Back to Journals » The Application of Clinical Genetics » Volume 16
Genetic Links to Episodic Movement Disorders: Current Insights
Authors Garg D ![]() , Mohammad S, Shukla A, Sharma S
, Mohammad S, Shukla A, Sharma S ![]()
Received 9 November 2022
Accepted for publication 24 February 2023
Published 1 March 2023 Volume 2023:16 Pages 11—30
DOI https://doi.org/10.2147/TACG.S363485
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Martin Maurer
Divyani Garg,1 Shekeeb Mohammad,2,3 Anju Shukla,4 Suvasini Sharma5
1Department of Neurology, All India Institute of Medical Sciences, New Delhi, India; 2Kids Neuroscience Centre, The Children’s Hospital at Westmead, Faculty of Medicine and Health, University of Sydney, Sydney, New South Wales, Australia; 3TY Nelson Department of Neurology and Neurosurgery, The Children’s Hospital at Westmead, The University of Sydney, Westmead, New South Wales, Australia; 4Department of Medical Genetics, Kasturba Medical College and Hospital, Manipal, India; 5Department of Pediatrics (Neurology Division), Lady Hardinge Medical College and Kalawati Saran Hospital, New Delhi, India
Correspondence: Suvasini Sharma, Department of Pediatrics (Neurology Division), Kalawati Saran Children’s Hospital, New Delhi, 110001, India, Tel +91 9910234344, Email [email protected]
Abstract: Episodic or paroxysmal movement disorders (PxMD) are conditions, which occur episodically, are transient, usually have normal interictal periods, and are characterized by hyperkinetic disorders, including ataxia, chorea, dystonia, and ballism. Broadly, these comprise paroxysmal dyskinesias (paroxysmal kinesigenic and non-kinesigenic dyskinesia [PKD/PNKD], paroxysmal exercise-induced dyskinesias [PED]) and episodic ataxias (EA) types 1– 9. Classification of paroxysmal dyskinesias has traditionally been clinical. However, with advancement in genetics and the discovery of the molecular basis of several of these disorders, it is becoming clear that phenotypic pleiotropy exists, that is, the same variant may give rise to a variety of phenotypes, and the classical understanding of these disorders requires a new paradigm. Based on molecular pathogenesis, paroxysmal disorders are now categorized as synaptopathies, transportopathies, channelopathies, second-messenger related disorders, mitochondrial or others. A genetic paradigm also has an advantage of identifying potentially treatable disorders, such as glucose transporter 1 deficiency syndromes, which necessitates a ketogenic diet, and ADCY5-related disorders, which may respond to caffeine. Clues for a primary etiology include age at onset below 18 years, presence of family history and fixed triggers and attack duration. Paroxysmal movement disorder is a network disorder, with both the basal ganglia and the cerebellum implicated in pathogenesis. Abnormalities in the striatal cAMP turnover pathway may also be contributory. Although next-generation sequencing has restructured the approach to paroxysmal movement disorders, the genetic underpinnings of several entities remain undiscovered. As more genes and variants continue to be reported, these will lead to enhanced understanding of pathophysiological mechanisms and precise treatment.
Keywords: paroxysmal movement disorders, episodic ataxia, PRRT2, PNKD, KCNA1
Introduction
Episodic or paroxysmal movement disorders (PxMD) are conditions, which occur episodically, are transient, usually have normal interictal periods, and are characterized by hyperkinetic disorders, including ataxia, chorea, dystonia, and ballism.
Broadly, these include two groups: paroxysmal dyskinesias (PxD), which are characterized by the occurrence of transient hyperkinetic movements (Table 1), and episodic ataxias (EA), characterized by recurrent attacks of cerebellar dysfunction (Tables 2 and Table 3).1 Broad classification schemes have categorized PxMD based on the age of onset, duration of episodes, interictal abnormalities, underlying pathophysiology, genetics and type of movement disorder. The term “dyskinesia” to describe the hyperkinetic movement disorder was proposed by Demirkiran and Jankovic in 1995.2 Etymologically, the term ‘paroxysmal dyskinesias’ has been argued to be restrictive in definition by many authors.3 However, as this term continues to prevail in the literature, we have used it in this article. In PxD, the phenomenology usually consists of chorea, dystonia or ballism. Other movement disorders, including tics, myoclonus, startle syndrome and tremors, are not included in the definition of PxD.
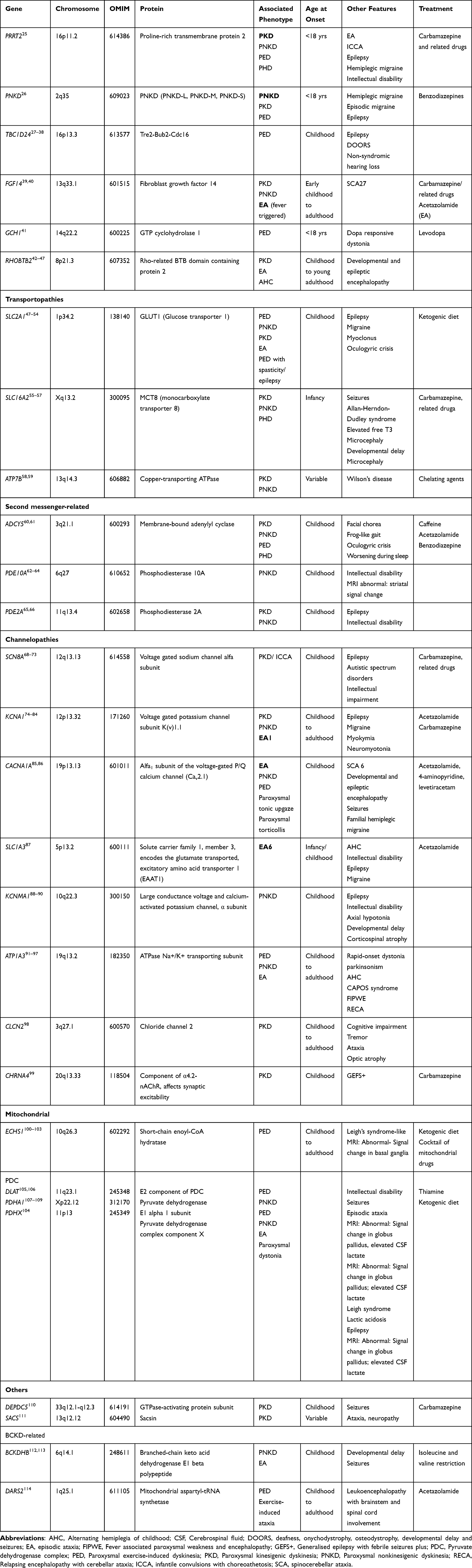 |
Table 1 Summary Table of Genes Associated with Paroxysmal Dyskinesia |
 |
Table 2 Summary Table of Genes Associated with Episodic Ataxia |
 |
Table 3 Features of Episodic Ataxia Syndromes |
Based on etiology, PxMD may be genetic or acquired. The pattern of inheritance in genetic conditions is usually autosomal dominant, either sporadic or familial.3,4 The acquired PxMD may arise from structural, metabolic, vascular, immune-mediated, or degenerative etiologies (Table 4).3 Clues for secondary causes include age at onset above 18 years, negative family history, variable duration of symptoms and triggers, or associated clinical features.5 With the advent of next-generation sequencing, a paradigm shift has emerged in the classification of episodic movement disorders. It is now recognized that variants that cause PxD can also be associated with epilepsy, ataxia, pyramidal signs, developmental delay, and other neurological features. While phenotypic recognition guides treatment, molecular diagnosis may be imperative to streamline therapy in certain disorders. For example, carbamazepine/oxcarbazepine is used to elicit excellent response in paroxysmal kinesigenic dyskinesia (PKD). However, PxD due to glucose transporter 1 (GLUT1)-deficiency syndrome responds to a ketogenic diet.
 |
Table 4 Acquired Paroxysmal Movement Disorders |
As these disorders are phenotypically and genotypically complex, episodic movement disorders remain underrecognized by clinicians. In this review, we aim to explore the current genetic links of episodic movement disorders.
Pathophysiology
PxD are considered to be network disorders, with both basal ganglia and cerebellum being implicated in pathophysiology.5 The aberration may involve either a primary striatal dysfunction or striatal dysfunction secondary to altered outflow from the cerebellum to the basal ganglia. Striatal cAMP plays a critical role in several hyperkinetic disorders, and it may play a pivotal role in the generation of PxMDs as well.
