Back to Journals » Clinical Interventions in Aging » Volume 15
Genetic Analysis of Chinese Patients with Early-Onset Dementia Using Next-Generation Sequencing
Authors Han LH, Xue YY, Zheng YC, Li XY, Lin RR, Wu ZY, Tao QQ ![]()
Received 13 July 2020
Accepted for publication 1 September 2020
Published 2 October 2020 Volume 2020:15 Pages 1831—1839
DOI https://doi.org/10.2147/CIA.S271222
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Richard Walker
Li-Hong Han,1,2,* Yan-Yan Xue,1,* Yi-Cen Zheng,3 Xiao-Yan Li,1 Rong-Rong Lin,1 Zhi-Ying Wu,1 Qing-Qing Tao1
1Department of Neurology and Research Center of Neurology in Second Affiliated Hospital, and Key Laboratory of Medical Neurobiology of Zhejiang Province, Zhejiang University School of Medicine, Hangzhou, People’s Republic of China; 2Department of Neurology, Second People’s Hospital of Luqiao District, Taizhou, People’s Republic of China; 3Department of Psychology, Tulane University School of Science and Engineering, New Orleans, LA, USA
*These authors contributed equally to this work
Correspondence: Zhi-Ying Wu; Qing-Qing Tao Email [email protected]; [email protected]
Objective: Early-onset dementia (EOD) is a relatively uncommon form of dementia that afflicts people before age 65. Only a few studies analyzing the genetics of EOD have been performed in the Chinese Han population. Diagnosing EOD remains a challenge due to the diverse genetic and clinical heterogeneity of these diseases. The aim of this study was to investigate the genetic spectrum and clinical features of Chinese patients with EOD.
Materials and Methods: A total of 49 EOD patients were recruited. Targeted next-generation (NGS) analyses were performed to screen for all of the known genes associated with dementia. Possible pathogenic variants were confirmed by performing Sanger sequencing. The genetic spectrum and clinical features of the EOD patients were analyzed.
Results: Seven previously reported pathogenic variants (p.I213T and p.W165C in PSEN1; p.D678N in APP; c.1349_1352del in TBK1; p.P301L and p.R406W in MAPT; p.R110C in NOTCH3) and two novel variants of uncertain significance (p.P436L in PSEN2; c.239-11G>A in TARDBP) were identified.
Conclusion: Our study demonstrated the genetic spectrum and clinical features of EOD patients, and it reveals that genetic testing of known causal genes in EOD patients can help to make a precise diagnosis.
Keywords: Chinese, genetic analysis, early-onset dementia, next-generation sequencing
Introduction
Early-onset dementia (EOD) usually refers to dementia that develops before the patient is 65 years old.1 It is speculated that genetic factors play a more important role in the pathogenesis of EOD than in late-onset dementia (LOD). EOD is a condition with a high heterogeneity that can be caused by several distinct diseases with different etiology and pathophysiology, including Alzheimer’s disease (AD), frontotemporal dementia (FTD), dementia with Lewy bodies (DLB), and other neurodegenerative brain disorders.2–5
AD is the most common type of dementia. Hallmarks of AD include the extracellular deposition of amyloid beta (Aβ) peptides and the formation of intraneuronal neurofibrillary tangles (NFTs), which leads to irreversible neuron loss and loss of memory.6 The pathogenesis of AD remains unclear, and both genetic and environmental factors contribute to its risk.7 Approximately 95% of AD patients are sporadic while the remaining are familial AD patients (FAD). Pathogenic variants in amyloid precursor protein (APP), presenilin 1 (PSEN1) and presenilin 2 (PSEN2) have been previously identified as causes of FAD.8 Several studies have been performed to determine how pathogenic variants in these three genes contribute to AD, thus providing insight into the pathogenesis of AD.9
FTD is another type of dementia that has neuropathological and clinical crossover with AD. FTD primarily affects the frontal and temporal lobes of the brain, and it is generally associated with dysfunction of the personality, behavior and language use.10 DLB is also a common cause of dementia. The characteristic clinical symptoms of DLB include fluctuating cognition, recurrent visual hallucinations, and Parkinsonism.11 Other neurologic diseases, such as amyotrophic lateral sclerosis (ALS), Parkinson’s disease (PD), and prion diseases, can also present with EOD. With an increasingly aged population, more attention has been given to dementia.12 However, diagnosing EOD remains a challenge due to the diverse genetics and clinical heterogeneity of these diseases.
Next-generation sequencing (NGS) has been used to examine EOD patients with an unknown diagnosis.13–15 However, genetic studies on EOD have been rarely performed in the Chinese population.16 In this study, NGS of 70 genes associated with dementia was performed for 49 Chinese patients with EOD, and we identified 2 novel variants of uncertain significance (VUS) and 7 previously reported pathogenic variants.
