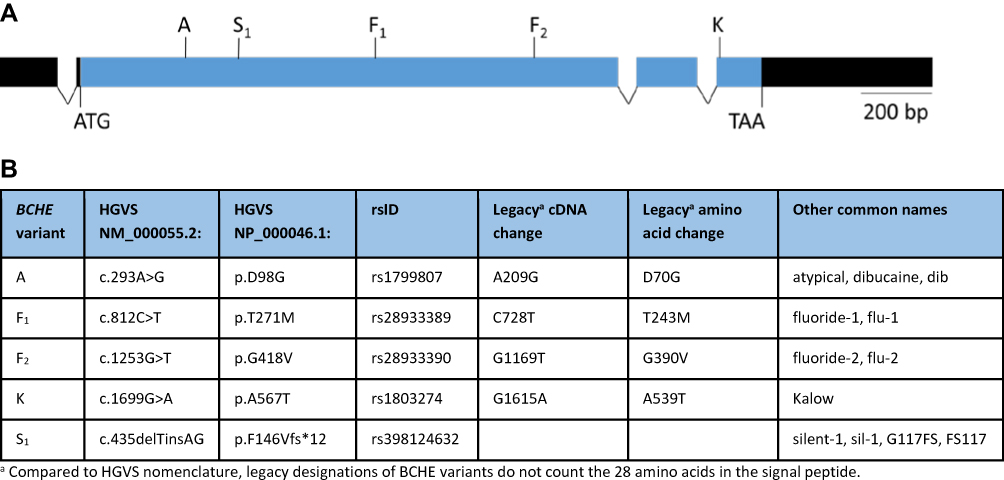 |
Figure 1 Functional BCHE variants and location. (A) Schematic diagram of the human BCHE gene. The coding region is shown in blue. Introns are not drawn to scale. Untranslated regions are shown in black. Sites of the five tested variants are shown above the exons. (B) Designations of the five tested variants according to various nomenclature systems. |
BCHE genotype designations used in this study list all variants which were detected. For example, the genotype AKK is used to identify an individual who has one copy of the A variant and two copies of the K variant. In this case, one BCHE allele contains both A and K, while the other allele contains K only. However, the PCR-based assay is not able to determine the phase of detected variants, and as such, for some genotypes it is not possible to determine which variants are on the same allele. For example, the genotype AK could have both A and K variants on the same allele, or one allele with the A variant and the other with the K variant.
1000 Genomes Data
Population genetics data for BCHE variants were extracted from the 1000 Genomes Project Phase 3 database (http://www.internationalgenome.org).
Statistical Analysis
Data analysis was performed using Microsoft Excel and R (R Foundation for Statistical Computing) version 3.6.1. The R package “HardyWeinberg” version 1.6.3 was used to perform two-sided Fisher’s exact tests for Hardy-Weinberg equilibrium (HWE). The R package “genetics” version 1.3.8.1.2 was used to calculate pairwise linkage disequilibrium (LD) estimates, including the Chi-squared p-value.
Results
Development of a Clinical Test for BCHE Genotyping
In order to develop a BCHE genotyping test, we selected 5 variants for which PCR-based testing had been shown to have a 85% detection rate for patients with prolonged post-succinylcholine apnea attributable to BCHE variation.13 These 5 variants are located at different areas of the coding region (Figure 1), and have been shown to be associated with reduced enzyme activity.9 While the S1 variant is a frameshift mutation resulting in no enzyme activity,8 the A and F variants are associated with a severe, 60–70% decrease in enzyme activity.9 In contrast, the K variant is associated with a milder, 30% decrease in enzyme activity compared to wild-type.14
Among the four missense variants (A, F1, F2, and K), the A and F variants are considered “qualitative” variants because they impair catalytic properties of the enzyme, while the K variant is “quantitative” because it affects the plasma BChE concentration and does not qualitatively change the encoded enzyme.15,16 The K variant is in LD with variants in noncoding regions of the gene, which may cause reduced BChE expression.14,17,18
BChE Phenotype Assignment
Translation of genotype to phenotype is recommended practice for implementation of pharmacogenetic testing, since clinical interpretations are provided based on phenotype.19 To translate BCHE genotypes to actionable phenotypes, we reviewed published associations of genotypes with both BChE enzyme activity and duration of succinylcholine-induced paralysis (Supplementary Tables 1 and 2, and references therein). These included studies in which genotypes were assigned indirectly based on biochemical results as well as DNA-based genotyping studies.