Evidence for striatal dysfunction emanates from various sources. Patients with stroke in the striatal regions may manifest with PxD. Conditions like PARKIN-related genetic Parkinson’s disease,6 characterized by striatal dopaminergic deficiency, may manifest with PED. The globus pallidus interna (Gpi) is a metabolically highly active region and is vulnerable to insults resulting in depletion of cerebral energy, as seen in pyruvate dehydrogenase deficiency and ECHS1 deficiency, which are associated with PxMD. Deep brain stimulation of the Gpi region is associated with improvement in PxD associated with GNAO1 and ADCY5-related disorders and PNKD.7,8
Abnormal cerebellar output has been associated with PKD due to monoallelic PRRT2 variants. In patients with biallelic PRRT2 variants, which are considerably rare, episodic ataxia may also be associated. PRRT2, which is highly expressed in cerebellar granule cells, may modulate and alter cerebellar output in patients with PRRT2-related PKD. PxD and cerebellar features occur concomitantly in patients with ATP1A2 and FGF14-related disorders. Similarly, PxD can be present in disorders associated with episodic ataxia, as in CACNA1A and KCNA1-related variants.
Cyclic AMP (cAMP) plays a role in modulating the balance between direct and indirect pathways, which, respectively, facilitate and inhibit execution of movement. cAMP-related pathways may play a role in the pathogenesis of PxD related to ADCY5, PDE10A, PDE2A, GNAO variants.5 Overall, the literature favors roles of both the striatum and the cerebellum in the pathogenesis of PxD. The cerebellar nuclei communicate with the striatum via pathway involving the central thalamic nucleus, and this bidirectional influence is likely to play a significant role in PxD.
Classification of Paroxysmal Disorders
In 1977, Lance classified kinesigenic PxD clinically into familial and sporadic forms.9 Demirkiran and Jankovic, in 1995, suggested a classification scheme based on triggering factors, independent of the duration of attacks. This scheme classified PxD into paroxysmal kinesigenic dyskinesia (PKD), paroxysmal non-kinesigenic dyskinesia (PNKD), paroxysmal exercise-induced dyskinesia (PED), and paroxysmal hypnogenic dyskinesia (PHD). PKD was triggered by sudden movements, PED by sustained exercise, and triggers in PNKD were heterogeneous but it was neither precipitated by sudden movement or sustained exercise. PHD was later determined to be a form of autosomal dominant frontal lobe epilepsy (ADFLE). This classification scheme continues to remain popularly used in the literature.
With the recognition of genetic underpinnings of PxMDs, certain classical notions have been challenged. For example, PRRT2 variants, which are the most frequent cause of primary PKD, are characterized by very brief episodes triggered by sudden movements. Hence, both the triggering factor and the duration of attacks are important in phenotypic understanding, bolstered by the molecular variant involved.
Another paradigm is to consider these disorders as either “isolated” or associated with other forms of neurological dysfunction. Yet another paradigm is to consider that these disorders are either primary (“familial”) or secondary (“acquired”), as proposed by Goodenough et al10 “Primary” disorders generally indicate an underlying genetic cause, whereas “secondary” disorders indicate an acquired disorder. Acquired disorders, largely considered to be “treatable”, affect the same circuitry, and manifest as paroxysmal movements. Some of these disorders include demyelination (multiple sclerosis,11–13 neuromyelitis optica spectrum disorder,14 acute disseminated encephalomyelitis15), immune-mediated (systemic lupus erythematosus/antiphospholipid antibody syndrome16), metabolic (hypoglycemia/hyperglycemia,17,18 calcium abnormalities, hyperthyroidism19,20), and limb-shaking transient ischemic attacks (internal carotid artery stenosis21,22/ Moyamoya disease23,24) (Table 4). Clues that point towards a primary etiology include age at onset below 18 years, presence of family history and fixed triggers and attack duration. Secondary disorders, on the other hand, may have age at onset above 18 years, absence of family history and variable triggers and duration of attacks. The term “primary” is controversial as it suggests absence of etiology, when, in fact, these disorders are “secondary” to specific genetic abnormalities. Another connotation was that primary disorders should lack interictal abnormalities, which may be prevalent in secondary disorders. It is now well-established that this may not be used as a discerning feature, as interictal abnormalities may be seen in several so-called primary disorders, as in SLC2A1 variants.
The other large group of PxMD is episodic ataxia. So far, nine subtypes of EA have been described (EA 1 to 9), of which EA1 and EA2 are the most frequent (Table 2). EAs are characterized by brief episodes of sudden-onset ataxia, which last seconds to minutes. Patients may also demonstrate interictal myokymia and/or neuromyotonia (Table 3).
Paroxysmal Dyskinesias
PxD are movement disorders that involve recurrent episodes of dystonia, chorea, athetosis or ballism without loss of consciousness. PKD is the most frequently occurring PxMD, with an incidence of 1 per 150,000. It is characterized by attacks of chorea/dystonia, which are less than 1 min in duration, and are triggered by sudden motion. Chen et al identified that variants in the proline-rich transmembrane protein 2 (PRRT2) gene were associated with most cases of PKD and related disorders.25 Since then, several other mutations have been identified to cause PKD, and include SCN8A, ADCY5 and SLC16A2 mutations (Table 2).
PNKD is rare, with a prevalence of 1 per 100,000. It is inherited in an autosomal dominant pattern. The responsible gene is PNKD, earlier known as myofibrillator regulator-1 (MR-1) gene. The disease usually originates in childhood, and the usual triggers are alcohol, coffee, stress and fatigue. The usual duration is minutes to hours. The condition may regress with age. Compared to PKD, PNKD episodes are rather less frequent. Individuals with variants in the PNKD gene have usual age at onset in infancy or early childhood, nearly universal precipitation by caffeine and alcohol, and respond to sleep and benzodiazepines, which abort attacks.
PED differs from PKD and PNKD in that attacks are triggered by sustained exercise and usually consist of episodes of dystonia lasting from minutes to hours. SLC2A1 variants account for 30–40% of patients with PED. Other important genes include GCH1, ECHS1, pyruvate dehydrogenase complex-related, and genetic Parkinson’s disease.
Genetic PxMD may be pathophysiologically categorized into synaptopathies, transportopathies, channelopathies, second-messenger related disorders and mitochondrial disorders (Figure 1).
 |
Figure 1 Pathophysiology and classification of paroxysmal dyskinesia. |
Synaptopathies
PRRT2 (OMIM 614386)
Proline-rich transmembrane protein 2 (PRRT2) modulates neurotransmitter release by its interaction with presynaptic proteins, SNAP25 and synaptotagmin. Additionally, it influences Nav1.2/Nav1.6 channels and modulates neural transmission.
In patients with PKD, there may be an autosomal dominant family history of PKD or a form of epilepsy called benign familial infantile seizure (BFIS). Both PKD and BFIS may co-exist, termed infantile convulsions with choreoathetosis (ICCA) syndrome. The term ICCA has been replaced with PKD/infantile convulsions (PKD/IC). Patients with PKD/IC develop epilepsy within the first 2 years of life and subsequently develop PKD. These attacks have a kinesigenic trigger but may also be precipitated by exercise and emotions. These attacks are very brief (<1 min) and occur several times a day. While between 70% and 90% of patients with PKD have autosomal dominant inheritance, the remaining have de novo mutations. Penetrance is incomplete (60–90%). Other phenotypes include PNKD, PED, PHD, paroxysmal torticollis, episodic ataxia, hemiplegic migraine, childhood absence epilepsy and intellectual disability. Nearly 30% of PRRT2-related patients with PKD have PKD/IC. Truncating mutations in PPRT2 as a cause of PKD were first identified in 2011.25 Loss-of-function (LOF) variants are mostly observed in PKD. The most frequent is the frameshift mutation, c.649dupC (p.Arg217ProfsTer8), which leads to a premature stop codon. Treatment is by carbamazepine and oxcarbazepine. Attacks tend to decrease with advancing age.
PNKD (OMIM 609023)
PNKD was earlier known as the myofibrillogenesis regulator-1 (MR-1) gene. It accounts for nearly 70% of PNKD.3 The PNKD encodes three alternate splice proteins of 385, 361 and 142 amino acids. The long isoform, PNKD-L, is enriched in the central nervous system, while the intermediate (PNKD-M) and short (PNKD-S) isoforms are more widespread. The PNKD-L and PNKD-M forms are homologous to hydroxyacylglutathione hydrolase (HAGH), which detoxifies methylglyoxal, a compound present in coffee and ethanol. This may explain why coffee and alcohol precipitate attacks in PNKD. PNKD also interacts with RAB-interacting molecule (RIM1) AND RIM2 and affects nigrostriatal release of dopamine. PNKD-L has also been associated with Tourette syndrome.26 Attacks in PNKD may be triggered by alcohol, coffee, stress and emotions. These usually last from 10 min to an hour but may continue up to 12 h and are infrequent. Treatment of PNKD comprises benzodiazepines, levetiracetam and valproic acid.
TBC1D24 (OMIM 613577)
TBC1 domain family member 24 (TBC1D24) is a member of the GTPase activating proteins. It is needed for normal brain development due to its role in synaptic function and vesicle traffic. Variants in TBC1D24 gene have been associated with diverse phenotypes, of which epilepsy is predominant. Epilepsy types usually include pharmaco-resistant myoclonic, focal, multifocal27 and early-onset epileptic encephalopathy, epilepsia partialis continua (EPC) and familial infantile myoclonic epilepsy (FIME).28–31 Other syndromes include DOORS (deafness, onychodystrophy, osteodystrophy, developmental delay and seizures)32 and non-syndromic hearing loss.33 Missense and loss-of-function variants are spread throughout the protein. Rolandic epilepsy - exercise induced dystonia phenotype - has been reported in one family. In this family, epilepsy was self-limited but dystonia persisted into adulthood.34 Exercise-induced paroxysmal dystonia was reported in two patients.35 Other phenotypes associated with TBC1D24 variants include alternating hemiplegia of childhood (AHC),36 AHC and EPC combination,37 and paroxysmal facial and limb myoclonus in infancy.38 Treatment includes carbamazepine, benzodiazepine and acetazolamide.
