Back to Journals » Infection and Drug Resistance » Volume 7
Genetic relatedness and host specificity of Pseudomonas aeruginosa isolates from cystic fibrosis and non-cystic fibrosis patients
Authors AbdulWahab A ![]() , Taj-Aldeen SJ, Ibrahim EB, Abdulla S, Muhammed R, Ahmed I, Abdeen Y, Sadek O, Abu-Madi M
, Taj-Aldeen SJ, Ibrahim EB, Abdulla S, Muhammed R, Ahmed I, Abdeen Y, Sadek O, Abu-Madi M
Received 1 August 2014
Accepted for publication 2 September 2014
Published 20 November 2014 Volume 2014:7 Pages 309—316
DOI https://doi.org/10.2147/IDR.S72112
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Suresh Antony
Atqah AbdulWahab,1 Saad J Taj-Aldeen,2 Emad Ibrahim,2 Shaikha H Abdulla,3 Ramees Muhammed,3 Irshad Ahmed,3 Yasmine Abdeen,4 Omnia Sadek,4 Marawan Abu-Madi4
1Department of Pediatrics, Hamad Medical Corporation, 2Microbiology Division, Department of Laboratory Medicine and Pathology, Hamad Medical Corporation, 3Molecular Biology Unit, Central Food Laboratories, Supreme Council of Health, 4Department of Health Sciences, College of Arts and Sciences, Qatar University, Doha, Qatar
Background: Pseudomonas aeruginosa is one of the primary pathogens isolated more frequently in cystic fibrosis (CF) and it exhibits innate resistance to a wide range of antibiotics.
Purpose: We sought to determine whether the highly prevalent genotypes of P. aeruginosa are specifically linked to CF patients and have any related multidrug antibiotic resistance. Isolates from hospitalized non-CF patients and from environmental sources were also genotypically analyzed.
Methods: Collections of P. aeruginosa from lower respiratory secretions (n=45) were genotyped using pulsed-field gel electrophoresis (PFGE). Phenotypic screening for antibiotic susceptibility was performed for the common antimicrobial agents by E-test and automated Phoenix method.
Results: P. aeruginosa isolates from CF (n=32), hospitalized non-CF patients (n=13), and environment sources (n=5) were analyzed. The population structure of P. aeruginosa is highly diverse and population-specific. All PFGE results of P. aeruginosa isolates fall among four major clusters. Cluster 1 contained 16 P. aeruginosa isolates from CF patients and two from environmental sources; cluster 2 contained 11 P. aeruginosa isolates from CF and one each from non-CF and environmental sources; cluster 3 contained 12 P. aeruginosa isolates from hospitalized non-CF patients and two P. aeruginosa isolates from one CF patient and one environmental source; and cluster 4 consisted of three isolates from CF patients and one from the environment. The majority of multidrug-resistant P. aeruginosa isolates were in clusters 3 and 4. P. aeruginosa isolates from CF patients were resistant to ciprofloxacin (34.4%) followed by resistance to amikacin and gentamicin (each 28%), whereas the majority of isolates from non-CF patients were resistant to meropenem (69%) and were grouped in cluster 3.
Conclusion: PFGE of P. aeruginosa isolates from CF patients shows a high degree of similarity, suggesting specific adaptation of these clones to CF-affected lungs. The hospitalized non-CF cluster has a different clonal origin, indicating specific clustering in a specific location, suggesting hospital-acquired P. aeruginosa infections.