Materials and Methods
Subjects
All of the patients in our study were recruited from the Department of Neurology, second affiliated Hospital of the Zhejiang university school of medicine from May 2015 to July 2019. A total of 49 EOD patients satisfied the following inclusion criteria for our study: 1) had early disease onset (<65 years old) and 2) had a phenotype of dementia. The exclusion criteria for our study: 1) had a disease or impairment of the central nervous system, including active or past subdural hematoma, active or past subarachnoid hemorrhage, and primary or metastatic brain cancer; or 2) had other systemic diseases that may result in cognitive impairment, including neurosyphilis, addiction to alcohol, drug use, vitamin deficiency and thyroid dysfunction. Subjects who had non-neurological diseases that may result in cognitive impairment were also excluded. Clinical evaluations and neurological examinations were performed by two senior neurologists. The criterion for a probable AD diagnosis was set according to the National Institute on Aging-Alzheimer’s Association (NIAAA).17 The criterion for a probable FTD and DLB diagnosis was set according to previous studies.18,19 This study was approved by the Ethics Committee of Second Affiliated Hospital, Zhejiang University School of Medicine. Written informed consent for participation in this study and publication of their clinical details was obtained from all of the participants or their legally authorized caregivers.
Targeted Next-Generation (NGS) Panel Analysis Screening
Blood samples were collected from all patients. Genomic DNA was extracted from the peripheral blood using a Blood Genomic DNA Extraction Kit (Qiagen, Hilden, Germany) according to standard procedures. A customized multigene panel that contained genes associated with dementia was designed (Table S1). NGS panel screening was performed using the Illumina HiSeq2000 platform (XY Biotechnology Co Ltd, Hangzhou, China). Publicly available databases including the Single Nucleotide Polymorphism Database (dbSNP) (https://www.ncbi.nlm.nih.gov/snp/), the 1000 Genomes Project (https://www.ncbi.nlm.nih.gov/variation/tools/1000genomes/), and the gnomAD browser (https://gnomad.broadinstitute.org/) were used to extract information about the detected variants. Three software programs, SIFT, PolyPhen2 and Mutation Taster, were used to predict the possible protein functional changes.
Sanger Sequencing and APOE Genotyping
