Back to Journals » Neuropsychiatric Disease and Treatment » Volume 21
Genetic Polymorphisms and Gene-Environment Interactions in Persistent Post-Stroke Depression
Authors Lan Y ![]() , Li X, Zhao X, Liang W, Pan C
, Li X, Zhao X, Liang W, Pan C ![]() , Qiu X, Miao J, Li G, Zhu Z, Zhu S
, Qiu X, Miao J, Li G, Zhu Z, Zhu S
Received 13 August 2025
Accepted for publication 16 December 2025
Published 25 December 2025 Volume 2025:21 Pages 2881—2894
DOI https://doi.org/10.2147/NDT.S560475
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Taro Kishi
Yan Lan,* Xianxian Li,* Xin Zhao, Wenwen Liang, Chensheng Pan, Xiuli Qiu, Jinfeng Miao, Guo Li, Zhou Zhu, Suiqiang Zhu
Department of Neurology, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, 430030, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Zhou Zhu, Department of Neurology, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, 1095 Jiefang Avenue, Qiaokou District, Wuhan, Hubei, 430030, People’s Republic of China, Tel +86-18171081029, Email [email protected] Suiqiang Zhu, Department of Neurology, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, 1095 Jiefang Avenue, Qiaokou District, Wuhan, Hubei, 430030, People’s Republic of China, Tel +86-13035101141, Email [email protected]
Purpose: Post-stroke depression (PSD) is the most common psychiatric complication after stroke, and its persistent form carries greater symptom burden and poorer long-term outcomes. The mechanisms of persistent PSD remain unclear. We investigated genetic variants associated with persistent PSD and evaluated prespecified gene–environment (G×E) interactions with modifiable stroke risk factors (lifestyle, diet, and common biomarkers) to test whether genotype modifies susceptibility across different environmental exposures.
Patients and Methods: Patients with first-onset acute ischemic stroke who met the inclusion criteria were recruited from three hospitals in Central China between May 2018 and October 2023. A nested case–control study from May 2018 to December 2020 was conducted for initial screening of PSD-associated single nucleotide polymorphisms (SNPs) via whole-exome sequencing (WES). Validation of risk SNPs was performed in a subsequent cohort enrolled between December 2020 and October 2023. Further, risk SNPs for persistent PSD were identified, and a G×E interaction model was applied to explore how environmental exposures modulate genetic risk in persistent PSD pathogenesis. Sensitivity analyses confirmed the robustness of the results.
Results: Through WES association analysis and validation, nine SNPs potentially related to PSD onset were identified: rs1055851, rs12647814, rs11108643, rs2481880, rs9965081, rs846791, rs4434123, rs1390318, and rs824695. Among these, rs9965081 showed a significant correlation with persistent PSD. This variant interacts with serum low-density lipoprotein cholesterol (LDL-C) levels in the development of persistent PSD and was validated by subgroup analysis.
Conclusion: rs9965081 may be a persistent PSD–associated SNP that interacts with serum LDL-C levels. Carriers of the rs9965081 risk allele are more sensitive to LDL-C fluctuations and therefore have greater susceptibility to persistent PSD.
Keywords: rs9965081, whole‑exome sequencing, low‑density lipoprotein cholesterol, ischemic stroke cohort
Introduction
Stroke ranks first globally in disability rate and second in mortality rate,1 with China being the developing country most burdened by stroke.2 According to the Guidelines for Primary Prevention of Ischemic Cerebrovascular Disease in China, the modifiable risk factors for acute ischemic stroke include hypertension, hypercholesterolemia (especially low-density lipoprotein cholesterol level, LDL-C), diabetes mellitus, hyperhomocysteinemia, smoking, alcohol consumption, body mass index (BMI), and diet and nutrition.50 Post-stroke depression (PSD) is the most common psychiatric complication after stroke, with an incidence rate of 18.8–41.8% at different time points after stroke.3–6 Previous studies had indicated that PSD can be transient, persistent, or maybe recurrent.3,7 Researchers have found that persistent PSD after stroke is more likely to lead to disability compared to other types of depression, with more severe depression levels, potentially leading to poor long-term functional prognosis and significantly decreased quality of life after a stroke.3,8,9
Persistent PSD is a complex condition influenced by sociopsychological, biological, and neuroimaging factors and also exhibits a certain genetic predisposition.10 According to the PRIOD study, persistent PSD may be associated with age, gender, stroke severity, and history of hypertension.8 Numerous studies have indicated that the left frontal lobe and left basal ganglia play a crucial role in the risk of PSD onset and the severity of depression.11,12 With the advent of the human connectome/brain network era, the exploration of PSD etiology is no longer limited to a single lesion or region of interest (ROI). In the previous research, we identified characteristic structural network disconnection patterns associated with PSD through voxel-based structural disconnection analysis.13 Furthermore, most researchers believe that PSD is also influenced by genetic factors. However, the pathogenesis of persistent PSD has not been fully explored and requires more scientifically rigorous research.
Guided by the monoamine imbalance and neuroinflammation hypotheses, related research mainly focuses on the following aspects: the serotonin transporter-linked polymorphic region (5-HTTLPR) S allele and serotonin transporter intron 2 variable number tandem repeat (STin2 VNTR) 9/12 or 12/12 genotypes, which markedly elevate PSD risk;14 serotonin (5-hydroxytryptamine, 5-HT) receptor polymorphisms that impair 5-HT signaling;15 interleukin-10 (IL-10) −1082A/A and interleukin-4 (IL-4) +33C/C variants linked to reduced anti-inflammatory activity;16 and brain-derived neurotrophic factor (BDNF) and apolipoprotein E (APOE) polymorphisms associated with PSD.17,18 To date, most studies have been limited to a single genetic locus or single nucleotide polymorphism (SNP). Such single-gene effects offer limited predictive value and fail to account for the impact of external environmental factors; therefore, a broader range of genetic polymorphisms should be incorporated for further investigation. Whole‐exome sequencing (WES) covers just 1–2% of the genome yet harbors approximately 85% of pathogenic variants, and its breadth, depth and cost‐effectiveness have driven widespread clinical adoption.
However, contemporary medical research recognizes that disease etiology rarely stems from genetic or environmental factors in isolation, but rather from their complex interplay. Investigating gene-environment interactions (G×E) provides critical insights into disease pathogenesis, particularly for mood and affective disorders.19 G×E research elucidates population-level variations in disease susceptibility and reveals how genetic vulnerabilities may manifest differentially across environmental contexts. For example, studies demonstrate divergent depression risk among individuals exposed to comparable stressors.20 Importantly, G×E models can uncover novel exposure effects in genetically susceptible subgroups that conventional analyses might miss. This was exemplified when protein intake showed no overall association with cognitive decline, but significant effects emerged in APOE ε4 carriers upon G×E analysis.21
Accordingly, the present study aims to systematically investigate G×E interactions in persistent PSD within a multicenter prospective cohort. Specifically, leveraging WES combined with subsequent validation, we seek to identify key genetic polymorphisms associated with persistent PSD. Focusing on the aforementioned modifiable acute ischemic stroke risk factors—ie, lifestyle, dietary habits, and common biological markers—we will further assess whether persistent PSD susceptibility varies across patient subgroups with distinct genetic profiles and varying environmental exposures, and ultimately develop a comprehensive G×E interaction model to clarify the combined genetic and environmental contributions to persistent PSD pathogenesis.