Keywords: cystic fibrosis, drug susceptibility testing, Pseudomonas aeruginosa, pulsed-field gel electrophoresis
Introduction
Pseudomonas aeruginosa is the main bacterial pathogen that infects the lungs of patients with cystic fibrosis (CF).1 CF patients chronically infected with P. aeruginosa show a greater loss of lung function and a higher overall morbidity and mortality in comparison with patients with intermittent or no colonization.1–4
Initial pulmonary infection with P. aeruginosa in CF is thought to occur through the acquisition of unique environmental strains of P. aeruginosa, which persist and undergo phenotypic changes over time in the lungs of CF patients.5 However, P. aeruginosa can spread between people with CF.6 Some so-called transmissible or epidemic strains are associated with a worse clinical outcome and can be associated with a more rapid annual loss of lung function, worse nutritional status, and even an increased rate of death or lung transplantation.7 An outbreak of colistin-resistant P. aeruginosa in a UK pediatric CF clinic was reported, describing the transmission of a genotypically indistinguishable P. aeruginosa strain between three patients in a nosocomial setting.8
Typing of strains of P. aeruginosa is important for specific characterization of the species, the choice of antibiotic regimen, the detection of unusual traits, and the recognition of a potential cluster of a single clone within patients.9
Several comprehensive molecular typing techniques for discriminating among P. aeruginosa strains have been developed, based either on DNA banding patterns such as restriction fragment length polymorphism and pulsed-field gel electrophoresis (PFGE), DNA sequencing (eg, multilocus sequence typing and genome sequencing), or on DNA hybridization (DNA macro- and microarrays).10 PFGE typing is considered a common genotyping method for P. aeruginosa fingerprinting and is sometimes mentioned as the gold standard method for this bacterium, useful for hospital epidemiologists who need to monitor the effectiveness of infection control measures.11–13 A recent study by Waters et al14 demonstrated the highest level of concordance between PFGE and multilocus sequence typing results during the genotyping of a blinded sample of well-characterized P. aeruginosa isolates from CF patients across multiple laboratories. The aim of this study is to determine whether the highly prevalent genotypes of P. aeruginosa are specifically linked to CF patients and related to multidrug antibiotic resistance. Isolates from hospitalized non-CF patients and environment sources were also genotypically analyzed.
Materials and methods
Patients and sampling
Sputum samples or deep-pharyngeal swabs were prospectively collected and cultured over a period of 6 months from CF patients attending the CF clinic or hospitalized at the Hamad Medical Corporation, Doha, Qatar. The diagnosis of CF was based on one or more clinical features consistent with CF, positive family history of CF in siblings and close relatives, elevated sweat chloride (>60 mmol/L) on two separate occasions, in addition to the presence of two disease-causing mutations in the CFTR gene. Patients with P. aeruginosa isolates cultured from sputum or deep-pharyngeal swab samples were enrolled in the study (18 CF patients with chronic P. aeruginosa and 3 CF patients with intermittent P. aeruginosa infections). Chronic P. aeruginosa infection was defined as persistent presence of P. aeruginosa for at least 6 consecutive months.15 P. aeruginosa isolates from the lower respiratory secretions of hospitalized non-CF patients during the same period and P. aeruginosa isolates from environmental samples obtained from a water basin were also included in the study.
Sputum samples or deep-pharyngeal swabs from patients not producing sputum were inoculated onto MacConkey agar, Columbia blood agar, and Columbia chocolate agar (Mast Diagnostics Ltd, Bootle, UK). Blood and chocolate agar media were incubated in a 5% CO2 atmosphere. After 24 hours of incubation at 37°C, lactose-negative and oxidase-positive colonies were selected from among the different colony morphology types – mucoid, nonmucoid, and characteristic-pigment-producing colonies – subcultured on 5% Columbia sheep blood agar, and identified up to the species level using the Phoenix 100 (Becton, Dickinson and Co, Franklin Lakes, NJ, USA). Mucoid colonies were further confirmed by growth on nutrient agar, including a control reference P. aeruginosa strain at 42°C. Phenotypically different isolates were preserved in cryotubes at −70°C until analysis. This study was reviewed and approved by the local ethics committee at Hamad Medical Corporation (number 13103/13).