FGF14 (OMIM 601515)
Fibroblast growth factor 14 (FGF 14) is a regulator of Cav2.1 presynaptic channel and modulates vesicular transport and synaptic transmission. Mutations in FGF14 are associated with EA.39 EA episodes present in childhood, are triggered by fever, and may be associated with vomiting and headache. These may last for several days. It is inherited in an autosomal dominant fashion. PKD and PNKD in isolation or in association with EA have also been reported.40 It is also a cause of the rare autosomal dominant spinocerebellar ataxia type 27 (SCA 27). Treatment is with acetazolamide.
GCH1 (OMIM 600225)
GTP cyclohydrolase 1, a catalyst in the formation of tetrahydropterin, is encoded by GCH1. Tetrahydropterin is required for the manufacture of dopamine, phenylalanine and serotonin. The characteristic phenotype associated with GCH1 variants is dopa-responsive dystonia. However, these may manifest with PED. Autosomal dominant familial PED, with exercise-induced foot posturing, has been reported in a family, with heterozygous stop codon variant in exon 1, c.411G>T.41 Other features in this family included restless leg syndrome, depression, migraine and atypical parkinsonism. PED responded to low-dose levodopa.
RHOBTB2 (OMIM 607352)
RHOBTB2 encodes an atypical Rho-related BTB-containing protein 2, a GTPase. Heterozygous variants have been reported to lead to developmental and epileptic encephalopathies,42,43 postnatal microcephaly, intellectual impairment and Rett-like phenotype.44 Paroxysmal movement disorders, including chorea, dystonia and dyskinesias, were reported in a series of 13 patients.42 Stereotypies were observed in three patients. Another patient with developmental delay had status epilepticus at 3 months of age, followed by paroxysmal dystonia at the age of one year due to a de novo missense variant in the RHOBTB2 gene, c.1532G>A [p.Arg511Gln].45 The paroxysmal movements responded to carbamazepine. Severe paroxysmal choreodystonia, along with aplasia cutis congenita, without epilepsy, was reported in another patient with a heterozygous missense variant, c.1448G>A [p.Arg483His].46
In 2021, TMEM151A variants were also recognized as a cause of PKD.47
Transportopathies
SLC2A1 (OMIM 138140)
SLC2A1 gene encodes the glucose transporter type 1 (GLUT1) on the blood–brain barrier, responsible for transport of glucose across the barrier and astrocytic membrane. Mutations lead to a wide spectrum of neurological disorders, including GLUT1 deficiency syndrome (GLUT1-DS), PED, progressive spastic paraparesis combined with PED48,49 and epilepsy. PKD and PNKD with SLC2A1 variants have also been described.50 Familial PED due to SLC2A1 mutation is an autosomal dominant condition.51 Most of the GLUT1-DS cases with PED are due to missense variants in SLC2A1. On the other hand, splice site, nonsense, insertion and deletion mutations are associated with severe phenotypes, including epilepsy, developmental delay and spasticity.
PED, induced by fasting or exercise, may be a main feature of GLUT1 deficiency.52 The movement disorder may include chorea-athetosis, dystonia, or ataxia. Often the lower limbs are involved. The movement disorder is provoked by sustained exercise.53 Paroxysmal ocular movements, described as “aberrant gaze saccades”, have also been reported with GLUT1-DS.54
CSF glucose below the 10th percentile, CSF: serum glucose below the 25th percentile, and CSF lactate levels below the ninetieth percentile are highly suggestive of GLUT1-DS.55
Recognition is imperative, as the institution of the ketogenic diet is beneficial in this condition.
SLC16A2 (OMIM 300095)
SLC16A2 encodes the monocarboxylate transporter type 8 (MCT8), a thyroid hormone transporter in the brain. Variants in SLC16A2 gene lead to Allan-Herndon-Dudley (AHD) syndrome, characterized by severe developmental delay and peripheral thyrotoxicosis. It is an X-linked recessive disorder. Thyroid hormone abnormalities include raised free T3, low reverse T3, low total/free T4 and normal or slightly elevated TSH level.
Axial hypotonia is a central feature.56 Eventually, spasticity may develop. Other features include muscle weakness, torsional nystagmus, contractures, skeletal abnormalities and central nervous system hypomyelination. p.R271H and p.G564R variants may result in a severe clinical phenotype. P.G564E variant has been associated with a relatively mild phenotype.57 PKD in association with AHD syndrome has been reported with a missense variant (c.1535T>C [p.Leu512Pro]) and a frameshift stop codon.58 It can be evoked by passive movement.
ATP7B (OMIM 606882)
Wilson’s disease (WD), due to mutations in ATP7B gene, is an uncommon cause of PxMD. PKD was reported in a 22-year-old male with WD, which was completely remitted with oxcarbazepine.59 PNKD has also been reported in a patient with WD, which responded to trientine, whose attacks lasted for seconds, and were ameliorated by smoking.60
Second-Messenger Related
ADCY5 (OMIM 600293)
Adenylyl cyclase 5 (ADCY5)-related disorders comprise a spectrum of hyperkinetic and often paroxysmal disorders that include chorea, dystonia, and myoclonus.61 Adenylyl cyclase is required for the conversion of ATP to cyclic adenosine-3’,5’-monophosphate (cAMP), which is an important second messenger in several intracellular processes. ADCY5 is the most common isoform of adenylyl cyclase, which is present in the striatum, and through the cAMP signaling pathways, prevents involuntary movements. ADCY5 comprises 1261 amino acids and is encoded by a gene located on chromosome 3p21.1. It has two transmembrane helical domains (M1 and M2) which bind to two intracellular catalytic domains (C1 and C2).
The p.A726T variant seems to harbor a milder phenotype. Somatic mosaicism, which may be seen in nearly 43% of de novo cases, may lead to milder phenotypes.62 Autosomal dominant inheritance prevails, although autosomal recessive inheritance has also been reported. Intercurrent illness, fatigue and stress may trigger these attacks. It was originally described as “Essential” or “benign” chorea or “familial dyskinesia and facial myokymia.”
Prominent facial dyskinesia is a hallmark feature and includes a combination of chorea and myoclonus. Upper limb involvement is also observed. Axial hypotonia, with frog-like adaptive gait are other features. Bouts last minutes to hours and worsen in the third decade of life. Thereafter, they either plateau or resolve. “Ballistic bouts” are frequently painful, truncal dystonia flexion-extension movements, which occur during drowsiness or sleep. Other phenotypes associated with ADCY5 mutations include familial myoclonus-dystonia, childhood-onset chorea, and alternating hemiplegia of childhood.
ADCY-related have been observed to respond to caffeine. Other drugs include benzodiazepine such as clonazepam and acetazolamide.
PDE10A (OMIM 610652)
Phosphodiesterase 10A (PDE10A) is richly present in the striatum.63 While striatal cAMP is synthesized by ADCY5, it is degraded by PDE10A. Biallelic variants in the PDE10A gene lead to loss of striatal cAMP, and hyperkinetic movement disorders.64 De novo mutations may lead to chorea in childhood. Bilateral T2-weighted symmetrical and bilateral striatal hyperintensities may be seen.65
PDE2A (OMIM 602658)
PDE2A is enriched in the striatal medium spiny neurons. It encodes phosphodiesterase 2A that catalyzes cAMP and cyclic guanosine monophosphate (cGMP). Loss-of-function homozygous mutations in PDE2A gene have been associated with early onset chorea.66 In these patients, PxD preceded development of chorea. Additionally, the child had intellectual impairment and EEG abnormalities. Biallelic PDE2A mutations were reported in three patients (two were siblings).67 Two patients presented with refractory paroxysmal dyskinesia, which was misdiagnosed as epilepsy. One patient had epilepsy at the age of 4 months. All patients also had cognitive impairment or developmental delay.