Materials and Methods
Patients
This prospective multicenter cohort study was conducted at three tertiary hospitals in Central China (Chinese Clinical Trial Registry: ChiCTR-ROC-17013993; registration date: 16 December 2017) and approved by the Ethics Committee of Tongji Medical College, Huazhong University of Science and Technology (approval No.: TJ-IRB20171108). Written informed consent was obtained from all participants in accordance with the Declaration of Helsinki. Between May 2018 and October 2023, 882 consecutive first-ever acute ischemic stroke patients were recruited from Tongji Hospital, Wuhan First Hospital, and Wuhan Central Hospital. Inclusion criteria: (1) age ≥18 years; (2) admission within 7 days of stroke onset; (3) ischemic stroke confirmed by computed tomography (CT) or magnetic resonance imaging (MRI); and (4) informed consent. Exclusion criteria: (1) nonvascular neurological dysfunction; (2) previous depression or other psychiatric disorders; (3) inability to complete assessments due to aphasia, dysarthria, blindness, deafness, or severe cognitive impairment precluding completion of study assessments, as determined clinically by attending neurologists; (4) other neurological diseases (eg, Parkinson’s disease, epilepsy); and (5) transient ischemic attacks (TIA).
Clinical Variables
Within 48 hours of admission, the following were completed: (1) baseline data collection via case report form (CRF) covering demographics and comorbidities, along with neuropsychological and cognitive assessment by two trained raters using the 17-item Hamilton Depression Rating Scale (HDRS-17) at admission and follow-up; (2) fasting blood sampling before 06:00 the next day to measure blood biochemical indicators.
WES was performed by Novogene Co., Ltd. on the Illumina NovaSeq 6000 platform (paired-end 150 bp [PE150] reads). Libraries were constructed using the Agilent SureSelect Human All Exon V6 kit, and variant calling was conducted with GATK HaplotypeCaller, followed by annotation using ANNOVAR. Subsequent genotype validation of candidate SNP loci was entrusted to Bomiao Biotechnology Co., Ltd. and performed using the massARRAY® system. The detection followed standardized protocols, and high-quality genotypic data were obtained for association validation analysis.
Depressive symptoms were evaluated via outpatient or video interview at 2 weeks, 3 months, 6 months, and 1 year (14±2 days, 84±7 days, 182±7 days, 360±7 days) after stroke, respectively. Patient baseline data were concealed from all raters during the study. We defined PSD according to the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-5) criteria and an HDRS-17 score ≥ 8, and defined persistent PSD as at least two consecutive positive assessments without remission or recurrence.3,8,22 Baseline covariates required for analysis were complete among included participants, outcome missingness (loss to follow-up or missing HDRS-17) was handled by exclusion, and no imputation was performed.
Statistical Analysis
This study comprised four phases: 1) Employing a nested case-control (NCC) study to conduct whole-exome association analysis for the initial screening of candidate SNPs associated with PSD; 2) Performing targeted genotyping of the candidate SNPs via massARRAY technology and utilizing PSM analysis to validate SNPs significantly associated with PSD in the subsequent cohort; 3) Systematically assessing the strength and independence of the validated SNPs associations with persistent PSD; 4) Exploring the G×E interactions between the risk SNPs and stroke-related modifiable risk factors (eg, metabolic indices, lifestyle factors) in the pathogenesis of persistent PSD. See Figure 1.
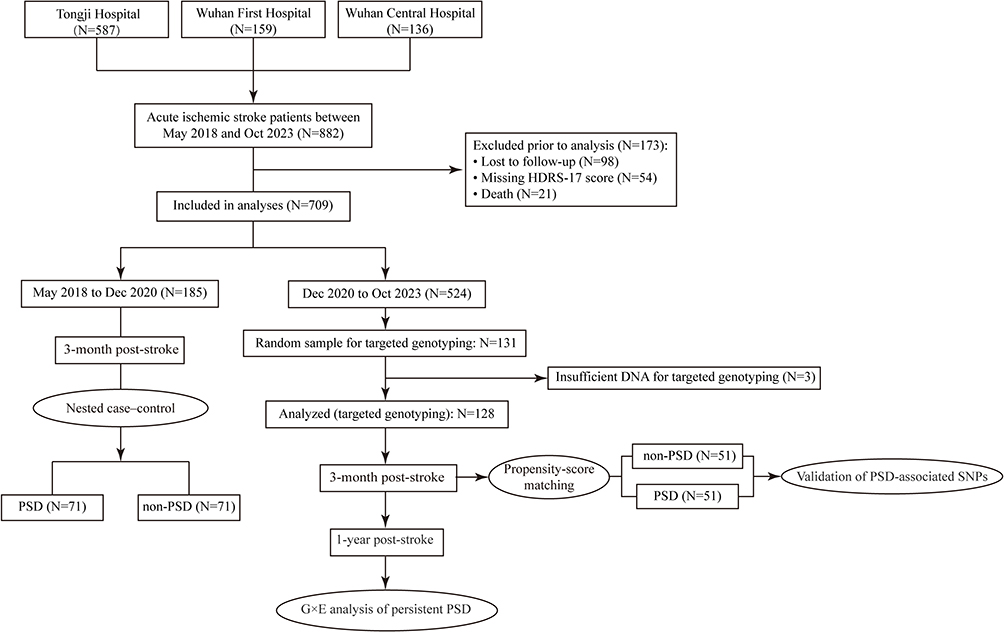 |
Figure 1 Selection flowchart. |
To screen for genetic polymorphisms associated with PSD, a NCC study was conducted within our research cohort—an approach that integrates the bias-mitigation strengths of prospective cohort studies with the efficiency of case-control designs. Between May 2018 and December 2020, patients who completed 3-month post-stroke follow-up and were definitively diagnosed with PSD were defined as cases. With age (±2 years) and gender as matching variables, 1:1 random nearest-neighbor matching was performed with non-PSD patients to establish the control group. Genomic DNA (gDNA) was extracted from baseline blood samples of both the case and control groups and subsequently subjected to WES. We corrected for population stratification in our nested cohort by principal component analysis (PCA) of the Novo-Zhonghua and 1000 Genomes datasets. Following aggregation of SNP variants across all samples, a two-tailed Fisher’s exact test was used to compare variant distribution differences between the two groups. To retain reliable genetic variants, a stringent quality control (QC) process was implemented: ① Variants with a genotyping call rate > 0.9 in the case group were retained to ensure the completeness and reliability of variant data for cases; ② Based on annotations from the RepeatMasker and genomicSuperDups databases, only variants annotated as “.” (ie, non-genomic repeat regions or segmental duplication regions) were retained to exclude false-positive variant interference from repetitive sequences; ③ Bonferroni correction was used to determine the significance threshold, which was set at 0.05 / 100,000 = 5×10−7; ④ Odds ratio (OR) > 1 was specified to prioritize potential PSD risk SNPs with higher allele frequencies in the case group.