Possible pathogenic variants were confirmed by performing Sanger sequencing with an ABI 3500XL DX Genetic Analyzer (Applied Biosystems, Foster City, CA, USA) following the procedure described in our previous study.20 A co-segregation analysis was performed for all available familial members. The APOE genotypes were determined by multiplex amplification refractory mutation system PCR according as previously described.20
Results
Demographic Data of the EOD Patients
A total of 49 unrelated EOD patients were included in our study. The mean age at onset (AAO) of the EOD patients was 52.9 ± 7.8 years. Of these patients, 42.9% were male and 57.1% had a positive family history (at least 2 family members in the last three generations were affected by dementia). The mean AAO of the probands with familial dementia was 52.8 ± 8.8 years. A total of 30.6% carried one APOEε4 allele, and 18.4% carried two APOEε4 alleles. The personal and medical histories of all 49 EOD patients are summarized in Table 1.
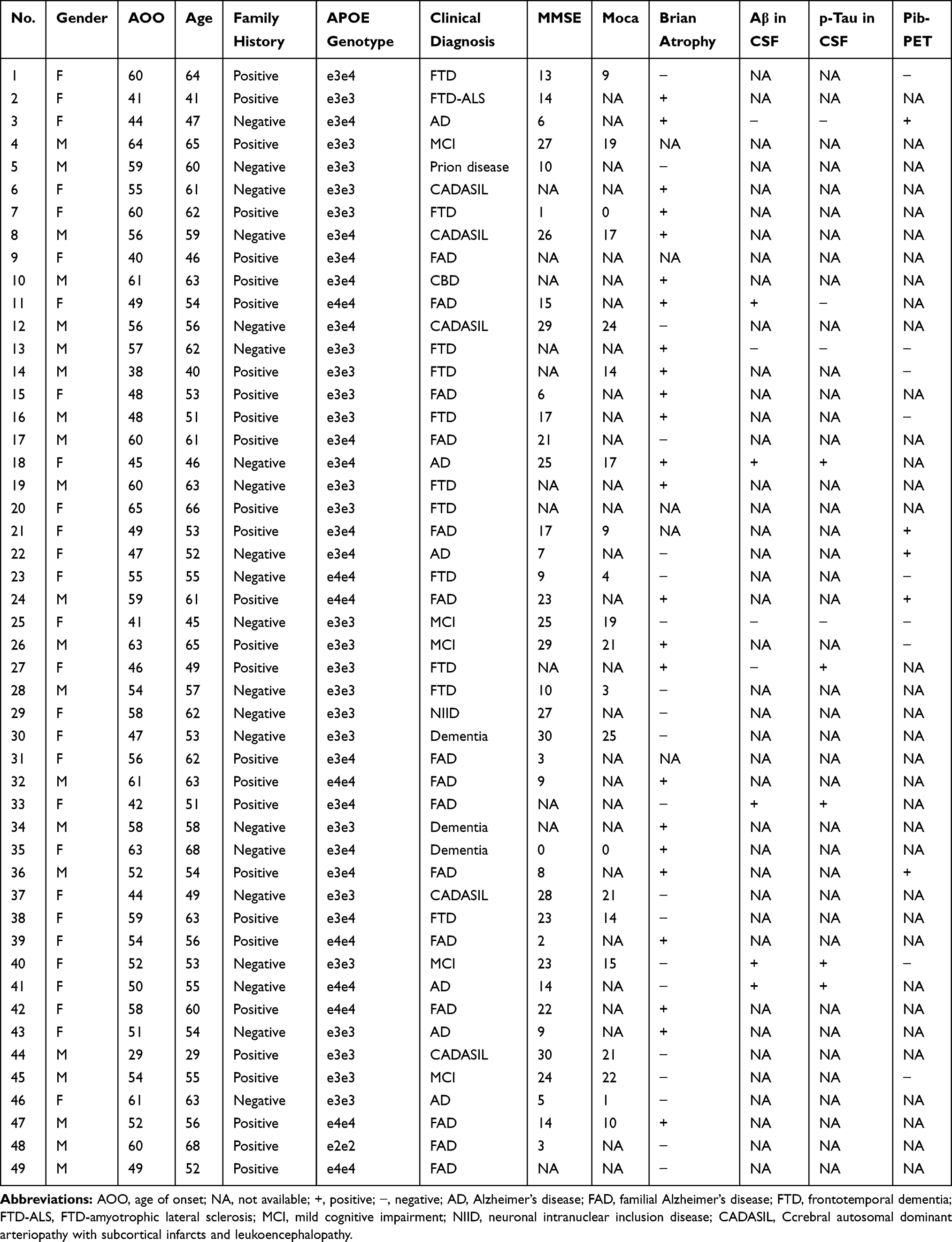 |
Table 1 The Personal and Medical Histories of EOD Patients in Present Study |
Identification of Variants
Targeted NGS was performed in this study. Coverage of the fraction of target bases indicated that 99.47% of the target bases had >30× coverage and that 98.83% of target bases had >50× coverage. The mean coverage of target bases ranged from 597.39 to 1313.36. After filtering, 9 variants in 7 known genes associated with EOD were identified in 9 index patients. Two variants (p.P436L in PSEN2; c.239-11G>A in TARDBP) were novel and 7 variants (p.I213T and p.W165C in PSEN1; p.D678N in APP; c.1349_1352del in TBK1; p.P301L and p.R406W in MAPT; p.R110C in NOTCH3) were previously reported.19–22 The presence of these variants was confirmed by Sanger sequencing and subsequent segregation analysis. Neither the 2 novel variants were detected in 500 healthy Chinese subjects. According to the American College of Medical Genetics and Genomics (ACMG) standards,21 the 2 novel variants were classified as VUS variants. Of 49 EOD patients, 7 (14.28%) were found to carry disease-causing mutations and two were found to carry uncertain significance variants (Table 2).
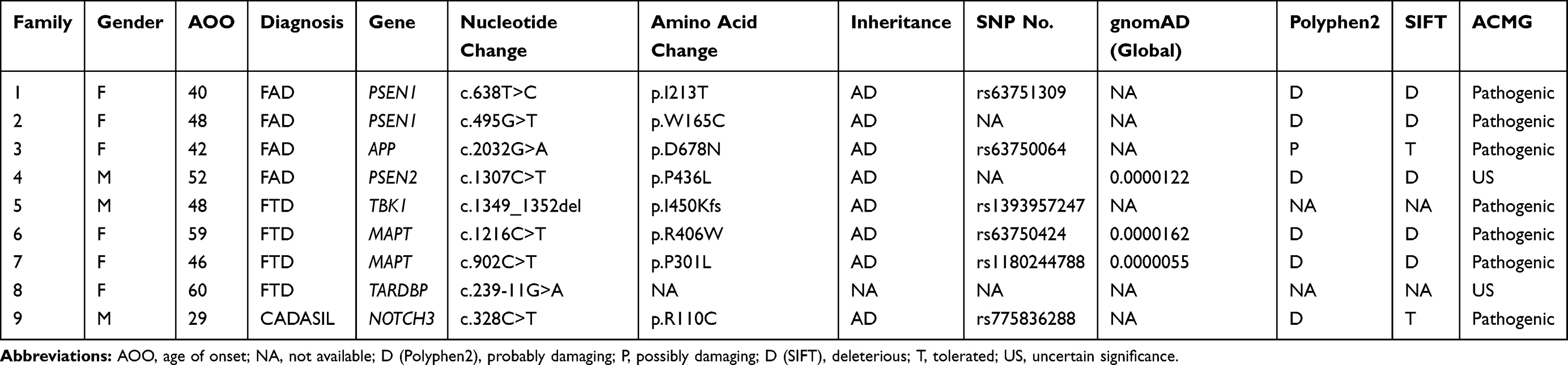 |
Table 2 Genetic and Clinical Information of EOD Patients Carrying Pathogenic and Uncertain Significance Variants |
Clinical Features of Patients Carrying PSEN1, APP and PSEN2 Variants
