Back to Journals » The Application of Clinical Genetics » Volume 14
Genetic Predictors of Mortality in Patients with Multiple Myeloma
Received 29 December 2020
Accepted for publication 31 March 2021
Published 29 April 2021 Volume 2021:14 Pages 241—254
DOI https://doi.org/10.2147/TACG.S262866
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Martin Maurer
Hamza Hassan,1 Raphael Szalat1,2
1Department of Hematology and Medical Oncology, Boston University Medical Center, Boston, MA, USA; 2Medical Oncology, Dana-Farber Cancer Institute, Harvard Medical School, Boston, MA, USA
Correspondence: Raphael Szalat
Department of Hematology and Oncology, FGH Building, One Boston Medical Center Pl, Boston, MA, 02118, USA
Tel +1 617 638 6428
Email [email protected]
Abstract: Multiple myeloma (MM) is a heterogeneous disease featured by clonal plasma cell proliferation and genomic instability. The advent of next-generation sequencing allowed unraveling the complex genomic landscape of the disease. Several recurrent genomic aberrations including immunoglobulin genes translocations, copy number abnormalities, complex chromosomal events, transcriptomic and epigenomic deregulation, and mutations define various molecular subgroups with distinct outcomes. In this review, we describe the recurrent genomic events identified in MM impacting patients’ outcome and survival. These genomic aberrations constitute new markers that could be incorporated into a prognostication model to eventually guide therapy at every stage of the disease.
Keywords: multiple myeloma, genomics, aneuploidy, copy number abnormalities, structural variants, translocations, mutations, overall survival
Introduction
Multiple myeloma (MM) is a hematologic malignancy characterized by the proliferation of clonal plasma cells. The disease is virtually always preceded by an asymptomatic stage named monoclonal gammopathy of undetermined significance (MGUS) that subsequently can progress to smoldering myeloma and eventually to symptomatic multiple myeloma.1 MM is a heterogeneous disease featured by various molecular subgroups with distinct outcomes. With the advent of many efficient therapeutic options in the past decade, including immunomodulators (IMiDs), proteasome inhibitors and monoclonal antibodies, patients’ outcome has significantly improved. The therapeutic decisions are based on patient and disease characteristics. Intensive therapies are avoided in frail patients, while more aggressive treatments are usually recommended for fit patients and patients with high-risk disease. High-risk MM defines patients with poor prognosis, early relapse or primary refractory disease and with shorter survival. Identifying this subgroup of patients is critical to define the best therapeutic strategy with currently available treatments and to develop new therapeutic strategies.2 Conversely, identifying standard-risk and good-prognosis patients is also very important as it refers to patients with prolonged overall survival that can potentially benefit from less intensive treatment. Therefore, risk stratification has become a major field of research in MM considering its potential impact on therapy. Current criteria defining high-risk myeloma include the revised international staging system (R-ISS) that combines high serum LDH and β2 microglobulin, low albumin levels and presence of any of 3 cytogenetic abnormalities (17p13 deletion, t(4;14) and t(14;16)).2 However, this classification is not accurate to identify all high-risk patients and does not identify good-prognosis patients. Next-generation sequencing approaches have recently unraveled the complex genomic landscape of MM and significantly changed our understanding of myelomagenesis.3 While few genomic events are recurrent, such as immunoglobulin (Ig) genes rearrangements along with other structural variants, no universal event has been identified differentiating MM from other malignancies like chronic myeloid leukemia4 or Waldenström macroglobulinemia5 that are featured by the Philadelphia chromosome t(9;22)(q34.1;q11.2)4 and MYD88 L265P5 mutation, respectively, for example. In MM, on the contrary, multiple recurrent genomic events have been discovered with distinct impact on disease outcome, further expanding the disease heterogeneity. The integration of DNA and RNA sequencing along with epigenomic profiling provides new critical information to improve current risk stratification in MM. We here review the genomic events involved in myelomagenesis and their impact on patients' overall survival.
Chromosome Abnormalities: Hyperdiploid and Non-Hyperdiploid MM
Conventional karyotyping and more recently fluorescent in situ hybridization (FISH) studies identified cytogenetic abnormalities in up to 70% of MM patients with two main groups: hyperdiploid (HDMM) and non-hyperdiploid myeloma (NHDMM).
HDMM is observed in 60% of MM and is characterized by trisomies of odd-numbered chromosomes 3, 5, 7, 9, 11, 15, 19 and/or 21.6,7 Duplication of odd chromosome is an early event in myelomagenesis and is observed at early stages of the disease, at MGUS and SMM stages.8 The mechanisms driving HDMM are largely unknown but may relate to a single catastrophic mitosis rather than serial gain of chromosomes over time.9 HDMM have been classically associated with standard risk and better outcome in comparison with NHDMM.10,11 However, recent studies have identified significant heterogeneity in this subgroup, with trisomy 3 and 5 being associated with significantly better overall survival, whereas trisomy 21 is associated with worse outcome.7 Moreover, the co-occurrence of hyperdiploidy with additional genomic events including immunoglobulin light chain gene translocations or focal deletions significantly impacts the prognosis of patients with HDMM and is discussed below.
NHDMM entails hypodiploid, pseudodiploid, near-tetraploid, tetraploid and hyperhaploid MM4,12,13 and constitutes about 40% of MM. NHDMM is associated with recurrent immunoglobulin heavy chain gene (IgH) translocations and is classically associated with worse outcome. Tetraploidy is observed in up to 6% of newly diagnosed MM and is an independent marker associated with significantly shorter overall survival.14 Hyperhaploidy is another subset of NHDMM where myeloma cells have 24–34 chromosomes with disomies of most odd number chromosomes 3, 5, 7, 9, 11, 15, 19, 21 and chromosome 18, and monosomies of all other autosomes resulting in clinically relevant monosomies of 1p, 6q, 13q and 16q and 17p.13,15,16 This last-mentioned aberration is considered to drive the poor prognosis observed in the setting of hyperhaploidy.
Both HDMM and NHDMM constitute two clear distinct entities, with distinct transcriptomic and mutational profiles.17,18 However, each subgroup is also heterogeneous and can be impacted by additional genomic events. For example, recurrent Ig heavy chain (IgH) translocations are mainly observed in context of NHDMM, and only in 10% of HDMM, and Ig Lambda light chain (IgL) and MYC translocations are mainly observed in HDMM.12
Copy Number Variations (CNV)
Copy number variations (CNVs) correspond to loss of heterozygosity (LOH), gain and loss of DNA at both a focal level or at an entire chromosome arm level. Similar to single nucleotide mutations, CNVs can be driver or passenger events, with highly recurrent CNVs likely to be drivers. CNVs are frequent in MM and are observed at an early stage of the disease.8,19 Several CNVs have been identified to be recurrent in MM with a frequency greater than 10%.20 The most frequent focal gains are 1q, 6p, 11q gains, while the most frequent deletions are 1p, 6q, 8p, 13q, 14q, 16q and 17p deletions. Although these CNVs are recurrent and involved in MM pathogenesis, their detection do not impact overall survival with the exception of 1q gain and 1p and 17p deletions, which have been independently shown to negatively impact patients’ outcome. As an example, del(13q) is found in about ~45–50% of cases, and more commonly in NHDMM, but is not associated with poor outcome.
Chromosome 1q Gain
Duplication of the 1q21 region of chromosome 1 is present in 35% of newly diagnosed MM patients.21 Three copies of chromosome 1q21 is classified as gain 1q21, while having 4 or more copies is classified as amplification of 1q21. The 1q gain is the most frequent recurrent chromosomal event that is an independent poor-prognosis factor impacting both period free survival (PFS) and overall survival (OS) as reported in several independent cohorts.21–24 Furthermore, presence of 4 copies or more of 1q is associated with the worst outcome.17 Several oncogenes located in this region have been involved in the mechanisms leading to poor outcome, such as MCL1, IL6R, BCL9, CSK1B, ILF2, ANP32E or ADAR1 that are amplified/overexpressed as a consequence of 1q gain. Resistance to proteasome inhibitor, apoptosis and DNA damage repair deregulation have been reported in that setting.25
Chromosome 1p Deletion
The 1p deletions are observed in 25% of newly diagnosed MM patients and are associated with poor prognosis. Two regions of the 1p arm are mainly affected, 1p12 and 1p32.3, and are seen in 15% and 8% of patients, respectively.20,26 These deletions are independently associated with poor outcome.26,27 Several tumor suppressor genes have been involved in the mechanisms driving poor outcome, including CDKN2C, FAF1, MTF2 and TMED5.