Based on literature review, the A, F, and S alleles were associated with severely reduced function, while the K allele was associated with mildly reduced function (Supplementary Table 1). Moreover, allele dosage analysis indicated that having more than one copy of these reduced function alleles had an additive effect. For example, individuals with two copies of the A allele (AA) had more reduction in enzyme activity compared to individuals with one copy (A) (References 2, 4, and 5 in Supplementary Table 1). Similarly, individuals with two copies of the K allele had more reduction in enzyme activity compared to individuals with one copy (References 1, 5, 6, and 7 in Supplementary Table 1). When more than one type of variant was present, the functional impact also appeared additive. For example, an individual with the AF or AS genotype had less enzyme activity than an individual with only one copy of A (Reference 2 in Supplementary Table 1).
We also reviewed in vivo impact of BCHE variants on neuromuscular blockade induced by succinylcholine or mivacurium, a neuromuscular blocking agent also metabolized by BChE (Supplementary Table 2). The A, F, and S variants were associated with longer paralysis compared to the K variant, consistent with their relative impact on enzyme activity. The AA genotype, which has a warning on the FDA drug label,10 was associated with longer paralysis than No Variants Detected (NVD), but a single copy of A (the A genotype) was also associated with longer paralysis, albeit intermediate between AA and NVD. In addition, AF and FS genotypes had more blockade compared to the A genotype, and AS compound heterozygotes had similar effects as AA and SS homozygotes. In patients screened for the A and K variants, those who had one copy of the K variant had slightly longer paralysis than those who had no detected variants (References 6 in Supplementary Table 2).
Based on the biochemical and clinical data, a strategy was developed to assign BChE phenotypes based on genotype, and also to provide clinical interpretation on the laboratory test report for each phenotype (Table 2). Since BChE deficiency is a recognized genetic condition,20 we used it as the basis for the phenotype nomenclature. The phenotype assignments were: severe BChE deficiency, moderate BChE deficiency, mild BChE deficiency and normal BChE activity (wild-type).
 |
Table 2 Assignment of BCHE Genotypes to Phenotypes |
Homozygotes of the A variant (AA), which were identified in the warning on the succinylcholine label, were predicted to have severe BChE deficiency. Since A, F1, F2, and S1 variants were all associated with severely reduced function, severe BChE deficiency was also assigned when two or more A, F1, F2, or S1 variants were detected. Patients with one copy of the A, F1, F2, or S1 variants were assigned to moderate BChE deficiency, consistent with lower impact of a single copy of these variants compared to two or more copies. Because the K variant confers only a mild reduction in enzyme activity, it had a different impact on phenotype assignment than the other four variants. Patients with one copy of the K variant and no other variants were assigned to mild BChE deficiency. While these individuals would likely have nearly normal enzyme activity, they would be at risk of prolonged neuromuscular blockade if other risk factors leading to BChE deficiency (such as severe burns, malnutrition, renal failure, malignancy, and/or pregnancy) were present.9 The K variant did not change phenotype assignment if the patient was also positive for A, F1, F2, or S1. Individuals with two copies of K, when negative for other variants, were assigned to moderate BChE deficiency. Finally, individuals with no reduced function variants detected were assigned to normal BChE activity.
