Back to Journals » International Journal of Nanomedicine » Volume 11
GE11 peptide modified and reduction-responsive hyaluronic acid-based nanoparticles induced higher efficacy of doxorubicin for breast carcinoma therapy
Authors Hu DR, Mezghrani O, Zhang L, Chen Y, Ke X, Ci TY
Received 24 May 2016
Accepted for publication 27 July 2016
Published 7 October 2016 Volume 2016:11 Pages 5125—5147
DOI https://doi.org/10.2147/IJN.S113469
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Linlin Sun
Danrong Hu, Omar Mezghrani, Lei Zhang, Yi Chen, Xue Ke, Tianyuan Ci
Department of Pharmaceutics, State Key Laboratory of Natural Medicines, China Pharmaceutical University, Nanjing, People’s Republic of China
Abstract: Novel breast carcinoma dual-targeted redox-responsive nanoparticles (NPs) based on cholesteryl-hyaluronic acid conjugates were designed for intracellular delivery of the antitumor drug doxorubicin (DOX). A series of reduction-responsive hyaluronic acid derivatives grafted with hydrophobic cholesteryl moiety (HA-ss-Chol) and GE11 peptide conjugated HA-ss-Chol (GE11–HA-ss-Chol) were synthesized. The obtained conjugates showed attractive self-assembly characteristics and high drug loading capacity. GE11–HA-ss-Chol NPs were highly stable under conditions mimicking normal physiological conditions, while showing a fast degradation of the vehicle’s structure and accelerating the drug release dramatically in the presence of intracellular reductive environment. Furthermore, the cellular uptake assay confirmed GE11–HA-ss-Chol NPs were taken up by MDA-MB-231 cells through CD44- and epidermal growth factor receptor-mediated endocytosis. The internalization pathways of GE11–HA-ss-Chol NPs might involve clathrin-mediated endocytosis and macropinocytosis. The intracellular distribution of DOX in GE11–HA-ss-Chol NPs showed a faster release and more efficient nuclear delivery than the insensitive control. Enhanced in vitro cytotoxicity of GE11–HA-ss-Chol DOX-NPs further confirmed the superiority of their dual-targeting and redox-responsive capacity. Moreover, in vivo imaging investigation in MDA-MB-231 tumor-bearing mice confirmed that GE11–HA-ss-Chol NPs labeled with 1,1'-dioctadecyl-3,3,3',3'-tetramethylindotricarbocyanine iodide, a near-infrared fluorescence dye, possessed a preferable tumor accumulation ability as compared to the single-targeting counterpart (HA-ss-Chol NPs). The antitumor efficacy showed an improved therapy efficacy and lower systemic side effect. These results suggest GE11–HA-ss-Chol NPs provide a good potential platform for antitumor drugs.
Keywords: self-assembled nanoparticles, redox responsive, dual-ligand targeting, hyaluronic acid, cancer therapy
Introduction
Doxorubicin (DOX), an anthracycline antibiotic possessing broad-spectrum antineoplastic activity, is one of the most active anticancer agents in the treatment of breast cancer.1 However, the clinical utility of DOX is hampered by cumulative, dose-limiting cardiotoxicity, myelosuppression, and the development of drug resistance.2 With the emergence of various kinds of drug delivery systems,3–13 interesting and meaningful efforts have been made to improve the drug efficacy and minimize adverse effects.14–20 Hydrophobized polysaccharide nanoparticles (NPs) are considered to be a promising approach.21 Self-assembled hydrophobized polysaccharide NPs are usually applied as promising carriers of water-insoluble drugs with various advantages, including high loading capacity, sustained release, prolonged circulation, and enhanced cellular uptake and tumor targeting.22
Hyaluronic acid (HA), a biocompatible polyanionic polysaccharide, has been the subject of many previous reviews focusing on its biological functions and medical applications.23,24 HA has a strong affinity with cell-specific surface receptors such as CD44 and RHAMM, which are abundantly overexpressed on the surface of many malignant tumors.25,26 In recent years, much attention has been paid to hydrophobically modified HA as the drug carrier, in which HA can be used as both hydrophilic shell and active targeting constituent.
Our previous work has demonstrated that HA-based NPs display predominant intracellular delivery of DOX for hepatocellular carcinoma therapy.27 However, the ligand–receptor mediated endocytosis interactions which enhance the cellular uptake efficiency might be limited due to either receptor saturation28 or downregulation.29 Therefore, a novel dual receptor targeting strategy, transporting the pharmaceuticals to the tumor more effectively, has been utilized in the field of targeted drug delivery systems.30 Epidermal growth factor receptor (EGFR) is significantly upregulated on the surface of many human malignancies, and it has been regarded as a significant target for cancer therapy.31 A novel small peptide GE11 (sequence YHWYGYTPQNVI) obtained by the phage display screening was shown to have high affinity toward EGFR-overexpressed cancer cells, but much lower mitogenic activity than the native ligand EGF, making it a promising targeting candidate for EGFR-targeted therapy.32
On the other hand, considering that the conventional polymeric carriers usually exhibit sustained release of drug resulting in reduced therapeutic efficacy,33 the nanocarriers with stimuli-responsive drug release after entering the cells are pursued. Nowadays, redox-responsive nanocarriers, engineered by integrating disulfide groups as linkages between the hydrophilic shell and the hydrophobic core, are generating more and more interest.34–38 These “smart” drug carriers are sensitive to the internal biological stimulus called glutathione. Also, the detachable carriers can remain stable under nonreductive physiological conditions and present a sudden burst of pharmaceuticals through structure degradation, leading to fast intracellular drug release.
Thus, in this study, we designed and synthesized the GE11 peptide functionalized and reduction-responsive NPs for triggered intracellular delivery of DOX for breast cancer (Figure 1). The redox-responsive NPs were based on hyaluronic acid derivatives grafted with hydrophobic cholesteryl moiety (HA-ss-Chol) using cystamine as the detachable linkage. EGFR-targeted peptide was further surface-conjugated to HA-ss-Chol (GE11–HA-ss-Chol) to achieve dual active targeting. Meanwhile, the reduction-insensitive counterparts HA-Chol and GE11–HA-Chol were synthesized for comparison, with nondetachable ethanediamine as conjunctions. Then, the chemotherapeutic agent DOX was successfully encapsulated by the polymeric NPs. The physicochemical characterization, in vitro cellular uptake and cytotoxity, in vivo targeting ability, and antitumor efficacy were evaluated in detail.
 | Figure 1 Illustration of dual-targeted reduction-responsive NPs based on GE11–HA-ss-Chol conjugates as a multifunctional platform for tumor-targeting drug delivery. |
Materials and methods
Materials
The EGFR-targeted peptide GE11, GGGGGYHWYGYTPQNVI, was custom-synthesized by GL Biochem Ltd. (Shanghai, People’s Republic of China), with the “GGGGG” sequence as a spacer and the N-terminal amino of the peptide reacting with the carboxyl group of HA. Sodium hyaluronate (molecular weight [MW] 10 kDa) was purchased from Shandong Freda Biochem Co., Ltd. (Shandong, People’s Republic of China). Cholesteryl chloroformate, 1-ethyl-3 (3-dimethylaminopropyl) carbodiimide hydrochloride (EDC), 1-hydroxybenzotriazole monohydrate (HOBT), N-hydroxysulfosuccinimide (sulfo-NHS), and pyrene were obtained from Aladdin Reagent Co., Ltd. (Shanghai, People’s Republic of China). Cystamine dihydrochloride, DL-dithiothreitol (DTT), and coumarin-6 (C6) were purchased from Sigma-Aldrich (St, Louis, MO, USA). Doxorubicin hydrochloride (DOX·HCl) was obtained from Zhejiang Hisun Pharmaceutical Co., Ltd. (Taizhou, People’s Republic of China). All other chemicals were of analytical grade.
Cell culture and animals
Human breast carcinoma cell lines MDA-MB-231 cells (relatively higher expression of CD44 and EGFR) and MCF-7 (lower expression of CD44 and EGFR) were obtained from the cell bank of the Chinese Academy of Sciences (Shanghai, People’s Republic of China). According to previous literature reports,39,40 MDA-MB-231 and MCF-7 cells express 2×105 and 1×104 EGFR per cell and exhibit positive and negative expression of CD44, respectively. MDA-MB-231 cells were cultured in high-glucose Dulbecco’s Modified Eagle’s Medium (DMEM) with 10% fetal bovine serum and 1% penicillin–streptomycin at 37°C in 5% CO2 atmosphere. MCF-7 cells were grown in Roswell Park Memorial Institute (RPMI) 1640 medium with the same additives.
Female BALB/c nude mice (6–8 weeks old, initial weight 20–25 g) were purchased from KeyGEN BioTECH Co., Ltd. (Nanjing, People’s Republic of China). All experiments were carried out in compliance with the Animal Management Rules of the Ministry of Health of the People’s Republic of China and the guidelines for the Care and Use of Laboratory Animals of China Pharmaceutical University. All procedures involving animals and their care in this study were in strict accordance with protocols approved by the Ethics Committee of China Pharmaceutical University.
