Back to Journals » Cancer Management and Research » Volume 12
Genetic Characterization and Risk Stratification of Acute Myeloid Leukemia
Authors Pourrajab F, Zare-Khormizi MR, Hashemi AS, Hekmatimoghaddam S ![]()
Received 15 December 2019
Accepted for publication 22 February 2020
Published 25 March 2020 Volume 2020:12 Pages 2231—2253
DOI https://doi.org/10.2147/CMAR.S242479
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sanjeev K. Srivastava
Fatemeh Pourrajab,1,2 Mohamad Reza Zare-Khormizi,3 Azam Sadat Hashemi,4 Seyedhossein Hekmatimoghaddam4,5
1Department of Biochemistry and Molecular Biology, School of Medicine, Shahid Sadoughi University of Medical Sciences, Yazd, Iran; 2Nutrition and Food Security Research Centre, Shahid Sadoughi University of Medical Sciences, Yazd, Iran; 3School of Medicine, Shahid Sadoughi University of Medical Sciences, Yazd, Iran; 4Hematology & Oncology Research Center, Shahid Sadoughi University of Medical Sciences, Yazd, Iran; 5Department of Advanced Medical Sciences and Technologies, School of Paramedicine, Shahid Sadoughi University of Medical Sciences, Yazd, Iran
Correspondence: Fatemeh Pourrajab
Department of Biochemistry and Molecular Biology, School of Medicine, Shahid Sadoughi University of Medical Sciences, Yazd 8915173149, Iran
Email [email protected]
Abstract: The most common acute leukemia in adults is acute myeloid leukemia (AML). The pathophysiology of the disease associates with cytogenetic abnormalities, gene mutations and aberrant gene expressions. At the molecular level, the disease manifests as changes in both epigenetic and genetic signatures. At the clinical level, two aspects of AML should be taken into account. First, the molecular changes occurring in the disease are important prognostic and predictive markers of AML. Second, use of novel therapies targeting these molecular changes. Currently, cytogenetic abnormalities and molecular alterations are the common biomarkers for the prognosis and choice of treatment for AML. Finding a panel of multiple biomarkers is a crucial diagnostic step for patient classification and serves as a prerequisite for individualized treatment strategies. Furthermore, the most important way of identifying relevant targets for new treatment approaches is defining specific patterns or a spectrum of driver gene mutations occurring in AML. Then, an algorithm can be established by the use of several biomarkers, to be used for personalized medicine. This review deals with molecular alterations, risk stratification, and relevant therapeutic decision-making in AML.
Keywords: acute myeloid leukemia, AML, genetic characterization, risk stratification
Acute Myeloid Leukemia
Acute myeloid leukemia (AML) results from clonal expansion of transformed hematopoietic stem cells (HSCs) through the acquisition of genetic abnormalities including chromosomal rearrangements and multiple gene mutations whereby HSCs are conferred a proliferative and survival advantage and impaired hematopoietic differentiation. AML is a phenotypically and genetically heterogeneous disease. Biologically, different types of mutations which complement each other and cooperate in leukemogenesis should occur in an HSC. Acquired mutations enhance self-renewal and proliferation in an HSC wherein normal mechanisms of differentiation are impaired and eventually lead to a clonal expansion of abnormal immature leukemic blasts which accumulate in the bone marrow. They gradually replace normal hematopoietic tissue and interfere with the production of normal blood cells, resulting in impaired hematopoiesis and bone marrow failure1,2 manifested as cytopenia. Blasts then frequently spread to other parts of the body such as the lymph nodes, spleen, liver, testes and central nervous system.3 Different subtypes of AML are each associated with distinct genetic and molecular abnormalities. Due to heterogeneity in the genomic landscape of leukemia, there are great efforts to devise new-targeted therapies to personalize medicine in AML therapy.4,5
This is evident by the increasing number of compounds have been made available and combined with conventional chemotherapy during induction or consolidation therapy, novel interventions that are personalized to the host and tumor genotype.6,7
A well-known procedure to distinguishing tumor genotypes and classify different subgroups of AML was proposed by the World Health Organization (WHO), through using morphologic, immunologic, cytogenetic and molecular biologic classification techniques (MICM). The classification scheme was proposed to distinguish different genetic and molecular abnormalities in the diagnosis of AML and provides a framework for clinical management. It especially emphasizes the importance of genetic test results to define clinically relevant disease entities in conjunction with morphology, immunophenotype, and other clinicopathologic features.7–8
However, for any samples suspected for AML, karyotyping is first required; if it is normal, then the fluorescent in situ hybridization (FISH) or reverse transcriptase-polymerase chain reaction (RT-PCR) technique is essential to detect cryptic rearrangement of the relevant locus (Table 1). This genetic approach seems to be used in clinics for initial diagnosis and decision made on therapeutic choices. Then, at the time of first remission or relapse, additional testing is ordered to refine prognosis.9–11
 |
Table 1 Summary of Major Cytogenetic Abnormalities Observed in AML and Related Methods of Detection |
Genetic Abnormalities in AML (a Collaboration of at Least Three Types of Mutations)
Based on the cytogenetic and molecular analysis and according to the risk stratification, AML genetic aberrations are placed into three categories; 1) Non-random chromosomal aberrations including balanced translocations, inversions, deletions, monosomies, and trisomies (Tables 2 and 3), 2) Multiple gene mutations (Table 4) and 3) Epigenetic alterations (Table 5).6,12–14
 |
Table 2 Cytogenetic Abnormalities (Non-Random Chromosomal Rearrangements) Found in the Leukemic Blasts of 55% of Adults with AML |
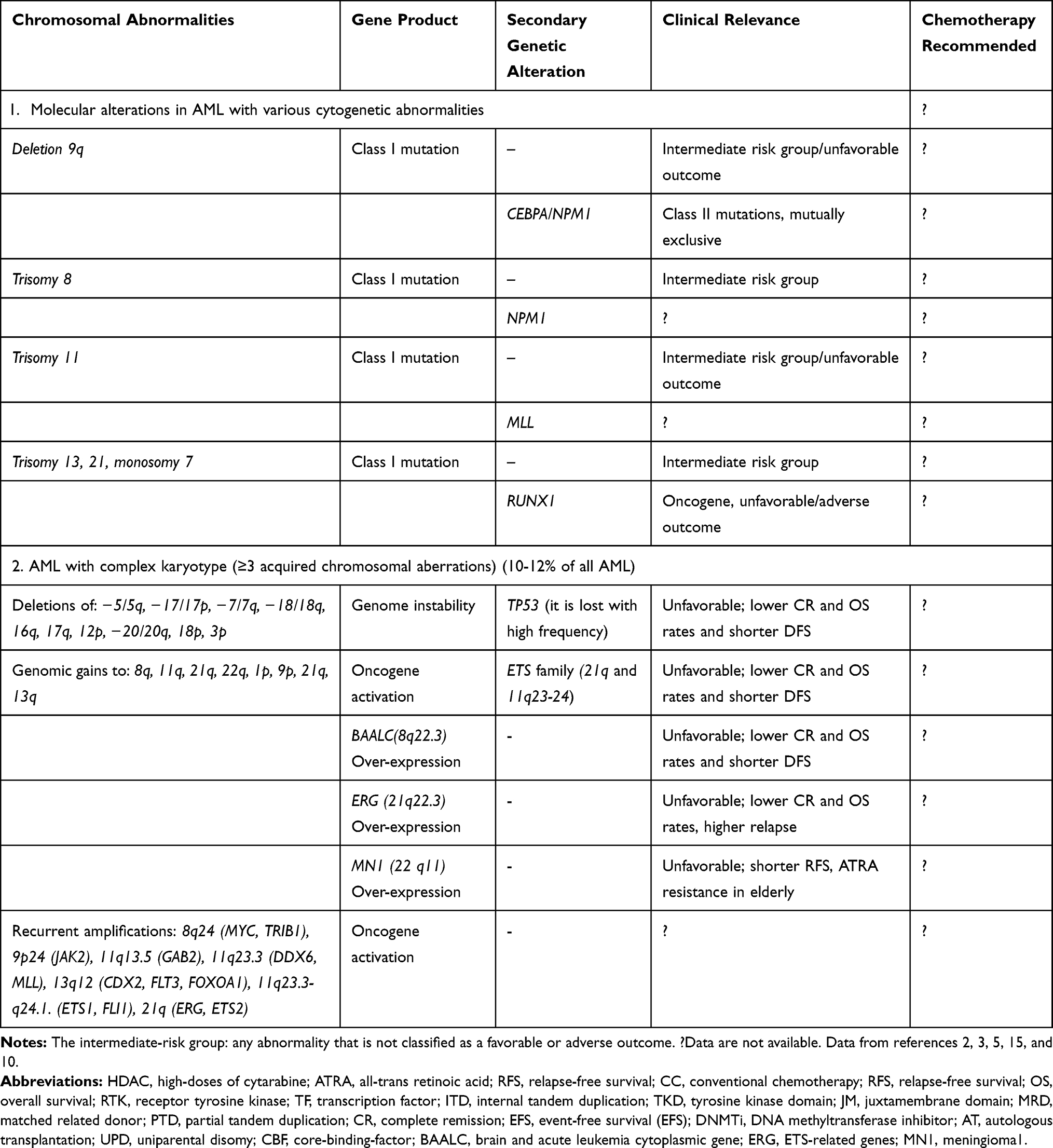 |
Table 3 Other Cytogenetic Abnormalities (Non-Random Chromosomal Rearrangements) Found in the Leukemic Blasts of 55% of Adults with AML |
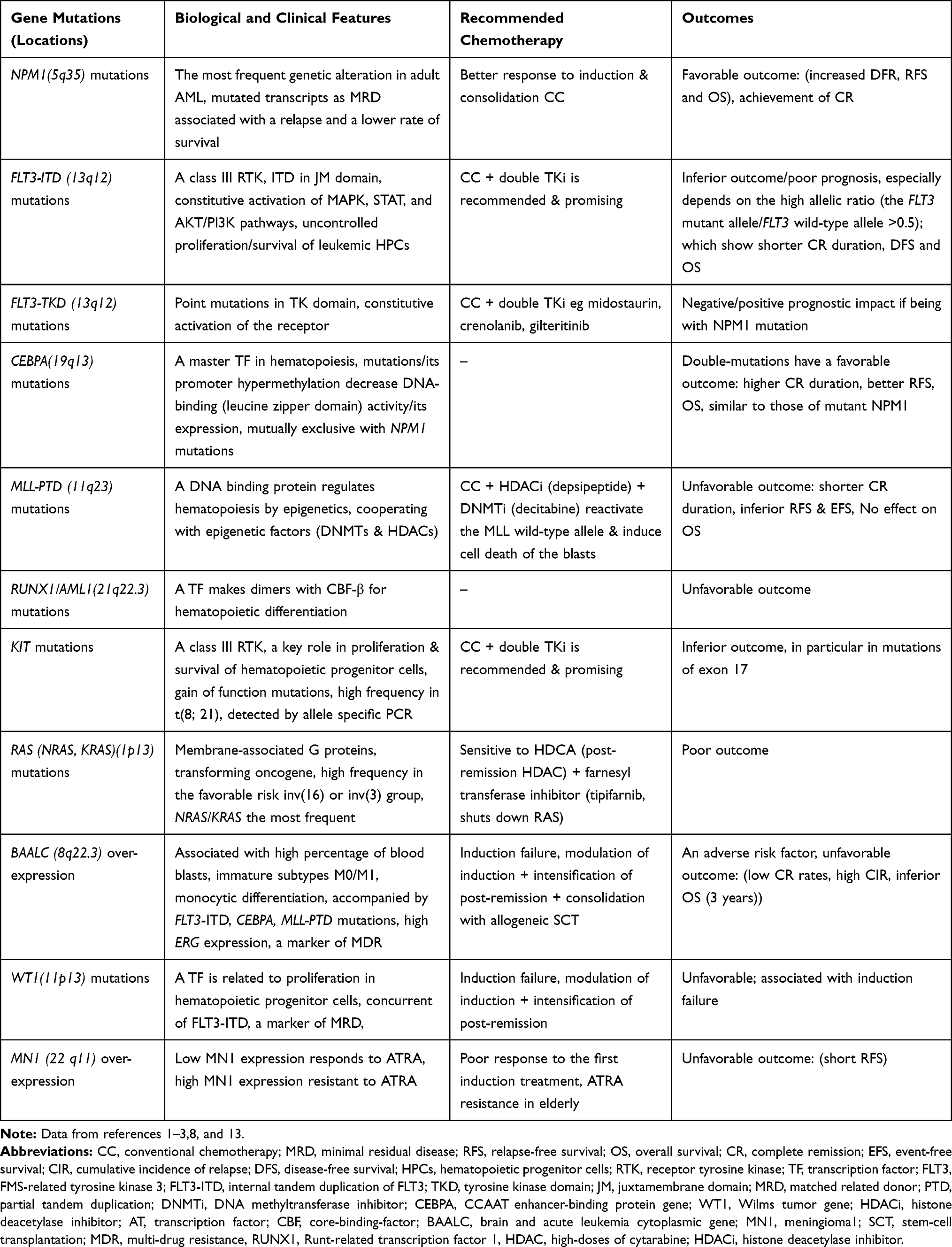 |
Table 4 Prototype of Gene Mutations Occurred in Cytogenetically Normal AML or in Cytogenetically Defined AML Subsets (~40–50% of Adult and 25% of Pediatric AML) |
 |
Table 5 Summary of the Most Common Epigenetic Mutations Occurred in AML |
Actually, aberrations should occur in genes relevant for pathogenesis, ie, master transcription factor fusions (18% of cases), NPM1 (27% of cases), tumor-suppressor genes (16%), DNA-methylation-related genes (44%), signaling genes (59%), chromatin-modifying genes (30%), myeloid-transcription genes (22%), cohesion complex genes (13%), and spliceosome complex genes (14%).1,3,7
At the molecular level, AML is the consequence of collaboration between at least three broad classes of gene alterations (Table 6 & Figure 1). Class I gene alterations are those aberrations that activating signal transduction pathways and enhancing proliferation with survival advantages of hematopoietic stem cells. This class of gene aberrations can involve and activate the receptor tyrosine kinase FLT3 and Kit or the RAS-associated signaling pathway. Class II gene alterations affect a master transcription factor or a protein involved in hematopoietic differentiation. This second aberration would impair differentiation of a hematopoietic progenitor cell (HPC) and aberrant acquisition of self-renewal properties whereby increase the likeliness of malignant transformation. Outstanding examples for this class of aberrations are the recurring gene fusions RUNX1-RUNX1T1 and PML-RARA resulting from t(8;21), inv(16)/t(16;16) and t(15;17) rearrangements, as well as mutations in CCAAT enhancer-binding protein A (CEBPA) and nucleophosmin 1 (NPM1) genes.15,16,17
 |
Table 6 Prototype of the Collaboration Between at Least Three Classes of Mutations (Class I, II, III) Associated with Appearance of AML |
Class 0/III are those alterations that promote epigenetic modifications of chromatin in a large area and affect further transcription factors or components of the transcriptional co-activation complexes whereby would confer malignant transformation to the HPCs and lead to overt AML (eg, DNMT3A and IDH1/2, involved in epigenetic regulation of chromatin and cellular processes). Class 0/III alterations, however, can also be happened before class I.2,6,12,13
 |