In the subsequent validation study, a non-overlapping cohort was selected using simple random sampling from participants enrolled between December 2020 and October 2023, who were stratified into PSD and non-PSD groups based on 3-month post-stroke assessments. Propensity Score Matching (PSM) was applied to mitigate confounding and sample bias. Target SNP genotyping was performed using the internationally standardized massARRAY® Analyzer 4 platform—a gold-standard matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry–based platform for genetic variant validation, characterized by high accuracy, sensitivity, and throughput. Strict QC was implemented during the assay: gDNA was qualified via NanoDrop2000 and 1.25% agarose gel electrophoresis (concentration >20 ng/μL, OD260/OD280=1.6–2.2, OD260/OD230>0.6, no OD230 absorption peak, intact gDNA); multiplex SNP primers were designed/optimized with Agena Assay Designer 4.0, followed by polymerase chain reaction (PCR) to enrich target fragments, and the amplified products underwent single-base extension and purification, were spotted on SpectroCHIP via MassARRAY Nanodispenser RS1000, detected by MALDI-TOF mass spectrometry, and analyzed with TYPER 4.0 software. For PSM, variables with a standardized mean difference >10% among common confounders were included as matching factors. Nearest-neighbor matching was performed at a 1:1 ratio with a caliper width of 0.2 standard deviations of the propensity score. Distributions of target SNP variants were compared between matched PSD and non-PSD groups, where associations were tested using χ2-test and multivariable logistic regression. Subsequently, we assessed the association of these validated SNPs with persistent PSD using χ2-tests.
Ultimately, we evaluated G×E interactions by including multiplicative terms between risk SNPs and modifiable acute ischemic stroke risk factors (lifestyle, dietary habits, common biomarkers) in logistic regression models, with subgroup linear regression of predicted risk scores visualized to illustrate genotype-specific environmental susceptibility.
Results
From May 2018 to October 2023, we enrolled 882 first-ever acute ischemic stroke patients at three hospitals in central China; over 12 months, 98 (11.1%) were lost to follow-up, 54 (6.1%) had missing outcome data, and 21 (2.4%) died, yielding 709 completers.
Whole-Exome Variant Association Analysis for PSD Phenotype
During the period from May 2018 to December 2020, a total of 185 patients were enrolled in this study, 71 patients who completed the 3-month follow-up after stroke were diagnosed with PSD, and 71 non-PSD patients were matched by gender and age and assigned to the control group in the NCC study. As shown in Figure 2, principal component analysis of PSD cases and non-PSD controls revealed tight clustering with no significant population stratification, and alignment with the Asian cohort of the 1000 Genomes Project, thereby excluding confounding by ancestry. WES was performed to compare genetic variant differences between