Frequency of BCHE Genotypes
We conducted a retrospective analysis of BCHE genotyping results in our laboratory database which contains test results from all 50 states in the United States.21 A total of 13,301 patients (2969 males, 4552 females, and 5780 of unknown gender) were tested for the five BCHE variants. Within this cohort, there were 893 African-Americans (7%), 58 Asians (0.4%), 6028 Caucasians (45%), and 300 Hispanics (2%) (Table 3). In addition, 251 patients (2%) had an ethnicity categorized as “Other” and 5771 patients (43%) did not have any ethnicity information.
 |
Table 3 Frequencies of BCHE Genotypes in Different Ethnic Groups in the United States |
Most patients (>60%) had no detected variants (Table 3). The next three most common genotypes were K, KK and AK. The highest number of variants in any tested individual was 4, which was found in 5 patients with the AAKK genotype. Approximately 5% of Caucasians were positive for A, F1, F2, and/or S1, while in African-Americans and Hispanics this percentage was lower at 0.90% and 3.33%, respectively. None of these four variants were detected in Asians in this study, although the number of Asian patients was low. Among these four variants, homozygosity and compound heterozygosity were rare. Homozygosity was detected only for the A variant in 5 patients, while compound heterozygotes were detected in 3 patients for A and F1, A and F2, and F1 and F2, respectively. No patient had 3 or more of these 4 variants.
For the K variant, we had sufficient data to evaluate HWE in specific ethnic populations; deviation from HWE can indicate selection bias or genotyping errors. The K variant was in HWE in African-American (p = 0.06), Asian (p = 0.33), Caucasian (p = 0.10), and Hispanic (p = 0.22) patients.
We were also able to evaluate LD for the K and A variants. These two variants were in LD in Caucasian (D′ = 0.834, r2 = 0.052, p < 2 x 10−16) and Hispanic (D′ = 0.544, r2 = 0.019, p = 7.6 x 10−4) patients, but significant LD of these two variants was not identified in African-Americans (p = 0.18). In Caucasians, K was also found to have LD with F2 (D′ = 0.635, r2 = 0.0006, p = 0.008).
The minor allele frequency (MAF) of each tested variant is listed in Table 4. The K variant was by far the most common variant in all 4 ethnic groups, while the S1 variant was the least frequently found. Of the remaining variants, the A allele was the most frequently found, and F2 was more frequently found than F1. Caucasians had the highest frequencies of A, F2, and K, while F1 and S1 were not detected in African-Americans. For patients with unknown ethnicity, the MAFs were most similar to Caucasians (data not shown).
 |
Table 4 Minor Allele Frequencies of BCHE Variants in Different Ethnic Groups |
We compared the MAFs to publicly available data. The MAFs of the tested variants from this study were largely consistent with the 1000 Genomes Project data for similar ethnic populations. Even though our study population had fewer Asians, the observed frequencies were similar to those of the East Asian cohort of 1000 Genomes. We observed S1 in Caucasian and Hispanic patients, but it was not found in smaller 1000 Genomes samples of similar ethnicities. We also detected A and F2 in African-Americans and F1 in Hispanics; these variants were also not found in smaller 1000 Genomes samples of similar ethnicities.
Frequency of Predicted BChE Phenotypes
We then analyzed the frequency of predicted BChE phenotypes (Table 5). Out of 13,301 patients, 8 (0.06%) were predicted to have severe BChE deficiency. Of these, 1 was Caucasian and 7 were of unknown ethnicity. Approximately 8% of patients were predicted to have moderate BChE deficiency, with highest frequency found in Caucasians, followed by Hispanics, African-Americans, and Asians. Finally, almost 30% of patients were predicted to have mild BChE deficiency. In total, approximately 40% of patients were predicted to have mild, moderate or severe BChE deficiency.
 |
Table 5 Frequencies of Predicted BChE Phenotype Categories in Different Ethnic Groups |
Discussion
In this study, we developed a BCHE genetic test to identify patients at risk of prolonged post-succinylcholine neuromuscular blockade. While the succinylcholine label indicates caution specifically for patients homozygous for the A variant,10 our test included the A variant and 4 additional variants. In our five-variant test, we identified 8 patients that were likely to have severe BChE deficiency. Of those, 3 patients would not have been identified by a test exclusively targeting homozygotes of the A variant.