Synthesis of reduction-sensitive and -insensitive conjugates
Neutralization of cystamine dihydrochloride
Cystamine base was prepared as previously described.41 Briefly, cystamine dihydrochloride (60.8 g, 0.27 mol) was neutralized by adding 40% aqueous solution of NaOH (4 mol) in an aqueous–organic solvent biphasic system (water/diethyl ether/tetrahydrofuran: 1:3.75:1.5 v/v/v) in an ice bath for 1 hour. After dessication over NaOH pellets and purification with diethyl ether and tetrahydrofuran (1:3 v/v), the organic solvents were evaporated under vacuum. The resulting product was stored at −20°C until further use. Cystamine base structure was characterized by 1H-nuclear magnetic resonance (1H-NMR) (Avace AV-500, 500 MHz; Bruker Optik GmbH, Ettlingen, Germany) using CDCl3 as the solvent: δ (ppm) 1.45 (m, 4H, 2NH2), 2.77–2.78 (t, 4H, 2CH2S), 3.00–3.03 (m, 4H, 2CH2N).
Synthesis of cholesteryl–cystamine (Chol–cys) and cholesteryl–ethylenediamine (Chol–eda) linkers
The modification of cholesteryl chloroformate with cystamine was carried out according to the following procedure. Precisely, 15.2 g (100 mmol) of cystamine and 695 μL (5 mmol) of triethylamine (TEA) were dissolved in 150 mL of anhydrous dichloromethane (DCM). To the mixture, 50 mL of anhydrous DCM containing cholesteryl chloroformate (2.25 g, 5 mmol) was added dropwise over 2 hours at 0°C. The reaction was then allowed to proceed for another 10 hours at room temperature. The previous operations were all carried out under a nitrogen atmosphere. The sediment was eliminated by filtration afterward. The filtrate was washed with water three times, dried over anhydrous magnesium sulfate, and then dried in vacuum. The crude product was then purified by column chromatography (SiO2, 180 g, DCM/methanol, 100/5 v/v) with a yield of 70%. The formation of the conjugate was confirmed by 1H-NMR in CDCl3 (Figure S1): δ (ppm) 5.36 (s, 1H, alkenyl), 5.05 (s, 1H, CONH), 4.48 (m, 1H, oxycyclohexyl), 3.48 (m, 2H, CH2NH), 3.00 (m, 2H, CH2N(H2)), 2.76–2.79 (m, 4H, 2CH2S), 0.66–2.33 (m, 45H, cholesteryl protons and NH2).
The nonreducible linker cholesteryl–ethylenediamine (Chol–eda) was obtained by reacting cholesteryl chloroformate with ethylenediamine in anhydrous DCM in the presence of TEA, according to a previously established method.42 The resulting product was characterized by 1H-NMR in CDCl3 (Figure S1C): δ (ppm) 5.37 (s, 1H, alkenyl), 4.99 (s, 1H, CONH), 4.50 (m, 1H, oxycyclohexyl), 3.21–3.23 (m, 2H, N(H2)CH2), 2.82–2.84 (m, 2H, CONCH2), 0.68–2.38 (m, 45H, cholesteryl protons, and NH2).
Synthesis and characterization of HA-ss-Chol conjugates and HA-Chol conjugates
HA-ss-Chol conjugates and HA-Chol conjugates were synthesized by chemically grafting Chol–cys and Chol–eda to HA, respectively, by amide bond formation. The carboxylic acid groups of HA were linked to the primary amino group of cystamine or ethanediamine. Briefly, 100 mg of HA (0.25 mmol) was dissolved in formamide (10 mL) at 50°C. After the solution was cooled down to room temperature, carboxylic acid group of HA was activated by EDC (143.8 mg, 0.75 mmol) and HOBT (101.6 mg, 0.75 mmol) for 15 minutes. Varying amounts of Chol–cys dissolved in 20 mL dimethyl formamide (DMF) were added, respectively. The molar feeding ratios of Chol–cys to HA’s sugar units were 0.03, 0.05, and 0.07, respectively, and the corresponding conjugates were referred to as HA-ss-Chol3, HA-ss-Chol5, and HA-ss-Chol7, respectively. The mixture was stirred for a further 24 hours and precipitated in excess cold acetone. The resulting precipitate was thoroughly washed with methanol and acetone, then dissolved in distilled water, and dialyzed against deionized water for 48 hours using a dialysis membrane with MW cut-off (MWCO) of 3,500. HA-Chol7 was prepared according to the method described earlier by grafting HA with Chol–eda linker using a molar feeding ratio of 0.07.
The structures of HA-ss-Chol and HA-Chol conjugate structures were determined by 1H-NMR spectroscopy. 1H-NMR spectra were acquired on a Bruker (Karlsruhe, Germany) AV-500 spectrometer using D2O as the solvent. The degree of substitution (DS), defined as the number of Chol–cys groups per 100 sugar units of HA, was determined through evaluation of disulfide bond content by Ellman’s method.43 Briefly, 250 μL of the polymer aqueous solution (5 mg/mL) was diluted with 250 μL of phosphate-buffered saline (PBS) 8.0 (0.1 M). Disulfide linkages were reduced by the addition of 500 μL of freshly prepared 2% aqueous solution of NaBH4. Following 1 hour stirring at 40°C, the excess amount of NaBH4 was discarded by the addition of 0.3 mL of 1 N HCl and the mixture was completed by 1.2 mL of PBS 8.0. The thiol content was determined by colorimetric method using 2.5 mL of 5,5′-Dithiobis-(2-nitrobenzoic acid) (DTNB) buffered solution (0.3%). The ultraviolet absorbance of the mixture was measured at λ=412 nm.
Synthesis and characterization of GE11-decorated HA-ss-Chol (GE11–HA-ss-Chol) and HA-Chol (GE11–HA-Chol)
The grafting of GE11 on HA-ss-Chol conjugate was achieved through amide bond formation between the amine group of GE11 and the carboxylic group of HA backbone. In brief, 20 mg of HA-ss-Chol7 dispersed in 5 mL of deionized water was activated with EDC (5 mg) and sulfo-NHS (5 mg) at room temperature for 4 hours, followed by addition of 2 mL of GE11 solution (1 mg/mL in deionized water). The pH was adjusted to 7.4. After 24 hours stirring at room temperature, the mixture was dialyzed against deionized water for 24 hours (MWCO 3,500) and then lyophilized. GE11–HA-Chol conjugate was prepared as per the same protocol (Supplementary materials).
The DS and the conjugation efficiency of GE11 to HA-ss-Chol were analyzed by 1H-NMR (D2O as solvent) and high-performance liquid chromatography (HPLC) under the following conditions: Promosil Column: C18 (4.6×250 mm, 5 μm); mobile phase: acetonitrile (solution A) and ultrapure water (solution B), both containing 0.1% trifluoroacetic acid; gradient elution mode: 0–20 minutes, 25%–50% A and 20.1–30 minutes, 50%–25% A; wavelength: 220 nm.
Preparation and physicochemical characterization of blank NPs
The redox-sensitive GE11–HA-ss-Chol and HA-ss-Chol NPs were obtained by subjecting an aqueous solution of the conjugates (1 mg/mL) to a sonication treatment for 30 minutes in an ice bath (200 W, 2 seconds pulse on, 2 seconds pulse off) using a probe-type ultrasonicator (JY 92-II; Ningbo Scientz Biotechnology Co., Ltd, Nanjing, People’s Republic of China). The resulting NP suspension was filtered through a 0.8 μm pore-sized microporous membrane. Reduction-insensitive GE11–HA-Chol and HA-Chol NPs were prepared following the same protocol. Particle size (PS), polydispersity index, and zeta potential of the NPs thus formed were acquired by dynamic laser scattering (DLS) with replication number of three. The morphology and size distribution were observed by transmission electron microscopy (TEM) using a Tecnai 12 (Philips, Amsterdam, the Netherlands) after negative staining with 2.5% sodium phosphotungstic acid solution.
Self-aggregation behavior of HA-ss-Chol amphiphilic conjugates with different DS values was evaluated by fluorescence spectroscopy using pyrene as a probe. Briefly, 1 mL of pyrene acetone solution (6.0×10−6 M) was taken in 10 mL volumetric flasks. After evaporation of acetone under vacuum, successive dilutions of HA-ss-Chol aqueous solution with concentrations ranging from 1.0×10−4 to 5×10−1 mg/mL were added. The samples were sonicated in a water bath for 1 hour and then incubated at 50°C for an additional hour. The flasks were kept in the dark overnight at room temperature to reach pyrene solubilization equilibrium. Pyrene emission spectrum was recorded from 350 to 450 nm using a fluorescence spectrophotometer with excitation wavelength 334 nm. To calculate the critical aggregation concentration (CAC), the intensity ratio of the first peak (I1, 371 nm) to the third peak (I3, 382 nm) (I371/I382) was plotted against the conjugate concentration’s logarithm.