Chromosome 17 Deletion
Hemizygous deletion of the whole p arm (del(17p)) is one of the CNVs that retained its adverse prognostic significance for both PFS and OS in the current R-ISS risk stratification2 and is observed in about 6% to 10% of NDMM patients.28,29 The incidence increases to 25–50% in primary plasma cell leukemia (PCL)/advanced stage disease, to as high as 75% in secondary PCL, and is associated with low hematological responses, early drug resistance/relapse, extramedullary disease and central nervous system involvement.28,30,31 The underlying mechanism is likely the loss of the guardian of genome gene, TP53. Multiple studies including phase 3 randomized controlled trials (RCTs) have confirmed the negative impact of del(17p) on overall survival while using different thresholds for the size of del(17p) clone ranging from a single cell in ELOQUENT-2 trial,32 1.5–7.5% in the SWOG S0777 trial,33 60% in the ASPIRE trial34 and 5–50% in TOURMALINE-MM trial.35 It seems that del(17p) is associated with worse prognosis (PFS and OS) irrespective of the therapy and % fraction of clone alteration,36 although a heightened risk population is identified with cutoff >55%.37
Complex Chromosomal Events
Macro-scale complex chromosomal events include chromothripsis, cyclo-templated insertions and chromoplexy. These events have been reported to occur in both early phases as well as at relapse potentially during one genetic catastrophic event.38 Chromothripsis, also known as chromosome shattering, is a complex process where large segments of a chromosome undergo massive oscillations by breaking into smaller pieces, rearranging and then randomly rejoining, ultimately leading to an erroneous new genomic configuration of a single or few chromosomes.39 This complex event has been captured in early stages of myeloma development and occurs in about 9–36% of cases.38,40 Chromothripsis was found to have an independent negative predictive effect on PFS (HR: 1.42) and OS (HR: 1.81) and also has a strong association with biallelic inactivation of TP53 (HR: 6.6).41 Chromoplexy includes copy number losses and mostly co-occur with cyclo-templated insertions. This leads to balanced double strand breaks, causing segmental rearrangements of multiple sites of different chromosomes (between 2 and 5) which have been implicated in myeloma relapse/drug resistance in about 10% of cases.38 Templated insertions have been reported to occur in about 19% of the cases and have been reported in hijacking the enhancer oncogenes like CCND1 and MYC.41 At least one driver CNV is present in 47% of all chromothripsis, 42% of chromoplexy occurrences and 28% of templated insertions.41 The causes of these complex chromosomic events and their impact on overall survival remain to be further investigated.
Recurrent Chromosomal Translocations
Normal B cells undergo affinity maturation and cell cycle arrest to evolve from a naïve B cell or centroblast to an antibody-secreting plasma cell in the germinal centers (GC). Affinity maturation is a complex mechanism that includes somatic hypermutation and class switch recombination that have been involved in lymphomagenesis and myelomagenesis. Activation-induced cytidine deaminase (AICDA or AID) is a major actor in these processes and generates double strand DNA-breaks at the variable region of both Ig heavy and light chain loci.42 Aberrant recombinations of these DNA breaks along with additional off-target DNA lesions can lead to translocations across the genome and drive tumorigenesis.42–45 Recurrent IgH translocations referred to as primary IgH translocations are detected in 40% of MM and are observed at early stages of the disease12,28 as they likely constitute primary driver events. Additional translocations involving Ig light chains and MYC locus have been more recently characterized and are also considered to be driver events (Table 1).
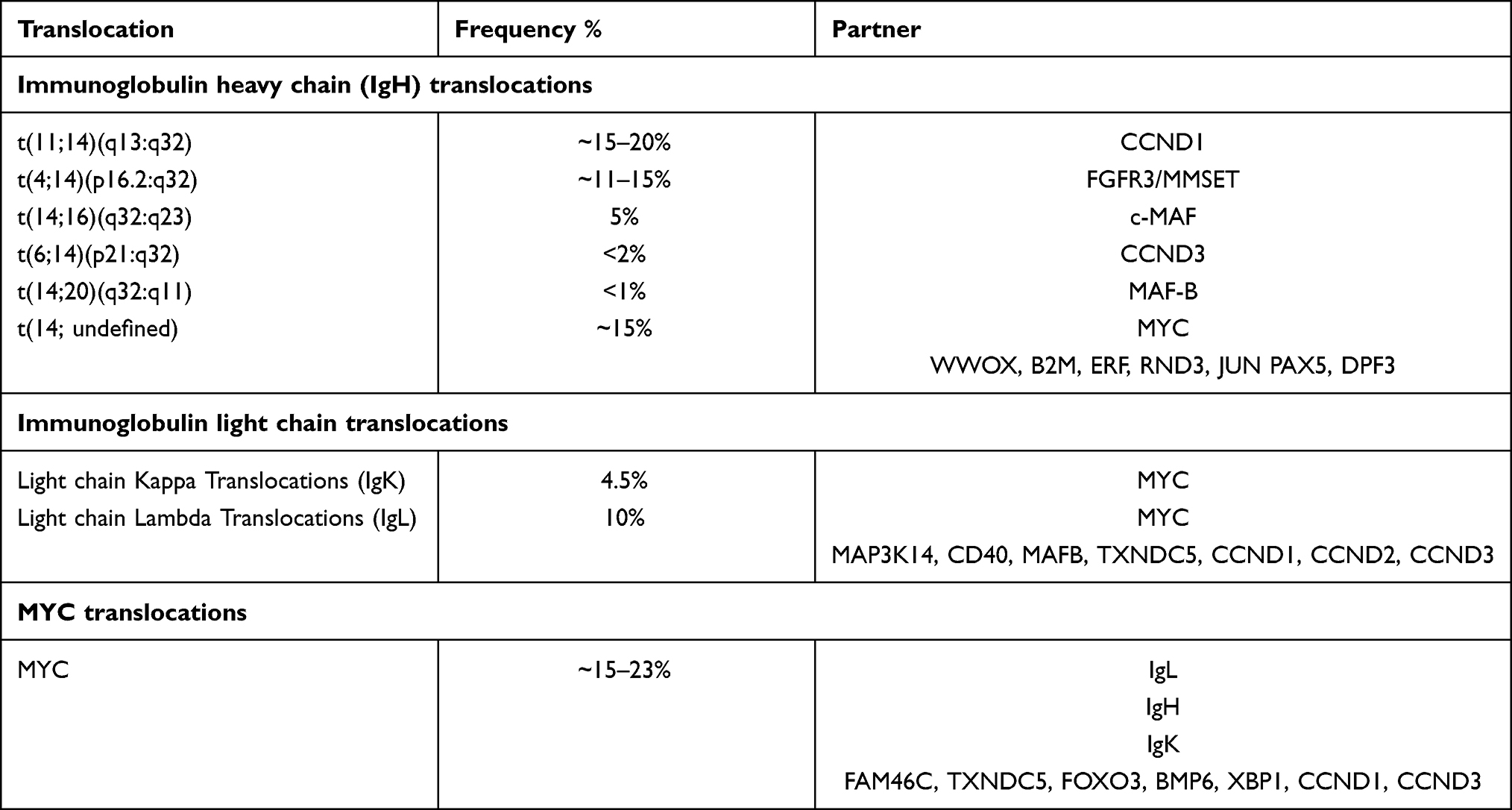 |
Table 1 Recurrent Translocations Observed in Multiple Myeloma |
Translocation t(11;14)(q13:q32) occurs in ~15–20% of MM patients and corresponds to the juxtaposition of CCND1 to the IgH enhancer region, leading to increased cyclin D1 production.46,47 Interestingly, t(11;14) is more frequent in IgM, IgE and non-secretory MM,48 and these patients are more likely to have lymphoplasmacytic morphology with CD20 expression and lambda light chain isotype.49 CCND1 is an oncogene encoding for cyclin D1, which is an activating regulatory subunit for cyclin-dependent kinase (CDK) 4 and 6.50 These activated cyclin complexes (D-CDK) phosphorylate and inactivate retinoblastoma (Rb) protein, which is a potent inhibitor of G1 to S phase progression. D-CDK complex also inhibits cyclin-dependent kinase inhibitor 1B (CDKI1B, also known as p27 and Kip1) that inhibits other CDK complexes required at later phases of cell cycle.51 The prognostic significance of t(11;14) is neutral; however, when found associated with an activating mutation of CCND1 (10%), it has been associated with poor prognosis.47 Studies have also shown inferior response to the proteasome inhibitor bortezomib and inferior outcomes in absence of expression of CD20, suggesting some heterogeneity in this subgroup.49,52,53 Nevertheless, patients with t(11;14) have a high expression of BCL-2 and have high sensitivity to BCL2 inhibitors such as venetoclax potentially through a TP53-independent mechanism.54–57
Translocation t(4;14)(p16.2:q32) is observed in ~11–15% of MM patients and leads to the juxtaposition of fibroblast growth factor receptor 3 (FGFR3) and multiple myeloma set domain (MMSET) to the IgH enhancer region. While MMSET is always overexpressed, FGFR3 is only overexpressed in 70% of t(4;14) MM depending on the breakpoint site. FGFR3 is a tyrosine kinase that acts as an oncoprotein activating the Ras-mitogen-activated protein kinase (MAPK) pathway.47 MMSET is a histone methyltransferase that methylates histones into H3K3658 and H4K2059 and significantly impacts expression of multiple genes. MMSET has also been involved in DNA damage repair responses by hindering the recruitment of p53 binding protein 1 (53BP1).59 Translocation t(4;14) is overall associated with adverse prognosis; however, this subgroup is heterogeneous. DNA breakpoint, transcriptomic changes and additional chromosomic events influence significantly patients' overall survival such as del(13q14), del(1p32), del(17p) and >30 chromosomal structural changes.60–62 Several studies have shown that use of proteasome inhibitor improves outcomes of t(4;14) MM patients.63
Translocation t(14;16)(q32:q23) is seen in 5% of MM patients and leads to overexpression of musculoaponeurotic fibrosarcoma (c-MAF),64,65 a known transcription factor that upregulates the expression of multiple genes including CCND2 by binding directly to its promoter.47 Translocation t(14;16) is associated with high mutational burden and a specific mutational signature linked to apolipoprotein B mRNA editing catalytic polypeptide-like (APOBEC) activity.47 Despite these consistent genomic patterns, the impact of t(14;16) on patient outcome has been controversial with distinct effect observed across distinct clinical trials.28,47,66,67 Nevertheless, t(14;16) is currently considered as a poor prognosis factor.