PFGE analysis
PFGE was performed according to a previously described protocol.16 PulseNet standard Salmonella enterica serovar Braenderup H9812 strain was used for running the gels in accordance with the PulseNet protocol.17 Two S. enterica serovar Braenderup H9812 precast plugs were digested with 50 IU of XbaI restriction enzyme (isolated from Xanthomonas badrii) alongside Pseudomonas plugs and loaded on the gel in the first and sixth lanes in order to normalize for analysis using BioNumerics software (Applied Maths, Sint-Martens-Latem, Belgium).
The electrophoresis was performed with the Bio-Rad CHEF-DR III system (Bio-Rad Laboratories Inc, Hercules, CA, USA). Electrophoresis run conditions were designed for a run time of 19 hours with 14°C cooling. The CHEF-DR III apparatus was then programmed to initial switch time 2.2 seconds, final switch time 54.2 seconds, gradient 6 V, and an angle of 120°. All other parameters remained identical with those of the standard procedure.
Following electrophoresis, the gels were stained for 45 minutes in 600 mL of sterile distilled water containing 50 μL of ethidium bromide (10 mg/mL) and destained in three washes of 30 minutes each in 600 mL of distilled water. The gels were analyzed using a gel documentation system (Bio-Rad) and saved as TIFF (tagged-image file format) files for further analysis. The same TIFF files were later imported to the BioNumerics software version 6.0 (Applied Maths). All the software settings were used as defaults, and the gel was analyzed to generate a dendrogram describing the relationship among the pulsotypes.
Drug susceptibility testing
Drug susceptibility testing and interpretation were performed according to Clinical and Laboratory Standards Institute guidelines using the E-test and the automated Phoenix method for nine antimicrobial agents, including amikacin, gentamicin, ceftazidime, cefepime, ciprofloxacin, imipenem, meropenem, piperacillin/tazobactam, and colistin. P. aeruginosa ATCC 27853 was used as a control.
Results
This study included 21 CF patients – 14 males and 7 females – from 15 families, with a median age of 15 years (range: 4–36 years). Thirteen CF families with CFTR I1234V mutation, belonging to a single large Arab kindred tribe, and constituted of two families with three CF siblings and two families with two CF siblings were enrolled. The remaining families contributed one sibling each. Three closely related families were cousins to each other. Two CF patients had mutations other than CFTR I1234V. Furthermore, 13 CF patients provided one respiratory sample, six CF patients gave two samples each, one CF patient had three samples, and one CF patient provided four samples in different periods of time. Six CF patients were hospitalized for acute chest exacerbation during the study period. Hospitalized non-CF patients included two patients on mechanical ventilation support, one with tracheostomy and the other with chest infections.
All the P. aeruginosa strains were isolated from 45 specimens (41 sputum samples and four deep-pharyngeal swabs) and five environmental sources over a 6-month period of the study. P. aeruginosa isolates from CF (n=32), hospitalized non-CF patients (n=13), and environment sources (n=5) were analyzed. All P. aeruginosa isolates were successfully typed by PFGE, and the clusters of isolates obtained thereby are illustrated in the resulting dendogram (Figure 1). The population structure of P. aeruginosa is highly diverse and population-specific. PFGE analysis showed that the levels of genetic variation within P. aeruginosa were different among different clusters, with genetic relatedness within the same cluster. All PFGE results of P. aeruginosa isolates fall among four major clusters. Cluster 1 contained 16 P. aeruginosa isolates from CF patients and two from environmental sources; cluster 2 contained 11 P. aeruginosa isolates from CF and one each from non-CF and environmental sources; cluster 3 contained 12 P. aeruginosa isolates from hospitalized non-CF patients and two P. aeruginosa isolates from one CF patient and one environmental source; and cluster 4 consisted of three isolates from CF patients and one from the environment (Table 1). P. aeruginosa isolates from one CF family with three siblings were displayed in cluster 1, two CF families with siblings and closely related patients had P. aeruginosa isolates displayed within PFGE cluster 2, and one CF family with two siblings having two respiratory samples had P. aeruginosa isolates displayed within PFGE clusters 2 and 4. The PFGE analysis of one CF patient with four respiratory samples showed genetic variation of P. aeruginosa, which was distributed in PFGE clusters 1 and 2 during different periods. One CF patient hospitalized with acute chest exacerbation, in whom both mucoid and nonmucoid P. aeruginosa isolates were evaluated, showed highly close genetic relatedness to hospitalized non-CF P. aeruginosa isolates in cluster 3.