DTT-triggered disassembly of redox-sensitive NPs
The disassembly behavior of redox-sensitive HA-ss-Chol7 NPs was evaluated under different reductive conditions mimicking different in vivo environments. Briefly, in response to various concentrations of DTT, the PS changes of NPs (1 mg/mL) suspended in PBS buffer (pH 7.4, 0.1 M) were monitored in time by DLS. Differing concentrations of DTT were added to obtain the specified concentration of 10 μM, 10 mM, and 20 mM DTT, respectively. The NP suspensions in different media were gently shaken at 37°C. As a control, the PS of HA-ss-Chol7 NPs was recorded in PBS in the absence of the reducing agent under the same conditions. The contrast study was carried out by monitoring the PS of reduction-nonresponsive HA-Chol7 NPs in response to 20 mM DTT.
Preparation of drug-loaded NPs (DOX-NPs)
DOX was chosen as a drug model and was loaded into NPs using a dialysis method. Briefly, 30 mg of DOX·HCl was dissolved in 1 mL of DMSO in the presence of TEA (molDOX·HCl:molTEA =1:3) followed by stirring for 24 hours to obtain DOX base. Thereafter, 330 μL of the solution was added to 3 mL of the conjugate solution in deionized water (6 mg/mL) in a dropwise manner. The mixture was further stirred for 4 hours and then sonicated at 200 W for 30 minutes. The organic solvent was discarded by dialysis against deionized water for 12 hours (MWCO 3,500). To remove the nonencapsulated DOX, the NPs solution was centrifuged at 3,000 rpm for 8 minutes and the supernatant filtered through a 0.45 μm pore-sized membrane. Finally, the filtrate was freeze-dried and kept at 4°C in a lightproof container.
The DOX content in the DOX-NPs was measured by ultraviolet–visible spectrophotometry. In brief, a known amount of freeze-dried DOX-NPs was dissolved in water, which was followed by dilution with methanol. Concentration of DOX in the resulting solution was measured at 481 nm. The drug loading content (DLC) and the entrapment efficiency (EE) were calculated as follows:
|
|
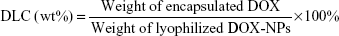
|
|
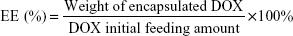
In vitro drug release
In vitro release of DOX from HA-ss-Chol7 DOX-NPs was investigated by a dialysis method. Lyophilized HA-ss-Chol7 DOX-NPs were dispersed in 1 mL of deionized water (100 μg DOX/mL) and sealed in a dialysis bag (MWCO 3,500). The bags were immersed in the release media containing 0 M, 10 μM, 10 mM, and 20 mM of DTT, respectively. The containers were placed in a shaking bed with a rotation speed of 100 rpm at 37°C. All the release media consisted of 100 mL PBS (pH 7.4, 0.1 M) containing 0.075% (w/v) Tween 80. At predetermined time intervals, 5 mL of samples were withdrawn and replaced by the same volume of fresh media. As reduction-nonresponsive control, drug release of HA-Chol7 DOX-NPs was evaluated in the presence of 20 mM DTT. The experiments were carried out with three repetitions for each group.
In vitro cell studies
In vitro cellular uptake study
The intracellular uptake of NPs in MDA-MB-231 cells and MCF-7 cells was evaluated by flow cytometry method. The fluorescence probe C6 was encapsulated in NPs according to DOX loading protocol using DMF as the organic solvent. The DLC was estimated to be 0.3%. Cells were seeded in a six-well plate during the exponential growth phase at a seeding density of 1×105 cells per well and incubated for 24 hours to allow cell attachment. Subsequently, MDA-MB-231 cells and MCF-7 cells were incubated with HA-ss-Chol7 C6-NPs and GE11–HA-ss-Chol7 C6-NPs (C6 concentration =200 ng/mL) at 37°C for 4 hours, respectively. Blank cells were used as negative control. Then the medium was aspirated, and the cells were washed with ice-cold PBS (pH 7.4) thrice, harvested by trypsinization, and suspended in PBS. The fluorescence intensity of C6 in cells was determined using FACSCalibur flow cytometer (Becton, Dickinson and Company, Franklin Lakes, NJ, USA).
In order to investigate the cellular internalization mechanism of GE11–HA-ss-Chol7 C6-NPs, competitive inhibitory studies were undertaken. To determine whether the NPs were internalized through CD44- and/or EGFR-mediated endocytosis, MDA-MB-231 and MCF-7 cells were incubated with 10 mg/mL of free HA polymer and 1 mg/mL GE11, respectively, for 1 hour prior to treatment with GE11–HA-ss-Chol7 C6-NPs. Cellular internalization efficiency was evaluated by flow cytometry as described earlier.
Furthermore, alternative cellular uptake pathways of the NPs were investigated using different endocytic inhibitors. Briefly, MDA-MB-231 and MCF-7 cells were separately pretreated with chloropromazine (CPZ) (20 μg/mL), amiloride (10 μM), and nystatin (30 μg/mL) at 37°C for 30 minutes. The cells were then incubated with GE11–HA-ss-Chol7 C6-NPs for 2 hours. Cellular uptake evaluation was carried out following the same protocol described earlier. Cells treated only with GE11–HA-ss-Chol7 C6-NPs were set as control, and their fluorescence intensities were expressed as 100%.
Intracellular drug distribution of DOX by confocal microscopy
Intracellular drug release of GE11–HA-ss-Chol7 and GE11–HA-Chol7 DOX-NPs was investigated by confocal laser scanning microscopy (CLSM). MDA-MB-231 and MCF-7 cells cultured 24 hours in glass bottom dishes were treated with DOX-NPs dispersed in serum-free cell culture medium (DOX concentration 10 μg/mL). After 1 or 4 hours of incubation at 37°C, the supernatant was removed; cells were washed three times with ice-cold PBS and fixed by 1 mL of immune staining fix solution for 20 minutes. The nuclei were strained with Hoechst 33342 (10 μg/mL) for 30 minutes and washed thoroughly with ice-cold PBS before fluorescent imaging. The release and transferring of DOX were observed using CLSM (FV1000; Olympus Corporation, Tokyo, Japan) with excitation and emission wavelengths set at 488 and 530 nm, respectively.
In vitro cytotoxicity assay
In vitro antitumor efficacy of DOX-NPs was evaluated by MTT assay. Briefly, MDA-MB-231 and MCF-7 cells seeded in 96-well plates at a density of 1×104 cells per well were allowed to attach for 24 hours. The cells were then treated with free DOX, HA-ss-Chol7 DOX-NPs, GE11–HA-Chol7 DOX-NPs, and GE11–HA-ss-Chol7 DOX-NPs, respectively. Serial dilutions of 200 μL of the sample solutions in serum-free cell culture medium were added and the cells were further incubated for 24 hours. Thereafter, MTT solution in PBS (20 μL, 5 mg/mL) was added to each well and cells were further incubated for 4 hours at 37°C. MTT solution was then withdrawn and 100 μL of DMSO was added to dissolve the formazan crystals. The optical density (OD) was measured at 570 nm with a microplate reader (SOFT maxTM PRO; Molecular Devices LLC, Sunnyvale, CA, USA). Cell viability (%) was calculated as (OD of test group/OD of control group) ×100. The median inhibitory concentration (IC50) was determined using the software GraphPad Prism 5 (GraphPad Software, Inc., La Jolla, CA, USA). The cytotoxicity of blank NPs was also evaluated for comparison.
In vivo and ex vivo imaging investigations
In vivo and ex vivo imaging experiments were carried out using a noninvasive near infrared fluorescence imaging method. NIR dye 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindotricarbocyanine iodide (DiR) was encapsulated in GE11–HA-ss-Chol7 NPs and HA-ss-Chol7 NPs following drug loading protocol using ethanol as the solvent (DLC =1.5%). Female BALB/c nude mice were used to prepare MDA-MB-231 tumor-bearing xenograft mouse model. For in vivo imaging analysis, DiR-labeled NPs (DiR-NPs) were systemically administered to tumor-bearing mice through tail vein at a dose of DiR 1 mg/kg. At predetermined time points, the mice were anesthetized by intraperitoneal injection of 10% chloral (3 μL/g) and near infrared fluorescence images were acquired on a Kodak FX-pro imaging station (Kodak, Rochester, NY, USA). Excitation and emission filters were set at λ720 and λ790 nm, respectively. Twenty-four hours after injection, the mice were sacrificed by cervical dislocation and ex vivo imaging was performed on the excised tumors and organs.