Channelopathies
SCN8A (OMIM 614558)
SCN8A encodes the alpha subunit of the Nav1.6 voltage-gated sodium channel, which is abundant in the brain and is pivotal in generation and propagation of action potentials. Heterozygous missense variant c.5302A>G [p.Asn1768Asp] was reported in epileptic encephalopathy, characterized by early onset seizures, autism and SUDEP.68 Heterozygous missense variant c.4423G>A [p.Gly1475Arg] has been reported to lead to early onset epileptic encephalopathy69 Missense variants lead to increased channel activity. De novo heterozygous missense mutation in c.4408C>A [p.Gln1470Lys] reported in a patient with possible antenatal onset of severe episodic tremulousness associated with hyperekplexia-like startle response, drug-refractory seizures and developmental regression, acquired microcephaly and gastroparesis.70
SCN8A mutation has been recognized as a cause of infantile convulsions and paroxysmal choreoathetosis (ICCA), which is a combination of benign familial infantile seizures (BFIS) and paroxysmal kinesigenic dyskinesia (PKD).71 Gain-of-function mutations have been associated with epileptic encephalopathy.68 Loss-of-function mutations have been associated with cognitive dysfunction.72
SCN8A missense mutation (c.4447G>A; p.E1483K) was reported in three families with infantile seizures and development of PKD in puberty, in the form of dystonia.71 However, some doubt was raised regarding the true PKD nature as one of the patients demonstrated cortical discharges on EEG during the PKD episode. Paroxysmal tonic upgaze (PTU) has also been described in one child associated with the SCN8A variant.73
KCNA1 (OMIM 171260)
KCNA1 encodes a voltage gated shaker-related family submember 1 potassium channel, Kv1.1 alpha subunit, which plays a role in presynaptic repolarization and modulation of inhibitory input to the cerebellum. Pathogenic variants are LOF and lead to reduced inhibitory input to the cerebellum. Inheritance is autosomal dominant, with reduced penetrance.
KCNA1 mutations have been primarily associated with episodic ataxia type 1 (EA1), with or without myokymia,74 epilepsy and severe dyskinesias with neonatal epilepsy.75 A heterozygous c.257G>A R86Q variant was reported with PNKD.76 Familial PKD is reported with c.956 T>G (p.319 L>R) and c.765 C>A (p.255 N>K) variants.77 In two patients, classical PKD was associated with p.Gly396Val and p.Gly396Arg variants.78 Among non-neurological manifestations, hypomagnesemia is also caused by mutations in KCNA1.79,80 Seizure-related variants cluster in S1/S2 domains of the transmembrane region and pore region of Kv1.1.81 Variants associated with EA1 occur along the entire length of the protein. Most mutations are missense, although frameshift mutations have also been reported.82
It has also been observed that individuals with KCNA1 variants at the C-terminus are more likely to suffer from seizures and developmental delay than those with variants at the N-terminus.83,84
CACNA1A (OMIM 601011)
CACNA1A encodes the alfa1 subunit of the voltage-gated P/Q calcium channel (Cav2.1). LoF variants in the CACNA1A gene disrupt calcium entry into the cerebellar Purkinje and granule cells, where these channels are richly present.85 The disease is autosomal dominant, with 80–90% penetrance.
Whereas GoF variants are associated with developmental and epileptic encephalopathy, epilepsy and familial hemiplegic migraine, LoF variants occur in PxMD, including EA2, PKD and PED.
EA2 is the most frequently occurring EA syndrome. The episodes are longer in comparison to EA1, and patients may also have vertigo, vomiting and dysarthria. Nearly 50% of patients may have migraine (hemiplegic), epilepsy or dystonia. These patients may also develop a progressive ataxia syndrome. Downbeat nystagmus may be observed. Paroxysmal tonic upgaze has also been reported in childhood, preceding the development of EA.86 EA2 is allelic with familial hemiplegic migraine type 1 (FHM1), CAG repeats in the CACNA1A gene may result in spinocerebellar ataxia type 6.
SLC1A3 (OMIM 600111)
The solute carrier family 1, member 3, encodes the glutamate transported, excitatory amino acid transporter 1 (EAAT1). Heterozygous variants in SLC1A3 are observed in EA type 6, which is inherited in an autosomal dominant pattern.87 Episodes of ataxia and epilepsy occur and are longer than CACNA1A-related disorder, lasting up to hours to days. Myokymia, nystagmus and tinnitus are not observed. Migraine may be associated additionally.
KCNMA1 (OMIM 300150)
KCNMA1 gene encodes the alfa subunit of “Big K+ (BK)” large conductance calcium and voltage-gated potassium channel (KCa1.1). This channel is enriched in the brain and modulates action potential and neurotransmitter release. Pathogenic GoF variants are associated with autosomal dominant PxD and epilepsy. LoF variants present with developmental delay/intellectual impairment, ataxia, axial hypotonia, and speech abnormalities.88 The p.Asp434Gly variant was associated with PNKD, epilepsy or both. P.Glu884Lys and p.Asn1053Ser variants were associated with early onset PNKD with developmental delay.89 Another variant, p.Arg458Ter, was associated with PNKD, epilepsy, developmental delay and cerebellar and corticospinal atrophy.90
ATP1A3 (OMIM 182350)
ATP1A3 is the alfa-three isoform of the Na+/K+ ATPase pump. Pathogenic variants may manifest with many neurological and non-neurological syndromes, including rapid-onset dystonia parkinsonism, alternating hemiplegia of childhood, cerebellar ataxia,91 optic atrophy and sensorineural hearing loss syndrome (CAPOS).92–94 ATP1A3 variants have been recognized as an important cause of AHC.95 The p.Asp923Asn variant has also been recognized as a cause of PED. In this case, AHC manifested first, followed by PED. R756H and R756L have been associated with fever-associated encephalopathy and generalized weakness, progressing to develop ataxia.96 This entity was termed “fever associated paroxysmal weakness and encephalopathy (FIPWE)” and “relapsing encephalopathy with cerebellar ataxia (RECA).”97
CLCN2 (OMIM 600570)
CLCN2 variants result in loss of function of chloride channel 2 and have been associated with leukoencephalopathy. Usually, these patients present with cognitive impairment, tremor, ataxia, and optic atrophy. A homozygous variant, p.Ser375CysTer6 in the CLCN2 gene, was associated with onset of paroxysmal kinesigenic dyskinesia since the age of 21 years.98 MRI brain showed characteristic signal change in the posterior limb of the internal capsule, cerebral peduncles, cerebellar peduncles, and cerebellar white matter. PKD was completely abolished with carbamazepine.
CHRNA4 (OMIM 118504)
Mutations in CHRNA4 have been associated with PKD or generalized epilepsy with febrile seizures plus (GEFS+). It is inherited in an autosomal dominant manner. It was identified in a family in which one individual had GEFS+ and two had PKD. A fully co-segregated mutation (NM_000744: c.979G>A) was identified.99
Mitochondrial
ECHS1 (OMIM 602292)
ECHS1 gene encodes for short-chain enoyl-CoA hydratase, which is a mitochondrial enzyme involved in valine and isoleucine pathways.100 Four main phenotypes have been described - a neonatal form with rapid progression, a severe infantile form with basal ganglia degeneration, a slowly progressive infantile form and paroxysmal exercise-induced dystonia, with a normal interictal period. It is also associated with Leigh’s syndrome.101 In a family of two siblings, the older sibling had a Leigh-like syndrome, with generalized dystonia and severe pallidal changes on MRI. The younger sibling developed only paroxysmal exercise-induced dystonia, with mild pallidal signal changes on MRI. Both siblings had compound heterozygous ECHS1 variants (c.232G>T [p.Glu78Ter] and c.518C>T [p.Ala173Val].102 Valine-restricted diet may be of potential benefit.103
Pyruvate Dehydrogenase Complex (PDC)
PDC deficiency necessitates prompt recognition so that a ketogenic diet may be initiated. It leads to ATP production deficits, and a host of neurological disorders, including microcephaly, epilepsy, hypotonia, developmental delay, and Leigh syndrome. Acute energy failure in infancy may lead to abnormalities in basal ganglia and PxMD.104
Homozygous missense variant (c.470T>G; p.Val157Gly) in the DLAT gene has been associated with PED.105 This patient presented with PED at the age of 3 years, which would last for 5–15 min. He had intellectual disability, dysconjugate gaze and pyramidal features. DLAT gene encodes for dihydrolipoamide acetyltransferase, the E2 component of the PDC. A ketogenic diet may be of benefit, as may thiamine replacement. Signal change in bilateral globus pallidus may be seen on T2-weighted MRI. DLAT gene variants may also lead to episodic dystonia and developmental delay.106
PDHA1 variants have also been associated with PED.107 One patient with a c.647T>C (p.Leu216Ser) was associated with reduced penetrance.107 This patient had abnormal MRI findings with pallidal signal change and was treated with thiamine. PED was reported in another patient with heat-associated dystonia, which was ameliorated with levodopa.108 PNKD has also been reported.109 Paroxysmal dystonia and episodic ataxia104 have also been reported in association with PDHA1 variants.
Variants in the PDHX gene have been associated with non-progressive encephalopathy (five cases).104 One patient had paroxysmal dystonia.
Others
DEPDC5 (OMIM 614191)
One patient with PKD associated with DEPDC5 variant was identified. The patient started having episodic bilateral limb posturing at the age of 13 years, with up to 30–40 attacks occurring per day. A variant c.3311C>T (p.S1104L) was identified in the patient and his mother, who also had similar attacks between 9 and 31 years of age.110
SACS (OMIM 604490)
Two patients with autosomal recessive spastic ataxia of Charlevoix-Saguenay have been reported to have PKD. In one patient, compound heterozygous mutations in the SACS gene were identified (p.P3007S and p.H3392fs). In the second patient, a homozygous truncating mutation (p.W1376X) was identified.111
BCKD Complex
Maple syrup urine disease (MSUD) is an autosomal recessive condition, due to mutations in the branched-chain alfa-ketoacid dehydrogenase (BCKD) complex.