Figure 1 A schematic representation of the collaboration between at least three classes of gene alterations (class I, II, III), that happen in a hematopoietic progenitor cell (HPC) and are associated with the appearance of AML. Class I alterations enhance proliferative signaling pathways and confer survival advantages. Class II alterations impair the processes of cell differentiation and apoptosis. Class 0/III, however, can also be an early event before class I, promotes epigenetic modifications which finally confer malignant transformation to the HPCs and lead to overt leukemia. Data from references 2, 6, 12, 13, 16, and 17. |
Cytogenetic and Chromosomal Rearrangements
Cytogenetic abnormalities, detectable in approximately 50–60% of newly diagnosed AML patients, are categorized as non-random chromosomal rearrangements. These aberrations are further classified into three categories; AML with balanced translocations/inversions (Table 2), AML with various cytogenetic abnormalities (un-balanced translocations, eg deletions, monosomies, and trisomies) and AML with complex karyotype (representing at least 3 acquired chromosomal aberrations) (Table 3).
Cytogenetically, AML has three prognostic categories: favorable, intermediate and poor-risk group. The first group includes balanced translocations with a favorable outcome. The poor risk group has a complex aberrant karyotype that confers a poor clinical outcome. The intermediate prognosis group includes normal karyotype and other karyotypic abnormalities (Table 1).6,7
Balanced Chromosomal Rearrangements (Translocations/Inversions)
Balanced translocations and inversions include inv(16)(p13.1q22)/t(16;16) (p13.1;q22), t(8;21)(q22;q22), inv(3)(q21q26.2)/t(3;3)(q21;q26.2), t(6;9)(p23;q34), t/inv (11q23) and t(15;17)(q24;q21). These lesions on their own are not sufficient to induce leukemia, and there is a requirement for additional secondary genetic lesions to accompany the balanced translocation/inversion. For example, trisomy 22 in AML with inv(16)/t(16;16), deletion 9q in AML with t(8;21), or monosomy 7 in AML with inv(3)/t(3;3) (Table 2).2,15
The results of t(8;21) and inv(16)/t(16;16) are, respectively, fusion proteins RUNX1-RUNX1T1 (AML1-ETO) and CBFβ-MYH11 which bock the transactivation of tumor-suppressor genes, and associate with favorable outcome in AML. Accordingly, the fusion proteins RUNX1/AML1 and CBFB-MYH11 impair myeloid differentiation and induce self-renewal properties of HPCs but on their own do not cause an overt leukemic phenotype. Rather, there should be further rare mutations in the germline of affected patients to predispose these persons for developing AML. More than one genetic change should occur in HPCs to show overt leukemia phenotype (Figures 2 and 3).15–9
 |
Figure 2 Percentage of the major cytogenetic subgroups of acute myeloid leukemia (AML) (excluding acute promyelocytic leukemia), and associated gene mutations. In the subgroup with various karyotypes, NPM1 mutations are frequently found in AML with 9q deletion and trisomy 8, CEBPA mutations in AML with 9q deletion, MLL mutations in AML with trisomy 11, and RUNX1 mutations in AML with trisomy 13 and trisomy 21 (frequencies of the cytogenetic subgroups are taken from Reference 36, derived from 2654 cytogenetically characterized adults (≥18 years) with de novo or secondary AML entered on AMLSG treatment trials). Note: Data from Chiaretti et al.1 |
 |
Figure 3 (A) Approximate distribution of gene mutations in cytogenetically normal acute myeloid leukemia (CN-AML) in all patients (class I & II gene mutations).8 Adapted from Zaidi SZ, Owaidah T, Al Sharif F, Ahmed SY, Chaudhri N, Aljurf M. The challenge of risk stratification in acute myeloid leukemia with normal karyotype. Data from Zaidi et al.9 (B): Distribution of class I/II gene mutations among CN-AML patients with NPM1 mutation. In about 28% of cases, NPM1 mutation is the only detectable genetic change, whereas in the majority of cases additional mutations are found in genes such as FLT3, NRAS and WT1 (in approximately 40%, 21%, 17% of NPM1 mutations, respectively). A small number of AML with NPM1 mutations have an additional hypothetical class II mutation, eg, NPM1 and concurrent CEBPA mutation. Data from Dohner and Döhner.2 |
The reciprocal t(8;21)(q22;q22) between the chromosomes 8 and 21 results in the fusion gene AML1-ETO (RUNX1-RUNX1T1) whose product is the fusion protein AML1-ETO. The RUNX protein family or core-binding factors (CBFs) constitutes of a group of heterodimeric transcription factors that are composed of an α-subunit (CBFα; encoded by 3 distinct genes: Runx1/Runx2/Runx3) and a β-subunit (CBFβ; encoded by CBFβ). This family is also known as the polyomavirus enhancer-binding protein-2 (PEB-2).10 At the molecular level, RUNX1 (AML1) is a DNA-binding factor, and a master regulator of HPCs, while RUNX1T1 (ETO) is a transcription factor with the repressor activity (an oncogene). The N-terminal domain of AML1 fuses to the 577 residues from the ETO C-terminal domain. The fusion protein AML1-ETO recognizes AML1 consensus binding sites and heterodimerizes with CBFβ, wherein harboring transcriptional repressor activities.1,2 The fusion protein acts as a transcriptional repressor to block AML1-dependent transactivation and transcription of tumor suppressors. In fact, the translocation interrupts AML1 protein between the N-terminal and transactivation domains, while leaving the transactivation domain of ETO protein intact.5,6 This type of translocation has the highest incidence in childhood AML (~12% of AML cases in children) (Figure 4), and profoundly associates with M2 FAB subtype (AML with maturation), but rarely seen with AML M1 or M4 subtypes. This kind of cytogenetic aberration is highly significant for diagnosis and therapy management, since on its own associates with favorable outcomes, high remission rates and long median survival, whereas c-Kit mutations occurring concurrently with t(8;21)(q22; q22), compose an independent adverse prognostic biomarker (Table 2, Figure 2). In addition, in pediatric AML M2 subtype with t(8;21), the X-chromosome loss may occur as the secondary cytogenetic events with no clinical significance, whereas the loss of the Y chromosome, as the secondary genetic event, forms a critical mutational event. Generally, conventional cytogenetic analysis FISH, or RT-PCR can readily predict AML1-ETO outcomes (Table 1).4–6 Patients with t(8;21) have a favorable prognosis with good response to conventional chemotherapy (the combination of an anthracycline and cytarabine, the “3 + 7” regimen) and show complete remissions (Tables 1 and 2). At least three or four cycles of intensive post-remission therapy with high doses of cytarabine (HDAC) are recommended for adults, improving substantially the outcome and maximizing the gain of chemotherapy (Table 2).6,15
However, in de novo AML cases, loss of function mutations (missense, nonsense or frameshift mutations) in RUNX1 gene are classified as intermediate-risk AML, rather than as the favorable risk group, and highly associate with AML M0. The RUNX1 mutations associate with higher chemotherapy resistance (where refractory rates are about 30%), and lower event-free, relapse-free, and overall survival rates.3
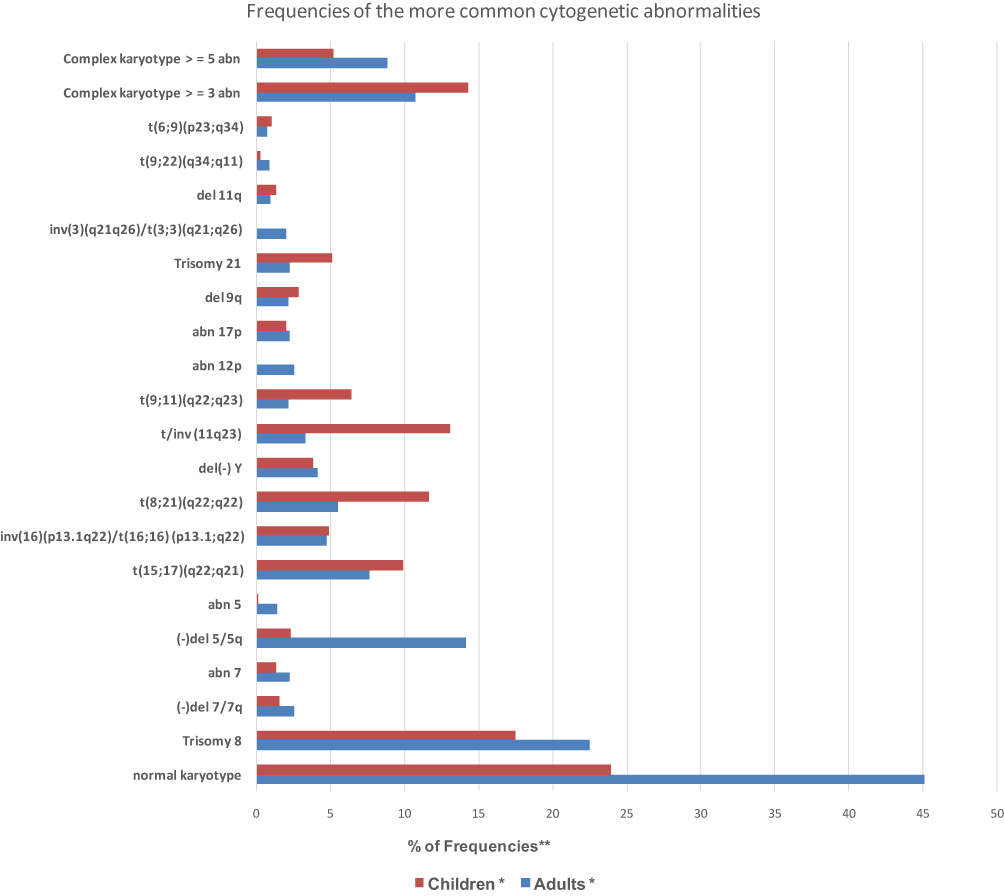 |
Figure 4 Frequencies of the more common cytogenetic abnormalities in adult and childhood AML. *Percentages for particular abnormalities were calculated using only those studies that provided relevant data where AML children aged from 2 months to 18 years and AML adults aged 18–86 years, diagnosed with de novo AML; data are derived from Mrozek et al8 and according to Cooperative Group Study of SWOG/ECOG, CALGB and MRC.Abbreviations: CALGB, Cancer and Leukemia Group B; MRC, United Kingdom Medical Research Council; SWOG/ECOG, Southwest Oncology Group/Eastern Cooperative Oncology Group; abn, abnormality. |
Inv(16)(p13;q22) leads to the formation of oncogene CBFβ-MYH11 whose product is the oncoprotein CBFβ-MYH11. The fusion protein acts as a transcriptional repressor cooperating with AML1 to repress transcription of tumor-suppressor genes PTEN, Bcl-2, CEBPA, ARF and PSGL-1. In the rearrangement, the C-terminal coiled-coil region of the smooth muscle myosin heavy chain 11 (MYH11) fuses with the first 165 residues of CBFβ.1,15 CBFβ-MYH11 fusion gene forms at least eight different transcripts in progenitor cells wherein fusion protein heterodimerizes with RUNX1 (CBFα).