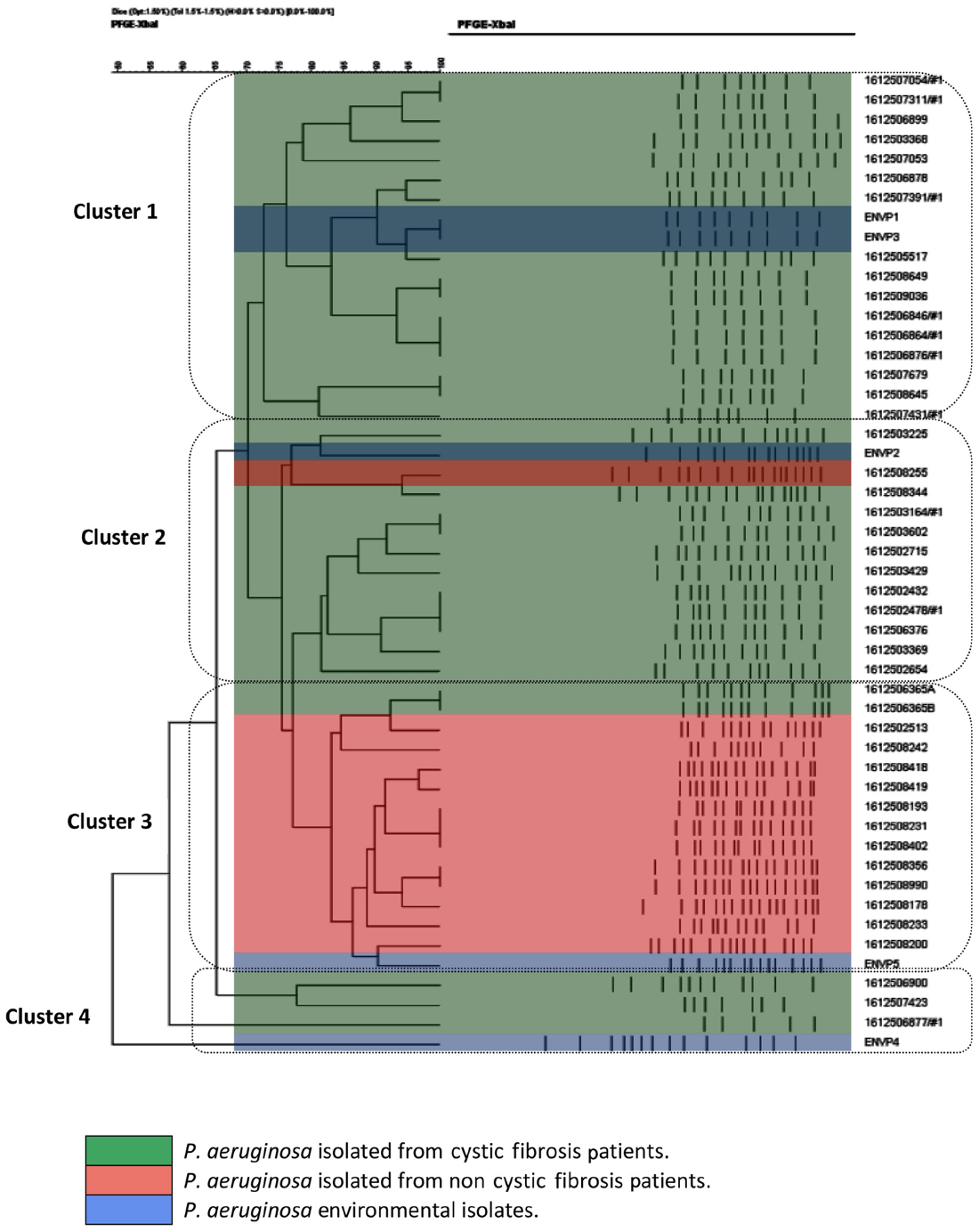 | Figure 1 Dendrogram based on percentage similarities among P. aeruginosa isolates obtained using PFGE fingerprinting. |
 | Table 1 Number of P. aeruginosa isolates and their respective clusters on PFGE typing |
P. aeruginosas isolates from CF patients showed high susceptibility to antipseudomonal agents and relatively low rates of antimicrobial resistance (Table 2). P. aeruginosa strains in CF patients were resistant to ciprofloxacin (34.4%), followed by resistance to amikacin and gentamicin (each 28%), whereas the majority of isolates from non-CF patients were resistant to meropenem (69%), followed by resistance to ciprofloxacin (30.7%), amikacin, and gentamicin (each 23%) and were grouped in cluster 3 (Table 1). None of the CF isolates were resistant to meropenem. The majority of multidrug-resistant P. aeruginosa (MRPA) samples were in clusters 3 and 4. Two CF siblings with advanced lung disease had the same P. aeruginosa multidrug resistance to fluoroquinolones, amikacin, gentamicin, and cefepime and were within the same cluster 4, indicating high P. aeruginosa genetic relatedness. Another