Translocation t(6;14)(p21:q32) is present in less than 2% of MM patients and leads to juxtaposition of CCND3 to the IgH enhancer region resulting in its upregulation.47,68,69 The prognostic significance of t(6;14) is neutral.70
Translocation t(14;20)(q32:q11) is seen in less than 1% of MM patients and leads to upregulation of the MAF gene paralogue, MAF-B (musculoaponeurotic fibrosarcoma oncology family, protein B) that increases the downstream CCND2 activity. The t(14;20) MM has a distinct APOBEC mutational signature driven by the upregulation of APOBEC4 expression.47 Translocation t(14;20) is classically associated with a poor prognosis when detected in newly diagnosed MM patients; however, some observation showed that its presence at early stage (MGUS or SMM) does not impact the time to progression to active disease, suggesting this single event is not enough to drive the disease.71
Translocation t(14; undefined): based on FISH analysis, 15% of IgH translocations happen with non-recurrent partners. The t(14; undefined) has been associated with a better outcome in comparison to t(11;14), t(4;14) and t(14;16) and might be associated with better response to bortezomib.49 These additional IgH partners, ahve been identified using NGS and include mainly MYC, and less frequently WWOX, B2M, ERF, RND3, JUN PAX5, DPF3 and MTMR1.72–74
Light Chain Translocations
Recurrent translocations involving kappa (IgK) and lambda (IgL) light chains locus at chromosome 2 and 22, respectively, are detected in 4.5% and 10% of newly diagnosed MM, respectively.73,75 Nearly half (41%) of IgL translocations and most of IgK translocations involve MYC locus. Other recurrent partners include MAP3K14, CD40, MAFB, TXNDC5, CCND1, CCND2 and CCND3 but with lower frequencies (1% to 7%). Importantly 80% of t(IgL) happen in HDMM and define a subgroup of HDMM with poor prognosis, with poorer overall survival and lower response to IMiDs.73 Translocation t(8;22) is often associated with IgL locus amplification seen in 80% of t(8;22).
MYC Translocations
The 8q24 region is often subject to complex structural variants including duplications, amplifications, templated insertions, chromoplexy, chromothripsis and translocations.73 This region includes the proto-oncogene MYC, which is a basic helix-loop-helix (bHLH) transcription factor that is involved in the pathogenesis of several cancers. MYC translocations are found in about 15–23% of newly diagnosed MM patients.40,73 The most frequently (range 16.5–3.5%) juxtaposed partner genes are IgL [t(8;22)], IgH [t(8;14)], FAM46C, TXNDC5, IgK [t(2;8)], FOXO3, BMP6 and more rarely XBP1, CCND1 and CCND3.73 MYC translocations are often sub-clonal and found in up to 20% of patients with SMM.76 In that setting, it is an independent risk factor of progression to symptomatic MM.19 Other MYC loci structural variants, such as duplications, are also common in SMM and MM. Both sub-clonality and increased frequency in more advanced stages of the disease suggest that MYC translocations and rearrangements contribute significantly to disease progression.47,73,76–78 MYC translocations in particular have been associated with kataegis which is a localized pattern of hypermutation linked to the deregulation of APOBEC-induced mutagenesis. It is hypothesized that clusters of APOBEC-induced hypermutations aggregate at the chromosomal rearrangement sites before the single stranded DNA (ssDNA) gets repaired.79 MYC translocations are more frequently observed in HDMM tumors (~65%)80 and are associated with a poor outcome, especially when they involve an IgL partner.47,73
Mutational Landscape
The advent of next-generation sequencing allowed deep DNA sequencing studies in large cohorts of MM patients. To date, more than one thousand myeloma genomes have been sequenced and reported in the literature using either targeted, whole-exome or whole-genome sequencing methods with distinct sequencing depth. While no universal driver of the disease has been identified, several recurrent mutations have been observed. KRAS, NRAS, DIS3, FAM46C, BRAF and TP53 have been found mutated in 20–23%, 19–20%, 1–11%, 6–11%, 6–12% and 8–12% of newly diagnosed MM, respectively40,80,81 (Table 2). More strikingly, pathway analysis revealed that RAS/MAPK and NF-kB pathways are recurrently mutated in 43% and 17% of MM patients, respectively.80 Interestingly, the distinct IgH translocation subgroups are enriched for specific mutations suggesting oncogenic dependencies. Thus, CCND1, IRF4, LTB and HUWE1 are almost exclusively mutated in t(11;14), while FGFR3, PRKD2, ACTG1, DIS3 on one hand, and ATM, BRAF, MAF, TRAF2, EP300 and DIS3 on the other hand are almost exclusively mutated in t(4;14) and t(14;16), respectively. However, the clinical impact of these mutations in each MM subgroup is unclear.17 The only recurrent mutations that significantly and negatively affect patients’ outcome are those observed in p53 pathway (10%; TP53, ATM, ATR). Mutations in IRF4 (3%) and PRDM1/BLIMP1 (5%) tend to be associated with a favorable outcome in patients treated with IMiDs based regimen. More consistently, the overall mutational load positively correlates with poorer outcome and is reported to be lowest in HDMM and highest in t(14;16).40,82
 |
Table 2 Recurrent Mutations Observed in Multiple Myeloma |
Mutational Signatures
Bayesian models analyzing the different 96 trinucleotide possible combinations related to nucleotide changes allowed determining mutational processes or signatures active across cancer genomes.83 In MM, an enrichment of C>T transitions at CpG dinucleotides, which reflect deamination of methylated cytosine to thymine, and the C>T transition associated with C>A and C>G transversion in TpC context has been consistently observed. C>T transitions at CpG dinucleotides are observed in various malignancies like breast, pancreatic, CLL, B cell lymphoma and myeloma and are thought to be ubiquitous in these malignancies.47,83 The very high frequency of C>T transitions observed in myeloma is a significant challenge to clearly evaluate its clinical impact. However, it is possible that these changes, when occurring at gene promoters, can impact methylation profile and contribute to disease progression.17,40,84 The latter process (C>T transition associated with C>A and C>G transversion in TpC context) was found to occur in clusters at specific intervals in a phenomenon known as kataegis. Most recent analysis of the mutational signature involved in myelomagenesis using whole-genome sequencing data in newly diagnosed MM samples revealed the role of age-related signature contributing to 25% of the total mutational burden, AID/APOBEC family of cytidine deaminases related signature involved in 5% of mutations, somatic hypermutation (9%), DNA damage related pathways (16%) and unknown processes (45%). These mutational signatures have different weights depending on the stage of the disease, with APOBEC related signature being more predominant at advanced stages of the disease. APOBEC and DNA damage related signatures predominantly contribute to sub-clonal mutations as opposed to AID related signature that is more frequently involved in clonal and driver mutations. This suggests that AID is a critical actor of myeloma initiation, while APOBEC contributes to the disease progression.78,82
Double Hit and Multiple Hit Myeloma