PNKD, involving curvature of the trunk to alternating sides, was reported in a 22-month child, diagnosed to have chronic intermediate MSUD, based on abnormal levels of branched chain amino acids and elevated alloisoleucine level.112 Paroxysmal spasticity was reported in two siblings with MSUD. These siblings exhibited compound heterozygous mutations (c.1076G>A [p.Arg359Lys] and c.705delT [p.Cys235Ter]) in the BCKDHB gene (OMIM 248611).113
DARS2 (OMIM 611105)
Variants in DARS2, which encodes a mitochondrial aspartyl-tRNA synthetase, are associated with leukoencephalopathy with brainstem and spinal cord involvement and brain lactate elevation (LBSL). One patient with paroxysmal exercise-induced ataxia and areflexia has been reported, who responded well to acetazolamide therapy.114
Genetic Approach to Paroxysmal Movement Disorders
Although the genetic understanding of PxMD has vastly expanded, clinical history and examination remain the cornerstone of initial evaluation. Presence of certain features may inform a secondary etiology of PxMD. These features include age at onset above 18 years, variable triggers, variable duration of attacks, absence of a family history, abnormal interictal examination, and abnormal neuroimaging. Family history of PxMD or associated conditions, such as epilepsy or migraine, may be obtained. However, an entirely clinical approach is insufficient due to low penetrance, phenotypic and genotypic pleiotropy.
Functional movement disorders (FMD) should be excluded, based on clinical features of entrainment, distractibility, and inconsistency. Other supportive features which may suggest FMD, but are not invariably present, include poor response to medication, onset in adult age, sudden onset, presence of a precipitating factor, stable or waxing/waning course, presence of other non-neurological functional symptoms, and improvement with placebo.115 Features related to the attack include poor responsiveness during the attack, uncommon triggering factors, variable attack duration and frequency, paroxysmal tremor, combination of multiple movement disorders during an attack, “huffing and puffing” vocalizations, specific motor patterns such as opisthotonos, rhythmic pelvic activity, side-to-side movement, isolated facial involvement, and very long duration of attacks.5 Treatable acquired conditions which can lead to PxMD such as hypocalcemia, hypoglycemia, demyelinating and vascular disorders etc. must be excluded. In patients in whom the suspicion for an acquired or secondary cause does not occur, one should proceed directly to genetic testing.
Genetic Evaluation
In children with associated developmental delay, dysmorphism, autism spectrum disorder or epilepsy, chromosomal microarray should be a first-tier diagnostic test.116 Pathogenic copy number variants (CNV), detected by microarray techniques, may not be picked up by next-generation sequencing (NGS) gene panels or whole-exome sequencing (WES).
Gene-panel testing is a second-tier investigation, in which multiple genes are sequences in parallel. The advantage of gene panels over WES or whole-genome sequencing (WGS) is that the former offers high-resolution coverage for exon deletions or duplications at exons, which may not be detected by the latter. Moreover, the possibility of detection of variants of unknown significance, which are unrelated to the phenotype, is reduced with gene panels, compared to WES/WGS.4
In patients where deep phenotyping is complex, WES/WGS is preferable as a second-tier investigation. WES/WGS should be performed if gene panel is negative. WGS has certain advantages over WES, including continuous coverage, intronic coverage, noncoding and intergenic variants, and ability to detect expanded repeats, and smaller CNVs. However, the technique to detect repeat expansions by WGS is available on only research basis. Hence, trinucleotide repeats need additional testing. The disadvantages of NGS techniques are their inability to detect CNVs and balanced translocations.
Mosaicism is reported in several PxMDs, including those related to ADCY5, ATP1A3, PDHA1 and SLC2A1. These require additional techniques for detection.
Future Directions
Underlying genetic diagnosis is present in only 50% of patients with PxMDs. A deeper understanding of the genetic basis of PxMD may guide future research and therapeutics. Targeting cerebellar outflows may be used in certain conditions, which demonstrate poor response to drugs, such as ATP1A3-related PxMD.5 Modulation of the cAMP signalling pathway may also be a promising therapeutic avenue and has already been harnessed in ADCY5-related PMD, which may respond to caffeine via effects on the adenosine A2A receptors. Whether genotype has a major impact on treatment remains to be seen, and there is a shift towards precision-based medicine in the treatment of PxMD. Examples include bypassing glucose transport defect in GLUT1-DS via the ketogenic diet and supplementing levodopa in GCH-1 related PMD. Advances in understanding of molecular mechanisms will help to guide future development of genetic and molecular therapies.
Conclusions
PxMD are a network disorder, with both the basal ganglia and the cerebellum implicated in its pathogenesis. Abnormalities in the striatal cAMP turnover pathway may also be implicated in PxMD. PxMD demonstrate great phenotypic pleiotropy, making molecular diagnosis challenging.
Although NGS has restructured its approach to PxMDs by uncovering the genetic architecture of many PxMDs, genetic underpinnings of several remain undiscovered. As more genes and variants continue to be reported in relation to PxMD, these will lead to enhanced understanding of pathophysiological mechanisms and precise treatment.
Acknowledgments
The authors thank their institutes for their support.
Disclosure
None of the authors report any conflict of interest.
References
1. Garone G, Capuano A, Travaglini L, et al. Clinical and genetic overview of paroxysmal movement disorders and episodic ataxias. Int J Mol Sci. 2020;21(10):E3603. doi:10.3390/ijms21103603
2. Demirkiran M, Jankovic J. Paroxysmal dyskinesias: clinical features and classification. Ann Neurol. 1995;38(4):571–579. doi:10.1002/ana.410380405
3. Erro R, Bhatia KP. Unravelling of the paroxysmal dyskinesias. J Neurol Neurosurg Psychiatry. 2019;90(2):227–234. doi:10.1136/jnnp-2018-318932
4. Harvey S, King MD, Gorman KM. Paroxysmal Movement Disorders. Front Neurol. 2021;12:659064. doi:10.3389/fneur.2021.659064
5. Delorme C, Giron C, Bendetowicz D, Méneret A, Mariani LL, Roze E. Current challenges in the pathophysiology, diagnosis, and treatment of paroxysmal movement disorders. Expert Rev Neurother. 2021;21(1):81–97. doi:10.1080/14737175.2021.1840978
6. Yoshimura K, Kanki R. Child-onset paroxysmal exercise-induced dystonia as the initial manifestation of hereditary Parkinson’s disease. Parkinsonism Relat Disord. 2018;49:108–109. doi:10.1016/j.parkreldis.2018.01.004
7. Yilmaz S, Turhan T, Ceylaner S, Gökben S, Tekgul H, Serdaroglu G. Excellent response to deep brain stimulation in a young girl with GNAO1-related progressive choreoathetosis. Childs Nerv Syst. 2016;32(9):1567–1568. doi:10.1007/s00381-016-3139-6
8. Dy ME, Chang FCF, Jesus SD, et al. Treatment of ADCY5-associated dystonia, chorea, and hyperkinetic disorders with deep brain stimulation: a multicenter case series. J Child Neurol. 2016;31(8):1027–1035. doi:10.1177/0883073816635749