CF patient with advanced lung disease had similar multidrug resistance to antipseudomonal agents in his four sputum samples over the 6-month period, with the samples clustered in two different clusters, ie, clusters 1 and 2, upon PFGE analysis for P. aeruginosa.
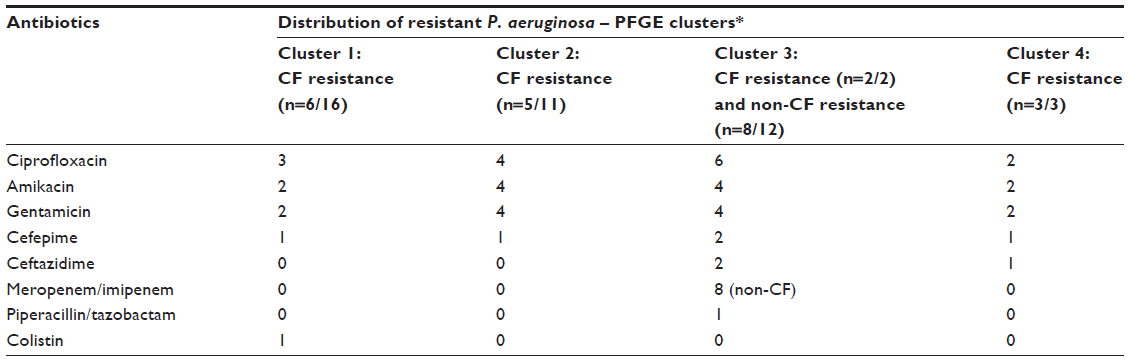 | Table 2 Number of P. aeruginosa isolates resistant to common antimicrobial agents in vitro and their respective clusters after PFGE typing |
Discussion
P. aeruginosa is ubiquitous in the environment, the principal respiratory pathogen in CF patients, and continues to be the principal cause of morbidity and mortality among CF patients. P. aeruginosa colonization and infection may either be community acquired or health care associated. We identified a considerable genetic heterogeneity of P. aeruginosa in our patient population, and this study demonstrates a unique cohort of CF and hospitalized non-CF patients from a single center, by exploring the genetic relatedness of P. aeruginosa isolates from respiratory secretion using PFGE, which is capable of discriminating between genotypes of different P. aeruginosa isolates.