The accumulation of genomic aberrations is a hallmark of cancer. In MM, the co-occurrence of few genomic events is significantly impacting patients’ outcome and define a very high-risk subgroup of patients. This is exemplified by the double hit of TP53 located on 17p chromosome (Figure 1). TP53 is a tumor suppressor gene that is mapped to 17p13.1 locus and encodes a 53 kDa transcriptional regulator protein, p53.85 P53 silencing role has been involved in several cancers, as it is involved in multiple vital cellular pathways including cell cycle regulation, cellular stress, DNA damage repair response and apoptosis. TP53 aberrations are seen in up to 10% of MM at diagnosis. It includes single hit with heterozygous deletion seen in ~6% of patients or monoallelic mutation in the DNA binding domain that are seen in less than 1% of patients. Bi-allelic or double hit events that include homozygous deletions, heterozygous deletions associated with TP53 mutation, bi-allelic mutation or mono-allelic mutation with LOH are seen in up to 4% of patients and are associated with worse clinical outcome.31 Of note, the vast majority of TP53 mutations occur in the context of heterozygous del(17p).86 Frequency of double hit lesions affecting TP53 increases with the stage of the disease and has been reported in up to 15% of relapsed and refractory MM patients.87 Other mechanisms of decreased expression of p53 in MM include overexpression of specific microRNAs, TP53 promoter methylation and overexpression of MDM2.88,89 TP53 aberrations are often sub-clonal, and clonality impacts on patients’ outcome.90 The impact of sub-clonal deletion 17p remains uncertain as contradictory results have been reported. A report from the myeloma XI trial that analyzed NDMM patients (n=1777) with multiplex ligation-dependent probe amplification (MLPA) showed that minor tumor subclones with TP53 deletion are independently associated with shorter OS. In this study, patients were divided into three subgroups: low sub-clonal 17p deletion identified by an MPLA <0.8 cutoff (equivalent to 10–20% sub-clonal tumor population), intermediate 17p deletion clonality identified by MPLA ≥0.55 but <7 (equivalent to >50% clonal tumor fraction) and clonal TP53 deletion identified by MPLA <0.55 (equivalent to dominant clonal deletion 95–100%). All three groups were individually associated with worse OS with HR of 1.8, 2.9 and 2.2, respectively, in comparison to non-del(17p) patients.90 Alternatively, in another large cohort study using whole-exome sequencing data, comparable OS and PFS were observed in del(17p) patients with cancer cell fraction (CCF) ≤0.55 in comparison to non-del(17p).37 The discordant results observed between the two studies may be related to the different methods and treatment received. Therefore, the sub-clonality threshold of myeloma cell population with del(17p) conferring a poor prognosis remains controversial at this time. Nevertheless, CCF or clonal content ≥55% is constantly associated with poor outcome.37
 |
Figure 1 Chromosome 17p13 and TP53 aberrations in multiple myeloma. Abbreviations: WT, wild type; NDMM, newly diagnosed multiple myeloma; LOH, loss of heterozygosity; Del 17p, deletion 17p arm; mut, mutant. |
In addition, co-occurrence of del(17p) with amplification 1q or t(4;14) significantly worsens patient outcome.17,62 Presence of more than one high-risk abnormality is seen in less than 5% of newly diagnosed MM including t(4;14), t(14;16), t(14;20), del(17p)/TP53 mutation, gain(1q), del(1p) and constitutes “double-hit” or “multi-hit“ myeloma, associated with significantly poorer prognosis.91,92 Similarly, coexistence of del(6q) and del(1p32) with del(17p) significantly worsens the prognosis of patients.60
Conversely, using a genomic scar score (GSS), whole-genome sequencing studies have also identified a subgroup of MM patients characterized by low GSS (low mutational burden, specific mutation signatures pattern and fewer structural variants) and a very good overall survival.82 GSS is a score calculated from an algorithm that evaluates allele-specific CNV by scarHRD-R-package93 as the sum of homologous recombination deficiency–loss of heterozygosity (HRD-LOH: number of 15-Mb-exceeding LOH regions which do not cover the whole chromosome), large-scale transitions (LST: chromosomal breaks between adjacent regions of at least 10 Mb, with a distance between them ≤3 Mb) and number of telomeric allelic imbalances (telomeAI: number AIs that extend to the telomeric end of a chromosome). MM patients with low GSS have a significantly superior outcome.82
Transcriptomic Profile
Multiple studies have identified several prognostic gene expression signatures in newly diagnosed MM.94 Most of these studies have identified a transcriptomic profile or gene expression profile (GEP) featuring high-risk patients as an independent prognostic factor. At least 8 studies from major myeloma research groups including the Dutch-Belgian Cooperative Trial Group for Hematology Oncology Group (HOVON), the Intergroupe Francophone du Myelome (IFM) and the University of Arkansas for Medical Sciences (UAMS) have validated distinct GEP as an independent prognosis factor in independent large cohort of patients.95–97 However, only very few or no genes overlap between these signatures, suggesting that each signature does not incorporate all high-risk patients. Incorporation of RNA splicing, and non-protein coding gene such as microRNA (miRNA) and long non-coding RNA (lncRNA) expression has also been shown to be useful to predict patient outcomes,98,99 even more accurately than ISS, standard cytogenetic studies and protein-coding gene expression profile alone.98 However, the lack of a uniform platform to perform transcriptomic profiling remains an important challenge to incorporate GEP as a prognosis marker in clinical practice to date.
Epigenomic Modifications
Epigenomic deregulation is a hallmark of MM at various levels. DNA methylation and histone modifications are essential mechanisms impacting on transcriptome regulation.100 The role of histone modification is highlighted in t(4;14) MM in which MMSET, a histone methyl transferase (HMT) involved in methylation of H3K36 and H4K20,59 is universally overexpressed and influences cell cycle progression, apoptosis, cell adhesion, oncogenesis and DNA damage response.101 In addition, up to 24% of MM patients harbor at least one potentially deleterious mutation in epigenomic regulator genes.102 Additional epigenetic modifiers including other overexpressed HMT have been shown to play critical roles in MM. Thus, the overexpression of the HMT PHD finger protein 19 (PHF19) and enhancer of zeste homolog 2 (EZH2) is a strong predictor of poor outcome.99,103 EZH2 is a component of the polycomb repressive complex 2 (PRC2), which tri-methylates histone H3 lysine residue 27 (H3K27me3) to repress gene transcriptome related to stem cell self-renewal, cell cycle checkpoints, metastasis and cellular differentiation. Aberrant activity of EZH2 is regulated at multiple levels driven by interleukin-6 especially in cell lines that harbor K- and N-RAS mutations,104 upregulation of NF-kB pathway105 and downregulation of several microRNA (miRNA).106 EZH2 also plays an important role in t(4;14) and in case of mutation or loss of expression of the histone acetyl transferase UTX/KDM6A, which occurs in up to 5% of MM patients, representing a possible therapeutic target in these settings. While EZH2 inhibitors are in the pre-clinical stages and include EPZ-6438, GSK126, UNC1999, OR-S2107–109 in myeloma, the recent FDA approval of tazemetostat for relapsed refractory EZH2-mutated positive follicular lymphoma is encouraging.