the two groups, and identified 11 potential risk SNPs associated with PSD (Figure 3). These SNPs—rs1055851 (ERO1B c.1395 C>G), rs12647814 (BMP3 c.1228 A>T), rs259136 (intronic STEAP2-AS1), rs11108643 (CFAP54 c.6503 T>C), rs2481880 (UFM1 c.112–1337 C>T), rs9965081 (CCDC178 c.1394 A>T), rs846791 (ASCC3 c.91–7 C>T), rs1109882 (CCDC152 c.262+77 G>T), rs4434123 (SLC9C1 c.1970 G>A), rs1390318 (BMP3 c.1228–1239 C>T) and rs824695 (GXYLT1 c.487–12 A>T)—are detailed in Table 1 and constitute potential genetic susceptibility markers for PSD.
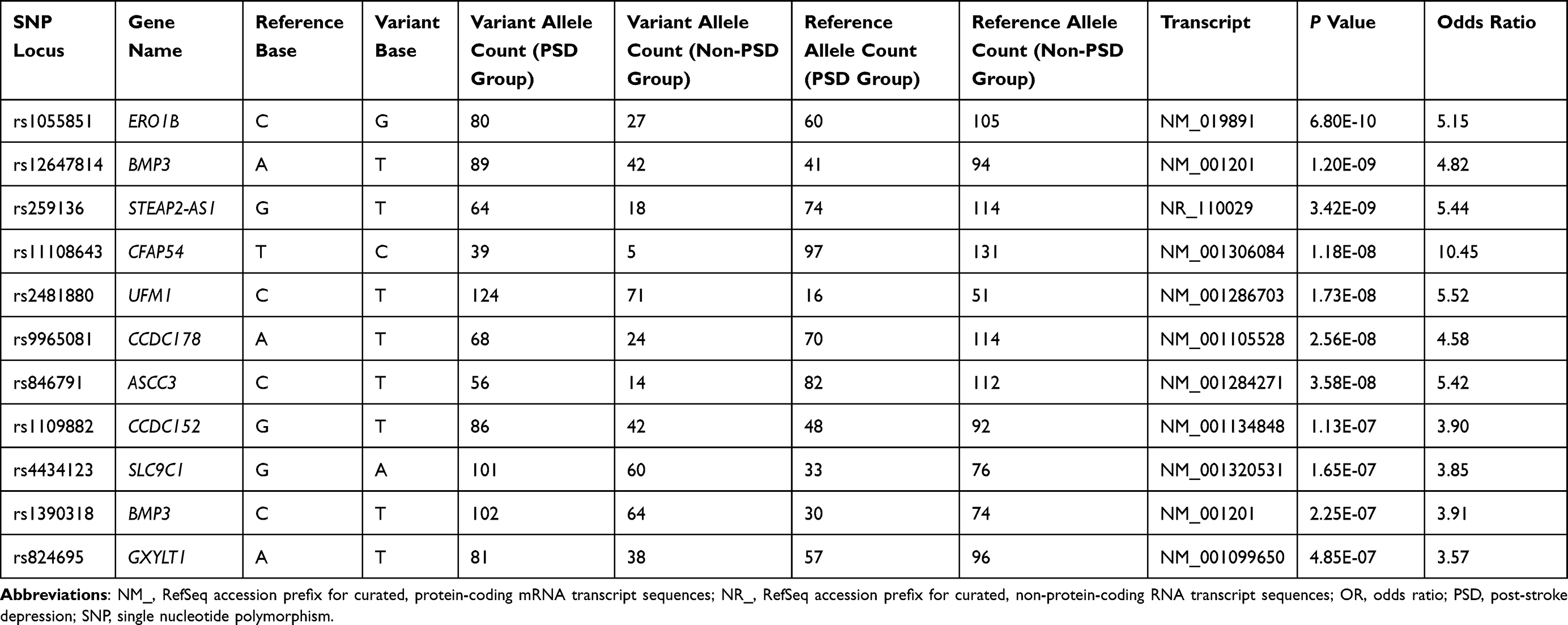 |
Table 1 Whole-Exome Association Analysis of the Post-Stroke Depression Phenotype |
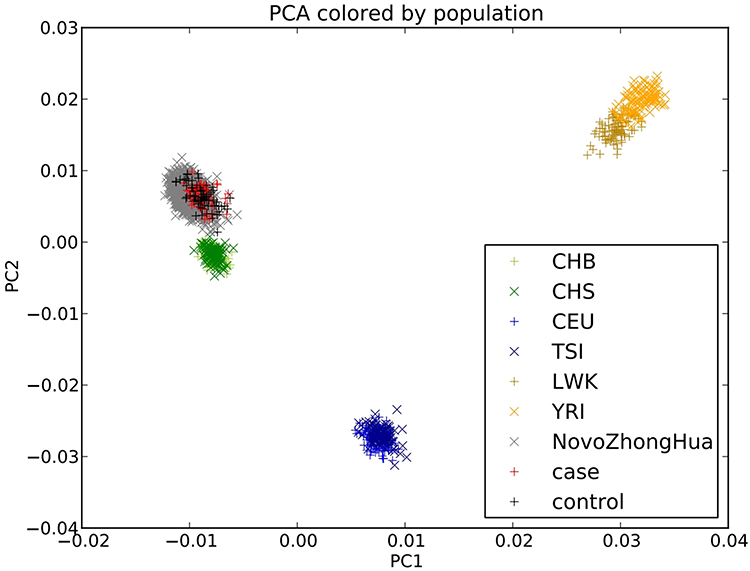 |
Figure 2 Principal component analysis plot. |
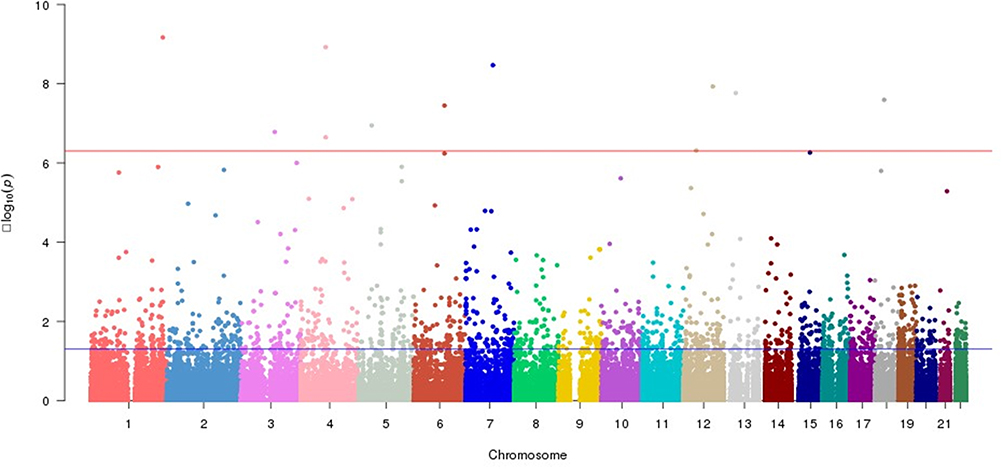 |
Figure 3 Manhattan plot of whole-exome association analysis. |
Validation of PSD‑Associated SNPs
In the validation cohort randomly sampled from patients enrolled after December 2020 (n=128, 131 patients (25%) were randomly selected, while 3 patients had insufficient baseline blood samples), PSM yielded 51 PSD cases and 51 controls (nine PSD cases unmatched). Post‐match kernel density plots (Figure S1) and absolute standardized mean differences (Figure S2) confirmed adequate balance—age, BMI and hypertension history all had absolute standardized mean differences (ASDs) <10%, and smoking history, although 12%, was deemed acceptable (Table S1). In this matched cohort, we evaluated 11 candidate SNPs and found 9 SNPs significantly enriched in PSD cases: rs1055851 (OR 3.39, 95% confidence interval [CI] 1.50–7.67, P = 0.003), rs12647814 (OR 2.61, 95% CI 1.17–5.80, P = 0.017), rs11108643 (OR 3.15, 95% CI 1.03–9.62, P = 0.038), rs2481880 (OR 4.78, 95% CI 1.88–12.15, P < 0.001), rs9965081 (OR 3.94, 95% CI 1.63–9.53, P = 0.002), rs846791 (OR 4.40, 95% CI 1.66–11.64, P = 0.002), rs4434123 (OR 2.27, 95% CI 1.02–5.10, P = 0.044), rs1390318 (OR 2.54, 95% CI 1.11–5.80, P = 0.025) and rs824695 (OR 2.64, 95% CI 1.18–5.89, P = 0.017), identifying them as candidate PSD susceptibility SNPs (Table 2).
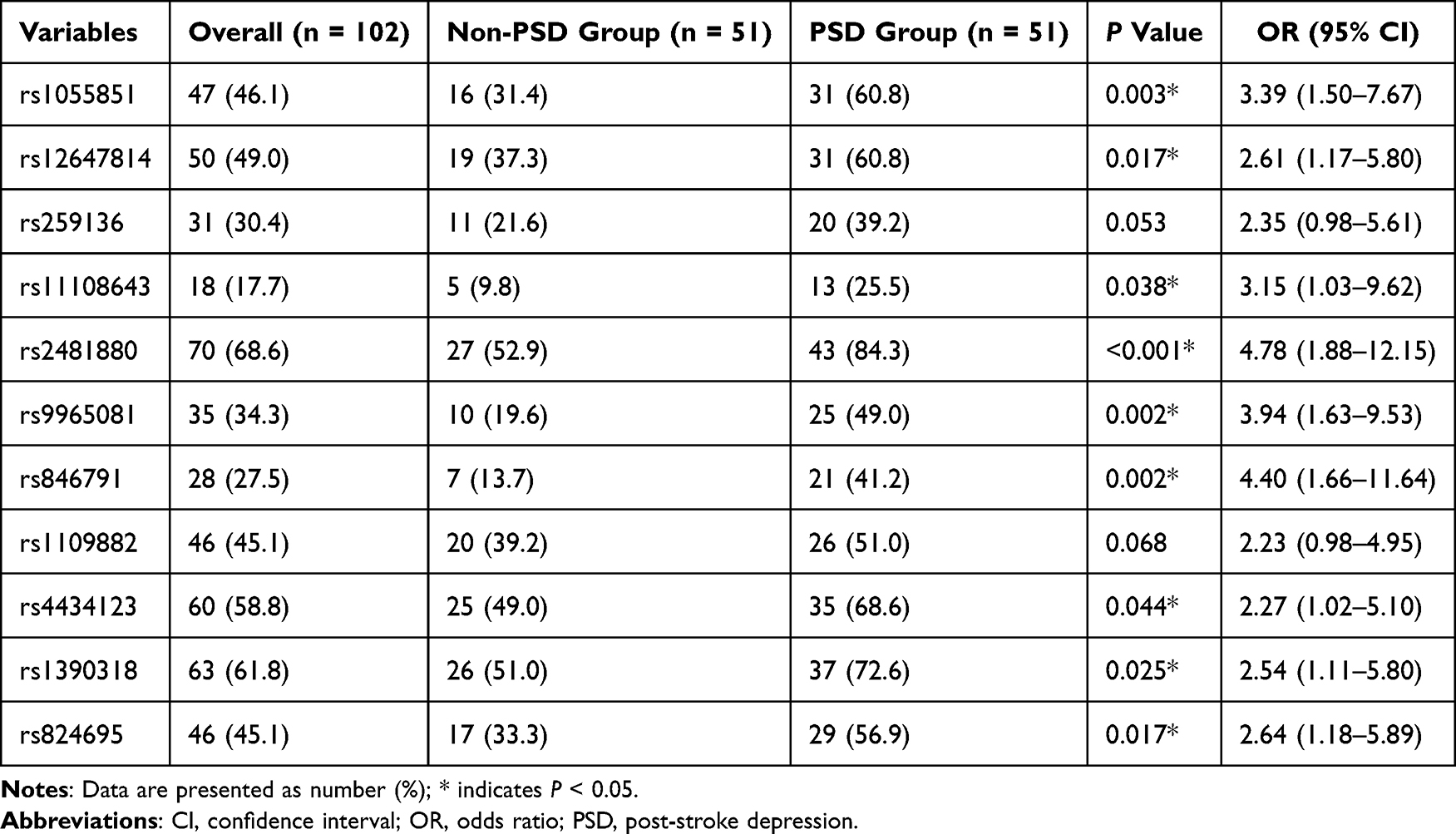 |
Table 2 Comparison of Target Locus Distribution Between Propensity-Matched Post-Stroke Depression and Non-Post-Stroke Depression Groups |
SNPs Associated with Persistent PSD
In the one‐year follow‐up of the validation cohort (n=128), 31 (24.2%) met criteria for persistent PSD. The median age was 58 years and 34.4% (44 patients) were female. As detailed in Table 3, genotype frequencies of 8 candidate SNPs—rs1055851 (P = 0.005), rs11108643 (P = 0.027), rs2481880 (P = 0.015), rs9965081 (P = 0.001), rs846791 (P = 0.007), rs4434123 (P = 0.004), rs1390318 (P = 0.043) and rs824695 (P = 0.005)—were significantly higher in the persistent PSD group than in non‐persistent cases. In multivariable logistic regression analysis adjusting for demographic, lifestyle and clinical covariates (Table 4), only rs9965081 remained independently associated with persistent PSD (Model 3: OR = 3.09, 95% CI 1.22–7.78, P = 0.017).