Interestingly, P. aeruginosa isolates from siblings of CF families were displayed within one PFGE cluster, suggesting that cross-transmission of P. aeruginosa among CF patients occurs more frequently when contact is more intense, especially among siblings. The ability of P. aeruginosa to be transmitted from person to person with CF has been recognized as early as the 1980s.18–26 By employing amplified fragment-length polymorphism molecular fingerprinting, a previous study has shown similar distribution of P. aeruginosa isolates among siblings and closely related families, indicating person-to-person transmission of P. aeruginosa within the same family or infection of P. aeruginosa possibly acquired from common environmental exposure.26 The PFGE of P. aeruginosa isolates was largely in agreement with the diversity found by amplified fragment-length polymorphism fingerprinting in several previous studies that showed that siblings with CF frequently carry identical P. aeruginosa isolates.18,27–29
One hospitalized CF patient with advanced lung disease had high similarity of genetic relatedness of both mucoid and nonmucoid P. aeruginosa isolates to the samples from hospitalized non-CF patients in cluster 3 indicating specific clustering in the specific location and suggesting hospital-acquired P. aeruginosa infection.
There were variations of the different clusters of P. aeruginosa among CF patients having more than one sample in different periods of time. These findings were similar to previously published studies demonstrating that the predominant clone of P. aeruginosa would vary over time, with one clone predominating in one period and another clone predominating in another period. This event suggests that P. aeruginosa infection of the CF patient’s lung is a very dynamic process, as proposed by Renders et al.30 P. aeruginosas isolates from CF patients showed high susceptibility to antipseudomonal agents and relatively low rates of antimicrobial resistance. Fluoroquinolones are the only available antibiotics for oral treatment of P. aeruginosa infections. However, P. aeruginosa easily becomes resistant to these drugs as we found in 31% among the CF population, followed by development of resistance to amikacin and gentamicin (each 24%). However, the carbapenems (imipenem and meropenem) remained very active against CF P. aeruginosa isolates, with susceptibility rates of 100%, whereas the majority of P. aeruginosa isolates from hospitalized non-CF patients were resistant to meropenem (69%) and were grouped in cluster 3; they were probably acquired from the hospital environment.
MRPA is a common respiratory pathogen found in CF patients, which routinely leads to chronic pulmonary infections.31,32 Multiple antibiotic-resistant P. aeruginosa is more likely to be a marker of more severe disease and more intensive therapy and is less likely to be contributing independently to more rapid lung function decline.33 In the present study, P. aeruginosa isolates from CF siblings with advanced lung disease and MRPA isolates had similar genetic relatedness on PFGE and were found within the same cluster, suggesting the transmission even of MRPA isolates among CF siblings. Clustering of P. aeruginosa isolate from another CF patient with advanced lung disease and MRPA isolates in different PFGE clusters 1, 2, and 4 suggests that the colonizing strain may occasionally be changed.
Polymyxin B sulfate and colistimethate, a prodrug of colistin, are administered to CF patients intravenously or by inhalation. Resistance to colistin has been rarely reported in CF patients, and there is potential for an emerging resistant strain.34 In the present study, we found that P. aeruginosa from one CF patient with a non-CFTR I1234V mutation showed in vitro resistance to colistin although the patient did not receive either nebulized or intravenous colistin.
We concluded that cross-transmission is probably common among CF siblings even with MRPA isolates. In addition, the presence of several isolates in the same patient with distinct genotypes suggests that the colonizing strain may occasionally be changed. Different patient populations have different PFGE clusters and different drug susceptibilities.
Acknowledgment
This publication was made possible by a generous grant from the Qatar National Research Fund (UREP 14-026-3-010). The statements made herein are solely the responsibility of the authors.
Disclosure
The authors report no conflicts of interest in this work.