Future Perspective
High-throughput technologies have been increasingly explored and include DNA-based studies (WGS, WES, array comparative genomic hybridization, high density SNP arrays) and RNA-based studies (RNA sequencing and microarrays). These techniques have allowed identification of recurrent mutations and affected pathways, mutational signatures, clonal evolution, CNVs, protein-coding gene (fusions, mutations, splicing, isoform expression, gene expression) and non-coding gene expressions. Some of these have already been shown to have clinical utility for both risk stratification and personalized medicine (Table 3). Detection of recurrent mutations may have therapeutic implications for targeted therapies. NRAS mutations in relapsing myeloma are associated with lower response rate to bortezomib,110 while IRF4 mutations are associated with better response to IMiD therapy.80 Presence of V600E BRAF mutation can be specifically targeted by vemurafenib.111 More accurate risk stratification, based on transcriptomic studies, mutational signatures and clonal shift harboring a high-risk mutation or low GSS can be captured and guide clinicians for utilizing more or less intensive treatment at induction, consolidation and maintenance stages.112 With the advent of efficient quadruplet regimens, BCMA targeting agents including monoclonal antibodies and chimeric antigen receptor T cells therapies in myeloma, identification of accurate prognosis markers will be extremely important. Major limitations for incorporating NGS to clinical practice currently relate to its availability and cost. The development of standardized and widely available genomic investigation methods is mandatory. Furthermore, the dynamic nature of cancer genome requires serial genomic evaluations over time to accurately prognosticate and identify potential therapeutic implications.
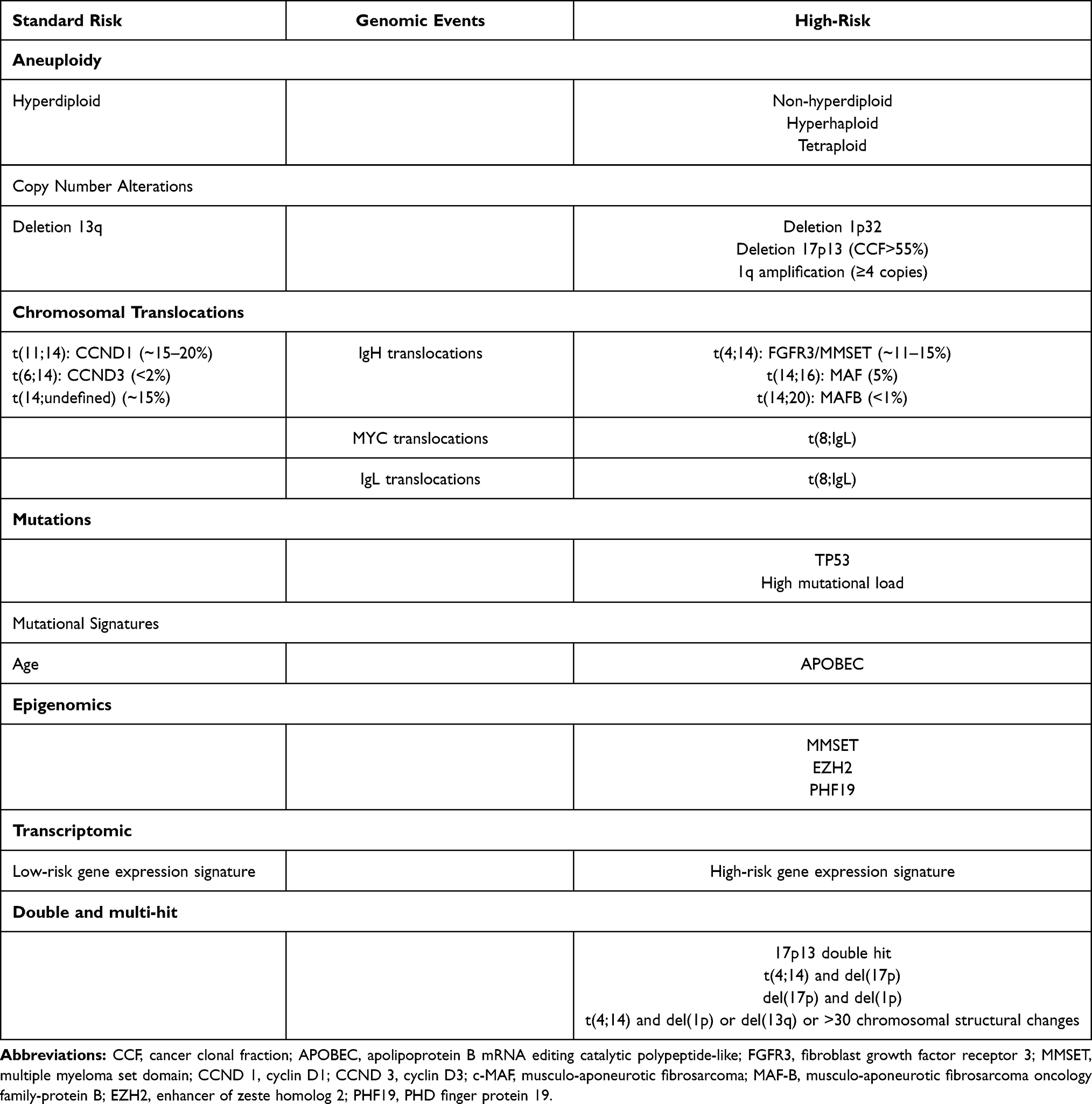 |
Table 3 Recurrent Genomic Events Impacting Overall Survival in Newly Diagnosed Multiple Myeloma Patients |
Conclusion and Perspectives
Multiple myeloma is a complex and heterogeneous disease. Genomic studies have identified various molecular subgroups and recurrent events involved in myelomagenesis and impacting patients’ outcome. While current risk stratification only include presence of del(17p), t(14;14) and t(14;16), new genomic markers can be incorporated to improve risk stratification and potentially to guide therapy. These genomic markers include high-risk markers (IgL and MYC translocations, high mutational burden and detection of double and multi-hit myeloma) and good-prognosis hallmarks (low mutational burden, low genomic scar score) at diagnosis. In addition, integrating genomic alterations associated with early progression at an early stage of the disease (MYC rearrangements, DNA damage and repair gene pathways abnormalities) will also be important. The inclusion of these markers can significantly improve patients’ management in the near feature. However, important challenges exist as it is necessary to develop a broadly available platform using high-throughput sequencing technologies to capture such genetic complexity and to take into account the dynamic evolution of the disease which requires serial evaluations to adjust therapy.
Acknowledgment
The authors thank their colleagues at Boston University School of Medicine and at the Dana-Farber Cancer Institute, patients and patients' families.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Landgren O, Kyle RA, Pfeiffer RM, et al. Monoclonal gammopathy of undetermined significance (MGUS) consistently precedes multiple myeloma: a Prospective Study. Blood. 2009;113(22):5412–5417. doi:10.1182/blood-2008-12-194241
2. Palumbo A, Avet-Loiseau H, Oliva S, et al. Revised international staging system for multiple myeloma: a report from International Myeloma Working Group. J Clin Oncol. 2015;33(26):2863–2869. doi:10.1200/jco.2015.61.2267
3. Corre J, Munshi NC, Avet-Loiseau H. Risk factors in MM: is it time for a revision? Blood. 2020;135(1). doi:10.1182/blood.2019004309
4. Faderl S, Talpaz M, Estrov Z, et al. The biology of chronic myeloid leukemia. N Engl J Med. 1999;341(3):164–172. doi:10.1056/nejm199907153410306
5. Treon SP, Xu L, Yang G, et al. MYD88 L265P somatic mutation in Waldenström’s macroglobulinemia. N Engl J Med. 2012;367(9):826–833. doi:10.1056/NEJMoa1200710
6. Kumar SK, Rajkumar SV. The multiple myelomas - current concepts in cytogenetic classification and therapy. Nat Rev Clin Oncol. 2018;15(7):409–421. doi:10.1038/s41571-018-0018-y
7. Chretien ML, Corre J, Lauwers-Cances V, et al. Understanding the role of hyperdiploidy in myeloma prognosis: which trisomies really matter? Blood. 2015;126(25):2713–2719. doi:10.1182/blood-2015-06-650242
8. Samur AA, Minvielle S, Shammas M, et al. Deciphering the chronology of copy number alterations in multiple myeloma. Blood Cancer J. 2019;9:1–10.
9. Onodera N, McCabe NR, Rubin CM. Formation of a hyperdiploid karyotype in childhood acute lymphoblastic leukemia. Blood. 1992;80(1):203–208. doi:10.1182/blood.V80.1.203.203
10. Smadja NV, Bastard C, Brigaudeau C, Leroux D, Fruchart C. Hypodiploidy is a major prognostic factor in multiple myeloma. J Am Soc Hematol. 2001;98:2229–2238.