In progenitor cells, RUNX1 (CBFα) and CBFβ form a complex regulating gene expression, at the Runx-binding sites on DNA. In a dominant negative manner by competing for heterodimerization and/or for DNA binding at Runx-binding sites, CBFβ-MYH11 and RUNX1-ETO fusion proteins inhibit the function of normal RUNX1 (CBFα)/CBFβ complex. CBFs are required in the hematopoietic ontogeny as the key regulators of different steps of hematopoiesis; therefore, the abnormal fused products disrupt their function and inhibit hematopoietic differentiation.3,4 The balanced rearrangements t(8;21)(q22;q22) and inv(16)(p13.1q22)/t(16;16)(p13.1;q22) are assigned as core-binding transcription factor leukemia (CBF-AML), and are the most common events found in AML, representing approximately 15–25% of all AML cases.3,6
Now, RQ-PCR and nested PCR techniques are sensitive and efficient approaches for the detection of these rearrangements (sensitivities ranging from 10 −4–10 −6).5,10 CBFβ-MYH11 has been linked with the AML M4Eo and a higher incidence in adults (Figure 4).3,4
Patients with inv(16)/t(16;16) have a favorable prognosis, good response to conventional chemotherapy (the combination of an anthracycline and cytarabine, the “3 + 7” regimen) and complete remissions (Tables 1 and 2). Likewise, t(8;21), receiving 3–4 cycles of HDAC is efficient and recommended for these patients (Table 2). The improved outcome with repeated therapy with cytarabine is addressed to the incorporation of cytarabine into the genomic DNA where increased sensitivity of the leukemic cells to cytarabine and inducing apoptosis.6,15
Patients with CBF-AML are consistently cytarabine-anthracycline responders with favorable outcomes (Table 2). The substantial heterogeneities observed in the outcome of CBF-AML patients are associated with the co-occurrence of other mutations occurred in specific genes, eg, FLT3, KIT and RAS (Figure 2 & Table 2). Therefore, for right diagnosis and proper management of the cancer, specific mutations occurred in FLT3, NPM1, CEBPA, RAN, and KIT genes should be taken into account, besides the cytogenetic alterations.3,15,10
The t(15;17)(q22;q12) is classified as a balanced translocation which fusing the retinoic acid receptor-α (RARA) gene on chromosome 17(q21) with the PML gene on chromosome 15(q24) and leading to the expression of oncogene PML-RARA in hematopoietic myeloid cells. In patients with acute promyelocytic leukemia (APL), the presence of the PML-RARA rearrangement predicts a favorable response to treatment with retinoic acid (RA). APL accounts for about 10–15% of AML cases. The t(15;17)(q24;q21) and its fusion protein PML-RARα are present in about 98% of APL cases, accounted as the biomarker of APL.1–3
Notably, low-risk APL patients can attain 100% complete remission (CR) and 2-year event-free survival (EFS) of 97%, just by a combination of arsenic trioxide and all-trans retinoic acid (ATRA), with no additional chemotherapy. Now, ATRA is considered as a proper treatment of leukemia or as a part of remission induction therapy, both in adults and in children.8,10 RARα is a member of the nuclear hormone-receptor superfamily, which activating transcription in the presence of retinoic acids (RAs) (ATRA or 9-cis retinoic acid, CTRA) and inducing many target genes required for the hematopoietic differentiation.5,6 At the molecular level, RARα and retinoid X receptor-α (RXRα) form the heterodimer RAR-RXR that binds DNA at retinoic acid-response elements (RARE). RAR-RXR forms a transcription activator complex required for promyelocytic differentiation. In the absence of RAs, RAR-RXR heterodimer acts as a transcription repressor by recruiting corepressors DNMT1, DNMT3A, histone deacetylases (HDACs) and histone methyltransferases, all of which remodeling chromatin. In the presence of RAs, conformational changes occur in RAR-RXR, whereby causing dissociation of co-repressor complexes from RAR-RXR, and transcriptional de-repression and activation of genes required for the differentiation of promyelocytes.6,8
PML-RARA competes with RARα to form a heterodimer with RXRα, then represses target promoters of the signaling chain in the same manner as RARA when it is not bound to its ligand. However, unlike the wild variant, it requires a greater concentration of ligand to eliminate the repression because it maintains a more stable interaction with the corepressor complex and some methylases such as DNMT1 and DNMT3A. Furthermore, PML-RARA has important effects on apoptosis because in a negatively dominant manner it interferes with the function of wild PML and its regulation of p53.15,10 The pharmacological concentrations of ATRA can activate transcription by inducing dissociation of corepressors from PML-RARα which is then degraded by a proteasome-dependent manner. Immature leukemic promyelocytes require ATRA to differentiate into mature granulocytes wherein bypassing the co-repressor activity of PML-RARα and differentiating into the myeloid lineage.1,3 There are t(15;17) variants that all lead to the formation of PML-RARA fusion genes and occur in AML, but are absent in patients with other leukemia types. To detecting this translocation successfully, the RT-PCR and FISH are reliable techniques that link approaches to a favorable prognosis (Table 1). Notably, the PML locus in 15(q24) encodes transcripts through alternative splicing generating more than 20 different tumor-suppressor isoforms. These tumor suppressors participate in nuclear structures called the PML-nuclear bodies (PML-NBs) which sequestrates and release proteins from the nucleus. PML-NBs mediate posttranslational modifications and promote nuclear events in response to cellular stressors.5,6
The balanced translocation; t(9;22)(q34;q11) occurs between chromosomes 9 and 22, and produces the fusion oncogene BCR-ABL1. The 3ʹ-sequences of proto-oncogene ABL, a tyrosine kinase on chromosome 9, is fused to the 5ʹ-sequences of the BCR, a gene on chromosome 22. This translocation leads to the formation of Philadelphia chromosome (Ph) generally occurring in childhood ALL and chronic myelogenous leukemia (CML). Predominantly, the Ph+ chromosome causes an early B-cell phenotype. The Ph+ patients are mostly adolescence or young adulthood, usually older than 10 years, who show elevated leukocyte counts and frequently central nervous system (CNS) disease. The Ph + chromosome may occur late as a secondary aberration in CML, B-ALL, T-ALL, and primarily AML. There are two variants of the BCR-ABL1 oncogene, “minor” (m-bcr, encoding p190 kDa protein) and “major” (M-bcr, encoding p210 kDa protein), as a result of two different breakpoint in cluster regions of the BCR gene. The interphase FISH, RT-PCR, or RQ-PCR methods are reliable techniques to detect this rearrangement (Table 1).6,15,10
The mixed-lineage myeloid/lymphoid leukemia (MLL) gene is mapped to 11q23 covering a genomic DNA region of approximately 100 kb. The chromosomal rearrangements at 11q23 include translocations, deletions, and duplications. The 11q23 rearrangement most often results in gene hybridization between the 5ʹ-sequences of the MLL gene and the 3ʹ-sequences of the other partner gene. Exons 5–11 on 11q23, cluster the majority of breakpoints, in a region extended about 8.3 kb long.8,10
The hybrid proteins harbor the N-terminal residues of MLL and the C-terminal residues of the partner protein. The hybrid proteins localize in the nucleus of the hematopoietic stem cell wherein exhibiting transforming activity. MLL is a master regulator of hematopoiesis in hematopoietic stem cells wherein regulating the homeobox genes (HOX). This group of genes consecutively influences hematopoietic stem-cell renewal and leukemogenesis. In addition to mixed-lineage leukemia, the rearrangement also occurs in myeloid and lymphoid leukemic cells, thereby exhibiting its origination from a stem cell or an early progenitor cell. The prognostic significance of 11q23 abnormalities has been variably assigned between intermediate-risk and adverse-risk groups (Tables 1 and 7).1,2
 |
Table 7 Risk Stratification of the Most Common Chromosomal Aberrations Detected in AML |
The most common genetic events occurring in ALL or AML children aged <12 months are the MLL rearrangements. It is dominant in 43–58% of infants aged less than 12 months, in 39% of children aged between 13 and 24 months, and in 8-9% of children who older than 24 months. Generally, the chance of MLL rearrangement decreases with age where it has 4 times more incidence in children than in adults (Figure 4). Approximately 5-10% of MLL rearrangements occur as a secondary event or therapy-related leukemia that arise in patients treated with topoisomerase II inhibitors for other malignancy.3