References
Gibson RL, Burns JL, Ramsey BW. Pathophysiology and management of pulmonary infections in cystic fibrosis. Am J Respir Crit Care Med. 2003;168(8):918–951. | |
Collins J, Farrell PM. Acceleration of lung disease in children with cystic fibrosis after Pseudomonas aeruginosa acquisition. Pediatr Pulmonol. 2001;32(4):277–287. | |
Henry RL, Mellis CM, Petrovic L. Mucoid Pseudomonas aeruginosa is a marker of poor survival in cystic fibrosis. Pediatr Pulmonol. 1992;12(3):158–161. | |
Li Z, Kosorok MR, Farrell PM, et al. Longitudinal development of mucoid Pseudomonas aeruginosa infection and lung disease progression in children with cystic fibrosis. JAMA. 2005;293(5):581–588. | |
Hogardt M, Heesemann J. Adaptation of Pseudomonas aeruginosa during persistence in the cystic fibrosis lung. Int J Med Microbiol. 2010;300(8):557–562. | |
Jones AM, Dodd ME, Morris J, Doherty C, Govan JR, Webb AK. Clinical outcome for cystic fibrosis patients infected with transmissible Pseudomonas aeruginosa: an 8-year prospective study. Chest. 2010;137(6):1405–1409. | |
Al-Aloul M, Crawley J, Winstanley C, Hart CA, Ledson MJ, Walshaw MJ. Increased morbidity associated with chronic infection by an epidemic Pseudomonas aeruginosa strain in CF patients. Thorax. 2004;59(4):334–336. | |
Denton M, Kerr K, Mooney L, et al. Transmission of colistin-resistant Pseudomonas aeruginosa between patients attending a pediatric cystic fibrosis center. Pediatr Pulmonol. 2002;34(4):257–261. | |
Logan C, Habington A, Lennon G, et al. Relatedness of Pseudomonas aeruginosa isolates among a paediatric cystic fibrosis patient cohort in Ireland. J Med Microbiol. 2012;61(pt 1):64–70. | |
van Mansfeld R, Willems R, Brimicombe R, et al. Pseudomonas aeruginosa genotype prevalence in Dutch cystic fibrosis patients and age dependency of colonization by various P. aeruginosa sequence types. J Clin Microbiol. 2009;47(12):4096–4101. | |
Johnson JK, Arduino SM, Stine OC, Johnson JA, Harris AD. Multilocus sequence typing compared to pulsed-field gel electrophoresis. J Clin Microbiol. 2007;45(11):3707–3712. | |
Gautom RK. Rapid pulsed-field gel electrophoresis protocol for typing of Escherichia coli O157:H7 and other gram-negative organisms in 1 day. J Clin Microbiol. 1997;35(11):2977–2980. | |
Kersulyte D, Struelens MJ, Deplano A, Berg DE. Comparison of arbitrarily primed PCR and macrorestriction (pulsed-field gel electrophoresis) typing of Pseudomonas aeruginosa strains from cystic fibrosis patients. J Clin Microbiol. 1995;33(8):2216–2219. | |
Waters V, Zlosnik JE, Yau YC, Speert DP, Aaron SD, Guttman DS. Comparison of three typing methods for Pseudomonas aeruginosa isolates from patients with cystic fibrosis. Eur J Clin Microbiol Infect Dis. 2012;31(12):3341–3350. | |
Brett MM, Simmonds EJ, Ghonheim ATM, Littlewood JM. The value of serum IgG titres against Pseudomonas aeruginosa in the management of early infection in cystic fibrosis. Arch Dis Child. 1992;67:1086–1088. | |
Pirnay JP, De Vos D, Cochez C, et al. Molecular epidemiology of Pseudomonas aeruginosa colonization in a burn unit: persistence of a multidrug-resistant clone and a silver sulfadiazine-resistant clone. J Clin Microbiol. 2003;41(3):1192–1202. | |
Ribot EM, Fair MA, Gautom R, et al. Standardization of pulsed-field gel electrophoresis protocols for the subtyping of Escherichia coli O157:H7, Salmonella, and Shigella for PulseNet. Foodborne Pathog Dis. 2006;3(1):59–67. | |