Two previously reported pathogenic variants in PSEN1 were identified in the two probands from Family 1 and Family 2. Case 1 (II-6 in Family 1) (Figure 1A), carrying the pathogenic variant p.I213T (c.638T>C) (Figure 1C) in PSEN1, is a 46-year-old woman who showed progressive memory impairment that began six years ago. At that time, she struggled to remember the way home and often forgot the location of frequently used items. As her cognitive abnormality progressed, she became irritable. She could not finish the mini-mental state examination (MMSE), Montreal cognitive assessment (MoCA) or CDR due to un-cooperativity. Her mother (Family 1, I-2) and elder brother (Family 1, II-1 and II-3) had similar clinical presentations. However, none of them was available for genetic evaluation. A pathogenic variant p.W165C (c.495G>T) in PSEN1 was identified in case2 (II-4 in Family 2) (Figure 1B and D). Case 2 is a 53-year-old woman who developed progressive memory impairment that began five years ago. The symptoms of memory decline became worse over the last few years. Over the past year, she started to exhibit emotional instability and gradually developed poor language expression, slow movement and poor execution. Her MMSE score was 6/30, and her MoCA was difficult to obtain due to a lack of cooperativity. Brain MRI analysis revealed global brain atrophy without vascular lesions. Similar clinical characteristics were reported for her father who died at 55 years old (Family 2, I-2).
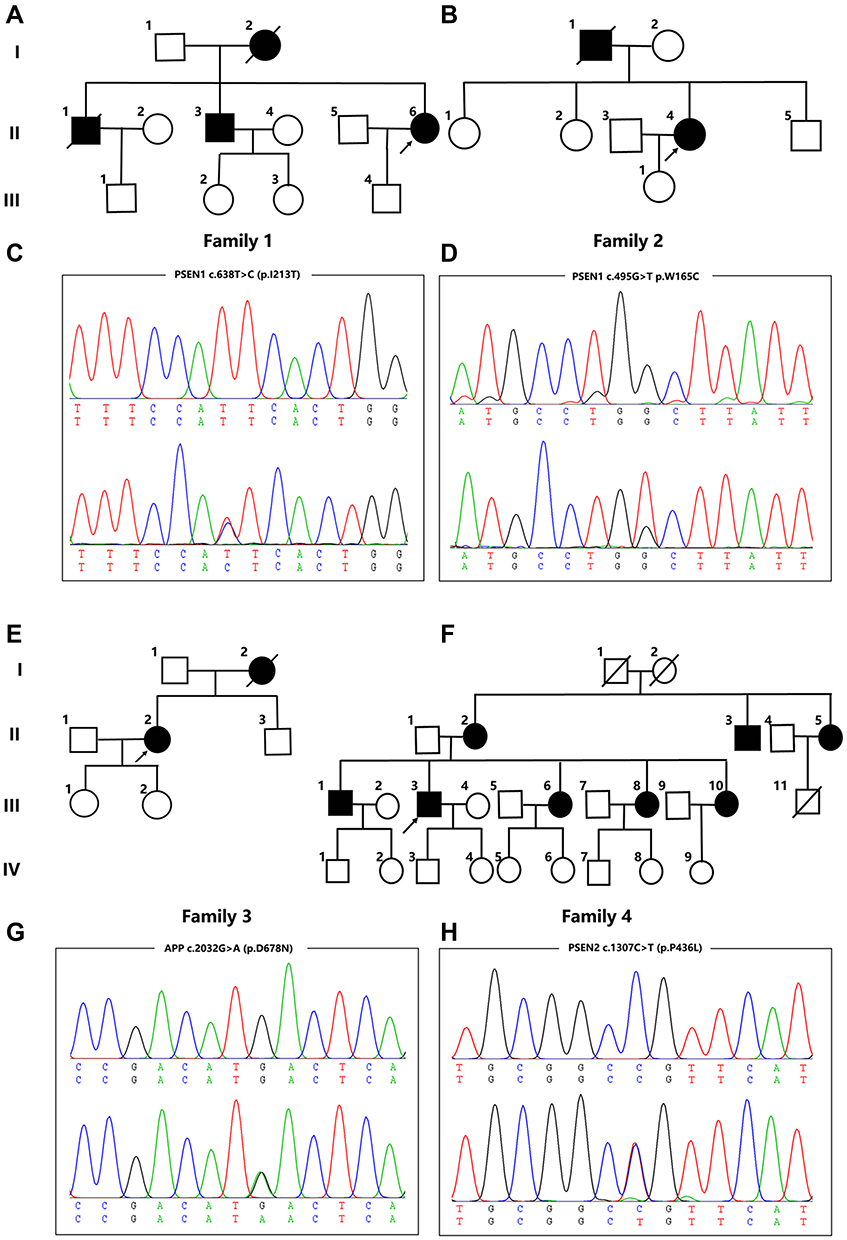 |
Figure 1 Pedigree charts and variants identified in Family 1-4. (A, B, E, F) Pedigree charts of Family 1-4; (C, D, G, H) Sequencing chromatograms of the PSEN1 variants (p.I213T and p.W165C), APP variants (p.D678N) and PSEN2 variants (p.P436L). |
The pathogenic variant p.D678N (c.2032G>A) in APP was identified in a 51-year-old woman (Family 3, II-2, case 3) (Figure 1E and G). Her initial symptom was a progressive decline of memory deficits that began at age 42 years. She sought care at a local hospital, was diagnosed with AD, and began treatment with donepezil. Over the next 9 years, her symptoms progressively worsened. She started to exhibit emotional instability and was reluctant to take part in social activities. Now, she has lost the ability to take care of herself in daily life. She could not cooperate to complete the MMSE or the MoCA. MRI revealed an abnormally high signal from the white matter in T2-weighted images as well as global brain atrophy. The patient’s mother (Family 3, I-2) had similar clinical presentations at 62 years old and died one year later.