MLL is a large protein with multi-domains and DNA-binding activity interacting directly with DNA and other DNA-binding proteins. As a histone methyltransferase belonging to the trithorax-group family, hematopoietic cells including stem and progenitor populations ubiquitously express MLL. The fusion protein binds DNA constitutively and sequentially activates HOX genes such as HoxA9 and HoxA10 which commonly up-regulated in leukemia. At the molecular level, MLL participates to methylate histone H3 on lysine residue 4 (H3K4), which positively regulates gene expression in embryogenesis and hematopoiesis.6
Profoundly, MLL gene fusion to a wide array of partner genes, including AF4, AF9, ENL, AF10 and ELL has leukemogenic effects. MLL rearrangements involve several partner genes including t(6;11)(q27;q23), in which MLL is fused to AF6; t(9;11)(p22; q23), in which MLL is fused with AF9; and t(10;11)(p12;q23), in which MLL (on 11q, the long arm of chromosome 11) is fused to AF10 or MLLT10 (on 10p, the short arm of chromosome 10). MLL-AF10, MLL-ABI1, and CALM-AF10 are fusion genes identified in leukemic patients with t(10;11)(p12;q23). The MLL fusion proteins attain a dominant gain-of-function trait enhancing the transcriptional activity and giving rise to the poor prognosis of patients with MLL translocations. Herein, RT-PCR method is a reliable technique to identify these subtle translocations, besides cytogenetic analysis. The fusion transcripts are readily detectable by RT-PCR, which is especially useful in tracking the minimal residual disease. Southern blotting with cytogenetic analysis is able to detect the majority of MLL rearrangements and iFISH in a complementary fashion for the accurate identification of MLL status.15,10
Patients with 11q23 rearrangements (MLL mutations) show intermediate to the adverse prognosis that depends on the second or partner genetic aberrations (Table 1). The 11q23 rearrangement with the partner t(9;11)(p22;q23) shows a more favorable prognosis that places it in the intermediate group (Tables 1 and 7). Patients with 11q23 rearrangements have an intermediate response to conventional chemotherapy (the combination of an anthracycline and HDAC for induction, then 1–2 cycles of HDAC-based consolidation followed by HSCT) (Tables 1 and 2).6,15,10
The hybrid gene OTT-MAL is a result of t(1;22)(p13;q13) translocation occurring mainly in young children, especially in those younger than 24 months, and is highly correlated with acute megakaryoblastic leukemia (AML M7). The strong correlation existing between t(1;22) and AML M7 implies that there is an existence of prenatal genetic factors involved in the incidence of this particular form of disease. Specifically, this translocation occurs primarily in girls and exhibits an inferior outcome.6,15
The meningioma 1 (MN1) gene, localized on human chromosome 22, is disrupted by the balanced translocation t(12;22) which results in the fusion protein TEL-MN1. Incooperation with HOXA9, TEL-MN1 promotes the growth of primitive HPCs. HOXA9 is over-expressed in AML. MN1 as a unique oncogene involving in hematopoiesis participates in both aspects of self-renewal; by promoting proliferation and blocking differentiation. The MN1 expression level is a predictive marker in AML treatment, wherein MN1 high-expression levels correlate with resistance to ATRA treatment. These predictive levels are particularly important in elderly patients.8
AML with Complex Karyotype (Secondary Cytogenetic Aberrations)
AML without the balanced chromosomal rearrangements (eg, t(8;21), inv(16)/t(16;16), and t(15;17)), but with at least 3 acquired chromosomal aberrations is defined as complex karyotype AML. There may be a low frequency of NPM1, FLT3, CEBPA, RAS and KIT gene mutations. There is an occurrence of chromosomal imbalances, with chromosomal losses occurring more frequently than chromosomal gains, and regarded as the worst prognostic group (Table 3). Patients with monosomal karyotype (MSK) abnormalities show an inferior OS. Incidence of AML with complex karyotype is increasing with age and constitutes 10–12% of all AML. This kind of AML seems to be induced by the deregulation of molecular pathways involved in genome stability.6,7,9 For instance, two-thirds of complex karyotype AML are identified with TP53 mutations and loss of p53 function which resulting in genetic instability (Table 3). Another contributing mechanism is chromosomal deletions and inactivation of a single allele, eg, deletion of 5q32 (RPS14 gene) identified for the −5q myelodysplastic syndromes. Genomic gain or amplifications associated with oncogene activation is another mechanism common in this type of AML. This mechanism includes members of the ETS gene family: eg, ERG/ETS2 located on 21q, and ETS1/FLI1 mapped to band 11q23.3-q24.1 (Table 3).2,6
The rearrangements of Nucleoporin 98 (NUP98) occur in a wide range of hematopoietic malignancies which including both ALL and AML. NUP98 has several different partners comprising two groups: the HOX partner genes (HOXA9, HOXA11, HOXA13, HOXC11, HOXC13, HOXD11, HOXD13, PRRX1/PMX1, and PRRX2) and the non-HOX partner genes (FN1, RAP1GDS1, NSD1, WHSC1L1/NSD3, PSIP2/LEDGF, ADD3, DDX10, and TOP1). The rare but recurrent translocation t(7;11)(p15;p15.5) causes fusion of NUP98 gene to the HOXA9 gene. The fusion occurs at the gly-leu-phe-gly (GLFG)-repeated domains of NUP98. In inv(11)(p15.5;q31), the acidic domain of a putative helicase encoded by the DDX10 gene fuses to the NUP98-GLFG repeat domains. This inversion frequently occurs in therapy-related AML (t-AML). Moreover, 5q deletion associated with the recurrent t(5;11)(q35;p15.5), frequently occurs in pediatric AML and disrupt NUP98 at 11p15.5. In translocation t(5;11), the conserved finger domains SET, SAC, and PHD of the NSD1 gene can fuse to the NUP98-GLFG repeat domains. Clinically, the incidence of NUP98 rearrangements is quite low in childhood AML, which appear with an aggressive clinical course and unfavorable therapeutic outcomes.5,6,10
ETS-related gene (ERG) at 21q22 is a cryptic amplification and frequently amplified in AML patients with a complex karyotype. ERG is a member of the ETS gene family (which has more than 30 members). Generally, the ETS gene family members are downstream nuclear targets of signal transduction pathways promoting cell proliferation and tissue invasion. Herein, recurrent amplification of ERG leads to its over-expression in complex karyotype AML, wherein contributing to an aggressive malignant phenotype. Additionally, ERG over-expression increases RR and short survival in AML with normal karyotype. High ERG expression in presence of MLL-PTD predicts a worse cumulative incidence of relapse (CIR).15
The incidence of AML with monosomy 7 and deletion of 7q [(del(7q)] is approximately 5% to 7%. AML patients exhibit del(7q) would experience an unfavorable outcome. Additionally, myelodysplastic syndrome (MDS), Fanconi anemia, congenital neutropenia, and neurofibromatosis are conditions characterized by del(7q) as a sole cytogenetic abnormality which are frequently followed by secondary AML. Individuals with these constitutional disorders are predisposed to myeloid leukemia. The balanced rearrangement of 7q22 or 7q32-q35 leads to the loss of chromosome 7 and AML. These regions contain the gene of a putative tumor suppressor that controls myeloid growth and regulates differentiation, whose loss of function leads to leukemic transformation. The deletion of a single allele leads to abnormal growth due to the reduced level of the tumor-suppressor protein, while inactivation of both alleles transformed immature myeloid cells to the leukemic phenotype.9,10
Generally, the incidence of chromosomal aberrations is higher in pediatric patients than in adults (approximately 76% of childhood AML, compared to 55% of adult AML) (Figure 4). For example, balanced rearrangements at 11q23 are on average four times more common in children than in adults. The frequency of 11q23 rearrangements in AML decreases appreciably with age. Likewise, the incidence of t(8;21) is twice as common in pediatric as in adult AML. In contrast, −5, del(5q) and other unbalanced structural abnormalities resulting in loss of material from 5q are much more frequent in adult than in childhood AML. Likewise, both inv(3)(q21q26)/t(3;3)(q21;q26), which are found in 2% of adults, are extremely rare in children and have so far never been detected in a patient with de novo AML younger than 12 years (Figure 4).8,16 The varying proportions of specific chromosomal aberrations existing between pediatric and adult AML can be attributed to the molecular reason and biological differences.