9. Lance JW. Familial paroxysmal dystonic choreoathetosis and its differentiation from related syndromes. Ann Neurol. 1977;2(4):285–293. doi:10.1002/ana.410020405
10. Goodenough DJ. Familial and acquired paroxysmal dyskinesias. A proposed classification with delineation of clinical features. Arch Neurol. 1978;35(12):827–831. doi:10.1001/archneur.1978.00500360051010
11. de Seze J, Stojkovic T, Destée M, Destée A, Vermersch P. Paroxysmal kinesigenic choreoathetosis as a presenting symptom of multiple sclerosis. J Neurol. 2000;247(6):478–480. doi:10.1007/s004150070184
12. Zittel S, Bester M, Gerloff C, Münchau A, Leypoldt F. Symptomatic paroxysmal kinesigenic choreoathetosis as primary manifestation of multiple sclerosis. J Neurol. 2012;259(3):557–558. doi:10.1007/s00415-011-6188-5
13. Freiha J, Riachi N, Chalah MA, Zoghaib R, Ayache SS, Ahdab R. Paroxysmal symptoms in multiple Sclerosis—A review of the literature. J Clin Med. 2020;9(10):E3100. doi:10.3390/jcm9103100
14. Kim S-M, Go MJ, Sung -J-J, Park KS, Lee K-W. Painful tonic spasm in neuromyelitis optica: incidence, diagnostic utility, and clinical characteristics. Arch Neurol. 2012;69(8):1026–1031. doi:10.1001/archneurol.2012.112
15. Chaudhry N, Puri V, Patidar Y, Khwaja GA. Pathological laughter associated with paroxysmal kinesigenic dyskinesia: a rare presentation of acute disseminated encephalomyelitis. Epilepsy & Behavior Case Reports. 2013;1:14–19. doi:10.1016/j.ebcr.2012.11.001
16. Merchut MP, Brumlik J. Painful tonic spasms caused by putaminal infarction. Stroke. 1986;17(6):1319–1321. doi:10.1161/01.str.17.6.1319
17. Tan NCK, Tan AKY, Sitoh YY, Loh KC, Leow MKS, Tjia HTL. Paroxysmal exercise-induced dystonia associated with hypoglycaemia induced by an insulinoma. J Neurol. 2002;249(11):1615–1616. doi:10.1007/s00415-002-0876-0
18. Ryan C, Ahlskog JE, Savica R. Hyperglycemic chorea/ballism ascertained over 15 years at a referral medical center. Parkinsonism Relat Disord. 2018;48:97–100. doi:10.1016/j.parkreldis.2017.12.032
19. Yen DJ, Shan DE, Lu SR. Hyperthyroidism presenting as recurrent short paroxysmal kinesigenic dyskinesia. Mov Disord. 1998;13(2):361–363. doi:10.1002/mds.870130231
20. Drake ME. Paroxysmal kinesigenic choreoathetosis in hyperthyroidism. Postgrad Med J. 1987;63(746):1089–1090. doi:10.1136/pgmj.63.746.1089
21. Salah Uddin ABM. Limb shaking transient ischemic attack--an unusual presentation of carotid occlusive disease. A case report and review of the literature. Parkinsonism Relat Disord. 2004;10(7):451–453. doi:10.1016/j.parkreldis.2004.04.006
22. Kumral E, Bayam FE, Erdoğan CE. Limb shaking transient ischemic attacks: a follow-up of 28 patients. Rev Neurol. 2020;176(7–8):587–591. doi:10.1016/j.neurol.2019.12.001
23. Gonzalez-Alegre P, Ammache Z, Davis PH, Rodnitzky RL. Moyamoya-induced paroxysmal dyskinesia. Mov Disord. 2003;18(9):1051–1056. doi:10.1002/mds.10483
24. Lyoo CH, Kim DJ, Chang H, Lee MS. Moyamoya disease presenting with paroxysmal exercise-induced dyskinesia. Parkinsonism Relat Disord. 2007;13(7):446–448. doi:10.1016/j.parkreldis.2006.07.014
25. Chen WJ, Lin Y, Xiong ZQ, et al. Exome sequencing identifies truncating mutations in PRRT2 that cause paroxysmal kinesigenic dyskinesia. Nat Genet. 2011;43(12):1252–1255. doi:10.1038/ng.1008
26. Sun N, Nasello C, Deng L, et al. The PNKD gene is associated with Tourette disorder or tic disorder in a multiplex family. Mol Psychiatry. 2018;23(6):1487–1495. doi:10.1038/mp.2017.179
27. Shao Q, Shi X, Ma B, Zeng J, Zheng A, Xie W. TBC1D24-related familial infantile multifocal myoclonus: description of a new Chinese pedigree with a 20 year follow up. Epilepsy Res. 2022;182:106923. doi:10.1016/j.eplepsyres.2022.106923
28. Balestrini S, Milh M, Castiglioni C, et al. TBC1D24 genotype-phenotype correlation: epilepsies and other neurologic features. Neurology. 2016;87(1):77–85. doi:10.1212/WNL.0000000000002807
29. Salemi M, Cali’ F, Giambirtone M, Elia M, Romano C. TBC1D24 gene mRNA expression in a boy with early infantile epileptic encephalopathy-16. Acta Neurol Belg. 2020;120(2):381–383. doi:10.1007/s13760-017-0818-3
30. Zhou Q, Lin Y, Ye J, et al. Homozygous TBC1D24 mutation in a case of epilepsia partialis continua. Front Neurol. 2017;8:750. doi:10.3389/fneur.2017.00750
31. Poulat AL, Ville D, de Bellescize J, et al. Homozygous TBC1D24 mutation in two siblings with familial infantile myoclonic epilepsy (FIME) and moderate intellectual disability. Epilepsy Res. 2015;111:72–77. doi:10.1016/j.eplepsyres.2015.01.008
32. Campeau PM, Kasperaviciute D, Lu JT, et al. The genetic basis of DOORS syndrome: an exome-sequencing study. Lancet Neurol. 2014;13(1):44–58.
33. Tona R, Lopez IA, Fenollar-Ferrer C, et al. Mouse models of human pathogenic variants of TBC1D24 associated with non-syndromic deafness dfnb86 and dfna65 and syndromes involving deafness. Genes. 2020;11(10):E1122. doi:10.3390/genes11101122
34. Lüthy K, Mei D, Fischer B, et al. TBC1D24-TLDc-related epilepsy exercise-induced dystonia: rescue by antioxidants in a disease model. Brain. 2019;142(8):2319–2335. doi:10.1093/brain/awz175
35. Steel D, Heim J, Kruer MC, et al. Biallelic mutations of TBC1D24 in exercise-induced paroxysmal dystonia. Mov Disord. 2020;35(2):372–373. doi:10.1002/mds.27981
36. Cordani R, Pisciotta L, Mancardi MM, et al. Alternating hemiplegia of childhood in a child harboring a novel TBC1D24 mutation: case report and literature review. Neuropediatrics. 2022;53(1):69–74. doi:10.1055/s-0041-1739132
37. Ragona F, Castellotti B, Salis B, et al. Alternating hemiplegia and epilepsia partialis continua: a new phenotype for a novel compound TBC1D24 mutation. Seizure. 2017;47:71–73. doi:10.1016/j.seizure.2017.03.003
38. Zimmern V, Riant F, Roze E, et al. Infantile-onset paroxysmal movement disorder and episodic ataxia associated with a TBC1D24 mutation. Neuropediatrics. 2019;50(5):308–312. doi:10.1055/s-0039-1688410
39. Piarroux J, Riant F, Humbertclaude V, et al. FGF14-related episodic ataxia: delineating the phenotype of episodic ataxia type 9. Ann Clin Transl Neurol. 2020;7(4):565–572. doi:10.1002/acn3.51005
40. Shimojima K, Okumura A, Natsume J, et al. Spinocerebellar ataxias type 27 derived from a disruption of the fibroblast growth factor 14 gene with mimicking phenotype of paroxysmal non-kinesigenic dyskinesia. Brain Dev. 2012;34(3):230–233. doi:10.1016/j.braindev.2011.04.014
41. Dale RC, Melchers A, Fung VSC, Grattan-Smith P, Houlden H, Earl J. Familial paroxysmal exercise-induced dystonia: atypical presentation of autosomal dominant GTP-cyclohydrolase 1 deficiency. Dev Med Child Neurol. 2010;52(6):583–586. doi:10.1111/j.1469-8749.2010.03619.x
42. Belal H, Nakashima M, Matsumoto H, et al. De novo variants in RHOBTB2, an atypical Rho GTPase gene, cause epileptic encephalopathy. Hum Mutat. 2018;39(8):1070–1075. doi:10.1002/humu.23550
43. Defo A, Verloes A, Elenga N. Developmental and epileptic encephalopathy related to a heterozygous variant of the RHOBTB2 gene: a case report from French Guiana. Mol Genet Genomic Med. 2022;10(6):e1929. doi:10.1002/mgg3.1929
44. Lopes F, Barbosa M, Ameur A, et al. Identification of novel genetic causes of Rett syndrome-like phenotypes. J Med Genet. 2016;53(3):190–199. doi:10.1136/jmedgenet-2015-103568
45. Spagnoli C, Soliani L, Caraffi SG, et al. Paroxysmal movement disorder with response to carbamazepine in a patient with RHOBTB2 developmental and epileptic encephalopathy. Parkinsonism Relat Disord. 2020;76:54–55. doi:10.1016/j.parkreldis.2020.05.031