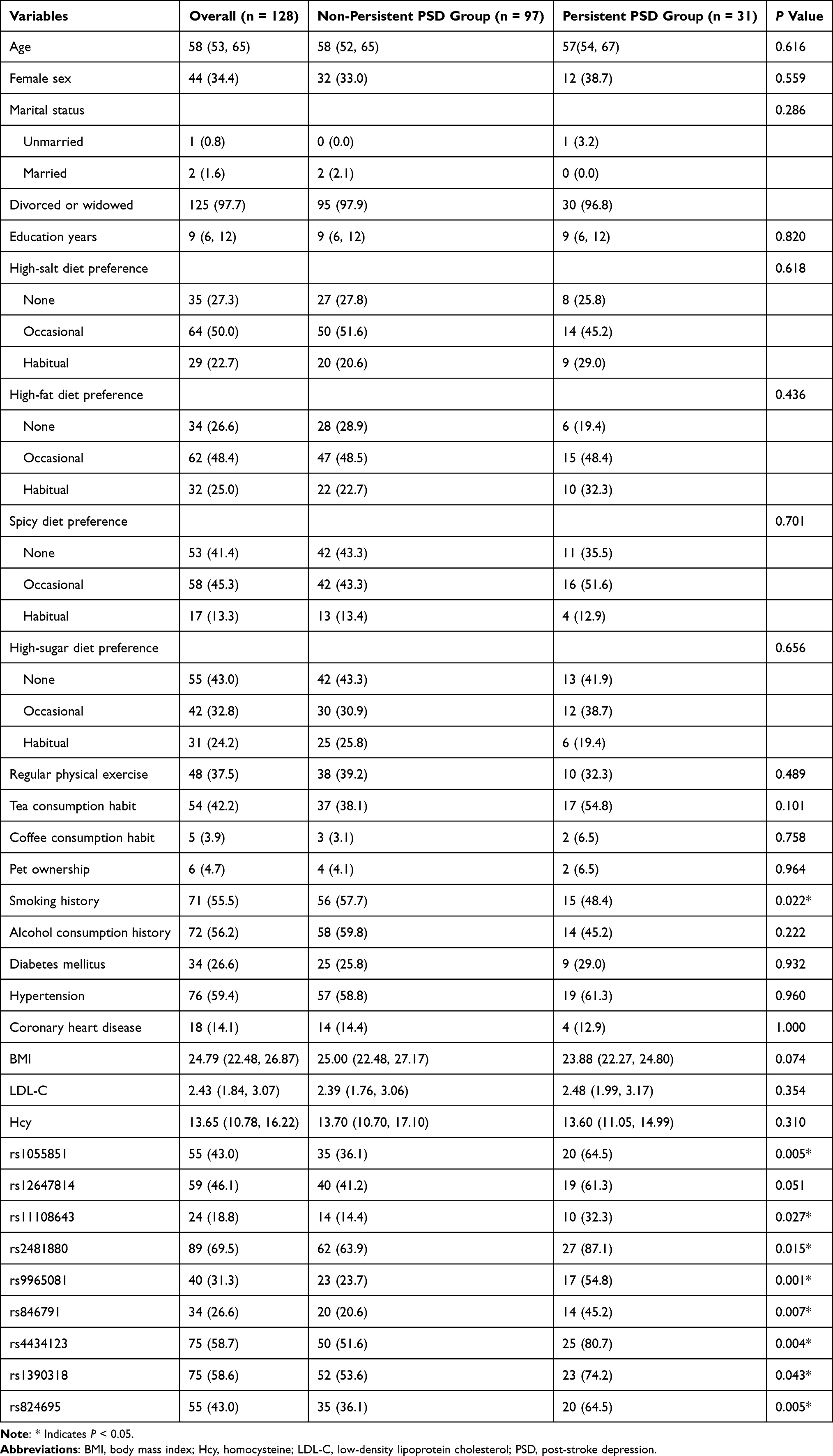 |
Table 3 Baseline Characteristics of the Study Cohort |
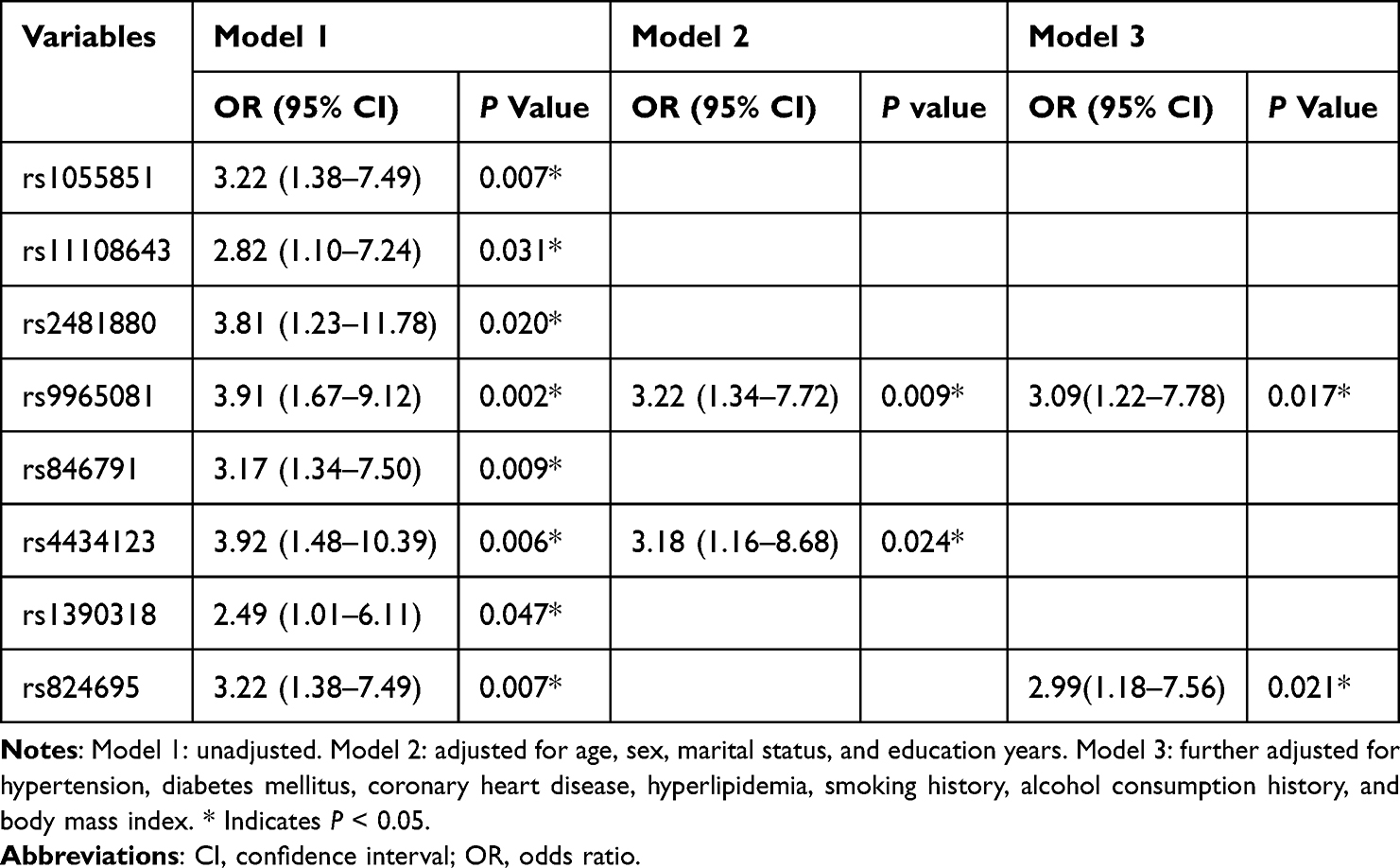 |
Table 4 Multivariate Regression Analysis of Locus Variants Associated with Persistent Post-Stroke Depression |
Gene–Environment Interaction Analysis in Persistent PSD
We assessed G×E interactions between rs9965081 and modifiable acute ischemic stroke risk factors for persistent PSD (Table S2), with analyses adjusted across multiple models incorporating distinct confounder sets. While no significant association was detected between serum LDL-C levels and persistent PSD in initial unadjusted models, the integration of the rs9965081 × serum LDL-C interaction term uncovered a robust association between this interaction and persistent PSD. Importantly, the rs9965081×LDL-C interaction was the only term consistently significant across all models (Model 6: OR 1.75, 95% CI 1.27–2.41, P = 0.001), whereas the rs9965081×spice-preference term lost significance once fully adjusted. Subgroup analysis (Figure 4) demonstrated a stronger association between elevated serum LDL-C and increased persistent PSD risk in rs9965081 risk allele carriers vs non-carriers, reflecting greater genetic susceptibility and enhanced vulnerability to LDL-C-related adverse effects in carriers.