AML with Various Cytogenetic Abnormalities
Non-random chromosomal rearrangements with unbalanced translocations may result in monosomies (monosomy 7), deletions of part or all chromosome 5 or 7 (−5/−7 AML) and trisomies (eg 8, 11, 13 and 21 trisomies).9,10 The most common trisomies in de novo AML are, in decreasing order of frequency, +8, +22, +13, +21 and +11.15 Notably, in this cytogenetic subset of AML, gene mutations such as CEBPA and NPM1 are occurring, which acting as class II mutations in the collaboration with class I alterations occurred in the chromosomal abnormality.1,2
An enormous clinical heterogeneity is seen among patients whose leukemic cells exhibit trisomy 8 that can be associated with an intermediate or poor prognosis.1,2,19 The trisomy 8 is likely to be a primary event modulating underlying cryptic translocations, deletions, or mutations.19 This aberration represents a mechanism to achieve the amplification of oncogenes required for leukemogenesis. Trisomy 8 causes an increasing dosage of oncogenes, including c-myc (8q24), c-mos (8q22), and ETO (8q22), significance in leukemogenesis.1,19
Trisomy 11 is a rare event in AML associated with FAB-M1 morphologic features and shows an intermediate to poor prognosis. Trisomy 11 represents a class I aberration conferring proliferation and apparent cell survival advantages to the clone, whereas a class II mutation impairs cellular differentiation. Cases of MDS with +11 show a high risk of progression to AML. A quantitative increase in the production of oncogenes including Cyclin D1 gene (11q13, regulating cellular transition from G 1 to S phase), and MLL gene (11q23, over-expressed in both myeloid and lymphoid leukemic cells, and in an elevated proportion of mixed-lineage leukemia) is observed. MLL plays a key role in hematopoiesis by regulating HOX genes, which sequentially involved in hematopoietic stem-cell renewal and leukemogenesis.15,10 The well-known proto-oncogene WT1 expressed in various cell types to promoting proliferation is mapped to 11p13 band.2,8
This trisomy is a rare chromosomal abnormality cooperated with a high frequency of mutations in RUNX1 and spliceosome genes and characterized with poor prognosis. Trisomy 13 represents a class I alteration associated with the amplification of oncogenes and over-expression of FOX1 and FLT3 (13q12), a potential mechanism for leukemogenesis. The frequent morphology of AML+13 cells is associated with FAB M0 and showing a high frequency (80% to 100%) of RUNX1 mutations. As the master regulator of hematopoietic differentiation, AML1 (RUNX1) mutations function as a transcriptional repressor that blocks AML1-dependent HPC differentiation.2,20
The pathogenesis of MDS/AML associated with monosomy 7 is mediated by gene dosage effects as class I alterations, identified by microarray analysis. Monosomy 7 is one of the most frequent chromosomal abnormalities observed in patients with MDS/AML and associated with poor clinical outcome, both in MDS and in AML. This aberration represents a primary event superimposed to the Philadelphia chromosome in chronic myelocytic leukaemia (CML) and in a variety of Mendelian and non-Mendelian predisposing disorders wherein subjects developing MDS/AML. The methods to monitor the monosomic clone and the parental origin of the chromosome 7 loss are FISH and QR-PCR.5,10,21
Cytogenetically Normal (CN) AML with Gene Mutations
A large subset of AML (~40–50% of adult and 25% of pediatric AML) (Figure 4) is cytogenetically normal (CN-AML), but a number of gene mutations, as well as deregulated expression of genes, have been identified in this subset (Table 4). AML patients with a normal karyotype (CN-AML) are considered to be at intermediate risk associated with 5-year survival rates between 24% and 42%.3,8 In CN-AML, mutations in specific genes are identified: the nucleophosmin 1 (NPM1) gene, the fms-related tyrosine kinase 3 (FLT3) gene, the CCAAT/enhancer-binding protein alpha (CEBPA) gene, the myeloid-lymphoid or mixed-lineage leukemia (MLL) gene, the neuroblastoma RAS viral oncogene homolog (NRAS) gene, the Wilms tumor 1 (WT1) gene, and the runt-related transcription factor 1 (RUNX1) gene (Figure 3A). These gene mutations are the most prevalent in CN-AML; however, these also occur in AML with abnormal karyotypes as secondary abnormalities (Figure 2) (Tables 2 and 4).11,18
Interestingly, class I mutations in KIT are exclusively associated with inv(16) and t(8;21), being rare in other AML subtypes and are associated with unfavorable outcomes.1,2
Mutations in NPM1 Gene
NPM1 mutations (duplication and insertion) are the most frequent AML-related mutation occurred in adults and found in one third of all adult cases of AML (~30%) (Figures 2 and 3). NPM1 mutations are enriched in CN-AML (about 45–64% of CN-AML cases) (Figure 3A), wherein exhibit good response to conventional induction chemotherapy (based on a combination of anthracycline and cytarabine) and favorable outcomes are achieving in CN-AML subtype without the FLT3-ITD mutation (high CR rate ~ 85%, EFS ~ 50–60%, OS rates ~ 50%).11,16,18
In pediatric AML, however, findings indicate that NPM1 mutations generally confer an independent favorable prognostic impact despite FLT3-ITD mutations. In addition, pediatric AML patients with both NPM1 and FLT3-ITD mutations appear to have favorable prognoses and may not need hematopoietic stem-cell transplantations. Incidence of NPM1 mutations cannot be found below the age of 3 years, but above this age increases with age. NPM1 mutations occur frequently in adult CN-AML and confer favorable outcome. The studies on CN-AML in adults have found that the prognosis of NPM1 mutations positive for FLT3-ITD is unfavorable. The European LeukemiaNet (ELN) scheme proposes that NPM1 mutations with FLT3-ITD allele ratio (AR) <0.5 (low AR) has a favorable prognosis, and allogeneic hematopoietic stem-cell transplant (HSCT) in the first complete remission (CR1) period is not actively recommended.22–24
Then in adults, CN-AML with isolated NPM1 mutations (ie the NPM1+FLT3-subgroup) exhibits a good response to chemotherapy, relapse-free survival (RFS), and improved OS. This favorable effect is however lost in the presence of an FLT3-ITD. The survival of double-positive subtype NPM1+/FLT3+ will be similar to the NPM1-/FLT3-ITD+ subtype. NPM1+/FLT3+ mutations show an intermediate prognosis where there is no survival difference between NPM1+/FLT3+ and NPM1-/FLT3+. Postulating, treatment of NPM1+/FLT3+ patients with recently developed FLT3 inhibitors would be effective by converting NPM1+/FLT3+ to a more chemotherapy-sensitive status (Table 4).11,16,18
NPM1 mutation is the only detectable genetic mutation in approximately 28% of CN-AML cases, whereas in the majority of cases, additional mutations exist in class I genes such as FLT3, NRAS and WT1, (approximately in 40%, 21% and 17.5% of cases, respectively) (Figure 3B).2,11 Additionally, NPM1 mutation can also coincide with secondary chromosomal abnormalities, such as trisomy 8 and 4 and del(9q).