In vivo antitumor activity
The in vivo antitumor activity of the DOX-NPs was evaluated in the subcutaneous MDA-MB-231 tumor-bearing xenograft mouse model. When the tumor volume reached approximately 100 mm3, the mice were randomly divided into five groups (n=5 for each group): saline group, free DOX group, HA-ss-Chol7 DOX-NPs group, GE11–HA-Chol7 DOX-NPs group, and GE11–HA-ss-Chol7 DOX-NPs group. Each mouse received a dose of 4 mg/kg DOX by tail vein injection every 3 days. The tumor sizes and body weights were measured for 21 days. Tumor sizes were calculated according to the equation: V = a × b2/2, where V (mm3) represents the tumor volume and a (mm) and b (mm) represent the tumor length and width, respectively. Mice were sacrificed on the 21st day after administration, and the tumors were weighed with the inhibition ratio (IR) calculated as follows: IR (%) = [(x − y)/x] ×100%, where x and y stand for the average tumor weight for the control group and treatment group, respectively.
Statistical analysis
Statistical analysis was performed by a Student’s t-test for pairwise comparisons and one-way analysis of variance for multiple groups. All results were expressed as mean ± standard deviation (SD) unless otherwise noted. P<0.05 was considered statistically significant.
Results
Synthesis and characterization of reduction-sensitive and -insensitive conjugates
The synthetic routes of the GE11–HA-ss-Chol are briefly summarized in Figure 2. The synthesis process included three steps. First, the amino group of cystamine reacted with the acyl chloride moiety of cholesteryl chloroformate to form the cholesteryl-amine linkers (Chol–cys or Chol–eda). Second, in order to prepare the amphiphilic conjugates, the purified hydrophobic cholesteryl-amine linkers were conjugated to HA’s carboxyl groups by carbodiimide chemistry. Third, the N-terminus of the peptide GE11 was attached to HA’s unmodified carboxyl groups. Same procedures were employed for the synthesis of GE11–HA-Chol while replacing cystamine with ethylenediamine in the first step to obtain the reduction-insensitive linker of Chol–eda. The synthesis scheme for GE11–HA-Chol is shown in Figure S2.
 | Figure 2 Schematic procedure of the synthesis of GE11–HA-ss-Chol with catalyst of (i) TEA/DCM, (ii) EDC/HOBT, (iii) EDC/sulfo-NHS. |
Figure 3 shows the 1H-NMR spectra of HA-Chol, HA-ss-Chol, and GE11–HA-ss-Chol conjugates. The characteristic peaks of HA (Figure 3A–C) appeared at 1.99 ppm (methyl proton of −NCOCH3 group) and 3.31–4.54 ppm (methylene and hydroxyl groups). The peaks in HA-ss-Chol spectra (Figure 3B) and GE11–HA-ss-Chol spectra (Figure 3C) at 0.66–1.50 ppm, 2.82 ppm, and 3.11 ppm were ascribed to typical cholesteryl groups (angular methyl protons) and cystamine (CH2S and CH2N), respectively, indicating that the Chol–cys was successfully linked to the carboxyl group of HA. Effective grafting of Chol–eda on HA backbone was confirmed by the peaks of cholesteryl protons at 0.66–1.50 ppm and ethylenediamine spacer at 2.85–3.12 ppm, as shown in Figure 3A. Furthermore, the specific signals of benzene on GE11 peptide at 6.7–7.6 ppm revealed that the average number of GE11 on HA-ss-Chol was 1.14 per 100 sugar units of HA.
 | Figure 3 1H-NMR spectra of HA-Chol conjugate (A), HA-ss-Chol conjugate (B), and GE11–HA-ss-Chol conjugate (C). |
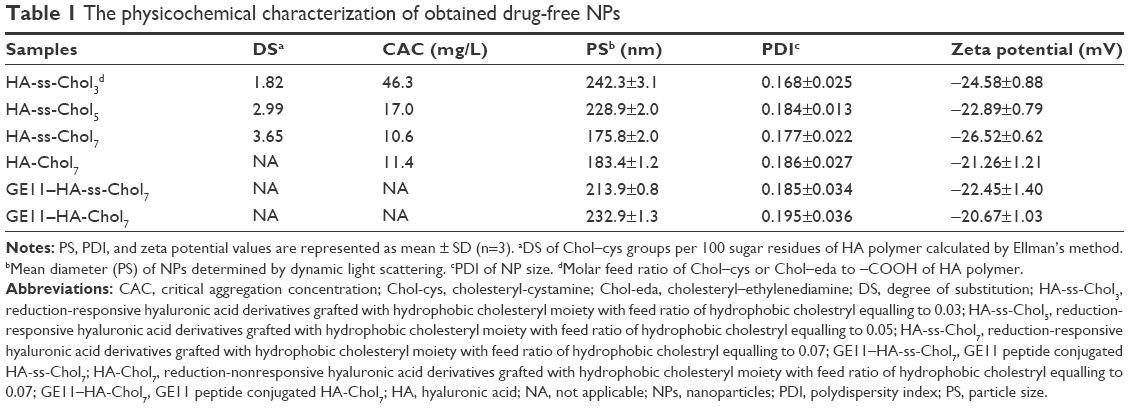
Ellman’s method was applied to determine the amount of Chol–cys in the conjugates by quantitatively analyzing the disulfide bonds between the hydrophilic HA main chain and the hydrophobic cholesteryl group. The cholesteryl group’s DS values are summarized in Table 1. As expected, the DS augmented as the molar feed ratio of Chol–cys to the sugar units in HA increased.
 | Table 1 The physicochemical characterization of obtained drug-free NPs |
HPLC was used to determine the DS and conjugation efficiency of GE11 peptide in conjugates. The retention time of GE11 peptide was approximately 8.2 minutes, while HA-ss-Chol did not show distinct characteristic absorption peak under similar conditions (Figure S3, curve a). According to the integration ratio of the GE11 peptide peak before and after reaction (Figure S3, curves b and c), the DS of GE11 in GE11–HA-ss-Chol was 1.35% and the conjugation efficiency was approximately 59.5%.
Preparation and characterization of blank NPs
The synthesized conjugates self-assembled into core/shell structured NPs after ultrasonication in aqueous media. Physicochemical properties of blank NPs are listed in Table 1. DLS measurements revealed a polydispersity index inferior to 0.2, indicating a narrow size distribution of the NPs. The mean vesicle size of HA-ss-Chol declined from 242 to 175 nm, as the DS of Chol–cys increased. The NPs were negatively charged with a zeta potential inferior to −20 mV. In addition, HA-Chol7 NPs presented a slightly increased PS compared to HA-ss-Chol7 NPs. Modification of the NPs with GE11 peptide resulted in an enlargement of PS and a minor variation of zeta potential. CAC values of HA-ss-Chol conjugates decreased from 46.3 to 10.6 mg/L, as the feed ratio of hydrophobic segment (Chol–cys) increased from 0.03 to 0.07 (Table 1). CAC value of HA-Chol7 conjugate (11.4 mg/L) was nearly as low as that of HA-ss-Chol7 conjugate.
The morphology and size distribution of HA-ss-Chol7 and GE11–HA-ss-Chol7 NPs were visualized using TEM. As shown in Figure 4, the NPs exhibited a spherical shape with narrow distribution. Moreover, the size estimated from TEM was smaller than that obtained from DLS in an aqueous phase, which might be attributed to the shrinkage of hydrophilic shell during the air-drying process in TEM sample preparation and the system error resulting from the determination principles between DLS (hydrated radius) and TEM.
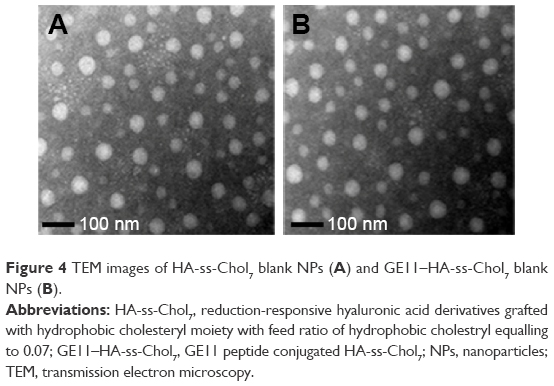 | Figure 4 TEM images of HA-ss-Chol7 blank NPs (A) and GE11–HA-ss-Chol7 blank NPs (B). |
The redox-induced degradation of HA-ss-Chol7 NPs was determined by monitoring the variation in size and size distribution in the presence of various concentrations of DTT using DLS. As shown in Figure 5, the size change of HA-ss-Chol7 is negligible (<20 nm) when exposed for 24 hours to 10 μM DTT which is similar to the bloodstream environment.44 Similar behavior was observed in the absence of DTT under otherwise the same conditions. Nevertheless, the presence of 10 and 20 mM DTT, mimicking the intracellular level of normal tissues44 and tumor cells,45 respectively, induced a sharp increase in size of HA-ss-Chol7 NPs from 177.8 to 374.7 nm and to 402.3 nm, respectively, at 24 hours. Also, a simultaneous broadening of HA-ss-Chol7 NPs size distribution was recorded (data not shown). In the control study, reduction-insensitive HA-Chol7 NPs were constant in size when exposed to 20 mM DTT.