A novel VUS variant p.P436L (c.1307C>T) in PSEN2 was identified in a 56-year-old man (Family 4, III-3, case 4) (Figure 1F and H) who presented with short-term memory impairment, a decline of visual construction abilities, deficits in calculation abilities, and mild personality changes at the age of 52 years. His MMSE and MoCA scores were 14/30 and 10/30, respectively. Brain MRI indicated atrophy of the bilateral temporal lobe and hippocampus. Similar clinical characteristics were reported for his mother (Family 4, II-2), mother’s brother (Family 4, II-3) and mother’s sister (Family 4, II-5), as well as his own elder brother (Family 4, III-1) and three sisters (Family 4, III-6, 8, and 10). This variant was absent in the 500 healthy Chinese subjects that were tested. His family members, however, refused genetic evaluation. This variant is classified as VUS according to the ACMG standards.
Clinical Features of Patients Carrying TBK1, MAPT and TARDBP Variants
Case 5 (II-6 in Family 5) (Figure 2A) was a 51-year-old man who was referred to our hospital following a three-year history of progressive memory impairment, language deficits, slowness in reaction time and changes in personality. He had a family history of dementia, as both his mother and brother had similar clinical characteristics. A pathogenic variant p.I450Kfs (c.1349_1352del) in TBK1 was identified by NGS analysis and confirmed with Sanger sequencing (Figure 2B). During his last physical examination, the patient showed less cooperation, and amnesic aphasia. There was no muscle atrophy; muscle strength, muscle tone and deep tendon reflexes were normal. Cerebral MRI showed bilateral atrophy of the frontal lobe, temporal lobes and hippocampus, and no obvious Aβ deposition was found by [11C]-PIB PET imaging (Figure 2C and D).
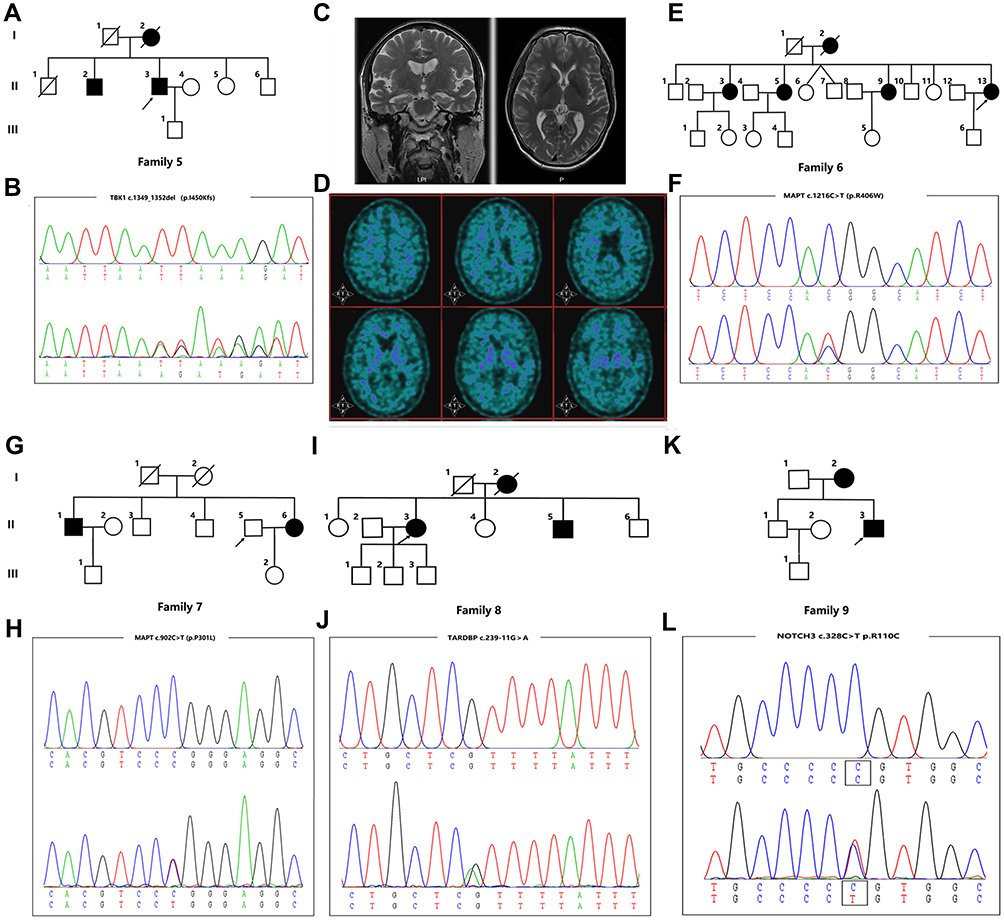 |
Figure 2 Pedigree charts and variants identified in Family 5-9. (A, E, G, I, K) Pedigree charts of Family 5-9; (C) Cerebral MRI of case 5; (D) [11C]-PIB PET imaging of case 5; (B, F, H, J, L) Sequencing chromatograms of the variants in TBK1 (p.I450Kfs), variants in MAPT (p.R406W and p.P301L), variants in TARDBP (c.239-11G>A) and variants in NOTCH3 (p.R110C). |
Both Case 6 (II-13 in Family 6) (Figure 2E) and case 7 (II-6 in Family 7) (Figure 2G) carried a pathogenic variant in MAPT. Case 6, carrying a pathogenic variant p.R406W (c.1216C>T) (Figure 2F), was a 63-year-old woman who came to our Neurology clinic with a complaint of mild memory impairment over the past four years. Her MMSE and MoCA scores were 23/30 and 14/30, respectively. Similar clinical characteristics were reported for her mother (Family 6, I-2) and three sisters (Family 6, II-3, II-5, and II-9). Case 7, carrying a