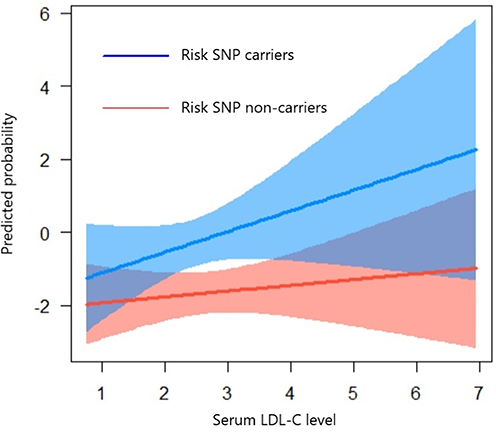 |
Figure 4 Subgroup differences in low-density lipoprotein cholesterol susceptibility. |
Discussion
In this multicenter prospective cohort study, we performed initial screening via whole-exome SNP association analysis, followed by validation using massARRAY-based targeted genotyping. Through this approach, several SNPs associated with PSD were identified and validated. Notably, rs9965081 was independently associated with persistent PSD and exhibited a significant multiplicative interaction with serum LDL-C levels, suggesting it as a potential risk SNP for persistent PSD. Specifically, among carriers of the rs9965081 risk allele, the risk of developing persistent PSD increased more significantly with elevated serum LDL-C levels.
rs9965081, located in CCDC178 (a protein-coding gene), localizes predominantly to the cytoskeleton, cytosol, and nucleus. Genotype-Tissue Expression (GTEx) data show that CCDC178 mRNA is most abundant in testis and visceral adipose tissue. GWAS Catalog reports an association between rs9965081 and BMI,23 suggesting it may influence metabolic phenotypes by modulating the expression or function of the gene harboring this variant, albeit no functional studies have been reported for this SNP, nor are there relevant ClinVar entries. This genetic association implies that rs9965081 could contribute to PSD susceptibility through a BMI-mediated pathway: the variant may alter BMI homeostasis, which in turn affects stroke risk and post-stroke depressive outcomes. To validate this hypothesis, we analyzed BMI differences between rs9965081 carriers and non-carriers in our Chinese cohort. Although carriers of the rs9965081 risk allele tended to have lower BMI, the difference did not reach statistical significance (Table S3). We attribute this non-significant finding to the relatively small sample size of the study, which may limit statistical power to detect subtle BMI variations. Nevertheless, this trend aligns with the GWAS-reported association between rs9965081 and BMI, providing preliminary evidence for a potential genetic-metabolic pathway in the Chinese population.
Both the 2024 AHA/ASA Guidelines for the Primary Prevention of Stroke and the Chinese Guidelines for the Primary Prevention of Ischemic Cerebrovascular Disease explicitly classify overweight/obesity (assessed by BMI) as a modifiable risk factor for ischemic stroke.24,50 Additionally, BMI is a key factor in depression pathogenesis and effectively reflects obesity status. Obesity’s link to depression has been attributed to genetic predisposition, hypothalamic–pituitary–adrenal axis dysregulation, inflammatory activation, energy-metabolism alterations, and gut-microbiota changes,25–27 as well as metabolic-syndrome–related circadian disruption, dyslipidemia, and insulin resistance.28–30 Metabolic health status further modulates depression risk independent of BMI,27 and both underweight and overweight status correlate with depressive symptoms.31 In this study, BMI may act as a “common mediator” connecting stroke risk and PSD susceptibility.
Extensive evidence links LDL-C to coronary heart disease and ischemic stroke, with elevated LDL-C increasing stroke risk,32–34 and prior studies suggest a bidirectional relationship with depression: most report that higher LDL-C associates with affective disorders and that lowering LDL-C alleviates symptoms,35–37 although some find low LDL-C increases depression risk.38 Depression itself can reduce LDL-C via BMI-mediated mechanisms,29,39 and LDL-C has been implicated in PSD pathogenesis.40 In our overall cohort, LDL-C showed no independent association with persistent PSD, implying interindividual variability in LDL-C sensitivity. However, in multivariable models incorporating the rs9965081×LDL-C interaction, this term remained significant (Model 6: OR 1.75, 95% CI 1.27–2.41, P = 0.001), indicating LDL-C contributes to PSD risk in a genetically susceptible subset. Subgroup analysis (Figure 4) confirmed that while PSD probability rose with LDL-C in all patients, rs9965081 carriers exhibited markedly greater sensitivity to LDL-C fluctuations.
Given the limited functional data on rs9965081, we consider BMI’s relationships with LDL-C and depressive symptoms to infer potential mechanisms. Population analyses—including the Spanish National Health and Nutrition Examination Survey—report that LDL-C rises with BMI in lean individuals but plateaus or declines at higher BMI levels,41 a pattern recently confirmed in a Chinese cohort.42 Both LDL-C and BMI reflect lipid-metabolic status, and post-stroke oxidative stress–induced dyslipidemia can provoke neurotoxicity and neurodegeneration,40,43 potentially precipitating persistent PSD. Lipid metabolism may also modulate depression via inflammatory pathways: LDL-C’s pro-inflammatory activation of innate immunity triggers NLRP3 inflammasome formation,44–46 while BMI is a major driver of systemic low-grade inflammation.26,47,48 The inflammation hypothesis in depression is well established, and our prior work implicates systemic low-grade inflammation as a key risk factor for late PSD.49 Although lipid-lowering therapy post-stroke reduces both LDL-C and inflammation, residual inflammatory activity often persists,51 which may underlie the association between high baseline LDL-C and persistent PSD.