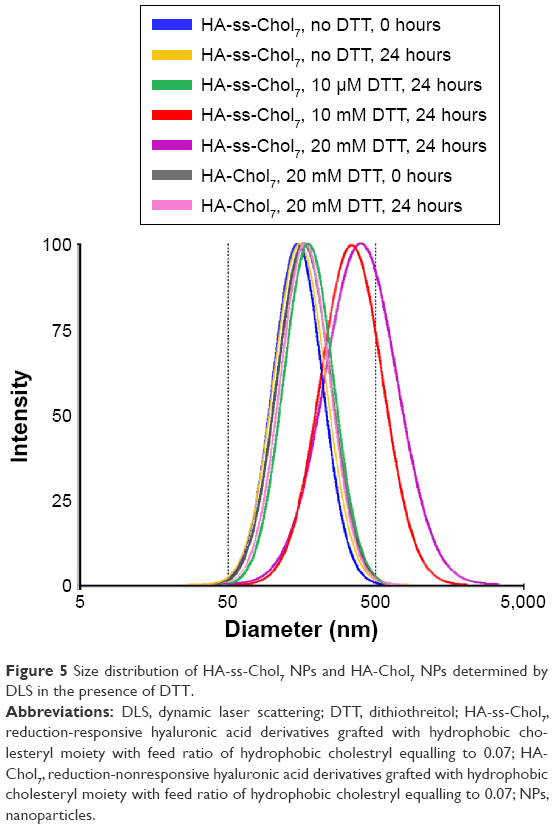 | Figure 5 Size distribution of HA-ss-Chol7 NPs and HA-Chol7 NPs determined by DLS in the presence of DTT. |
The results of redox-induced degradation assay of HA-ss-Chol7 NPs showed that HA-ss-Chol7 NPs treated with 10 μM DTT maintained their initial structure in the environment mimicking that of bloodstream. Conversely, when the HA-ss-Chol7 NPs were exposed to 20 mM DTT analogous to the tumor cell, the dramatic size swelling with a broadening of size distribution indicated rapid disassembly due to cleavage of the disulfide linkages between the hydrophilic groups and the hydrophobic groups.
Preparation and characterization of DOX-NPs
DOX was physically incorporated into NPs using a dialysis method. The characteristics of DOX-NPs are listed in Table 2. Both HA-ss-Chol7 and HA-Chol7 NPs showed considerable loading capacities for the hydrophobic model drug DOX. The DLC and EE of HA-ss-Chol7 DOX-NPs were in the range of 27.4%–33.6% and 67.8%–91.2%, respectively. The sizes of DOX-NPs were smaller than those of their corresponding drug-free samples. Zeta potential values (approximately −20 mV) remained essentially unchanged, compared to the blank NPs. HA-Chol7 DOX-NPs showed similar properties (PS, DLC, and EE) as those of HA-ss-Chol7 DOX-NPs. Moreover, modification with GE11 had a small influence on the DLC and EE of the corresponding NPs (DLC: 30.5%, EE: 78.8%).
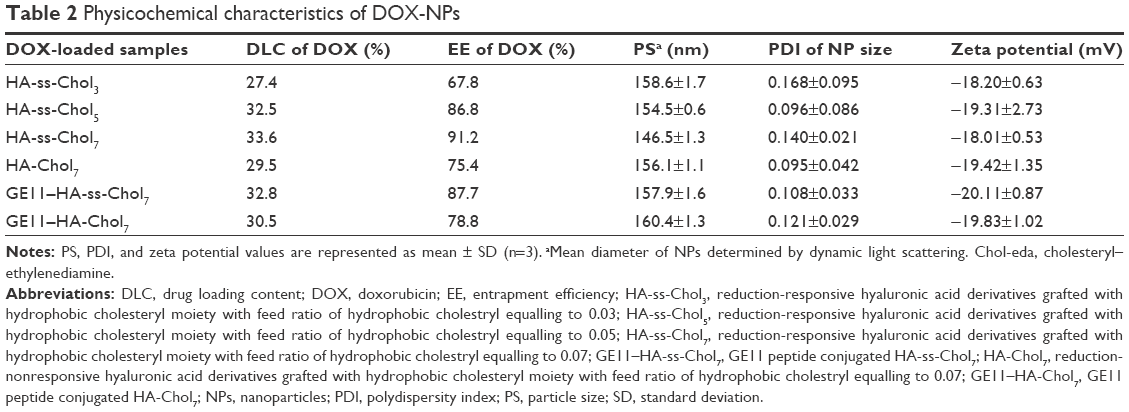 | Table 2 Physicochemical characteristics of DOX-NPs |
In vitro drug release
The in vitro drug release profile of HA-ss-Chol7 DOX-NPs was carried out in PBS (pH 7.4) containing different concentrations of DTT. As illustrated in Figure 6, the release behavior of HA-ss-Chol7 DOX-NPs sensitively depend on the concentration of DTT. In the presence of 10 μM DTT mimicking the extracellular environment, only 34.97% of DOX was released from the NPs within 48 hours. However, as the DTT concentration reached 10 mM, the DOX release curve dramatically showed an increase to 76.29% after 48 hours, representing 2.2-fold over that with 10 μM DTT. Moreover, at a higher DTT concentration (20 mM), corresponding to the intracellular reduction levels in tumor cells, 40.08% of DOX was released in the first 4 hours and 79.67% was released after 48 hours. In contrast, HA-Chol7 NPs slowly released the drug in the medium containing 20 mM DTT (35.05% over 48 hours), which had no significant difference with the release of DOX from HA-ss-Chol7 NPs in 0 M DTT (31.37% over 48 hours) or 10 μM DTT (34.97% over 48 hours).
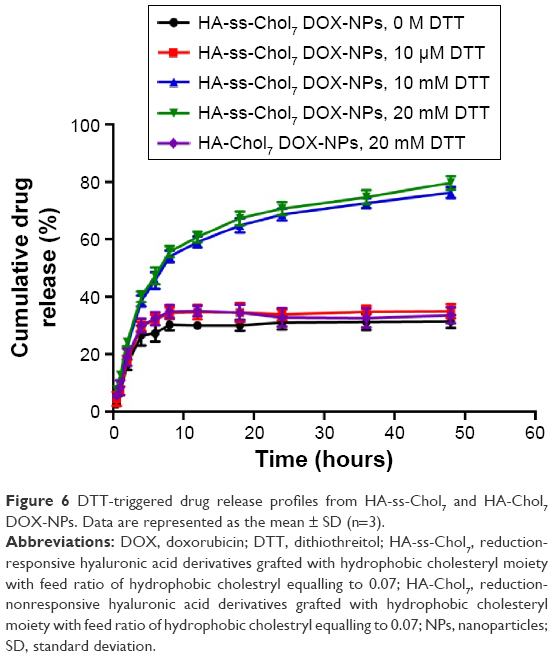 | Figure 6 DTT-triggered drug release profiles from HA-ss-Chol7 and HA-Chol7 DOX-NPs. Data are represented as the mean ± SD (n=3). |
In vitro cellular uptake study
The cellular uptake efficiency of GE11–HA-ss-Chol7 C6-NPs and HA-ss-Chol7 C6-NPs was quantitatively evaluated by flow cytometry, as illustrated in Figure 7. For MDA-MB-231 and MCF-7 cells, after 4 hours incubation, the mean fluorescence intensity (MFI) of cells treated with GE11–HA-ss-Chol7 C6-NPs was 1.43-fold and 1.13-fold of those treated with HA-ss-Chol7 C6-NPs, respectively. Moreover, it showed a distinct difference in the cellular uptake performance of GE11–HA-ss-Chol7 C6-NPs between MDA-MB-231 and MCF-7 cells, wherein the cellular level of MFI of the former cells was over 2.28-fold of the latter, indicating a stronger NP internalization capability of MDA-MB-231 cells compared to MCF-7 cells.