46. Necpál J, Zech M, Valachová A, et al. Severe paroxysmal dyskinesias without epilepsy in a RHOBTB2 mutation carrier. Parkinsonism Relat Disord. 2020;77:87–88. doi:10.1016/j.parkreldis.2020.06.028
47. Li HF, Chen YL, Zhuang L, et al. TMEM151A variants cause paroxysmal kinesigenic dyskinesia. Cell Discov. 2021;7(1):83. doi:10.1038/s41421-021-00322-w
48. Weber YG, Kamm C, Suls A, et al. Paroxysmal choreoathetosis/spasticity (DYT9) is caused by a GLUT1 defect. Neurology. 2011;77(10):959–964. doi:10.1212/WNL.0b013e31822e0479
49. Schneider SA, Paisan-Ruiz C, Garcia-Gorostiaga I, et al. GLUT1 gene mutations cause sporadic paroxysmal exercise-induced dyskinesias. Mov Disord. 2009;24(11):1684–1688. doi:10.1002/mds.22507
50. Pons R, Collins A, Rotstein M, Engelstad K, De Vivo DC. The spectrum of movement disorders in Glut-1 deficiency. Mov Disord. 2010;25(3):275–281. doi:10.1002/mds.22808
51. Tacik P, Loens S, Schrader C, Gayde-Stephan S, Biskup S, Dressler D. Severe familial paroxysmal exercise-induced dyskinesia. J Neurol. 2014;261(10):2009–2015. doi:10.1007/s00415-014-7441-5
52. Mongin M, Mezouar N, Dodet P, Vidailhet M, Roze E. Paroxysmal exercise-induced dyskinesias caused by GLUT1 deficiency syndrome. Tremor Other Hyperkinet Mov. 2016;6:371. doi:10.7916/D89W0F96
53. Klepper J, Leiendecker B, Eltze C, Heussinger N. Paroxysmal nonepileptic events in Glut1 deficiency. Mov Disord Clin Pract. 2016;3(6):607–610. doi:10.1002/mdc3.12387
54. Reis S, Matias J, Machado R, Monteiro JP. Paroxysmal ocular movements - an early sign in Glut1 deficiency Syndrome. Metab Brain Dis. 2018;33(4):1381–1383. doi:10.1007/s11011-018-0225-3
55. Leen WG, Klepper J, Verbeek MM, et al. Glucose transporter-1 deficiency syndrome: the expanding clinical and genetic spectrum of a treatable disorder. Brain. 2010;133(Pt3):655–670. doi:10.1093/brain/awp336
56. Remerand G, Boespflug-Tanguy O, Tonduti D, et al. Expanding the phenotypic spectrum of Allan-Herndon-Dudley syndrome in patients with SLC16A2 mutations. Dev Med Child Neurol. 2019;61(12):1439–1447. doi:10.1111/dmcn.14332
57. Novara F, Groeneweg S, Freri E, et al. Clinical and molecular characteristics of SLC16A2 (MCT8) mutations in three families with the Allan-Herndon-Dudley syndrome. Hum Mutat. 2017;38(3):260–264. doi:10.1002/humu.23140
58. Dumitrescu AM, Liao XH, Best TB, Brockmann K, Refetoff S. A novel syndrome combining thyroid and neurological abnormalities is associated with mutations in a monocarboxylate transporter gene. Am J Hum Genet. 2004;74(1):168–175. doi:10.1086/380999
59. Micheli F, Tschopp L, Cersosimo MG. Oxcarbazepine-responsive paroxysmal kinesigenic dyskinesia in Wilson disease. Clin Neuropharmacol. 2011;34(6):1. doi:10.1097/WNF.0b013e3182348964
60. Kim HJ, Yoon JH. A case of Wilson’s disease presenting with paroxysmal dystonia. Neurol Sci. 2017;38(10):1881–1882. doi:10.1007/s10072-017-3008-4
61. Ferrini A, Steel D, Barwick K, Kurian MA. An update on the phenotype, genotype and neurobiology of ADCY5-related disease. Mov Disord. 2021;36(5):1104–1114. doi:10.1002/mds.28495
62. Chen DH, Méneret A, Friedman JR, et al. ADCY5-related dyskinesia: broader spectrum and genotype-phenotype correlations. Neurology. 2015;85(23):2026–2035. doi:10.1212/WNL.0000000000002058
63. Erro R, Mencacci NE, Bhatia KP. The emerging role of phosphodiesterases in movement disorders. Mov Disord. 2021;36(10):2225–2243. doi:10.1002/mds.28686
64. Diggle CP, Sukoff Rizzo SJ, Popiolek M, et al. Biallelic mutations in PDE10A lead to loss of striatal PDE10A and a hyperkinetic movement disorder with onset in infancy. Am J Hum Genet. 2016;98(4):735–743. doi:10.1016/j.ajhg.2016.03.015
65. Mencacci NE, Kamsteeg EJ, Nakashima K, et al. De novo mutations in PDE10A cause childhood-onset chorea with bilateral striatal lesions. Am J Hum Genet. 2016;98(4):763–771. doi:10.1016/j.ajhg.2016.02.015
66. Salpietro V, Perez-Dueñas B, Nakashima K, et al. A homozygous loss-of-function mutation in PDE2A associated to early-onset hereditary chorea. Mov Disord. 2018;33(3):482–488. doi:10.1002/mds.27286
67. Doummar D, Dentel C, Lyautey R, et al. Biallelic PDE2A variants: a new cause of syndromic paroxysmal dyskinesia. Eur J Hum Genet. 2020;28(10):1403–1413. doi:10.1038/s41431-020-0641-9
68. Veeramah KR, O’Brien JE, Meisler MH, et al. De novo pathogenic SCN8A mutation identified by whole-genome sequencing of a family quartet affected by infantile epileptic encephalopathy and SUDEP. Am J Hum Genet. 2012;90(3):502–510. doi:10.1016/j.ajhg.2012.01.006
69. Xiao Y, Xiong J, Mao D, et al. Early-onset epileptic encephalopathy with de novo SCN8A mutation. Epilepsy Res. 2018;139:9–13. doi:10.1016/j.eplepsyres.2017.10.017
70. Pons L, Lesca G, Sanlaville D, et al. Neonatal tremor episodes and hyperekplexia-like presentation at onset in a child with SCN8A developmental and epileptic encephalopathy. Epileptic Disord. 2018;20(4):289–294. doi:10.1684/epd.2018.0988
71. Gardella E, Becker F, Møller RS, et al. Benign infantile seizures and paroxysmal dyskinesia caused by an SCN8A mutation. Ann Neurol. 2016;79(3):428–436. doi:10.1002/ana.24580
72. Trudeau MM, Dalton JC, Day JW, Ranum LPW, Meisler MH. Heterozygosity for a protein truncation mutation of sodium channel SCN8A in a patient with cerebellar atrophy, ataxia, and mental retardation. J Med Genet. 2006;43(6):527–530. doi:10.1136/jmg.2005.035667
73. Solazzi R, Castellotti B, Canafoglia L, et al. Paroxysmal tonic upgaze in a child with SCN8A-related encephalopathy. Epileptic Disord. 2021;23(4):643–647. doi:10.1684/epd.2021.1305
74. Lee H, Wang H, Jen JC, Sabatti C, Baloh RW, Nelson SF. A novel mutation in KCNA1 causes episodic ataxia without myokymia. Hum Mutat. 2004;24(6):536. doi:10.1002/humu.9295
75. Verdura E, Fons C, Schlüter A, et al. Complete loss of KCNA1 activity causes neonatal epileptic encephalopathy and dyskinesia. J Med Genet. 2020;57(2):132–137. doi:10.1136/jmedgenet-2019-106373
76. Set KK, Ghosh D, Huq AHM, Luat AF. Episodic ataxia type 1 (K-channelopathy) manifesting as paroxysmal nonkinesogenic dyskinesia: expanding the phenotype. Mov Disord Clin Pract. 2017;4(5):784–786. doi:10.1002/mdc3.12518
77. Yin XM, Lin JH, Cao L, et al. Familial paroxysmal kinesigenic dyskinesia is associated with mutations in the KCNA1 gene. Hum Mol Genet. 2018;27(4):625–637. doi:10.1093/hmg/ddx430
78. Kegele J, Krüger J, Koko M, et al. Genetics of paroxysmal dyskinesia: novel variants corroborate the role of KCNA1 in paroxysmal dyskinesia and highlight the diverse phenotypic spectrum of KCNA1- and SLC2A1-related disorders. Front Neurol. 2021;12:4. doi:10.3389/fneur.2021.701351
79. van der Wijst J, Konrad M, Verkaart SAJ, et al. A de novo KCNA1 mutation in a patient with tetany and hypomagnesemia. Nephron. 2018;139(4):359–366. doi:10.1159/000488954
80. Glaudemans B, van der Wijst J, Scola RH, et al. A missense mutation in the Kv1.1 voltage-gated potassium channel-encoding gene KCNA1 is linked to human autosomal dominant hypomagnesemia. J Clin Invest. 2009;119(4):936–942. doi:10.1172/JCI36948
81. Paulhus K, Ammerman L, Glasscock E. Clinical spectrum of KCNA1 mutations: new insights into episodic ataxia and epilepsy comorbidity. Int J Mol Sci. 2020;21(8):E2802. doi:10.3390/ijms21082802
82. Lee GB, Kim GY, Jeong IH, Kim N, Kim JW, Novel A. KCNA1 mutation in an episodic ataxia type 1 patient with asterixis and falls. J Clin Neurol. 2021;17(2):333–335. doi:10.3988/jcn.2021.17.2.333