This study is the first to explore genetic polymorphism and gene–environment interactions in persistent PSD, with important public‐health implications. Identifying the rs9965081 × LDL‑C interaction may enable targeted interventions: carriers of the rs9965081 risk allele could derive greater benefit from intensive LDL‑C lowering to reduce persistent PSD risk. However, key limitations include the modest sample size—both the WES association and subsequent validation were conducted in relatively small cohorts, potentially undermining statistical power. The use of PSM in the validation cohort bolsters methodological rigor, but replication in larger, independent samples is needed to confirm and generalize these findings. Additionally, the lack of in vitro or in vivo functional studies limits mechanistic insight. Future studies are warranted to conduct targeted basic experiments to elucidate the underlying molecular mechanisms.
Conclusion
WES association analysis, followed by massARRAY-based genotyping validation, identified multiple candidate SNPs associated with PSD. Subsequent analyses of genetic polymorphisms and gene-environment interactions in persistent PSD confirmed rs9965081 as a risk SNP for this phenotype, which displayed a significant multiplicative interaction with LDL-C. Specifically, carriers of the rs9965081 risk allele demonstrated enhanced susceptibility to fluctuations in LDL-C levels and a substantially higher risk of developing persistent PSD. These findings highlight rs9965081 as a potential genetic marker for persistent PSD susceptibility, particularly in individuals with abnormal LDL-C metabolism. The identified G×E interaction provides novel insights into the pathogenic mechanisms underlying persistent PSD, suggesting that targeted lipid management (eg, LDL-C control) may mitigate PSD risk in genetically susceptible individuals.
Data Sharing Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Ethics Approval and Informed Consent
This study was approved by the Ethics Committee of Tongji Medical College, Huazhong University of Science and Technology (ID: TJ-IRB20171108) and conducted in accordance with the Declaration of Helsinki. Written informed consent was obtained from all participants. The clinical trial was registered at Chinese Clinical Trial Registry (ChiCTR-ROC-17013993; https://www.chictr.org.cn/showproj.html?proj=23653; 16 December 2017).
Acknowledgments
We are deeply grateful to the patients and caregivers for their participation in this project, and to the clinical staff for their unwavering support and vital contributions.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising, or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the National Key Research and Development Program of China (Grant No. 2017YFC1310000) and the Hubei Technological Innovation Special Fund (Grant No. 2019ACA132). The funders had no role in study design, data collection, analysis, decision to publish, or preparation of the manuscript.
Disclosure
The authors declare that they have no competing interests.
References
1. Feigin VL, Nguyen G, Cercy K, et al. Global, regional, and country-specific lifetime risks of stroke, 1990 and 2016. N Engl J Med. 2018;379(25):2429–2437.
2. Wang W, Jiang B, Sun H, et al. Prevalence, incidence, and mortality of stroke in China: results from a nationwide population-based survey of 480 687 adults. Circulation. 2017;135(8):759–771. doi:10.1161/CIRCULATIONAHA.116.025250
3. Zhang N, Wang CX, Wang AX, et al. Time course of depression and one‐year prognosis of patients with stroke in Mainland China. CNS Neurosci Ther. 2012;18(6):475–481. doi:10.1111/j.1755-5949.2012.00312.x
4. Aben I, Verhey F, Strik J, Lousberg R, Lodder J, Honig A. A comparative study into the one year cumulative incidence of depression after stroke and myocardial infarction. J Neurol Neurosurg Psychiatry. 2003;74(5):581–585. doi:10.1136/jnnp.74.5.581
5. Bour A, Rasquin S, Aben I, Boreas A, Limburg M, Verhey F. A one-year follow-up study into the course of depression after stroke. J Nutr Health Aging. 2010;14(6):488–493. doi:10.1007/s12603-010-0033-x
6. Kouwenhoven SE, Kirkevold M, Engedal K, Kim HS. Depression in acute stroke: prevalence, dominant symptoms and associated factors. A systematic literature review. Disability Rehabil. 2011;33(7):539–556. doi:10.3109/09638288.2010.505997
7. Whyte EM, Mulsant BH, Vanderbilt J, Dodge HH, Ganguli M. Depression after stroke: a prospective epidemiological study. J Am Geriatr Soc. 2004;52(5):774–778. doi:10.1111/j.1532-5415.2004.52217.x
8. Li LJ, Yao XM, Guan BY, Chen Q, Zhang N, Wang CX. Persistent depression is a predictor of quality of life in stroke survivors: results from a 5-year follow-up study of a Chinese cohort. Chin Med J. 2019;132(18):2206–2212. doi:10.1097/CM9.0000000000000400
9. Shi YZ, Xiang YT, Wu SL, et al. The relationship between frontal lobe lesions, course of post-stroke depression, and 1-year prognosis in patients with first-ever ischemic stroke. PLoS One. 2014;9(7):e100456. doi:10.1371/journal.pone.0100456
10. Guo J, Wang J, Sun W, Liu X. The advances of post-stroke depression: 2021 update. J Neurol. 2022;269(3):1236–1249. doi:10.1007/s00415-021-10597-4
11. Robinson RG, Shoemaker WJ, Schlumpf M, Valk T, Bloom FE. Effect of experimental cerebral infarction in rat brain on catecholamines and behaviour. Nature. 1975;255(5506):332–334. doi:10.1038/255332a0
12. Robinson RG, Kubos KL, Starr LB, Rao K, Price TR. Mood disorders in stroke patients. Importance of location of lesion. Brain. 1984;107(Pt 1):81–93. doi:10.1093/brain/107.1.81
13. Pan C, Li G, Jing P, et al. Structural disconnection-based prediction of poststroke depression. Transl Psychiatry. 2022;12(1):461. doi:10.1038/s41398-022-02223-2
14. Kohen R, Cain KC, Mitchell PH, et al. Association of serotonin transporter gene polymorphisms with poststroke depression. Arch Gen Psychiatry. 2008;65(11):1296–1302. doi:10.1001/archpsyc.65.11.1296
15. Fuying Z, Yingying Y, Shining Z, et al. Novel susceptibility genes were found in a targeted sequencing of stroke patients with or without depression in the Chinese Han population. J Affective Disorders. 2019;255:1–9. doi:10.1016/j.jad.2019.05.023
16. Kim JM, Stewart R, Kim SW, et al. Associations of cytokine gene polymorphisms with post-stroke depression. World J Biol Psychiatry. 2012;13(8):579–587. doi:10.3109/15622975.2011.588247
17. Chatzistefanidis D, Giannopoulos S, Spengos K, et al. Apolipoprotein E polymorphisms and ischaemic stroke: a two-center Greek study. Eur J Neurol. 2014;21(8):1083–1088. doi:10.1111/ene.12365
18. Kim JM, Stewart R, Kang HJ, et al. A longitudinal study of BDNF promoter methylation and genotype with poststroke depression. J Affect Disord. 2013;149(1–3):93–99. doi:10.1016/j.jad.2013.01.008
19. Manuck SB, McCaffery JM. Gene-environment interaction. Annu Rev Psychol. 2014;65(1):41–70. doi:10.1146/annurev-psych-010213-115100