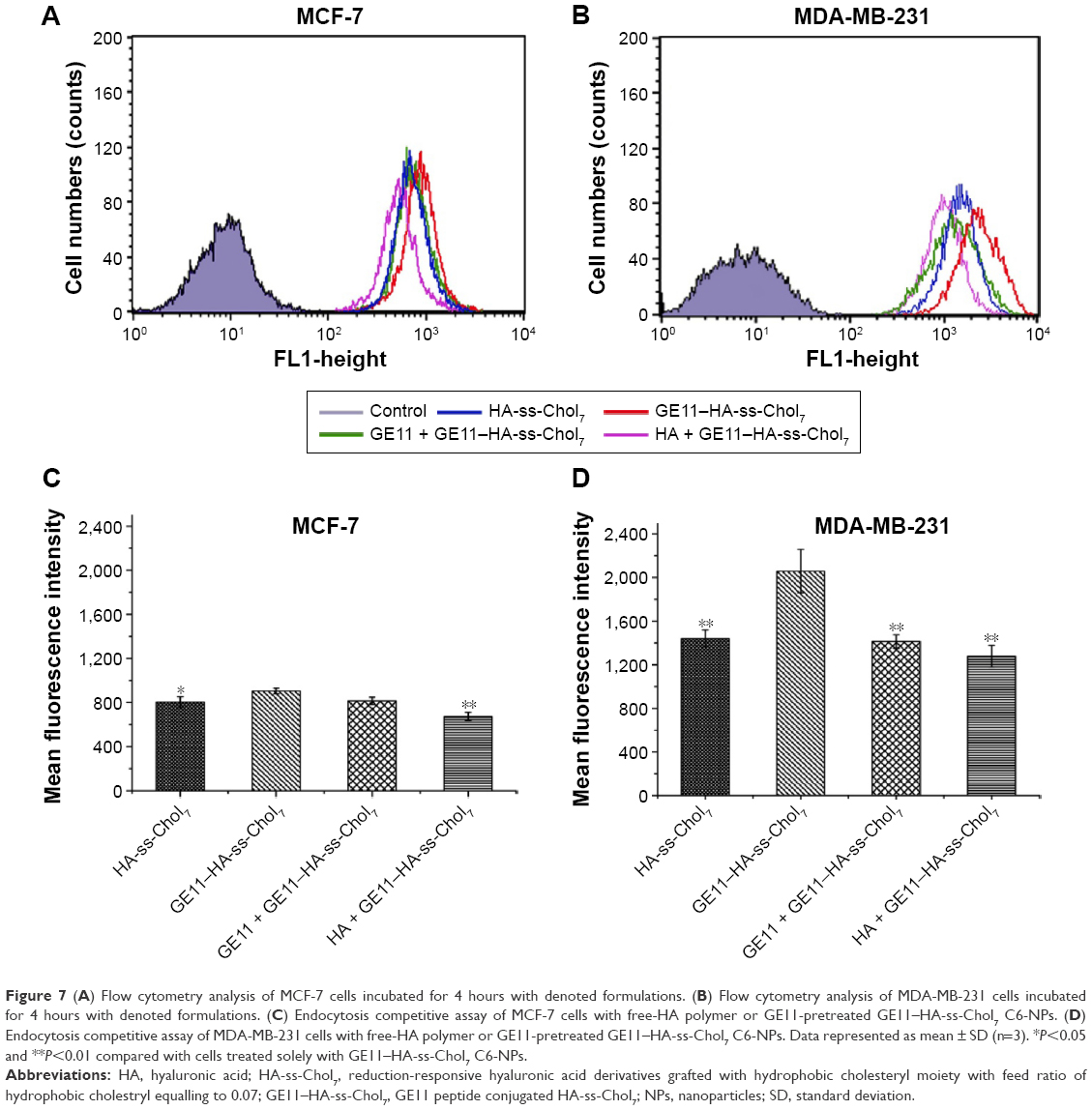 | Figure 7 (A) Flow cytometry analysis of MCF-7 cells incubated for 4 hours with denoted formulations. (B) Flow cytometry analysis of MDA-MB-231 cells incubated for 4 hours with denoted formulations. (C) Endocytosis competitive assay of MCF-7 cells with free-HA polymer or GE11-pretreated GE11–HA-ss-Chol7 C6-NPs. (D) Endocytosis competitive assay of MDA-MB-231 cells with free-HA polymer or GE11-pretreated GE11–HA-ss-Chol7 C6-NPs. Data represented as mean ± SD (n=3). *P<0.05 and **P<0.01 compared with cells treated solely with GE11–HA-ss-Chol7 C6-NPs. |
To further confirm whether GE11–HA-ss-Chol7 NPs were internalized by CD44- and/or EGFR-mediated endocytosis, competitive inhibitory experiments were performed. The MFI of cells pretreated with free HA or GE11 was inferior to that of untreated ones when incubated with GE11–HA-ss-Chol7 NPs. Specifically, a 40% and 30% decline in the fluorescence of MDA-MB-231 cells incubated with GE11–HA-ss-Chol7 NPs was observed in the presence of HA and GE11, respectively (Figure 7C). Meanwhile, pretreatment of MCF-7 cells with free HA or GE11, as illustrated in Figure 7D, resulted in a fluorescence decrease of 25% and 10%, respectively. Therefore, saturation of CD44 and EGFR leads to a less efficient cellular internalization of GE11–HA-ss-Chol7 NPs.
To further probe the cellular uptake mechanism of GE11–HA-ss-Chol7 NPs, the effect of several endocytosis inhibitors was evaluated. Treatment of MCF-7 cells with the caveolae-mediated endocytosis inhibitor nystatin did not significantly change the cellular uptake of GE11–HA-ss-Chol7 NPs (Figure S4). However, clathrin-mediated endocytosis pathway inhibitor, CPZ, and macropinocytosis pathway inhibitor, amiloride, reduced the cellular uptake by approximately 30% and 20%, respectively. The same results were obtained in MDA-MB-231 cells, except that the influence of CPZ to the NPs uptake efficiency was more critical, showing a 50% inhibition rate (Figure S4).
Intracellular drug distribution of DOX by confocal microscopy
As shown in Figure 8, confocal microscopy was applied to investigate the intracellular distribution of DOX. After 1 hour of incubation with GE11–HA-ss-Chol7 and GE11–HA-Chol7 DOX-NPs, weak fluorescence signals were detected in the cytoplasm closely around the nuclei in both MCF-7 (Figure 8a and b) and MDA-MB-231 cells (Figure 8e and f). The DOX fluorescence signals became much stronger as the incubation time increased to 4 hours (Figure 8c, d, g, and h). Interestingly, the DOX fluorescence of both cells tended to locate in nuclei rather than cytoplasm, when incubated with reduction-responsive GE11–HA-ss-Chol7 DOX-NPs. In contrast, cells treated with reduction-nonresponsive NPs exhibited a selective distribution in the cytoplasm. Furthermore, a markedly higher fluorescence was found in MDA-MB-231 cells compared to MCF-7 cells.
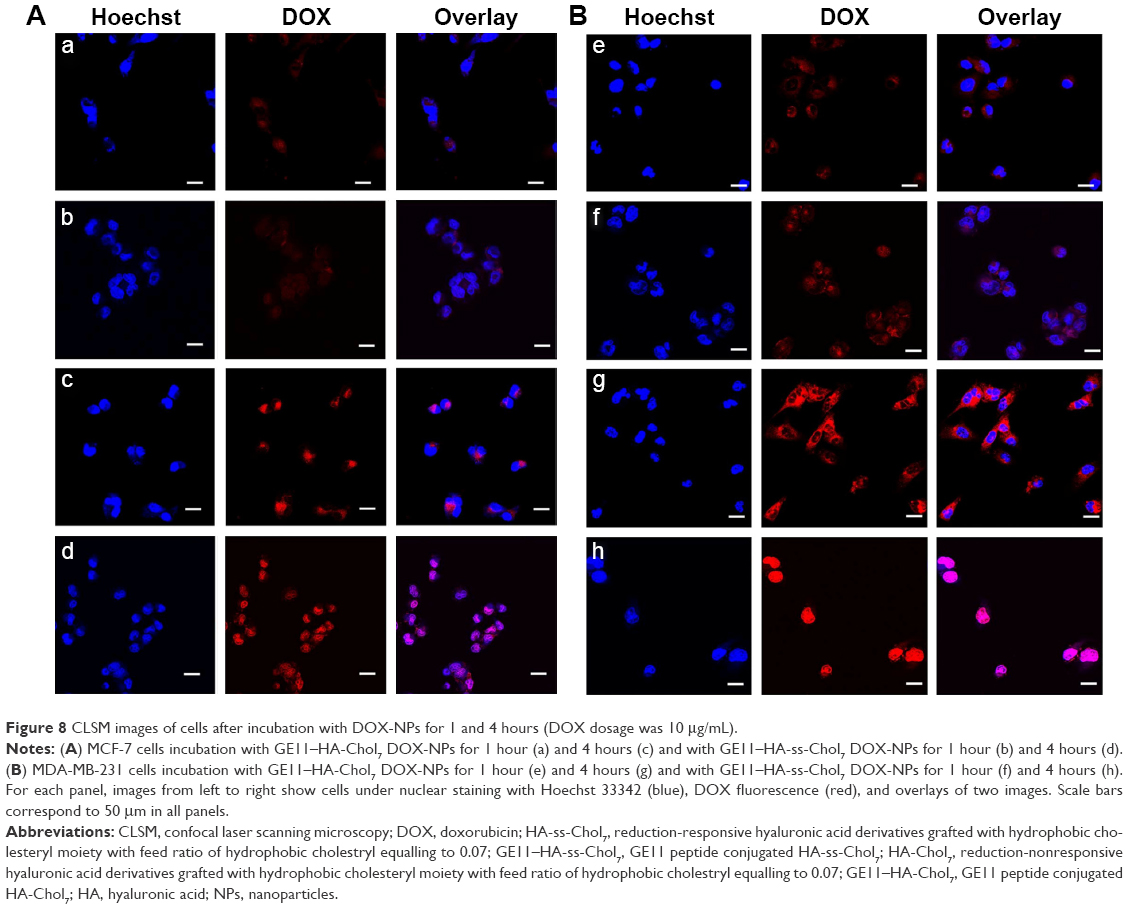 | Figure 8 CLSM images of cells after incubation with DOX-NPs for 1 and 4 hours (DOX dosage was 10 μg/mL). |
In vitro cytotoxicity assay
The antiproliferative effect of different DOX formulations against MDA-MB-231 and MCF-7 cells was evaluated using MTT assay. Preliminary evaluation of GE11–HA-ss-Chol7, GE11–HA-Chol7, and HA-ss-Chol7 blank NPs biocompatibility showed negligible cytotoxicity. After 24 hours treatment with drug-free NPs (10–500 μg/mL), the cell viability was above 90% (Figure S5).