pathogenic variant p.P301L (c.902C>T) (Figure 2H), was a 49-year-old woman who presented with memory impairment as well as a decline of language and calculation abilities at the age of 46. One year later, slowness in reaction times and changes in personality were observed by her husband. During her last physical examination, which was three years after the disease onset, the patient showed less cooperation, hyperactivity, and emotional instability. The patient could not cooperate to finish the Frontal Behavioral Inventory, MMSE or MoCA. Cerebral MRI showed bilateral atrophy of the hippocampus. Her brother and nephew had a similar clinical presentation.
A novel VUS variant c.239-11G>A in TARDBP was identified in a 60-year-old woman (Family 8, III-3, case 8) (Figure 2I and J) who presented with memory impairment. Her symptoms gradually got worse, and one year after disease onset, she could not remember the way home or how to prepare food. She also presented with a decline in calculation and language abilities as well as changes in personality. Her MMSE score was 3/30 and her MoCA score was 0. Brain MRI showed bilateral atrophy of the hippocampus. She has a positive family history as her mother and brother both have similar symptoms. However, her family members were unavailable for genetic evaluation. This variant is classified as VUS according to the ACMG standards.
In addition, a variant p.R110C (c.328C>T) in NOTCH3 was identified in a 29-year-old man (Family 9, II-3, case 9) (Figure 2K and L) who presented with mild memory impairment. His MoCA score was 21. Brain MRI showed bilateral frontoparietal lobe and paraventricular white matter lesions. His mother (Family 9, I-2) had experienced 20 year history of memory decline starting when she was 36 years old. She had a cerebral infarction in her 40s. This variant in NOTCH3 was reported previously to be pathogenic; therefore, this patient was diagnosed with cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL).
Discussion
EOD is a condition caused by several diseases with extensive genetic and clinical heterogeneity. Making a specific diagnosis of EOD remains a challenge. Many of EOD patients do not have a precise clinical diagnosis. The genetic analysis of patients with EOD has been investigated extensively in the Caucasian and Korean populations but rarely in the Chinese population. In the present study, we performed NGS and Sanger sequencing to identify the genetic etiology of 49 EOD patients. Two novel VUS variants (p.P436L in PSEN2; c.239-11G>A in TARDBP) and 7 previously reported pathogenic variants (p.I213T and p.W165C in PSEN1; p.D678N in APP; c.1349_1352del in TBK1; p.P301L and p.R406W in MAPT; p.R110C in NOTCH3) were identified in 9 EOD patients.