11. Smadja NV, Leroux D, Soulier J, et al. Further cytogenetic characterization of multiple myeloma confirms that 14q32 translocations are a very rare event in hyperdiploid cases. Genes Chromosomes Cancer. 2003;38(3):234–239. doi:10.1002/gcc.10275
12. Fonseca R, Debes-Marun CS, Picken EB, et al. The recurrent IgH translocations are highly associated with nonhyperdiploid variant multiple myeloma. Blood. 2003;102(7):2562–2567. doi:10.1182/blood-2003-02-0493
13. Sawyer JR, Tian E, Shaughnessy JD
14. Sidana S, Jevremovic D, Ketterling RP, et al. Tetraploidy is associated with poor prognosis at diagnosis in multiple myeloma. Am J Hematol. 2019;94(5):E117–e120. doi:10.1002/ajh.25420
15. Ashby C, Tytarenko RG, Wang Y, et al. Poor overall survival in hyperhaploid multiple myeloma is defined by double-hit bi-allelic inactivation of TP53. Oncotarget. 2019;10(7):732–737. doi:10.18632/oncotarget.26589
16. Peterson JF, Rowsey RA, Marcou CA, et al. Hyperhaploid plasma cell myeloma characterized by poor outcome and monosomy 17 with frequently co-occurring TP53 mutations. Blood Cancer J. 2019;9(3):20. doi:10.1038/s41408-019-0182-z
17. Walker BA, Mavrommatis K, Wardell CP, et al. A high-risk, double-hit, group of newly diagnosed myeloma identified by genomic analysis. Leukemia. 2019;33(1):159–170. doi:10.1038/s41375-018-0196-8
18. Samur MK, Shah PK, Wang X, et al. The shaping and functional consequences of the dosage effect landscape in multiple myeloma. BMC Genom. 2013;14(1):672. doi:10.1186/1471-2164-14-672
19. Bustoros M, Sklavenitis-Pistofidis R, Park J, et al. Genomic profiling of smoldering multiple myeloma identifies patients at a high risk of disease progression. J Clin Oncol. 2020;38(21):2380–2389. doi:10.1200/JCO.20.00437
20. Avet-Loiseau H, Li C, Magrangeas F, et al. Prognostic significance of copy-number alterations in multiple myeloma. J Clin Oncol. 2009;27(27):4585–4590. doi:10.1200/JCO.2008.20.6136
21. Avet-Loiseau H, Attal M, Campion L, et al. Long-term analysis of the IFM 99 trials for myeloma: cytogenetic abnormalities [t(4;14), del(17p), 1q gains] play a major role in defining long-term survival. J Clin Oncol. 2012;30(16):1949–1952. doi:10.1200/jco.2011.36.5726
22. Chang H, Qi X, Trieu Y, et al. Multiple myeloma patients with CKS1B gene amplification have a shorter progression-free survival post-autologous stem cell transplantation. Br J Haematol. 2006;135(4):486–491. doi:10.1111/j.1365-2141.2006.06325.x
23. Shaughnessy J. Amplification and overexpression of CKS1B at chromosome band 1q21 is associated with reduced levels of p27Kip1 and an aggressive clinical course in multiple myeloma. Hematology. 2005;10(Suppl 1):117–126. doi:10.1080/10245330512331390140
24. Fonseca R, Van Wier SA, Chng WJ, et al. Prognostic value of chromosome 1q21 gain by fluorescent in situ hybridization and increase CKS1B expression in myeloma. Leukemia. 2006;20(11):2034–2040. doi:10.1038/sj.leu.2404403
25. Marchesini M, Ogoti Y, Fiorini E, et al. ILF2 is a regulator of RNA splicing and DNA damage response in 1q21-amplified multiple myeloma. Cancer Cell. 2017;32(1):88–100.e106. doi:10.1016/j.ccell.2017.05.011
26. Hebraud B, Leleu X, Lauwers-Cances V, et al. Deletion of the 1p32 region is a major independent prognostic factor in young patients with myeloma: the IFM experience on 1195 patients. Leukemia. 2014;28(3):675–679. doi:10.1038/leu.2013.225
27. Ouyang J, Gou X, Ma Y, Huang Q, Jiang T. Prognostic value of 1p deletion for multiple myeloma: a meta-analysis. Int J Lab Hematol. 2014;36(5):555–565. doi:10.1111/ijlh.12189
28. Fonseca R, Blood E, Rue M, et al. Clinical and biologic implications of recurrent genomic aberrations in myeloma. Blood. 2003;101(11):4569–4575. doi:10.1182/blood-2002-10-3017
29. Tiedemann RE, Gonzalez-Paz N, Kyle RA, et al. Genetic aberrations and survival in plasma cell leukemia. Leukemia. 2008;22(5):1044–1052. doi:10.1038/leu.2008.4
30. Chang H, Sloan S, Li D, Keith Stewart A. Multiple myeloma involving central nervous system: high frequency of chromosome 17p13.1 (p53) deletions. Br J Haematol. 2004;127(3):280–284. doi:10.1111/j.1365-2141.2004.05199.x
31. Corre J, Roussel M. Del17p without TP53 mutation confers poor prognosis in intensively treated newly diagnosed multiple myeloma patients. Blood. 2020;135(1). doi:10.1182/blood.2020008346
32. Lonial S, Dimopoulos M, Palumbo A, et al. Elotuzumab therapy for relapsed or refractory multiple myeloma. N Engl J Med. 2015;373(7):621–631. doi:10.1056/NEJMoa1505654
33. Durie BGM, Hoering A, Abidi MH, et al. Bortezomib with lenalidomide and dexamethasone versus lenalidomide and dexamethasone alone in patients with newly diagnosed myeloma without intent for immediate autologous stem-cell transplant (SWOG S0777): a randomised, open-label, phase 3 trial. Lancet. 2017;389(10068):519–527. doi:10.1016/s0140-6736(16)31594-x
34. Avet-Loiseau H, Fonseca R, Siegel D, et al. Carfilzomib significantly improves the progression-free survival of high-risk patients in multiple myeloma. Blood. 2016;128(9):1174–1180. doi:10.1182/blood-2016-03-707596
35. Avet-Loiseau H, Bahlis NJ, Chng W-J, et al. Ixazomib significantly prolongs progression-free survival in high-risk relapsed/refractory myeloma patients. Blood. 2017;130(24):2610–2618. doi:10.1182/blood-2017-06-791228
36. Bolli N, Biancon G, Moarii M, et al. Analysis of the genomic landscape of multiple myeloma highlights novel prognostic markers and disease subgroups. Leukemia. 2018;32(12):2604–2616. doi:10.1038/s41375-018-0037-9
37. Thakurta A, Ortiz M, Blecua P, et al. High subclonal fraction of 17p deletion is associated with poor prognosis in multiple myeloma. Blood. 2019;133(11):1217–1221. doi:10.1182/blood-2018-10-880831
38. Maura F, Bolli N, Angelopoulos N, et al. Genomic landscape and chronological reconstruction of driver events in multiple myeloma. Nat Commun. 2019;10(1):3835. doi:10.1038/s41467-019-11680-1
39. Stephens PJ, Greenman CD, Fu B, et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell. 2011;144(1):27–40. doi:10.1016/j.cell.2010.11.055
40. Bolli N, Avet-Loiseau H, Wedge DC, et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat Commun. 2014;5(1):2997. doi:10.1038/ncomms3997
41. Rustad EH, Yellapantula VD, Glodzik D, et al. Revealing the impact of structural variants in multiple myeloma. Blood Cancer Discov. 2020;1(3):258–273. doi:10.1158/2643-3230.BCD-20-0132
42. González D, van der Burg M, García-Sanz R, et al. Immunoglobulin gene rearrangements and the pathogenesis of multiple myeloma. Blood. 2007;110(9):3112–3121. doi:10.1182/blood-2007-02-069625
43. Bergsagel PL, Kuehl WM. Critical roles for immunoglobulin translocations and cyclin D dysregulation in multiple myeloma. Immunol Rev. 2003;194(1):96–104. doi:10.1034/j.1600-065x.2003.00052.x
44. Hakim O, Resch W, Yamane A, et al. DNA damage defines sites of recurrent chromosomal translocations in B lymphocytes. Nature. 2012;484(7392):69–74. doi:10.1038/nature10909
45. Kyle RA, Remstein ED, Therneau TM, et al. Clinical course and prognosis of smoldering (asymptomatic) multiple myeloma. N Engl J Med. 2007;356(25):2582–2590. doi:10.1056/NEJMoa070389
46. Chesi M, Bergsagel PL, Brents LA, et al. Dysregulation of cyclin D1 by translocation into an IgH gamma switch region in two multiple myeloma cell lines. Blood. 1996;88(2):674–681. doi:10.1182/blood.V88.2.674.bloodjournal882674
47. Walker BA, Wardell CP, Murison A, et al. APOBEC family mutational signatures are associated with poor prognosis translocations in multiple myeloma. Nat Commun. 2015;6(1):6997. doi:10.1038/ncomms7997
48. Avet-Loiseau H, Garand R, Lodé L, Harousseau JL, Bataille R. Translocation t(11;14)(q13;q32) is the hallmark of IgM, IgE, and nonsecretory multiple myeloma variants. Blood. 2003;101(4):1570–1571. doi:10.1182/blood-2002-08-2436
49. Mao X-H, Zhuang J-L, Zhao -D-D, et al. IgH translocation with undefined partners is associated with superior outcome in multiple myeloma patients. Eur J Haematol. 2020;105(3):326–334. doi:10.1111/ejh.13440
50. Seto M, Yamamoto K, Iida S, et al. Gene rearrangement and overexpression of PRAD1 in lymphoid malignancy with t(11;14)(q13;q32) translocation. Oncogene. 1992;7(7):1401–1406.