We used our test to determine the frequency of BCHE variants in a cohort of 13,301 patients of different ethnicities from all 50 states. Unlike previously published studies of patients who had personal or family history of prolonged post-succinylcholine neuromuscular blockade, this study evaluated the frequency of BCHE variants in patients receiving pharmacogenetic testing who may or may not have been administered succinylcholine, much less experienced any adverse effects in response to the medication. As such, the observed frequencies may be representative of the general US population. In our cohort, the K variant was the most frequently found variant, followed by A, then F2, F1 and S1 variants. The observed frequencies of A, F2 and K variants in our Caucasian patients were similar to those reported in smaller Caucasian cohorts from other countries.22–25 Of the five tested variants, the S1 variant had the lowest MAF (0.0004) in our cohort and our study represents the first reliable frequency estimate for this variant.
We found BCHE reduced function variants in all four major ethnicities in the US. Indeed, this is the first study reporting BCHE variant frequencies in African-Americans and Hispanics. Among the 4 ethnic groups, Caucasians were found to have the highest frequency of individuals who were positive for at least one variant with severely reduced function (A, F1, F2 and/or S1). Accordingly, Caucasians had the highest frequency of predicted moderate and severe BChE deficiency, followed by Hispanics. It is tempting to speculate that African-Americans and Asians may have lower risk of prolonged neuromuscular blockade by succinylcholine. However, it is also possible that other genetic variants may be important in determining BChE activity in these ethnic groups. Further studies are needed.
In contrast to the current definition of BChE deficiency as a single autosomal recessive disorder,20 we defined three levels of BChE deficiency that classify patients into risk categories. Patients with genotypes containing ≥2 severely reduced function variants were classified at the highest risk category, severe BChE deficiency, for which a clinician could consider avoiding succinylcholine. Moderate BChE deficiency was defined for genotypes containing 1 severely reduced function variant. Published evidence indicates that patients with moderate BChE deficiency are also at risk of prolonged succinylcholine-induced neuromuscular blockade, and succinylcholine may be used cautiously. Finally, mild BChE deficiency phenotype allowed us to identify patients that are at low risk. For patients with mild and moderate BChE deficiency, the presence of additional risk factors could lead a clinician to consider avoiding succinylcholine. Some common non-genetic risk factors include pregnancy, cancer, kidney disease, and liver disease.9
The BChE deficiency phenotype categories are also associated with the extent of prolonged neuromuscular blockade by succinylcholine. Our literature review indicates that while individuals with severe BChE deficiency are more likely to have dramatically prolonged blockade, individuals with moderate and mild BChE deficiency may still exhibit longer blockade compared to individuals who do not have BCHE reduced function variants. For shorter procedures such as electroconvulsive therapy, a small increase in succinylcholine action may be clinically significant.26 In such scenarios, patients with mild and moderate BChE deficiency could be more sensitive to succinylcholine.
Using BCHE genetic testing to identify patients at risk of prolonged post-succinylcholine neuromuscular blockade may allow the clinician to employ alternative neuromuscular blocking agents. For example, rocuronium is an alternative that is not metabolized by BChE and whose neuromuscular blockade can be reversed by sugammadex.27 However, rocuronium may not be ideal for all patients since it is not as short-acting as succinylcholine and may be associated with hepatotoxicity.28 In addition, the use of rocuronium in general anesthesia in obstetrics has been associated with adverse outcomes for neonates compared to succinylcholine.29 Furthermore, reversal of block by sugammadex does not always succeed in myasthenic patients.30
Currently, biochemical BChE testing is only performed in patients who are undergoing surgery and have a personal or family history of prolonged post-succinylcholine neuromuscular blockade. The high frequency of patients predicted to have moderate or severe BChE deficiency suggests that genetic testing may be feasible for all patients who are undergoing surgery. A three to five-day turnaround time from specimen receipt to test results may allow for genetic testing to be performed after surgery is scheduled but before the surgery takes place. Genetic testing could also be incorporated into newer care models such as the perioperative surgical home.31 For emergency situations, a strategy of pre-emptive pharmacogenetic testing with test results available in electronic medical records would be ideal.32 Moreover, a BChE testing strategy that combines enzymatic and genetic testing may be more predictive.11