In our study, of the 49 indexed EOD patients, 14.28% carried pathogenic variants. In a recent study of 221 Belgian patients with EOD, 2.84% of the patients had a pathogenic variant in APP, MAPT, SOD1, TBK1, and C9orf72.14 A previous study performed genetic analysis of a cohort of Koreans with early-onset AD (EOAD) by NGS; pathogenic variants were identified in 6% of the 67 EOAD patients.22 Similarly, in another study that included 60 APOE ε4 non-carrying Korean patients with EOAD, 8.3% of the patients were found to carry likely pathogenic variants.23 Additionally, pathogenic variants have been identified in 5% of early-onset AD and FTD patients from Korea.13 Compared with these studies, the frequency of pathogenic variants (14.28%) was higher in our study. One of the explanations for this discrepancy is that the proportion of patients with a positive family history was relatively high (54%) in our study. In a recent study, PSEN1, PSEN2, and APP mutations were identified in a Chinese cohort that contained 1330 patients with AD or MCI in 404 pedigrees; 16.83% of these patients carried PSEN1, PSEN2, or APP mutations, comparable with the rate in the present study.24
Among all of the identified patients, three patients each had AD and FTD. AD and FTD are two common neurodegenerative diseases. There is a neuropathological and clinical crossover between these two diseases. For example, similar clinical presentations such as a decline in memory, personality changes, and phosphorylated tau protein deposition can be observed in both diseases. In this study, case 3, who carried a pathogenic variant in APP, exhibited personality changes and short-term memory impairment, whereas case 6, who carried a pathogenic variant in MAPT, demonstrated only mild memory impairment four years after disease onset. Similarly, a recent study has reported FTD patients with typical AD clinical presentations.25 It is difficult to discriminate between these two diseases if patients have an atypical clinical presentation. Thus, it is worthwhile to perform NGS in EOD patients, especially those with a positive family history, to help formulate a genetics-based diagnosis.
TBK1 is a causative gene of FTD-ALS that has been recently identified.26,27 Its encode protein, tumor necrosis factor receptor associated factor NF-kB activator-binding kinase 1 (TBK1), is a serine/threonine protein kinase that participates in multiple cellular pathways including neuroinflammation28 and selective autophagy degradation.29 A pathogenic variant in TBK1 p.I450Kfs (c.1349_1352del) was identified in case 5. This variant has been previously reported in patients with ALS.26 Very few studies have linked TBK1 with FTD in Chinese populations.25,30 To the best of our knowledge, this is the first study to link this variant with FTD in the Chinese population.
The majority of EOD cases in our study remain genetically unexplained, indicating additional causal and risk genes need to be identified. In this NGS study, 2 novel uncertain significance variants in the PSEN2 and TARDBP genes were identified. The role of these 2 variants needs to be determined in the future.
Conclusion
In conclusion, 7 previously reported pathogenic variants and 2 novel VUS variants were identified in the present study. All of the identified patients had early onset and a positive family history of dementia. Our study expanded the genetic spectrum of EOD and reveals that NGS is a great help in the definite diagnosis of EOD patients.
Acknowledgments
We would like to thank all participants for their supports and willingness to participate in this study. This work was supported by the grants from the National Natural Science Foundation of China to Qing-Qing Tao (81970998) and the Key Research and Development project of Zhejiang Province to Zhi-Ying Wu (2019C03039), and research foundation for distinguished scholar of Zhejiang University to Zhi-Ying Wu (188020-193810101/089).
Disclosure
The authors declare that they have no competing interests.
References
1. Masellis M, Sherborn K, Neto P, et al. Early-onset dementias: diagnostic and etiological considerations. Alzheimers Res Ther. 2013;5(Suppl 1):S7. doi:10.1186/alzrt197
2. Van der Flier WM. Clinical heterogeneity in familial Alzheimer’s disease. Lancet Neurol. 2016;15(13):1296–1298.
3. Benussi A, Padovani A, Borroni B. Phenotypic heterogeneity of monogenic frontotemporal dementia. Front Aging Neurosci. 2015;7:171.
4. Walker L, Stefanis L, Attems J. Clinical and neuropathological differences between Parkinson’s disease, Parkinson’s disease dementia and dementia with Lewy bodies – current issues and future directions. J Neurochem. 2019;150(5):467–474. doi:10.1111/jnc.14698
5. Qiu Y, Jacobs DM, Messer K, Salmon DP, Feldman HH. Cognitive heterogeneity in probable Alzheimer disease: clinical and neuropathologic features. Neurology. 2019;93(8):e778–e790.