51. Quintanilla-Martinez L, Davies-Hill T, Fend F, et al. Sequestration of p27Kip1 protein by cyclin D1 in typical and blastic variants of mantle cell lymphoma (MCL): implications for pathogenesis. Blood. 2003;101(8):3181–3187. doi:10.1182/blood-2002-01-0263
52. Lakshman A, Alhaj Moustafa M, Rajkumar SV, et al. Natural history of t(11;14) multiple myeloma. Leukemia. 2018;32(1):131–138. doi:10.1038/leu.2017.204
53. An G, Xu Y, Shi L, et al. t(11;14) multiple myeloma: a subtype associated with distinct immunological features, immunophenotypic characteristics but divergent outcome. Leuk Res. 2013;37(10):1251–1257. doi:10.1016/j.leukres.2013.06.020
54. Anderson MA, Deng J, Seymour JF, et al. The BCL2 selective inhibitor venetoclax induces rapid onset apoptosis of CLL cells in patients via a TP53-independent mechanism. Blood. 2016;127(25):3215–3224. doi:10.1182/blood-2016-01-688796
55. Kumar S, Kaufman JL, Gasparetto C, et al. Efficacy of venetoclax as targeted therapy for relapsed/refractory t(11;14) multiple myeloma. Blood. 2017;130(22):2401–2409. doi:10.1182/blood-2017-06-788786
56. Kumar SK, Harrison SJ, Cavo M, et al. Venetoclax or placebo in combination with bortezomib and dexamethasone in patients with relapsed or refractory multiple myeloma (BELLINI): a randomised, double-blind, multicentre, phase 3 trial. Lancet Oncol. 2020;21(12):1630–1642. doi:10.1016/S1470-2045(20)30525-8
57. Moreau P, Chanan-Khan A, Roberts AW, et al. Promising efficacy and acceptable safety of venetoclax plus bortezomib and dexamethasone in relapsed/refractory MM. Blood. 2017;130(22):2392–2400. doi:10.1182/blood-2017-06-788323
58. Martinez-Garcia E, Popovic R, Min D-J, et al. The MMSET histone methyl transferase switches global histone methylation and alters gene expression in t(4;14) multiple myeloma cells. Blood. 2011;117(1):211–220. doi:10.1182/blood-2010-07-298349
59. Pei H, Zhang L, Luo K, et al. MMSET regulates histone H4K20 methylation and 53BP1 accumulation at DNA damage sites. Nature. 2011;470(7332):124–128. doi:10.1038/nature09658
60. Hebraud B, Magrangeas F, Cleynen A, et al. Role of additional chromosomal changes in the prognostic value of t(4;14) and del(17p) in multiple myeloma: the IFM experience. Blood. 2015;125(13):2095–2100. doi:10.1182/blood-2014-07-587964
61. Zhan F, Huang Y, Colla S, et al. The molecular classification of multiple myeloma. Blood. 2006;108(6):2020–2028. doi:10.1182/blood-2005-11-013458
62. Lazareth A, Song X-Y, Coquin A, et al. MB4-2 breakpoint in MMSET combined with del(17p) defines a subset of t(4;14) multiple myeloma with very poor prognosis. Haematologica. 2015;100(11):e471–474. doi:10.3324/haematol.2015.127001
63. Avet-Loiseau H, Leleu X, Roussel M, et al. Bortezomib plus dexamethasone induction improves outcome of patients with t(4;14) myeloma but not outcome of patients with del(17p). J Clin Oncol. 2010;28(30):4630–4634. doi:10.1200/jco.2010.28.3945
64. Hanamura I, Iida S, Akano Y, et al. Ectopic expression of MAFB gene in human myeloma cells carrying (14;20)(q32;q11) chromosomal translocations. Jpn J Cancer Res. 2001;92(6):638–644. doi:10.1111/j.1349-7006.2001.tb01142.x
65. Hurt EM, Wiestner A, Rosenwald A, et al. Overexpression of c-maf is a frequent oncogenic event in multiple myeloma that promotes proliferation and pathological interactions with bone marrow stroma. Cancer Cell. 2004;5(2):191–199. doi:10.1016/s1535-6108(04)00019-4
66. Avet-Loiseau H, Malard F, Campion L, et al. Translocation t(14;16) and multiple myeloma: is it really an independent prognostic factor? Blood. 2011;117(6):2009–2011. doi:10.1182/blood-2010-07-295105
67. Maura F, Petljak M, Lionetti M, et al. Biological and prognostic impact of APOBEC-induced mutations in the spectrum of plasma cell dyscrasias and multiple myeloma cell lines. Leukemia. 2018;32(4):1044–1048. doi:10.1038/leu.2017.345
68. Shaughnessy J
69. Prideaux SM, Conway O’Brien E, Chevassut TJ. The genetic architecture of multiple myeloma. Adv Hematol. 2014;2014:864058. doi:10.1155/2014/864058
70. Weinhold N, Johnson DC, Chubb D, et al. The CCND1 c.870G>A polymorphism is a risk factor for t(11;14)(q13;q32) multiple myeloma. Nat Genet. 2013;45(5):522–525. doi:10.1038/ng.2583
71. Ross FM, Chiecchio L, Dagrada G, et al. The t(14;20) is a poor prognostic factor in myeloma but is associated with long-term stable disease in monoclonal gammopathies of undetermined significance. Haematologica. 2010;95(7):1221–1225. doi:10.3324/haematol.2009.016329
72. Cleynen A, Szalat R, Kemal samur M, et al. Expressed fusion gene landscape and its impact in multiple myeloma. Nat Commun. 2017;8(1):1893. doi:10.1038/s41467-017-00638-w
73. Barwick BG, Neri P, Bahlis NJ, et al. Multiple myeloma immunoglobulin lambda translocations portend poor prognosis. Nat Commun. 2019;10(1):1911. doi:10.1038/s41467-019-09555-6
74. Smadbeck J, Peterson JF, Pearce KE, et al. Mate pair sequencing outperforms fluorescence in situ hybridization in the genomic characterization of multiple myeloma. Blood Cancer J. 2019;9(12):1–18. doi:10.1038/s41408-019-0255-z
75. Affer M, Chesi M, Chen WD, et al. Promiscuous MYC locus rearrangements hijack enhancers but mostly super-enhancers to dysregulate MYC expression in multiple myeloma. Leukemia. 2014;28(8):1725–1735. doi:10.1038/leu.2014.70
76. Misund K, Keane N, Stein CK, et al. MYC dysregulation in the progression of multiple myeloma. Leukemia. Blood. 2020;34(1):322-326. Epub 2019 Aug 22. doi:10.1038/s41375-019-0543-4
77. Weinhold N, Kirn D, Seckinger A, et al. Concomitant gain of 1q21 and MYC translocation define a poor prognostic subgroup of hyperdiploid multiple myeloma. Haematologica. 2016;101(3):e116–119. doi:10.3324/haematol.2015.136929
78. Bolli N, Maura F, Minvielle S, et al. Genomic patterns of progression in smoldering multiple myeloma. Nat Commun. 2018;9(1):3363. doi:10.1038/s41467-018-05058-y
79. Haradhvala NJ, Polak P, Stojanov P, et al. Mutational strand asymmetries in cancer genomes reveal mechanisms of DNA damage and repair. Cell. 2016;164(3):538–549. doi:10.1016/j.cell.2015.12.050
80. Walker BA, Boyle EM, Wardell CP, et al. Mutational spectrum, copy number changes, and outcome: results of a sequencing study of patients with newly diagnosed myeloma. J Clin Oncol. 2015;33(33):3911. doi:10.1200/JCO.2014.59.1503
81. Lohr JG, Stojanov P, Carter S, et al. Widespread genetic heterogeneity in multiple myeloma: implications for targeted therapy. Cancer Cell. 2014;25(1):91–101. doi:10.1016/j.ccr.2013.12.015