The use of genetic test results to inform therapy in surgical patients is not a new idea. Testing for Factor V Leiden and Factor II G20210A is suggested as part of an assessment for venous thromboembolism risk, and carriers of these variants may be candidates for prophylaxis.33 There are also many pharmacogenetic associations with perioperative medications in addition to BCHE and succinylcholine response.34,35 For example, variants in RYR1 and CACNA1S are associated with increased risk of malignant hyperthermia with succinylcholine and potent volatile anesthetic agents.4 Response to opioids, which are frequently used during general anesthesia and for postoperative pain management, may be impacted by variation in cytochrome P450 metabolism genes such as CYP2D6 and CYP3A4 and pharmacodynamic genes such as OPRM1 and COMT.36 Testing for BCHE and other pharmacogenes could be performed with a single oral sample prior to surgery. This non-invasive, multigenic testing approach has the potential to reduce adverse events, improve therapeutic decision making, and decrease costs associated with perioperative care.34
There are several limitations to our study. 1) The number of Asian participants was small. However, our variant frequencies in Asians are consistent with those from the larger East Asian group in the 1000 Genomes Project (Table 4). 2) Our PCR-based test could not determine the phase of two variants when the patient was heterozygous for both. 3) Our genotype-phenotype strategy is limited by the current knowledge of BCHE variants and could be improved as more data become available. 4) Since the study was designed to assess genetic variant frequencies in a population unselected for succinylcholine response, this study did not include biochemically measured BChE activity or clinical response to succinylcholine such as duration of paralysis. 5) BChE activity can be affected by genetic and non-genetic factors that were not included in this study.9,10,14 For example, we did not test BCHE variants in noncoding regions, which may have functional impact.18
In conclusion, we have developed a clinical BCHE genotyping test and found that approximately 8% of patients in the US have genotypes associated with moderate or severe BChE deficiency and are at increased risk for prolonged post-succinylcholine neuromuscular blockade. BCHE genetic testing is feasible, as part of a pre-operative risk assessment, to identify patients who could be candidates for alternative neuromuscular blockers. Indeed, BCHE could be tested with other pharmacogenes that are associated with perioperative medication response. Future studies of the impact of BCHE genotyping on clinical care and patient outcomes are warranted.
Abbreviations
BCHE, butyrylcholinesterase; FDA, Food and Drug Administration; HGVS, Human Genome Variation Society; HWE, Hardy–Weinberg equilibrium; LD, linkage disequilibrium; MAF, minor allele frequency; PCR, polymerase chain reaction; qPCR, quantitative polymerase chain reaction.
Acknowledgments
The authors would like to thank Hua Fang, Bae Riley, Frank Espin, and Tanya Moreno for their help in development and validation of the BCHE genotyping test. This study was supported by Millennium Health, LLC.
Disclosure
Drs Zhu, Dawson, Huskey, and Del Tredici are employees of Millennium Health, LLC, San Diego, California. Dr Gordon is a consultant of Millennium Health and Kailos Genetics. The authors report no other conflicts of interest in this work.
References
1. World Health Organization. Model List of Essential Medicines, 21st List, 2019. Geneva: World Health Organization; 2019. Licence: CC BY-NC-SA 3.0 IGO. Available from: https://apps.who.int/iris/bitstream/handle/10665/325771/WHO-MVP-EMP-IAU-2019.06-eng.pdf?ua=1.
2. April MD, Arana A, Pallin DJ, et al. Emergency department intubation success with succinylcholine versus rocuronium: a National Emergency Airway Registry Study. Ann Emerg Med. 2018;72(6):645–653. doi:10.1016/j.annemergmed.2018.03.042
3. Bauld HW, Gibson PF, Jebson PJ, Brown SS. Aetiology of prolonged apnoea after suxamethonium. Br J Anaesth. 1974;46(4):273–281. doi:10.1093/bja/46.4.273
4. Gonsalves SG, Dirksen RT, Sangkuhl K, et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) guideline for the use of potent volatile anesthetic agents and succinylcholine in the context of RYR1 or CACNA1S genotypes. Clin Pharmacol Ther. 2019;105(6):1338–1344. doi:10.1002/cpt.1319
5. Avidan MS, Stevens TW. The diving bell and the butterfly. Br J Anaesth. 2015;115(Suppl 1):i8–i10. doi:10.1093/bja/aev143
6. Thomsen JL, Nielsen CV, Eskildsen KZ, Demant MN, Gatke MR. Awareness during emergence from anaesthesia: significance of neuromuscular monitoring in patients with butyrylcholinesterase deficiency. Br J Anaesth. 2015;115(Suppl 1):i78–i88. doi:10.1093/bja/aev096
7. Puura AIE, Rorarius MGF, Manninen P, Hoppu S, Baer GA. The costs of intense neuromuscular block for anesthesia during endolaryngeal procedures due to waiting time. Anesth Analg. 1999;88(6):1335–1339. doi:10.1213/00000539-199906000-00026
8. Lockridge O. Review of human butyrylcholinesterase structure, function, genetic variants, history of use in the clinic, and potential therapeutic uses. Pharmacol Ther. 2015;148:34–46. doi:10.1016/j.pharmthera.2014.11.011
9. Soliday FK, Conley YP, Henker R. Pseudocholinesterase deficiency: a comprehensive review of genetic, acquired, and drug influences. AANA J. 2010;78(4):313–320.