6. DeTure MA, Dickson DW. The neuropathological diagnosis of Alzheimer’s disease. Mol Neurodegener. 2019;14(1):32. doi:10.1186/s13024-019-0333-5
7. Dunn AR, O’Connell KMS, Kaczorowski CC. Gene-by-environment interactions in Alzheimer’s disease and Parkinson’s disease. Neurosci Biobehav Rev. 2019;103:73–80. doi:10.1016/j.neubiorev.2019.06.018
8. Kim JH. Genetics of Alzheimer’s disease. Dement Neurocogn Disord. 2018;17(4):131–136. doi:10.12779/dnd.2018.17.4.131
9. Li NM, Liu KF, Qiu YJ, Zhang HH, Nakanishi H, Qing H. Mutations of beta-amyloid precursor protein alter the consequence of Alzheimer’s disease pathogenesis. Neural Regen Res. 2019;14(4):658–665. doi:10.4103/1673-5374.247469
10. Bang J, Spina S, Miller BL. Frontotemporal dementia. Lancet. 2015;386(10004):1672–1682. doi:10.1016/S0140-6736(15)00461-4
11. Matar E, Ehgoetz Martens KA, Halliday GM, Lewis SJG. Clinical features of Lewy body dementia: insights into diagnosis and pathophysiology. J Neurol. 2019.
12. Shinan-Altman S, Werner P. Illness representations of dementia: a scoping review. Clin Interv Aging. 2019;14:179–193. doi:10.2147/CIA.S193316
13. Luukkainen L, Helisalmi S, Kytovuori L, et al. Mutation analysis of the genes linked to early onset Alzheimer’s disease and frontotemporal lobar degeneration. J Alzheimers Dis. 2019;69(3):775–782. doi:10.3233/JAD-181256
14. Perrone F, Cacace R, Van Mossevelde S, et al. Genetic screening in early-onset dementia patients with unclear phenotype: relevance for clinical diagnosis. Neurobiol Aging. 2018;69:292 e297–292 e214. doi:10.1016/j.neurobiolaging.2018.04.015
15. Bonvicini C, Scassellati C, Benussi L, et al. Next generation sequencing analysis in early onset dementia patients. J Alzheimers Dis. 2019;67(1):243–256. doi:10.3233/JAD-180482
16. Jiang B, Zhou J, Li HL, et al. Mutation screening in Chinese patients with familial Alzheimer’s disease by whole-exome sequencing. Neurobiol Aging. 2019;76:215 e215–215 e221. doi:10.1016/j.neurobiolaging.2018.11.024
17. McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):263–269. doi:10.1016/j.jalz.2011.03.005
18. Rascovsky K, Hodges JR, Knopman D, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain. 2011;134(Pt 9):2456–2477.
19. McKeith IG, Boeve BF, Dickson DW, et al. Diagnosis and management of dementia with Lewy bodies: fourth consensus report of the DLB Consortium. Neurology. 2017;89(1):88–100.
20. Tao -Q-Q, Chen Y, Liu Z-J, et al. Associations between apolipoprotein E genotypes and serum levels of glucose, cholesterol, and triglycerides in a cognitively normal aging Han Chinese population. Clin Interv Aging. 2014;9:1063–1067.
21. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. doi:10.1038/gim.2015.30
22. Giau VV, Bagyinszky E, Yang YS, Youn YC, An SSA, Kim SY. Genetic analyses of early-onset Alzheimer’s disease using next generation sequencing. Sci Rep. 2019;9(1):8368. doi:10.1038/s41598-019-44848-2
23. Park JE, Kim HJ, Kim YE, et al. Analysis of dementia-related gene variants in APOE epsilon4 noncarrying Korean patients with early-onset Alzheimer’s disease. Neurobiol Aging. 2020;85(155):e155–155 e158. doi:10.1016/j.neurobiolaging.2019.05.009
24. Jia L, Fu Y, Shen L, et al. PSEN1, PSEN2, and APP mutations in 404 Chinese pedigrees with familial Alzheimer’s disease. Alzheimers Dement. 2020;16(1):178–191. doi:10.1002/alz.12005
25. Ma L, Zhang J, Shi Y, et al. Gene mutations in a Han Chinese Alzheimer’s disease cohort. Brain Behav. 2019;9(1):e01180.
26. Freischmidt A, Wieland T, Richter B, et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat Neurosci. 2015;18(5):631–636. doi:10.1038/nn.4000
27. Gijselinck I, Van Mossevelde S, van der Zee J, et al. Loss of TBK1 is a frequent cause of frontotemporal dementia in a Belgian cohort. Neurology. 2015;85(24):2116–2125. doi:10.1212/WNL.0000000000002220
28. Xu D, Jin T, Zhu H, et al. TBK1 suppresses RIPK1-driven apoptosis and inflammation during development and in aging. Cell. 2018;174(6):1477–1491 e1419. doi:10.1016/j.cell.2018.07.041
29. Duan W, Guo M, Yi L, et al. Deletion of Tbk1 disrupts autophagy and reproduces behavioral and locomotor symptoms of FTD-ALS in mice. Aging. 2019;11(8):2457–2476. doi:10.18632/aging.101936
30. Yu H, Yu W, Luo SS, et al. Association of the TBK1 mutation p.Ile334Thr with frontotemporal dementia and literature review. Mol Genet Genom Med. 2019;7(3):e547. doi:10.1002/mgg3.547
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.