82. Samur MK, Aktas samur A, Fulciniti M, et al. Genome-wide somatic alterations in multiple myeloma reveal a Superior Outcome Group. J Clin Oncol. 2020;38(27):3107–3118. doi:10.1200/JCO.20.00461
83. Alexandrov LB, Nik-Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature. 2013;500(7463):415–421. doi:10.1038/nature12477
84. Kaiser MF, Johnson DC, Wu P, et al. Global methylation analysis identifies prognostically important epigenetically inactivated tumor suppressor genes in multiple myeloma. Blood. 2013;122(2):219–226. doi:10.1182/blood-2013-03-487884
85. Finlay CA, Hinds PW, Levine AJ. The p53 proto-oncogene can act as a suppressor of transformation. Cell. 1989;57(7):1083–1093. doi:10.1016/0092-8674(89)90045-7
86. Lode L, Eveillard M, Trichet V, et al. Mutations in TP53 are exclusively associated with del(17p) in multiple myeloma. Haematologica. 2010;95(11):1973–1976. doi:10.3324/haematol.2010.023697
87. Weinhold N, Ashby C, Rasche L, et al. Clonal selection and double-hit events involving tumor suppressor genes underlie relapse in myeloma. Blood. 2016;128(13):1735–1744. doi:10.1182/blood-2016-06-723007
88. Teoh G, Urashima M, Ogata A, et al. MDM2 protein overexpression promotes proliferation and survival of multiple myeloma cells. Blood. 1997;90(5):1982–1992. doi:10.1182/blood.V90.5.1982
89. Okada N, Lin CP, Ribeiro MC, et al. A positive feedback between p53 and miR-34 miRNAs mediates tumor suppression. Genes Dev. 2014;28(22):438–450. doi:10.1101/gad.233585.113
90. Shah V, Johnson DC, Sherborne AL, et al. Subclonal TP53 copy number is associated with prognosis in multiple myeloma. Blood. 2018;132(23):2465–2469. doi:10.1182/blood-2018-06-857250
91. Binder M, Rajkumar SV, Ketterling RP, et al. Prognostic implications of abnormalities of chromosome 13 and the presence of multiple cytogenetic high-risk abnormalities in newly diagnosed multiple myeloma. Blood Cancer J. 2017;7(9):e600. doi:10.1038/bcj.2017.83
92. Baysal M, Demirci U, Umit E, et al. Concepts of double hit and triple hit disease in multiple myeloma, entity and prognostic significance. Sci Rep. 2020;10(1):5991. doi:10.1038/s41598-020-62885-0
93. Sztupinszki Z, Diossy M, Krzystanek M, et al. Migrating the SNP array-based homologous recombination deficiency measures to next generation sequencing data of breast cancer. NPJ Breast Cancer. 2018;4(1):1–4. doi:10.1038/s41523-018-0066-6
94. Szalat R, Avet-Loiseau H, Munshi NC. Gene expression profiles in myeloma: ready for the real world? Clin Cancer Res. 2016;22:5434–5442. doi:10.1158/1078-0432.CCR-16-0867
95. Decaux O, Lodé L, Magrangeas F, et al. Prediction of survival in multiple myeloma based on gene expression profiles reveals cell cycle and chromosomal instability signatures in high-risk patients and hyperdiploid signatures in low-risk patients: a study of the intergroupe francophone du myelome. J Clin Oncol. 2008;26(29):4798–4805. doi:10.1200/JCO.2007.13.8545
96. Kuiper R, Broyl A, de Knegt Y, et al. A gene expression signature for high-risk multiple myeloma. Leukemia. 2012;26(11):2406–2413. doi:10.1038/leu.2012.127
97. Zhan FH, Barlogie B, John DS
98. Samur MK, Minvielle S, Gulla A, et al. Long intergenic non-coding RNAs have an independent impact on survival in multiple myeloma. Leukemia. 2018;32(12):2626–2635. doi:10.1038/s41375-018-0116-y
99. Manier S, Liu C-J, Avet-Loiseau H, et al. Prognostic role of circulating exosomal miRNAs in multiple myeloma. Blood. 2017;129(17):2429–2436. doi:10.1182/blood-2016-09-742296
100. Agirre X, Castellano G, Pascual M, et al. Whole-epigenome analysis in multiple myeloma reveals DNA hypermethylation of B cell-specific enhancers. Genome Res. 2015;25(4):478–487. doi:10.1101/gr.180240.114
101. Marango J, Shimoyama M, Nishio H, et al. The MMSET protein is a histone methyltransferase with characteristics of a transcriptional corepressor. Blood. 2008;111(6):3145–3154. doi:10.1182/blood-2007-06-092122
102. Pawlyn C, Kaiser MF, Heuck C, et al. The spectrum and clinical impact of epigenetic modifier mutations in myeloma. Clin Cancer Res. 2016;22(23):5783–5794. doi:10.1158/1078-0432.CCR-15-1790
103. Mason MJ, Schinke C, Eng CLP, et al. Multiple myeloma DREAM challenge reveals epigenetic regulator PHF19 as marker of aggressive disease. Leukemia. 2020;34(7):1866–1874. doi:10.1038/s41375-020-0742-z
104. Croonquist PA, Van Ness B. The polycomb group protein enhancer of zeste homolog 2 (EZH 2) is an oncogene that influences myeloma cell growth and the mutant ras phenotype. Oncogene. 2005;24(41):6269–6280. doi:10.1038/sj.onc.1208771
105. Neo WH, Lim JF, Grumont R, Gerondakis S, Su I-H. c-Rel regulates Ezh2 expression in activated lymphocytes and malignant lymphoid cells. J Biol Chem. 2014;289(46):31693–31707. doi:10.1074/jbc.M114.574517
106. Rastgoo N, Pourabdollah M, Abdi J, Reece D, Chang H. Dysregulation of EZH2/miR-138 axis contributes to drug resistance in multiple myeloma by downregulating RBPMS. Leukemia. 2018;32(11):2471–2482. doi:10.1038/s41375-018-0140-y
107. Tremblay-LeMay R, Rastgoo N, Pourabdollah M, Chang H. EZH2 as a therapeutic target for multiple myeloma and other haematological malignancies. Biomarker Res. 2018;6(1):1–10. doi:10.1186/s40364-018-0148-5
108. Harding T, Swanson J, Van Ness B. EZH2 inhibitors sensitize myeloma cell lines to panobinostat resulting in unique combinatorial transcriptomic changes. Oncotarget. 2018;9(31):21930. doi:10.18632/oncotarget.25128
109. Nylund P, Atienza Párraga A, Haglöf J, et al. A distinct metabolic response characterizes sensitivity to EZH2 inhibition in multiple myeloma. Cell Death Dis. 2021;12(2):1–16. doi:10.1038/s41419-021-03447-8
110. Mulligan G, Lichter DI, Di Bacco A, et al. Mutation of NRAS but not KRAS significantly reduces myeloma sensitivity to single-agent bortezomib therapy. J Am Soc Hematol. 2014;123:632–639.
111. Andrulis M, Lehners N, Capper D, et al. Targeting the BRAF V600E mutation in multiple myeloma. Cancer Discov. 2013;3(8):862–869. doi:10.1158/2159-8290.CD-13-0014
112. Lonial S, Boise LH, Kaufman J. How I treat high-risk myeloma. Blood. 2015;126(13):1536–1543. doi:10.1182/blood-2015-06-653261
113. Walker BA, Mavrommatis K, Wardell CP, et al. Identification of novel mutational drivers reveals oncogene dependencies in multiple myeloma. Blood. 2018;132(6):587–597. doi:10.1182/blood-2018-03-840132
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.