Generally, there are two prognoses for the outcome of NPM1 mutation without FLT3-ITD; 1) patients do not necessarily benefit from allogeneic stem-cell transplant following conventional induction chemotherapy (based on anthracycline and cytarabine), and 2) elderly patients might benefit from combination of ATRA to their chemotherapy regimen.1,3,5
At the molecular level, the nucleophosmin 1 (NPM1) gene encodes a multi-functional chaperone protein shuttling between nucleus and cytoplasm. NPM1 seems to be mainly involved in ribosomal protein assembly and biogenesis, stabilization of the tumor-suppressor p19ARF and p53 pathway, DNA-repair process, genomic stability, and finally regulation of DNA transcription through modulation of chromatin structure. Any mutation that disrupts NPM1 normal function as a tumor suppressor would lead to malignant transformation. Herein, NPM1 mutations prevent proper folding and lead to loss-of-function. Moreover, NPM1 single allele mutations perturb the function of WT protein through cytoplasmic localization of both mutant and WT protein.8,18
Other concordant conclusions are that NPM1 mutations are mostly present in adult AML with more preference in females, associated with increased blood leukocyte count, myelomonocytic phenotype and low CD34 expression. This highly prevalent mutation provides a suitable marker for monitoring minimal residual disease (MRD) of AML.11,16 QRT-PCR is an implement to quantify mutated transcripts and to evaluate the presence of MDR. Herein, the persistence of mutated transcripts in blood of patients would associate with a greater risk of relapse after chemotherapy cycle, for example, NPM1-mutated transcripts detected in the blood of 15% of patients after the second chemotherapy cycle, associates with a greater risk of relapse after 3 years of follow-up as well as a lower rate of survival.3,5
Mutations in FLT3 Gene
FLT3 (FMS-like tyrosine kinase) is a member of class III of receptor tyrosine kinases (RTK, the same family of FMS, KIT, PDGFR-α/β), whereby ligand binding signals intracellular pro-proliferative pathways. FLT3 mutations are the most common RTK mutation in AML (~30% of all cases). The most frequent FLT3 mutations (~25%) are the internal tandem duplication (ITD) within the cytoplasmic-juxtamembrane (JM) region (FLT3-ITD). Less frequent (~7%) are point mutations in the activation loop of the tyrosine kinase domain (FLT3-TKD mutation), such as the D835Y mutation. FLT3-ITD usually occurs in CN-AML wherein is associated with a dismal outcome (Figure 3) (Table 4). Both types of mutations are classified as class I mutations which provide cells constitutive-proliferative and survival advantages. FLT3 mutations result in ligand-independent kinase activation of FLT3 and its downstream signaling transducers STAT5, RAS/MAPK, PI3K, src homologous and collagen gene (SHC), and cytoplasmic tyrosine phosphatase SH2 domains (SHP2).5,6,8
Clinically, FLT3-ITD patients represent high blast counts and normal cytogenetics. The t(15;17) may coincide FLT3-ITD but rarely co-occurs in AML with complex karyotype or CBF-leukemia (CBF-MYH11 and AML1-ETO). The FLT3-ITD presence is a negative risk factor for OS, EFS and prognoses (Tables 3 and 4).10,11,16
Patients who are homozygous for FLT3-ITD or lose wild-type FLT3 allele would experience inferior outcomes or poor prognosis. By using DNA fragment analysis (PCR products sized by capillary electrophoresis which detecting ITD abnormally large amplicons), labs can quantify the relative level of the mutant allele and are able to define a cut-off value that distinguishes between prognostic subgroups.2,5
Unfavorable prognosis and inferior outcome depend, especially, on a high ratio of the expression of FLT3 mutant allele to FLT3 wild-type allele (the allelic ratio >0.78). The high level of the mutant allele (some studies, however, mentioned a mutant allele/wt allele ratio >0.5), is likely of importance which leads to shorter CR duration, death-free survival (DFS) and OS.2,15,8
Expression levels of FLT3 (CD135) are associated with certain subtypes of AML; with a minimum level in FAB M3 subtype and a maximum level in FAB M5 subtype.3,8 Small molecule tyrosine kinase inhibitors are remarkable strategies to inhibit FLT3 signaling and to specifically kill leukemic cells.4,8 From clinical trials, multi-targeted tyrosine kinase inhibitors showed a great benefit to the therapeutic anti-leukemia activity. Nevertheless, none of the designed inhibitors would lead to sustained therapeutic response on their own while in combination with other therapeutics make a preferred strategy for the treatment of AML patients. Some RTK inhibitors include midostaurin, lestaurtinib (CEP701) and sorafenib.3
Mutations in CEBPA
CEBPA encodes CCAAT-enhancer-binding protein-α (CEBP-α), a member of transcription factors with leucine-zipper motifs. Members of basic leucine-zipper (bZIP) transcription factors consist of C-terminal DNA-binding (basic region) and dimerization (leucine zipper) motifs and two less-conserved N-terminal transactivation domains.5,8 CEBP-α is a master transcription factor with a primary role in myeloid differentiation. About 10% of AML patients experience CEBPA mutations which mostly coincide with intermediate-risk karyotypes, and associate with a good prognosis.11,18 About 7-15% of CN-AML patients experience CEBPA mutations (Figure 3A) (Table 4), the majority of which being FAB-M2 subtype. CEBPA double-mutations show favorable outcomes (longer remission duration and higher OS ~ 8 years), and better prognostic impact (especially in patients aged 16–60 years).5,8 Importantly, aberrations such as FLT3 mutations and MLL-PTD rearrangement have no significant influence on the prognosis outcome of CEBPA double-mutation. This benefit is lost in the presence of CEBPA wild-type allele or CEBPA single-mutation which is an independent adverse prognostic marker affecting remission duration and OS rates.11,16 The coincidence of CEBPA double-mutation with FLT3-ITD mutation is rare, where CEBPA mutation is mutually exclusive with NPM1 mutation.3,11,18 At the molecular level, CEBPA has a single exon mapped to band 19q 13.1, with tumor-suppressor activity. CEBP-α induces a number of genes involved in granulocytic specific differentiation. Consecutive up-regulation of CEBPA gives rise to granulocytic differentiation, while its further conditional expression triggers neutrophil differentiation. CEBPA mutations result in loss-of-function trait contributing to an early block of granulocyte maturation. The t(8;21) is an alternative mechanism for CEBPA inactivation wherein the AML1-ETO fusion protein suppresses CEBPA transcription and hence blocks granulocyte differentiation.2,16,17
Mutations in CEBPA have a favorable prognosis and good response to the conventional chemotherapy and are associated with higher CR rate and better RFS and OS, comparable with that of CBF-AML (Tables 1 and 2).2,6,8
BAALC Over-Expression
The brain and acute leukemia, cytoplasmic (BAALC) gene is mapped to band 8q22.3. BAALC expression occurs in neural and hematopoietic stem cells. In HPCs, BAALC is a specific marker for the proliferation stage, as its down-regulation leads to cell differentiation. CD34+ progenitor cells from both normal and abnormal bone marrow express BAALC. Since all subtypes of CD34 + cells express BAALC, it would be present in early progenitor cells, as a novel marker common for the myeloid, lymphoid, and erythroid pathways. As patients with an elevated level of BAALC show a higher rate of primary resistant leukemia, a higher CIR and an inferior OS (3 years), its over-expression is an independent risk factor associated with chemotherapy resistance and unfavorable outcomes. To improve the unfavorable outcome for BAALC high-risk patients, modifying and adjusting induction therapy and intensifying post-remission therapy should be taken into account. Reports suggest that AML patients with a high level of BAALC would benefit from consolidation with allogeneic SCT, while therapy with autologous SCT seems unfavorable. Clinically, high-level BAALC patients exhibit an escalated percentage of blood blasts and immature FAB M0/M1 subtypes, while low BAALC expression associates with monocytic differentiation and FAB M5b subtype and gingival hyperplasia. BAALC over-expression may be accompanied by FLT3-ITD, CEBPA, MLL-PTD, and high ERG over-expression mutations. Importantly, over-expressed BAALC induces drug resistance gene (MDR1) and stem-cell markers (CD133, CD34, KIT).5,15,8
RAS Mutations
RAS (rat sarcoma) oncogenes encode a family of membrane-associated G proteins that regulate signal transduction by binding to a variety of membrane receptors. The RAS G proteins are involved in signal transduction pathways that induce proliferation. The genome contains three functional RAS genes: N-(neuroblastoma), K-(Kirsten), and H-(Harvey) RAS. Each RAS gene (H-RAS, K-ras, N-RAS) contains 4 exons can carry transforming mutations exclusively in codons 12/13/61, which leads to constitutive activation of RAS protein identified in many cancer types.3,8
Accordingly, RAS mutations are the most frequent mutations occurring in CBF-AML patients (45% N-RAS; 13% K-ras) (Figure 2) (Table 2) and N-RAS is prominent (11–30% in all AML) (Figures 2 and 3), especially in those under the 60 years of age. As mentioned, RAS mutations occur with a higher frequency in the favorable risk groups wherein lead to poor outcomes. Screening RAS mutations in AML patients are thereby proposed as a benefit guide for therapeutic decisions. AML patients with RAS mutations showed a good response to post-remission high-dose cytarabine therapy.8,16,18
WT1 Mutations
Wilms tumor (WT1) mutations (insertions or deletions) are detectable in 10% of AML cases, mainly clustered in exons 7 and 9, and show induction failure. WT1 mutations belong to the class I oncogene activating mutations which confer WT1 constitutive activation.2,16
The WT1 gene is mapped to band 11p13 which encodes the transcription factor WT1, an oncogene expressed in various cell types to promoting proliferation, as well as to blocking differentiation. At the molecular level, WT1 mutations mostly occur with FLT3-ITD mutation and are failure to standard induction chemotherapy. WT1 can be a reliable marker for MDR assessment in acute leukemia patients. After induction chemotherapy, the WT1 levels in blood samples of treated patients would allow to distinguish those with continuous CR from those who obtain only an “apparent” CR or who relapse within a few months. The level of WT1 helps identify patients at high risk of relapse soon after induction chemotherapy, those who need post-induction therapy being intensified. Moreover, the expression level of the WT1 gene in normal HPCs is approximately 10 times less than those in leukemic cells.2,8
Partial Tandem Duplication (PTD) of MLL Gene
Approximately 5–11% of CN-AML patients carry partial tandem duplications of the MLL gene (MLL-PTD). In MLL-PTD, duplication of genomic regions spanning exons 5–11 occurs where the duplicated segment is inserted into intron 4 of the gene. In contrast to MLL fusion protein, MLL-PTD retains all its functional domains. In the presence of MLL-PTD, the wild-type MLL allele is suppressed. This silencing is mediated through epigenetic mechanisms, wherein a combination of decitabine and depsipeptide, which inhibiting DNA methyltransferase and histone deacetylase, respectively, leads to transcriptional reactivation of the wild-type allele in MLL-PTD-positive blasts. Reactivation of wild-type MLL induces cell death in blasts. Clinically, MLL-PTD mutations associate with unfavorable outcomes (short-duration CR, high relapse rate and inferior EFS).6,16,18
MN1 Over-Expression
In CN-AML, high expression of MN1 strongly occurs in un-mutated NPM1 cases and is an independent prognostic marker for poor response to the first course of induction therapy, higher relapse rate, shorter RFS and less OS. In treatment with ATRA, patients who express low levels of MN1 would have prolonged EFS and more OS, but patients with high-expression level of MN1 would exhibit higher relapse rate, shorter RFS and less OS. HPCs express MN1 in high levels while differentiated CD34+cells down-regulate it.15,8
TP53 Mutations
Tumor-suppressor p53 is a DNA-binding protein and the master transcription factor activated in response to diverse cellular stresses to induce cell cycle arrest and DNA repair or apoptosis. TP53 mutations involve single nucleotide changes which are prevailing in other types of cancer but not predominant in blood cancers. TP53 mutations occur in 75–78% of AML patients with complex karyotype, but rare in other AML subtypes. TP53 mutation is the most important marker of poor prognosis in both CK-AML and therapy-related AML. TP53 alterations frequently occur in old patients who display decreases in CR, EFS, RFS, and OS.3,16
CN-AML and cytogenetically abnormal AML patients have the chance to get more chromosomes in rearrangements in their leukemic karyotype. Late rearrangements or secondary aberrations are common in both ALL and AML, prevailingly follow a primary change and lead to substantial variability in patient outcome. These include del(7q) (−7), trisomy 8 (+8), del(16)/t(1;16)(q12-23;q12-24) and trisomy 21 (+21), which have been observed in both ALL and AML (Table 2)10