Scott FW, Pitt TL. Identification and characterization of transmissible Pseudomonas aeruginosa strains in cystic fibrosis patients in England and Wales. J Med Microbiol. 2004;53(pt 7):609–615. | |
Aaron SD, Vandemheen KL, Ramotar K, et al. Infection with transmissible strains of Pseudomonas aeruginosa and clinical outcomes in adults with cystic fibrosis. JAMA. 2010;304(19):2145–2153. | |
Armstrong DS, Nixon GM, Carzino R, et al. Detection of a widespread clone of Pseudomonas aeruginosa in a pediatric cystic fibrosis clinic. Am J Respir Crit Care Med. 2002;166(7):983–987. | |
Cheng K, Smyth RL, Govan JR, et al. Spread of beta-lactam resistant Pseudomonas aeruginosa in a cystic fibrosis clinic. Lancet. 1996;348(9028):639–642. | |
Johansen HK, Moskowitz SM, Ciofu O, Pressler T, Høiby N. Spread of colistin resistant non-mucoid Pseudomonas aeruginosa among chronically infected Danish cystic fibrosis patients. J Cyst Fibros. 2008;7(5):391–397. | |
Jones AM, Govan JR, Doherty CJ, et al. Spread of a multiresistant strain of Pseudomonas aeruginosa in an adult cystic fibrosis clinic. Lancet. 2001;358(9281):557–558. | |
O’Carroll MR, Syrmis MW, Wainwright CE, et al. Clonal strains of Pseudomonas aeruginosa in paediatric and adult cystic fibrosis units. Eur Respir J. 2004;24(1):101–106. | |
Pedersen SS, Koch C, Høiby N, Rosendal K. An epidemic spread of multiresistant Pseudomonas aeruginosa in a cystic fibrosis centre. J Antimicrob Chemother. 1986;17(4):505–516. | |
Abdul Wahab A, Taj-Aldeen SJ, Hagen F, et al. Genotypic diversity of Pseudomonas aeruginosa in cystic fibrosis siblings in Qatar using AFLP fingerprinting. Eur J Clin Microbiol Infect Dis. 2014;33(2):265–271. | |
Speert DP, Campbell ME, Henry DA, et al. Epidemiology of Pseudomonas aeruginosa in cystic fibrosis in British Columbia, Canada. Am J Respir Crit Care Med. 2002;166(7):988–993. | |
Grothues D, Koopmann U, von der Hardt H, Tümmler B. Genome fingerprinting of Pseudomonas aeruginosa indicates colonization of cystic fibrosis siblings with closely related strains. J Clin Microbiol. 1988;26(10):1973–1977. | |
Wolz C, Kiosz G, Ogle JW, et al. Pseudomonas aeruginosa cross-colonization and persistence in patients with cystic fibrosis. Use of a DNA probe. Epidemiol Infect. 1989;102(2):205–214. | |
Renders NHM, Sijmons MAF, Van Belkum A, Overbeek SE, Mouton JW, Verbrugh HA. Exchange of Pseudomonas aeruginosa strains among cystic fibrosis siblings. Res Microbiol. 1997;148(5):447–454. | |
Annual Report of the European Antimicrobial Resistance Surveillance Network 7 (EARS-Net). Antimicrobial Resistance Surveillance in Europe 2011. Stockholm: 8 European Centre for Disease Prevention and Control; 2012. | |
Falagas ME, Koletsi PK, Bliziotis IA. The diversity of definitions of multidrug-resistant (MDR) and pandrug-resistant(PDR) Acinetobacter baumannii and Pseudomonas aeruginosa. J Med Microbiol. 2006; 55(pt 12):1619–1629. | |
Ren CL, Konstan MW, Yegin A, Scientific Advisory Group, Investigators, and Coordinators of the Epidemiologic Study of Cystic Fibrosis, et al. Multiple antibiotic-resistant Pseudomonas aeruginosa and lung function decline in patients with cystic fibrosis. J Cyst Fibros. 2012;11(4):293–299. | |
Moskowitz SM, Brannon MK, Dasgupta N, et al. PmrB mutations promote polymyxin resistance of Pseudomonas aeruginosa isolated from colistin-treatedcystic fibrosis patients. Antimicrob Agents Chemother. 2012;56(2):1019–1030. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.