20. Caspi A, Sugden K, Moffitt TE, et al. Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science. 2003;301(5631):386–389. doi:10.1126/science.1083968
21. Zhang Y, Jin X, Lutz MW, et al. Interaction between APOE epsilon4 and dietary protein intake on cognitive decline: a longitudinal cohort study. Clin Nutr. 2021;40(5):2716–2725. doi:10.1016/j.clnu.2021.03.004
22. Berg A, Lönnqvist J, Palomäki H, Kaste M. Assessment of depression after stroke. Stroke. 2009;40(2):523–529. doi:10.1161/STROKEAHA.108.527705
23. MacArthur J, Bowler E, Cerezo M, et al. The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog). Nucleic Acids Res. 2017;45(D1):D896–D901. doi:10.1093/nar/gkw1133
24. Bushnell C, Kernan WN, Sharrief AZ, et al. 2024 guideline for the primary prevention of stroke: a guideline from the American heart association/American stroke association. Stroke. 2024;55(12):e344–e424. doi:10.1161/STR.0000000000000475
25. Karageorgiou V, Casanova F, O’Loughlin J, et al. Body mass index and inflammation in depression and treatment-resistant depression: a Mendelian randomisation study. BMC Med. 2023;21(1):355. doi:10.1186/s12916-023-03001-7
26. Ambrosio G, Kaufmann FN, Manosso L, et al. Depression and peripheral inflammatory profile of patients with obesity. Psychoneuroendocrinology. 2018;91:132–141. doi:10.1016/j.psyneuen.2018.03.005
27. Malmir H, Mirzababaei A, Moradi S, Rezaei S, Mirzaei K, Dadfarma A. Metabolically healthy status and BMI in relation to depression: a systematic review of observational studies. Diabetes Metab Syndr. 2019;13(2):1099–1103. doi:10.1016/j.dsx.2019.01.027
28. Moreira FP, Jansen K, Mondin TC, et al. Biological rhythms, metabolic syndrome and current depressive episode in a community sample. Psychoneuroendocrinology. 2016;72:34–39. doi:10.1016/j.psyneuen.2016.06.007
29. Igna CV, Julkunen J, Vanhanen H, Keskivaara P, Verkasalo M. Depressive symptoms and serum lipid fractions in middle-aged men: physiologic and health behavior links. Psychosom Med. 2008;70(9):960–966. doi:10.1097/PSY.0b013e318189a942
30. Skilton MR, Moulin P, Terra JL, Bonnet F. Associations between anxiety, depression, and the metabolic syndrome. Biol. Psychiatry. 2007;62(11):1251–1257. doi:10.1016/j.biopsych.2007.01.012
31. Luo H, Li J, Zhang Q, et al. Obesity and the onset of depressive symptoms among middle-aged and older adults in China: evidence from the CHARLS. BMC Public Health. 2018;18(1):909. doi:10.1186/s12889-018-5834-6
32. Holme I, Aastveit AH, Hammar N, Jungner I, Walldius G. Relationships between lipoprotein components and risk of ischaemic and haemorrhagic stroke in the Apolipoprotein MOrtality RISk study (AMORIS). J Intern Med. 2009;265(2):275–287. doi:10.1111/j.1365-2796.2008.02016.x
33. Sacco RL. Newer risk factors for stroke. Neurology. 2001;57(5 Suppl 2):S31–34. doi:10.1212/WNL.57.suppl_2.S31
34. Kurth T, Everett BM, Buring JE, Kase CS, Ridker PM, Gaziano JM. Lipid levels and the risk of ischemic stroke in women. Neurology. 2007;68(8):556–562. doi:10.1212/01.wnl.0000254472.41810.0d
35. van Reedt Dortland AK, Giltay EJ, van Veen T, van Pelt J, Zitman FG, Penninx BW. Associations between serum lipids and major depressive disorder: results from the Netherlands study of depression and anxiety (NESDA). J Clini Psych. 2010;71(6):729–736. doi:10.4088/JCP.08m04865blu
36. Moreira FP, Jansen K, Cardoso TA, et al. Metabolic syndrome in subjects with bipolar disorder and major depressive disorder in a current depressive episode: population-based study: metabolic syndrome in current depressive episode. J Psychiatr Res. 2017;92:119–123. doi:10.1016/j.jpsychires.2017.03.025
37. Hummel J, Westphal S, Weber-Hamann B, et al. Serum lipoproteins improve after successful pharmacologic antidepressant treatment: a randomized open-label prospective trial. J Clini Psych. 2011;72(7):885–891. doi:10.4088/JCP.09m05853blu
38. Aijänseppä S, Kivinen P, Helkala EL, Kivelä SL, Tuomilehto J, Nissinen A. Serum cholesterol and depressive symptoms in elderly Finnish men. Int J Geriatric Psychiatry. 2002;17(7):629–634. doi:10.1002/gps.666
39. Ariyo AA, Haan M, Tangen CM, et al. Depressive symptoms and risks of coronary heart disease and mortality in elderly Americans. Cardiovasc Health Study Collab Res Group Circulation. 2000;102(15):1773–1779.
40. Shen H, Tu X, Luan X, Zeng Y, He J, Tang W. Serum lipid profiles and post-stroke depression in acute ischemic stroke patients. Neuropsychiatr Dis Treat. 2019;15:1573–1583. doi:10.2147/NDT.S204791
41. Laclaustra M, Lopez-Garcia E, Civeira F, et al. LDL cholesterol rises with BMI only in lean individuals: cross-sectional US and Spanish representative data. Diabetes Care. 2018;41(10):2195–2201. doi:10.2337/dc18-0372
42. Li H, Ma J, Zheng D, et al. Sex differences in the non-linear association between BMI and LDL cholesterol in middle-aged and older adults: findings from two nationally representative surveys in China. Lipids Health Dis. 2021;20(1):162. doi:10.1186/s12944-021-01591-w
43. Nabavi SF, Dean OM, Turner A, Sureda A, Daglia M, Nabavi SM. Oxidative stress and post-stroke depression: possible therapeutic role of polyphenols? Curr Med Chem. 2015;22(3):343–351. doi:10.2174/0929867321666141106122319
44. Duewell P, Kono H, Rayner KJ, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464(7293):1357–1361. doi:10.1038/nature08938
45. Stewart CR, Stuart LM, Wilkinson K, et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat Immunol. 2010;11(2):155–161. doi:10.1038/ni.1836
46. Jukema RA, Ahmed TAN, Tardif JC. Does low-density lipoprotein cholesterol induce inflammation? If so, does it matter? Current insights and future perspectives for novel therapies. BMC Med. 2019;17(1):197. doi:10.1186/s12916-019-1433-3
47. Benson S, Janssen OE, Hahn S, et al. Obesity, depression, and chronic low-grade inflammation in women with polycystic ovary syndrome. Brain Behav Immun. 2008;22(2):177–184. doi:10.1016/j.bbi.2007.07.003
48. Capuron L, Lasselin J, Castanon N. Role of adiposity-driven inflammation in depressive morbidity. Neuropsychopharmacology. 2017;42(1):115–128. doi:10.1038/npp.2016.123
49. Sun W, Miao J, Song Y, et al. Systemic low-grade inflammation and depressive symptomology at chronic phase of ischemic stroke: the chain mediating role of fibrinogen and neutrophil counts. Brain Behav Immun. 2022;100:332–341. doi:10.1016/j.bbi.2021.10.011
50. 中华医学会神经病学分会, Society 中HCSoNHCS [Chinese Society of Neurology, Chinese Medical Association. Chinese Guidelines for the Primary Prevention of Cerebrovascular Diseases]. 中国脑血管病一级预防指南. Chinese Journal of Neurology. 2019;52(9):684–709.
51. Ruscica M, Ferri N, Macchi C, Corsini A, Sirtori CR. Lipid lowering drugs and inflammatory changes: an impact on cardiovascular outcomes? Ann Med. 2018;50(6):461–484. doi:10.1080/07853890.2018.1498118
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.