Mutations in Epigenetic Genes
Class 0/III mutations impairing epigenetic regulation in HPCs and defined in AML can be the earliest alteration occurring in an HSC. Importantly, mutations in the genes that encode DNA-methylation regulators (eg, DNMT3A, IDH1/2, TET2) are often acquired early and with a high recurrence rate with NPM1 and CEBPA alterations. Particularly, 73% of the NPM1-mutated AML carry mutations in DNA-methylation genes (DNMT3A, IDH1, IDH2, and TET2). Furthermore, there is evidence confirming that TET2 with FLT3 mutations and IDH1 with NPM1 mutations cooperate to induce AML. Moreover, the epigenetic changes are reversible by small molecules and therapeutics that reactivate epigenetically silenced genes and improve outcomes of the disease (Table 5).12,13
DNMT3A Mutations
DNMT3A mutations, one of the most important epigenetic-related alterations, are classified as the earliest and recurrent aberration in myeloid malignancies, occurring in 20–22% of adults with de novo AML (rare in children). Almost all CN-AML patients harbor at least a single point mutation in one of DNMT3A alleles and about 30–37% of them show DNMT3A loss-of-function mutations. In this regard, the CN-AML group is categorized into two subtypes, ie, with or without DNMT3A mutations with prognosis significance.12–14 Clinically, DNMT3A mutations frequently accompany intermediate-risk AML, where they may correlate with inferior outcomes.12,16 DNMT3A mutations have a negative impact on patient clinical outcome (a significant decrease in OS in comparison to those with wild-type DNMT3A). In particular, DNMT3A-mutation effects persist in HPCs and mature cells after remission (Table 5).3
At the molecular level, DNA methyltransferase 3A gene (DNMT3A) encodes DNMT3A that catalyzes the de novo addition of a methyl group to the cytosine residue of CpG dinucleotide. DNMT3A mutations have a severe impact on DNA-methylation patterns whereby give rise to global shifts in gene expression accompanied by increased self-renewal of hematopoietic cells at the blocking of normal differentiation.12 Mutations mainly disrupt the catalytic domain of the enzyme, which are associated with a loss-of-function of the enzyme activity.12–14 DNMT3A loss-of-function mutations result in hypomethylation of HSC specific genes commonly over-expressed in AML (eg Runx1, Erg, Myc, Smad3) which consequently impair HSC differentiation.1,3 In particular, DNMT3A function is required for HSC self-renewal and myeloid differentiation; its mutations have been detected in preleukemic HSCs and considered as an early event in AML. DNMT3A mutations lead to myeloid transformation in vivo and promote myeloid malignancies in impaired HSCs. Decitabine treatment caused an improved response in patients harboring mutated DNMT3A. Additionally, patients achieved a higher clinical remission rate and a superior OS when compared to those bearing the wild-type DNMT3A (75% vs 34% and 15.2 vs 11 months, respectively). High levels of miR-29b (which targets DNMT3A) would be a good marker for response to decitabine treatment. Importantly, patients with myeloid malignancies who are harboring DNMT3A, IDH1/IDH2 mutations show a favorable response to decitabine and azacitidine, specific DNMT inhibitors and hypomethylating agents (HMAs).12–14
IDH1/2 (Isocitrate Dehydrogenase) Mutations
The IDH1/2 genes encode tumor-suppressor proteins IDH1/2 whose mutations appear to induce DNA hypermethylation at specific sequences. About 15–20% of all AML subtypes and 25–30% of CN-AML subtype harbor IDH1/2 mutations. This frequency is similar in adults and children. IDH1/2 mutations prevailingly accompany NPM1 mutations but not FLT3-ITD.12,13 Epigenetic alterations occurring in IDH1/2 mutations intensify HPC proliferation and increase its pool. IDH1/2 impairment suppresses the histone demethylation process whereby associates with DNA hypermethylation, differentiation blocking and clonal expansion of HSCs.1,3 Biochemically, IDH1/2 are, respectively, cytosolic and mitochondria NADP-dependent dehydrogenases, decarboxylating isocitrate into αKG. The produced molecule is used by TET enzyme when catalyzing histone demethylation. Mutations give rise to the new catalytic activity by the enzyme whereby converts αKG to 2-HG. The oncometabolite 2-HG is a putative inhibitor of histone demethylase TET2. Blocking histone demethylation is accompanied by DNA hypermethylation phenotype (Table 5). All IDH1/2 mutations have been reported heterozygous and mutually exclusive to TET2 mutations, possibly due to overlapping molecular effects.12,13 IDH1/2 mutations are related to an adverse clinical outcome with poor prognosis, particularly when accompanying other mutations like as NMP1 and FLT3-ITD, however, compared to DNMT3A, IDH1/2 mutation has a better prognosis, DFS and superior OS. In the favorable risk groups, the coincidence of IDH1/2 mutations with other aberrations would lead to a lower rate of RFS and 5-year OS duration. Similar to DNMT3A mutations, IDH 1/2s show significantly higher remission rate and favorable response to anti-leukemia therapeutic decitabine and azacitidine. The IDH1 and IDH2 inhibitors are, respectively, AG-220 and AG-221, which reported to show a clinical response, with a significant reduction in 2-HG levels (~50–90%). It is reported that IDH 1/2 inhibitors are being evaluated in Phase III clinical trials and show a prominent impact on AML prognosis.13,16
The α-Ketoglutarate-Dependent Dioxygenase, Ten-Eleven Translocation (TET) Proteins
Approximately 10–20% of AMLs carry TET2 mutations (deletions, nonsense and missense mutations). Clinically, TET2 mutations associate with intermediated risk and short OS. Biologically, these loss-of-function mutations induce DNA hypermethylation, however, mutually exclusive with IDH1/2 mutations. TET2 mutations are prevalent in MDS and myeloproliferative disorders and are highly frequent in chronic myelomonocytic leukemia, wherein associated with monocytosis and poor outcomes.1,12 As an early event present in HSCs, TET2 inactivation induces pre-leukemic HSCs, clonal expansion and leukemogenesis. TET2 mutations may act in the same pathway as IDH1/IDH2 (Table 5). Biochemically, TET activation leads to DNA demethylation in enhancer regions of tumor-suppressor genes through the sequential conversion of 5-methylcytosines (5mC) to 5-hydroxymethylcytosine (5hmC), then 5-formylcytosine to 5-carboxylcytosine which finally accompanied by DNA glycosylase and base excision repair system. Specifically, TET2 mutations cause a reduction in 5hmC levels and an induction in DNA methylation mainly in enhancer regions of nearby tumor suppressors. TET2 inactivation correlates with hypermethylation of tumor suppressors in AML.1,3,6
Epigenetic Biomarkers in AML
The epigenetic regulators DNMT3A, IDH1/IDH2 and TET1/2 are epigenetic markers useful for risk stratification, therapy decision-making and clinical predicting of response to treatment. The epigenetic biomarkers can also include changes in DNA-methylation patterns or expression profile of non-coding RNAs (miRNAs). These epigenome alterations can be a result of class 0/III mutations which contribute to the cancer molecular pathogenesis. It is also said that epigenetic changes can be relatively reversible in response to combinations of epigenetic small molecular agents such as inhibitors of methyltransferases (cytarabine, azacitidine and decitabine), in combination with other available inhibitors including histone deacetylase (HDAC) inhibitors, which are now tested in clinical trials.12,13
Herein, purin and deoxynucleoside antimetabolites (eg thioguanine/mercaptopurine and cytarabine/azacytidine) are inhibitors of DNA methyltransferases (DNMTi) wherein their basic mechanism is quite similar. These compounds enter the cells and are converted into their respective nucleotide analogues and incorporated into the genomic DNA strands. Their incorporation into the DNA inhibits enzymes critical for DNA synthesis, repair, and methylation (acting as hypomethylating agents (HMAs)). Examples of HDAC inhibitors (HDACi) are vorinostat and valproic acid which numerously administrated in clinical trials in combination with sorafenib for treating patients with advanced/metastatic solid malignancies and refractory/relapsed AML. HDACi target directly the histone deacetylases. In clinical trials, rational combinations of HDACi and DNMTi result in the transcriptional activation of the corresponding genes including tumor-suppressor genes often silenced in cancers. Rational combinations and therapy duration of HDACi/DNMTi with DNA damage-inducing therapies (eg anthracyclines/TOP inhibitors) synergistically enhance the irradiation, growth inhibition and apoptotic effects of therapy. DNMT/HDAC inhibitors increase DNA accessibility by DNA damage-inducing therapies. Several cycles of therapeutics are required and the duration of inhibitors is emphasized for the manifestations of hematologic responses to improve survival in AML patients.25,26
Alteration in the Expression Profile of Non-Coding RNA (miRNAs)
Gene expression profiling (GEP) of non-coding RNAs (miRNAs) can discriminate different cytogenetic subtypes or act as prognostic factors. These small RNAs have been proposed as potential biomarkers of integrated panels for prognosis and risk stratification of disease. For example, a panel of 12 miRNAs is proposed which can divide CN-AML into poor and intermediate-risk categories independently of FLT-ITD.5,6 In particular, elevated expression of miR-17-92, miR196, miR-29, miR-125, miR-142, miR-146 and miR-155 is a characteristic of AML (Table 8), addressed as pathogenesis biomarkers. Generally, during normal myelopoiesis, there is a low expression of miRNA clusters whereas highly expressed in leukemic conditions. For instance, miR-125 family, encoded by three conserved clusters mapped to chromosomes 19/21/11, is over-expressed in leukemic HSCs. Over-expression of miR-21/10/196 is reported in NMP1 mutants wherein associated with blockage of normal HSC differentiation and down-regulation of PDCD4 (Programmed Cell Death 4).1,3 Moreover, MLL rearrangements are characterized by a high expression of miR-17-92 cluster, as well as, miR-196b which is located at 7p15, between HOXA9 and HOXA10 genes. Also, FLT3-ITD is associated with increased expression of miR-155/miR-181 family whereby leading to leukemic blast expansion. Moreover, miR-181a/b over-expression is a marker of unfavorable outcomes, in particular in CN-AML CEBPA+/FLT3-IDT+/NPM1-.11,16,18 Even more, expression of miRNAs changes during cytarabine therapy which correlates with outcomes and OS. For example, high expression of miR-191 and miR-199a can be associated with worse OS while down-regulation of miR29b (targeting DNMT3A & DNMT3B) is a marker of a better response to treatment.1,3
 |
Table 8 Summary of Expression Profile of Prognostic miRNAs in AML |
Changes in DNA-Methylation Patterns
Each cytogenetic or molecular subtype is supposed to have a distinct DNA-methylation profile. For example, cases with t(8;21), inv(16) or t(16;16), t(15;17) or t(v;11q23) exhibit unique DNA-methylation signatures whereby define AML subtypes. Methylome analysis has indicated that specific regions of the genome have lower methylation levels in AML compared to the normal, such as gene bodies and repetitive sequences. Hypomethylation of repetitive sequences with a high content of methylated CpGs (eg SINEs (short interspersed nuclear elements) and LINEs (long interspersed nuclear elements)) are associated with rearrangements.1,12 Additionally, in methylome analysis of CN-AML, the most pronounced DNA hypermethylation is detected in CpG islands representing tumor-suppressor promoters.3 Recurrent mutations (eg NPM1, CEBPA, RUNX1), can also be defined by distinct DNA-methylation patterns. For example, NPM1 mutations can be defined by four distinct DNA-methylation clusters. AML subtypes can be further classified according to the methylation profiles related to prognosis and clinical outcomes. CEBPA double mutations show two distinct DNA-methylation signatures whereby patients could be split into two distinct prognosis subtypes: one hypermethylated and one hypomethylated.12,13 Herein, initiation, progression and maintenance of the tumor phenotype associate with distinct DNA-methylation patterns.3
The underlying mechanism of aberrant DNA-methylation induction in these AML subsets is attributed to the fusion genes in recruiting DNMTs to their binding sites, or to a secondary epigenetic dysregulation including binding of PML-RARα to genomic regions of epigenetic modifiers such as DNMT3A and/or DNA-methylation disruption of AML1-ETO target genes. For instance, methylation of target genes involved in the genome stability were found to be changed in APL patients at diagnosis time, which predispose them to a hypomethylation phenotype seen in CBFB-MYH11 fusion gene, CEBPA promoter and in the regulatory regions of MN1.1,12 Methylation assays suggest that global DNA hypomethylation occurred in AML patients during treatment can be a marker associated with a successful response, an increased CR rate and an OS improvement. Methylome analyses indicate that hypomethylation of LINE-1 may also associate with low blast counts (<45%), better CR rate during the first cycle of azacitidine, and hematological improvement.