83. Yuan H, Yuan H, Wang Q, et al. Two novel KCNA1 variants identified in two unrelated Chinese families affected by episodic ataxia type 1 and neurodevelopmental disorders. Mol Genet Genomic Med. 2020;8(10):e1434. doi:10.1002/mgg3.1434
84. Manville RW, Sidlow R, Abbott GW. Case report: a novel loss-of-function pathogenic variant in the KCNA1 cytoplasmic N-terminus causing carbamazepine-responsive type 1 episodic ataxia. Front Neurol. 2022;13:975849. doi:10.3389/fneur.2022.975849
85. Sintas C, Carreño O, Fernàndez-Castillo N, et al. Mutation spectrum in the CACNA1A gene in 49 patients with episodic ataxia. Sci Rep. 2017;7(1):2514. doi:10.1038/s41598-017-02554-x
86. Tantsis EM, Gill D, Griffiths L, et al. Eye movement disorders are an early manifestation of CACNA1A mutations in children. Dev Med Child Neurol. 2016;58(6):639–644. doi:10.1111/dmcn.13033
87. Chivukula AS, Suslova M, Kortzak D, Kovermann P, Fahlke C. Functional consequences of SLC1A3 mutations associated with episodic ataxia 6. Hum Mutat. 2020;41(11):1892–1905. doi:10.1002/humu.24089
88. Liang L, Li X, Moutton S, et al. De novo loss-of-function KCNMA1 variants are associated with a new multiple malformation syndrome and a broad spectrum of developmental and neurological phenotypes. Hum Mol Genet. 2019;28(17):2937–2951. doi:10.1093/hmg/ddz117
89. Zhang ZB, Tian MQ, Gao K, Jiang YW, Wu Y. De novo KCNMA1 mutations in children with early-onset paroxysmal dyskinesia and developmental delay. Mov Disord. 2015;30(9):1290–1292. doi:10.1002/mds.26216
90. Yeşil G, Aralaşmak A, Akyüz E, Içağasıoğlu D, Uygur Şahin T, Bayram Y. Expanding the phenotype of homozygous KCNMA1 mutations; dyskinesia, epilepsy, intellectual disability, cerebellar and corticospinal tract atrophy. Balkan Med J. 2018;35(4):336–339. doi:10.4274/balkanmedj.2017.0986
91. Sasaki M, Sumitomo N, Shimizu-Motohashi Y, et al. ATP1A3 variants and slowly progressive cerebellar ataxia without paroxysmal or episodic symptoms in children. Dev Med Child Neurol. 2021;63(1):111–115. doi:10.1111/dmcn.14666
92. Salles PA, Mata IF, Brünger T, Lal D, Fernandez HH. ATP1A3-Related disorders: an ever-expanding clinical spectrum. Front Neurol. 2021;12:637890. doi:10.3389/fneur.2021.637890
93. Sweney MT, Newcomb TM, Swoboda KJ. The expanding spectrum of neurological phenotypes in children with ATP1A3 mutations, alternating hemiplegia of childhood, rapid-onset dystonia-parkinsonism, CAPOS and beyond. Pediatr Neurol. 2015;52(1):56–64. doi:10.1016/j.pediatrneurol.2014.09.015
94. Vezyroglou A, Akilapa R, Barwick K, et al. The phenotypic continuum of ATP1A3-related disorders. Neurology. 2022;99(14):e1511–e1526. doi:10.1212/WNL.0000000000200927
95. Ulate-Campos A, Fons C, Artuch R, et al. Alternating hemiplegia of childhood with a de novo mutation in ATP1A3 and changes in SLC2A1 responsive to a ketogenic diet. Pediatr Neurol. 2014;50(4):377–379. doi:10.1016/j.pediatrneurol.2013.11.017
96. Yano ST, Silver K, Young R, et al. Fever-induced paroxysmal weakness and encephalopathy, a new phenotype of ATP1A3 mutation. Pediatr Neurol. 2017;73:101–105. doi:10.1016/j.pediatrneurol.2017.04.022
97. Biela M, Rydzanicz M, Szymanska K, et al. Variants of ATP1A3 in residue 756 cause a separate phenotype of relapsing encephalopathy with cerebellar ataxia (RECA)-Report of two cases and literature review. Mol Genet Genomic Med. 2021;9(9):e1772. doi:10.1002/mgg3.1772
98. Hanagasi HA, Bilgiç B, Abbink TEM, et al. Secondary paroxysmal kinesigenic dyskinesia associated with CLCN2 gene mutation. Parkinsonism Relat Disord. 2015;21(5):544–546. doi:10.1016/j.parkreldis.2015.02.013
99. Jiang Y-L, Yuan F, Yang Y, Sun X-L, Song L, Jiang W. CHRNA4 variant causes paroxysmal kinesigenic dyskinesia and genetic epilepsy with febrile seizures plus? Seizure. 2018;56:88–91. doi:10.1016/j.seizure.2018.02.005
100. Masnada S, Parazzini C, Bini P, et al. Phenotypic spectrum of short-chain enoyl-Coa hydratase-1 (ECHS1) deficiency. Eur J Paediatr Neurol. 2020;28:151–158. doi:10.1016/j.ejpn.2020.07.007
101. Marti‐Sanchez L, Baide‐Mairena H, Marcé‐Grau A, et al. Delineating the neurological phenotype in children with defects in theECHS1orHIBCHgene. J Inherit Metab Dis. 2021;44(2):401–414. doi:10.1002/jimd.12288
102. Olgiati S, Skorvanek M, Quadri M, et al. Paroxysmal exercise-induced dystonia within the phenotypic spectrum of ECHS1 deficiency. Mov Disord. 2016;31(7):1041–1048. doi:10.1002/mds.26610
103. Sato-Shirai I, Ogawa E, Arisaka A, et al. Valine-restricted diet for patients with ECHS1 deficiency: divergent clinical outcomes in two Japanese siblings. Brain Dev. 2021;43(2):308–313. doi:10.1016/j.braindev.2020.10.003
104. Barnerias C, Saudubray J-M, Touati G, et al. Pyruvate dehydrogenase complex deficiency: four neurological phenotypes with differing pathogenesis. Dev Med Child Neurol. 2010;52(2):e1–9. doi:10.1111/j.1469-8749.2009.03541.x
105. Friedman J, Feigenbaum A, Chuang N, Silhavy J, Gleeson JG. Pyruvate dehydrogenase complex-E2 deficiency causes paroxysmal exercise-induced dyskinesia. Neurology. 2017;89(22):2297–2298. doi:10.1212/WNL.0000000000004689
106. McWilliam CA, Ridout CK, Brown RM, McWilliam RC, Tolmie J, Brown GK. Pyruvate dehydrogenase E2 deficiency: a potentially treatable cause of episodic dystonia. Eur J Paediatr Neurol. 2010;14(4):349–353. doi:10.1016/j.ejpn.2009.11.001
107. Castiglioni C, Verrigni D, Okuma C, et al. Pyruvate dehydrogenase deficiency presenting as isolated paroxysmal exercise induced dystonia successfully reversed with thiamine supplementation. Case report and mini-review. Eur J Paediatr Neurol. 2015;19(5):497–503. doi:10.1016/j.ejpn.2015.04.008
108. Head RA, de Goede CGEL, Newton RWN, et al. Pyruvate dehydrogenase deficiency presenting as dystonia in childhood. Dev Med Child Neurol. 2007;46(10):710–712. doi:10.1111/j.1469-8749.2004.tb00986.x
109. Brown GK, Otero LJ, LeGris M, Brown RM. Pyruvate dehydrogenase deficiency. J Med Genet. 1994;31(11):875–879. doi:10.1136/jmg.31.11.875
110. Tian W-T, Huang X-J, Mao X, et al. Proline-rich transmembrane protein 2 - negative paroxysmal kinesigenic dyskinesia: clinical and genetic analyses of 163 patients. Mov Disord. 2018;33(3):459–467. doi:10.1002/mds.27274
111. Lu Q, Shang L, Tian WT, Cao L, Zhang X, Liu Q. Complicated paroxysmal kinesigenic dyskinesia associated with SACS mutations. Ann Transl Med. 2020;8(1):8. doi:10.21037/atm.2019.11.31
112. Temudo T, Martins E, Poças F, Cruz R, Vilarinho L. Maple syrup disease presenting as paroxysmal dystonia. Ann Neurol. 2004;56(5):749–750. doi:10.1002/ana.20288
113. Liu Y-D, Chu X, Liu R-H, Sun Y, Kong Q-X, Li Q-B. Paroxysmal spasticity of lower extremities as the initial symptom in two siblings with maple syrup urine disease. Mol Med Rep. 2019;19(6):4872–4880. doi:10.3892/mmr.2019.10133
114. Synofzik M, Schicks J, Lindig T, et al. Acetazolamide-responsive exercise-induced episodic ataxia associated with a novel homozygous DARS2 mutation. J Med Genet. 2011;48(10):713–715. doi:10.1136/jmg.2011.090282
115. Liao JY, Salles PA, Shuaib UA, Fernandez HH. Genetic updates on paroxysmal dyskinesias. J Neural Transm. 2021;128(4):447–471. doi:10.1007/s00702-021-02335-x
116. Battaglia A, Doccini V, Bernardini L, et al. Confirmation of chromosomal microarray as a first-tier clinical diagnostic test for individuals with developmental delay, intellectual disability, autism spectrum disorders and dysmorphic features. Eur J Paediatr Neurol. 2013;17(6):589–599. doi:10.1016/j.ejpn.2013.04.010
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.