10. Anectine® (Succinylcholine Chloride Injection, USP) [prescribing information]. Princeton, NJ: Sandoz Inc; 2018.
11. Parnas ML, Procter M, Schwarz MA, Mao R, Grenache DG. Concordance of butyrylcholinesterase phenotype with genotype: implications for biochemical reporting. Am J Clin Pathol. 2011;135(2):271–276. doi:10.1309/AJCPPI5KLINEKH7A
12. Jensen FS, Skovgaard LT, Viby-Mogensen J. Identification of human plasma cholinesterase variants in 6688 individuals using biochemical analysis. Acta Anaesthesiol Scand. 1995;39(2):157–162. doi:10.1111/j.1399-6576.1995.tb04035.x
13. Yen T, Nightingale BN, Burns JC, Sullivan DR, Stewart PM. Butyrylcholinesterase (BCHE) genotyping for post-succinylcholine apnea in an Australian population. Clin Chem. 2003;49(8):1297–1308. doi:10.1373/49.8.1297
14. Bartels CF, Jensen FS, Lockridge O, et al. DNA mutation associated with the human butyrylcholinesterase K-variant and its linkage to the atypical variant mutation and other polymorphic sites. Am J Hum Genet. 1992;50(5):1086–1103.
15. Alvarellos ML, McDonagh EM, Patel S, McLeod HL, Altman RB, Klein TE. PharmGKB summary: succinylcholine pathway, pharmacokinetics/pharmacodynamics. Pharmacogenet Genomics. 2015;25(12):622–630. doi:10.1097/FPC.0000000000000170
16. Primo-Parmo SL, Bartels CF, Wiersema B, van der Spek AF, Innis JW, La Du BN. Characterization of 12 silent alleles of the human butyrylcholinesterase (BCHE) gene. Am J Hum Genet. 1996;58(1):52–64.
17. Altamirano CV, Bartels CF, Lockridge O. The butyrylcholinesterase K-variant shows similar cellular protein turnover and quaternary interaction to the wild-type enzyme. J Neurochem. 2000;74(2):869–877. doi:10.1046/j.1471-4159.2000.740869.x
18. Jasiecki J, Zuk M, Krawczynska N, et al. Haplotypes of butyrylcholinesterase K-variant and their influence on the enzyme activity. Chem Biol Interact. 2019;307:154–157. doi:10.1016/j.cbi.2019.05.007
19. Hicks JK, Dunnenberger HM, Gumpper KF, Haidar CE, Hoffman JM. Integrating pharmacogenomics into electronic health records with clinical decision support. Am J Health Syst Pharm. 2016;73(23):1967–1976. doi:10.2146/ajhp160030
20. Bocchini CA. Butyrylcholinesterase deficiency; BCHED. OMIM (Online Mendelian Inheritance in Man) Web site. April 12, 2018 [Updated May 4, 2018]. Available from: https://www.omim.org/entry/617936.