According to the DNA methylation-shifted loci observed in AML, samples can be defined into three categories: shifted loci unique to diagnosis, loci unique to relapse, lastly loci can be seen at both diagnosis and relapse. Accordingly, dividing AML samples into these categories do not correlate with age, white blood cell count, or the French–American–British (FAB) classification. Finally, DNA methylation-shifted loci are expected to be an independent classification for AML. Therefore, identification of mutations associated with DNA methylation and evaluation of changes occurred in methylation signatures would contribute to individual therapy of AML patients.3,12,13
Conclusion
The AML development is a consequence of an accompaniment between genetic, epigenetic and proteomic alterations, causes of specific molecular mechanisms involved in. Nowadays, genetic mutations and cytogenetic alterations are taken into account as markers of great importance for risk stratification and therapeutic decision-making in clinical management. Due to the heterogeneity of mutations in AML, finding a panel of biomarkers would be of more importance for diagnosis, prognosis or monitoring of individual patients, in addition to facilitating individualized-therapeutic decision-making. For example, a panel of integrated biomarkers was recently proposed combining FLT3, NPM1, ERG, CEBPA, and BAALC mutations to place patients into one of the four categories respecting the risks and benefits of proper therapy. Other integrated panels combining ERG, BAALC, WT1, EVI1, MN1 mutations and microRNA expression profiles have been proposed which similarly respecting prognostic factors for risk stratification and therapeutic decision-making. Once an algorithm is established according to these panels, clinical practices can be much closer to achieving personalized medicine and the development of precision medicine. Therefore, identification of mutations and specific biomarkers in each patient and evaluation of the change in methylation signature would contribute to individualized-therapeutic decision-making in AML.5,12,16
Disclosure
The authors report no conflicts of interest in this work.
References
1. Chiaretti S, Gianfelici V, Ceglie G, Foà R. Genomic characterization of acute leukemias. Med Princ Pract. 2014;23(6):487–506. doi:10.1159/000362793
2. Dohner K, Döhner H. Molecular characterization of acute myeloid leukemia. Haematologica. 2008;93(7):976–982. doi:10.3324/haematol.13345
3. Prada-Arismendy J, Arroyave JC, Röthlisberger S. Molecular biomarkers in acute myeloid leukemia. Blood Rev. 2017;31(1):63–76. doi:10.1016/j.blre.2016.08.005
4. Hekmatimoghaddam S, Jebali A, Dargahi M. Folic acid-functionalized gold and silver nanoparticles: their cytotoxic effect on cancerous myeloid cells with microwave irradiation. Nano Life. 2013;3(2):
5. Gulley ML, Shea TC, Fedoriw Y. Genetic tests to evaluate prognosis and predict therapeutic response in acute myeloid leukemia. J Mol Diagn. 2010;12(1):3–16. doi:10.2353/jmoldx.2010.090054
6. Lagunas-Rangel FA, Chavez-Valencia V, Gomez-Guijosa MA, Cortes-Penagos C. Acute myeloid leukemia-genetic alterations and their clinical prognosis. Int J Hematol Oncol Stem Cell Res. 2017;11(4):328–339.
7. Masoumi-Dehshiri R, Hashemi AS, Neamatzadeh H, Zare-Zardeini H. a case report: acute myeloid leukemia (FAB M7). Iran J Ped Hematol Oncol. 2014;4(4):188–190.
8. Zaidi SZ, Owaidah T, Al Sharif F, Ahmed SY, Chaudhri N, Aljurf M. The challenge of risk stratification in acute myeloid leukemia with normal karyotype. Hematol Oncol Stem Cell Ther. 2008;1(3):141–158. doi:10.1016/S1658-3876(08)50023-9
9. Haferlach C, Alpermann T, Schnittger S, et al. Prognostic value of monosomal karyotype in comparison to complex aberrant karyotype in acute myeloid leukemia: a study on 824 cases with aberrant karyotype. Blood. 2012;119(9):2122–2125. doi:10.1182/blood-2011-10-385781
10. Braoudaki M, Tzortzatou-Stathopoulou F. Clinical cytogenetics in pediatric acute leukemia: an update. Clin Lymphoma Myeloma Leuk. 2012;12(4):230–237. doi:10.1016/j.clml.2012.04.004
11. Schlenk RF, Dohner K, Krauter J, et al; German-Austrian Acute Myeloid Leukemia Study Group. Mutations and treatment outcome in cytogenetically normal acute myeloid leukemia. N Engl J Med. 2008;358(18):1909–1918. doi:10.1056/NEJMoa074306
12. Li Y, Xu Q, Lv N, et al. Clinical implications of genome-wide DNA methylation studies in acute myeloid leukemia. J Hematol Oncol. 2017;10(1):41. doi:10.1186/s13045-017-0409-z
13. Wagner K, Damm F, Gohring G, et al. Impact of IDH1 R132 mutations and an IDH1 single nucleotide polymorphism in cytogenetically normal acute myeloid leukemia: SNP rs11554137 is an adverse prognostic factor. J Clin Oncol. 2010;28(14):2356–2364. doi:10.1200/JCO.2009.27.6899
14. Hekmatimoghaddam S, Dehghani Firoozabadi A, Zare-Khormizi MR, Pourrajab F. Sirt1 and Parp1 as epigenome safeguards and microRNAs as SASP-associated signals, in cellular senescence and aging. Ageing Res Rev. 2017;40:120–141. doi:10.1016/j.arr.2017.10.001
15. Mrozek K, Heerema NA, Bloomfield CD. Cytogenetics in acute leukemia. Blood Rev. 2004;18(2):115–136. doi:10.1016/S0268-960X(03)00040-7
16. Metzeler KH, Herold T, Rothenberg-Thurley M, AMLCG Study Group; et al. Spectrum and prognostic relevance of driver gene mutations in acute myeloid leukemia. Blood. 2016;128(5):686–698. doi:10.1182/blood-2016-01-693879
17. Green CL, Koo KK, Hills RK, Burnett AK, Linch DC, Gale RE. Prognostic significance of CEBPA mutations in a large cohort of younger adult patients with acute myeloid leukemia: impact of double CEBPA mutations and the interaction with FLT3 and NPM1 mutations. J Clin Oncol. 2010;28(16):2739–2747. doi:10.1200/JCO.2009.26.2501
18. Dufour A, Schneider F, Metzeler KH, et al. Acute myeloid leukemia with biallelic CEBPA gene mutations and normal karyotype represents a distinct genetic entity associated with a favorable clinical outcome. J Clin Oncol. 2010;28(4):570–577. doi:10.1200/JCO.2008.21.6010
19. Bakshi SR, Brahmbhatt MM, Trivedi PJ, et al. Trisomy 8 in leukemia: A GCRI experience. J Hum Genet. 2012;18(1):106–108.
20. Herold T, Metzeler KH, Vosberg S, et al. Isolated trisomy 13 defines a homogeneous AML subgroup with high frequency of mutations in spliceosome genes and poor prognosis. Blood. 2014;124(8):1304–1311. doi:10.1182/blood-2013-12-540716
21. Knop S, Hebart H, Gratwohl A, et al. Monosomy 7 in myeloid malignancies: parental origin and monitoring by real-time quantitative PCR. Leukemia. 2007;21:1833–1835. doi:10.1038/sj.leu.2404708
22. Sakaguchi M, Yamaguchi H, Najima Y, et al. Prognostic impact of low allelic ratio FLT3-ITD and NPM1 mutation in acute myeloid leukemia. Blood Adv. 2018;2(20):2744–2754. doi:10.1182/bloodadvances.2018020305
23. Xu LH, Fang JP, Liu YC, Jones AI, Chai L. Nucleophosmin mutations confer an independent favorable prognostic impact in 869 pediatric patients with acute myeloid leukemia. Blood Cancer J. 2020;10(1):1–10. doi:10.1038/s41408-019-0268-7
24. Hollink IHIM, Zwaan CM, Zimmermann M, et al. Favorable prognostic impact of NPM1 gene mutations in childhood acute myeloid leukemia, with emphasis on cytogenetically normal AML. Leukemia. 2009;23:262–270. doi:10.1038/leu.2008.313
25. Rostamian T, Pourrajab F, Hekmatimoghaddam SH. The effect of 6-thioguanine on proliferation, viability and expression of the genes DNMT 3A, DNMT 3B and HDAC3 in Lymphoid Cancer Cell Line Nalm6. Iran J Ped Hematol Oncol. 2019;10(1):28–37.
26. Suraweera A, O’Byrne KJ, Richard DJ. Combination therapy with histone deacetylase inhibitors (HDACi) for the treatment of cancer: achieving the full therapeutic potential of HDACi. Front Onco. 2018;8(92):1–15.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.