As presented in Figure 9, the inhibition of cell proliferation by free DOX and DOX-NPs showed a typical dose-dependent behavior. The IC50 values of free DOX and DOX-NPs are summarized in Table 3. For MDA-MB-231 cells, GE11–HA-ss-Chol7 DOX-NPs exhibited the lowest IC50. Specifically, the IC50 values of free DOX, HA-ss-Chol7 DOX-NPs, and GE11–HA-Chol7 DOX-NPs were 1.25-, 2.40-, and 3.35-fold over that of GE11–HA-ss-Chol7 DOX-NPs.
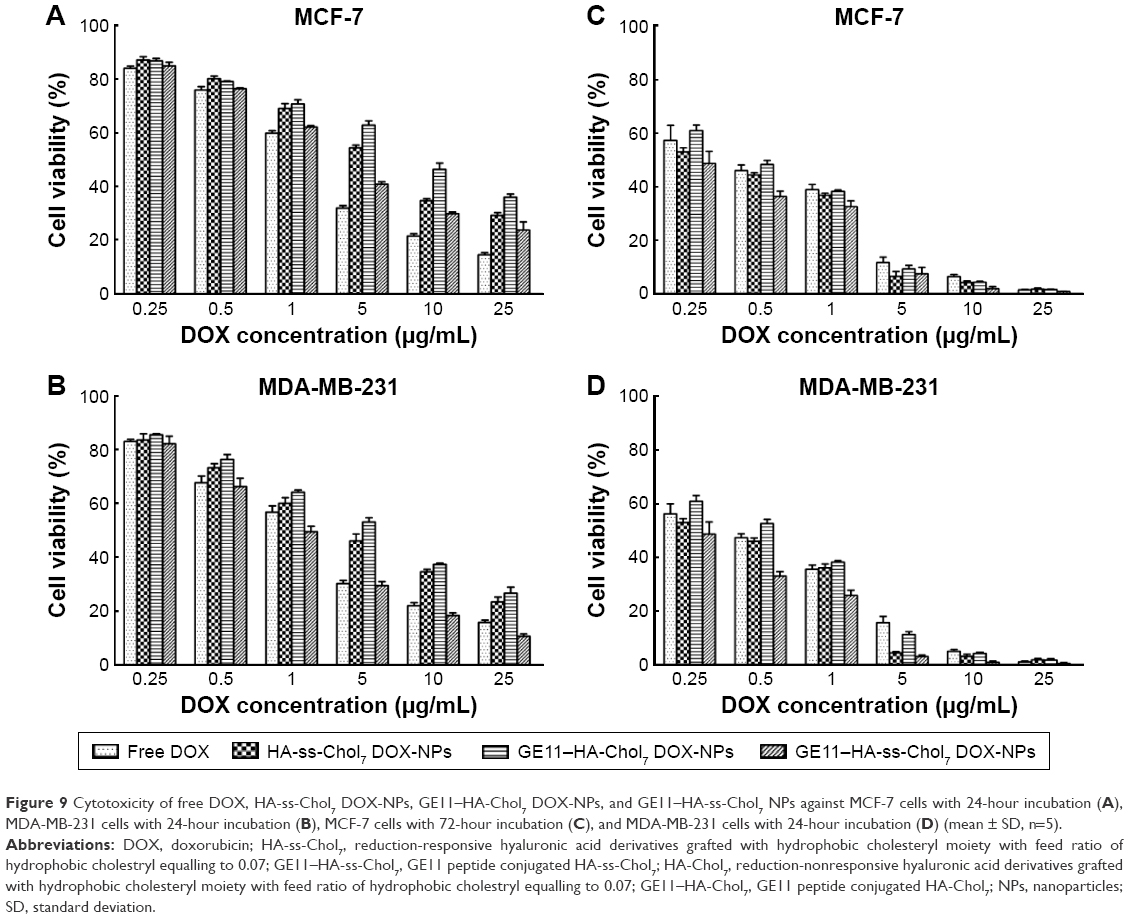 | Figure 9 Cytotoxicity of free DOX, HA-ss-Chol7 DOX-NPs, GE11–HA-Chol7 DOX-NPs, and GE11–HA-ss-Chol7 NPs against MCF-7 cells with 24-hour incubation (A), MDA-MB-231 cells with 24-hour incubation (B), MCF-7 cells with 72-hour incubation (C), and MDA-MB-231 cells with 24-hour incubation (D) (mean ± SD, n=5). |
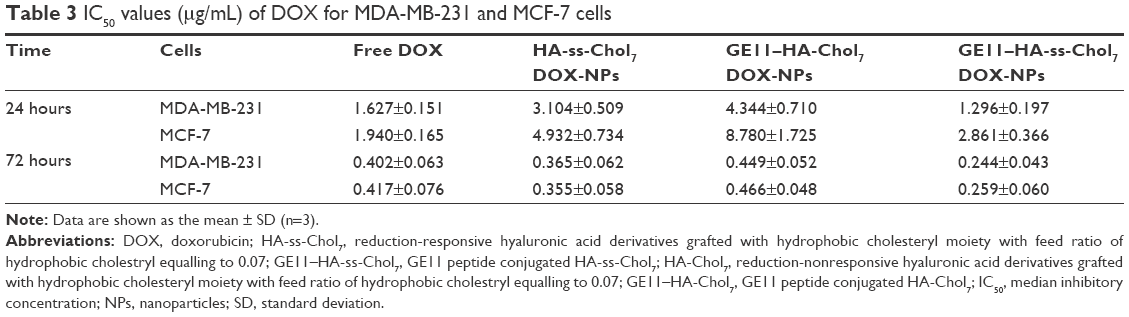 | Table 3 IC50 values (μg/mL) of DOX for MDA-MB-231 and MCF-7 cells |
MCF-7 cells, expressing CD44 and EGFR in an inferior grade, were also used for cytotoxicity assay. It was found that the IC50 values of HA-ss-Chol7 NPs and GE11–HA-Chol7 NPs were 1.72- and 3.07-fold over that of GE11–HA-ss-Chol7 DOX-NPs, while free DOX group showed the most efficient cytotoxicity.
However, at longer incubation time of 72 hours, all the drug groups exhibited stronger cytotoxicity, as shown in Table 3, with IC50 values far lower than those at 24 hours. The reason may be that longer time of incubation would lead to stronger interaction between the drug and cells.
In vivo and ex vivo imaging analysis
The tumor-targeting abilities of HA-ss-Chol7 and GE11–HA-ss-Chol7 NPs were assessed in MDA-MB-231 breast tumor xenograft mouse model. Time-dependent biodistribution of DiR-NPs and free DiR was visualized after intravenous administration using a noninvasive NIR imaging technique. As represented in Figure 10A, GE11–HA-ss-Chol7 DiR-NPs exhibited the strongest fluorescence signal at the tumor site, followed by HA-ss-Chol7 DiR-NPs and free DiR. Moreover, the fluorescence detected in free DiR group faded away rapidly, while it was sustained at the tumor location in DiR-NPs groups at all the time points postinjection. These observations suggest a tumor-specific accumulation of the NPs, particularly GE11–HA-ss-Chol7 DiR-NPs.