21. Del Tredici AL, Malhotra A, Dedek M, et al. Frequency of CYP2D6 alleles including structural variants in the United States. Front Pharmacol. 2018;9:305. doi:10.3389/fphar.2018.00305
22. Jasiecki J, Jonca J, Zuk M, et al. Activity and polymorphisms of butyrylcholinesterase in a Polish population. Chem Biol Interact. 2016;259(Pt B):70–77. doi:10.1016/j.cbi.2016.04.030
23. Bretlau C, Sorensen MK, Vedersoe AL, Rasmussen LS, Gatke MR. Response to succinylcholine in patients carrying the K-variant of the butyrylcholinesterase gene. Anesth Analg. 2013;116(3):596–601. doi:10.1213/ANE.0b013e318280a3f3
24. Souza RL, Castro RM, Pereira L, Freund AA, Culpi L, Chautard-Freire-Maia EA. Frequencies of the butyrylcholinesterase K mutation in Brazilian populations of European and African origin. Hum Biol. 1998;70(5):965–970.
25. Mikami LR, Wieseler S, Souza RL, et al. Five new naturally occurring mutations of the BCHE gene and frequencies of 12 butyrylcholinesterase alleles in a Brazilian population. Pharmacogenet Genomics. 2008;18(3):213–218. doi:10.1097/FPC.0b013e3282f5107e
26. Mollerup HM, Gatke MR. Butyrylcholinesterase gene mutations in patients with prolonged apnea after succinylcholine for electroconvulsive therapy. Acta Anaesthesiol Scand. 2011;55(1):82–86. doi:10.1111/j.1399-6576.2010.02316.x
27. Keating GM. Sugammadex: a review of neuromuscular blockade reversal. Drugs. 2016;76(10):1041–1052. doi:10.1007/s40265-016-0604-1
28. Sauer M, Piel I, Haubner C, et al. Rocuronium is more hepatotoxic than succinylcholine in vitro. Eur J Anaesthesiol. 2017;34(9):623–627. doi:10.1097/EJA.0000000000000666
29. Kosinova M, Stourac P, Adamus M, et al. Rocuronium versus suxamethonium for rapid sequence induction of general anaesthesia for caesarean section: influence on neonatal outcomes. Int J Obstet Anesth. 2017;32:4–10. doi:10.1016/j.ijoa.2017.05.001
30. Fernandes HDS, Ximenes JLS, Nunes DI, Ashmawi HA, Vieira JE. Failure of reversion of neuromuscular block with sugammadex in patient with myasthenia gravis: case report and brief review of literature. BMC Anesthesiol. 2019;19(1):160. doi:10.1186/s12871-019-0829-0
31. Elhassan A, Elhassan I, Elhassan A, et al. Perioperative surgical home models and enhanced recovery after surgery. J Anaesthesiol Clin Pharmacol. 2019;35(Suppl 1):S46–S50.
32. van der Wouden CH, van Rhenen MH, Jama WOM, et al. Development of the PGx-passport: a panel of actionable germline genetic variants for pre-emptive pharmacogenetic testing. Clin Pharmacol Ther. 2019;106(4):866–873. doi:10.1002/cpt.1489
33. Gould MK, Garcia DA, Wren SM, et al. Prevention of VTE in nonorthopedic surgical patients: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141(2 Suppl):e227S–e277S. doi:10.1378/chest.11-2297
34. Jhun EH, Apfelbaum JL, Dickerson DM, et al. Pharmacogenomic considerations for medications in the perioperative setting. Pharmacogenomics. 2019;20(11):813–827. doi:10.2217/pgs-2019-0040
35. Kaye AD, Mahakian T, Kaye AJ, et al. Pharmacogenomics, precision medicine, and implications for anesthesia care. Best Pract Res Clin Anaesthesiol. 2018;32(2):61–81. doi:10.1016/j.bpa.2018.07.001
36. Trescot AM. Genetics and implications in perioperative analgesia. Best Pract Res Clin Anaesthesiol. 2014;28(2):153–166. doi:10.1016/j.bpa.2014.03.004
37. Levano S, Ginz H, Siegemund M, et al. Genotyping the butyrylcholinesterase in patients with prolonged neuromuscular block after succinylcholine. Anesthesiology. 2005;102(3):531–535. doi:10.1097/00000542-200503000-00009
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.