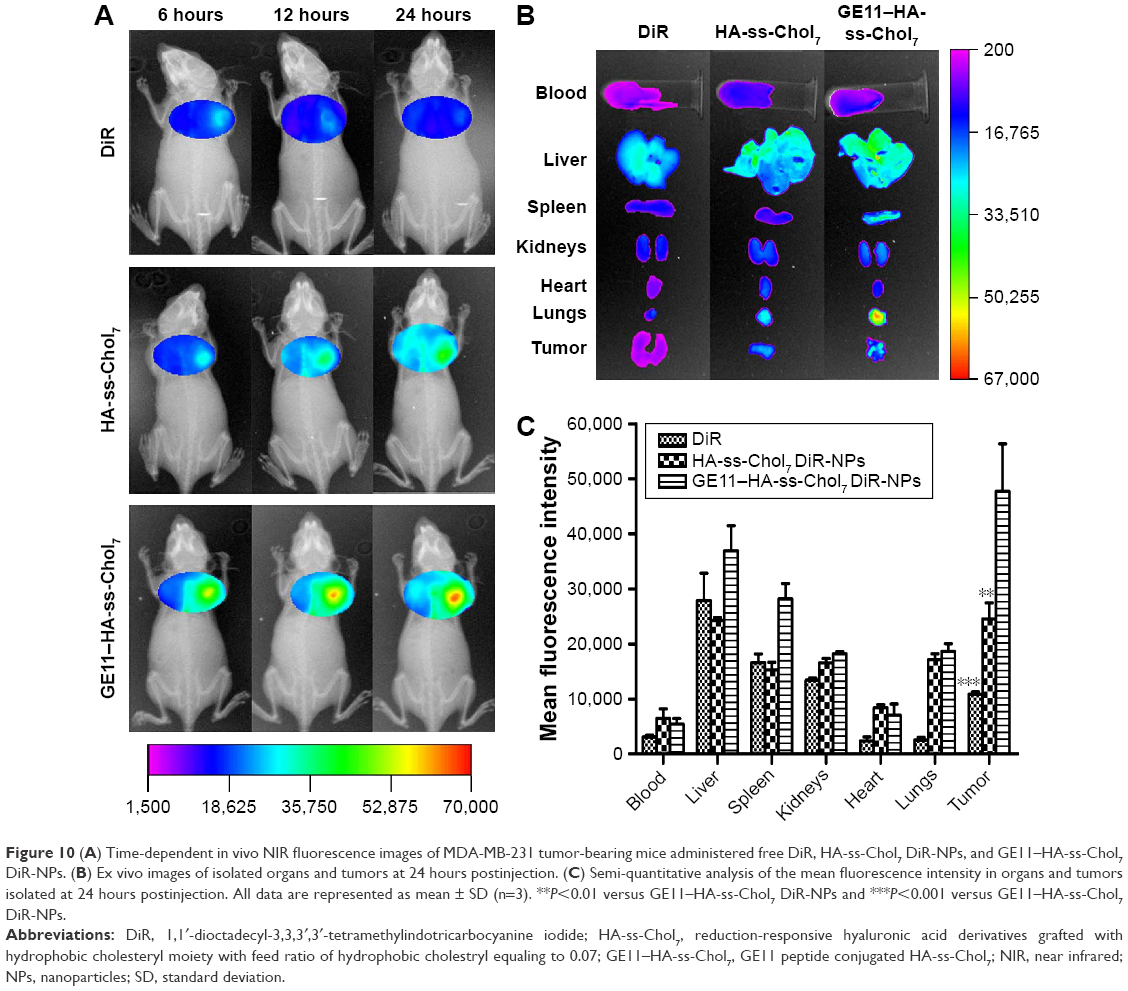 | Figure 10 (A) Time-dependent in vivo NIR fluorescence images of MDA-MB-231 tumor-bearing mice administered free DiR, HA-ss-Chol7 DiR-NPs, and GE11–HA-ss-Chol7 DiR-NPs. (B) Ex vivo images of isolated organs and tumors at 24 hours postinjection. (C) Semi-quantitative analysis of the mean fluorescence intensity in organs and tumors isolated at 24 hours postinjection. All data are represented as mean ± SD (n=3). **P<0.01 versus GE11–HA-ss-Chol7 DiR-NPs and ***P<0.001 versus GE11–HA-ss-Chol7 DiR-NPs. |
Twenty-four hours postadministration, ex vivo imaging was performed. Figure 10B shows the fluorescent images of the excised tumors and major organs of the sacrificed mice. GE11–HA-ss-Chol7 DiR-NPs accumulated in tumors more efficiently than HA-ss-Chol7 DiR-NPs, suggesting an enhanced tumor-specific targeting ability of the former vector. The lowest fluorescent signal in tumor was detected in free DiR group, and the accumulation in livers and spleen was higher than in other organs for free DiR group. The semiquantitative analysis of the fluorescent images (Figure 10C) was in good agreement with ex vivo imaging results. GE11–HA-ss-Chol7 DiR-NPs showed over 1.9-fold and 4.3-fold higher fluorescence intensity at the tumor than HA-ss-Chol7 DiR-NPs and free DiR, respectively.
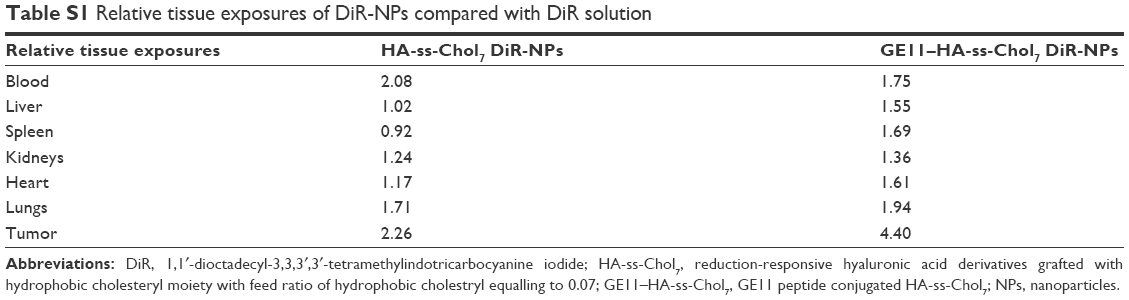
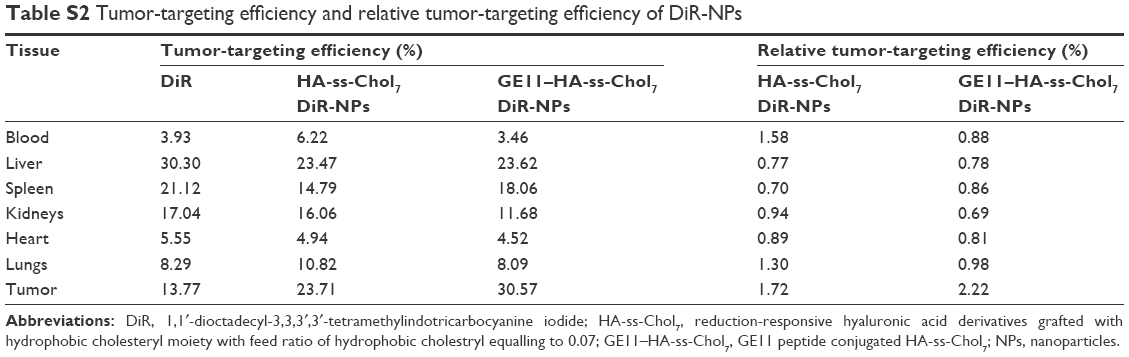
The targeting abilities of NPs were also analyzed by relative tissue exposure (Re), tumor targeting efficiency, and relative tumor targeting efficiency (RTe), as shown in Tables S1 and S2. All these data indicate the dual-targeted GE11–HA-ss-Chol7 NPs exhibited better targeting ability when compared with HA-ss-Chol7 NPs or free drug solutions.
In vivo antitumor assay
To provide in vivo evidence for the antitumor potential of HA-based DOX-NPs, the antitumor efficacy was evaluated using an MDA-MB-231 tumor-bearing mouse model. As shown in Figure 11A, compared to the saline solution group (negative control), the four treatment groups showed distinct tumor growth inhibition efficiency. Specifically, the group treated with free DOX, HA-ss-Chol7 DOX-NPs, and GE11–HA-Chol7 DOX-NPs showed smaller tumor volumes than the saline control group (P<0.05). Moreover, GE11–HA-ss-Chol7 DOX-NPs displayed a marked reduction in tumor size, when compared with other groups. The final tumor weight shown in Figure 11B was in good agreement with tumor volume assay. The results revealed that GE11–HA-ss-Chol7 DOX-NPs inhibited the tumor growth most efficiently and the IR calculated by tumor weight was 86.03%; they were followed by free DOX (76.41%), GE11–HA-Chol7 DOX-NPs (66.46%), and HA-ss-Chol7 DOX-NPs (35.03%). The body weight investigation was applied to evaluate systemic toxicity in mice. Compared with the saline group, the mice in free DOX group showed a significant decrease in body weight, while no notable change was found in HA-based DOX-NPs groups (Figure 11C).
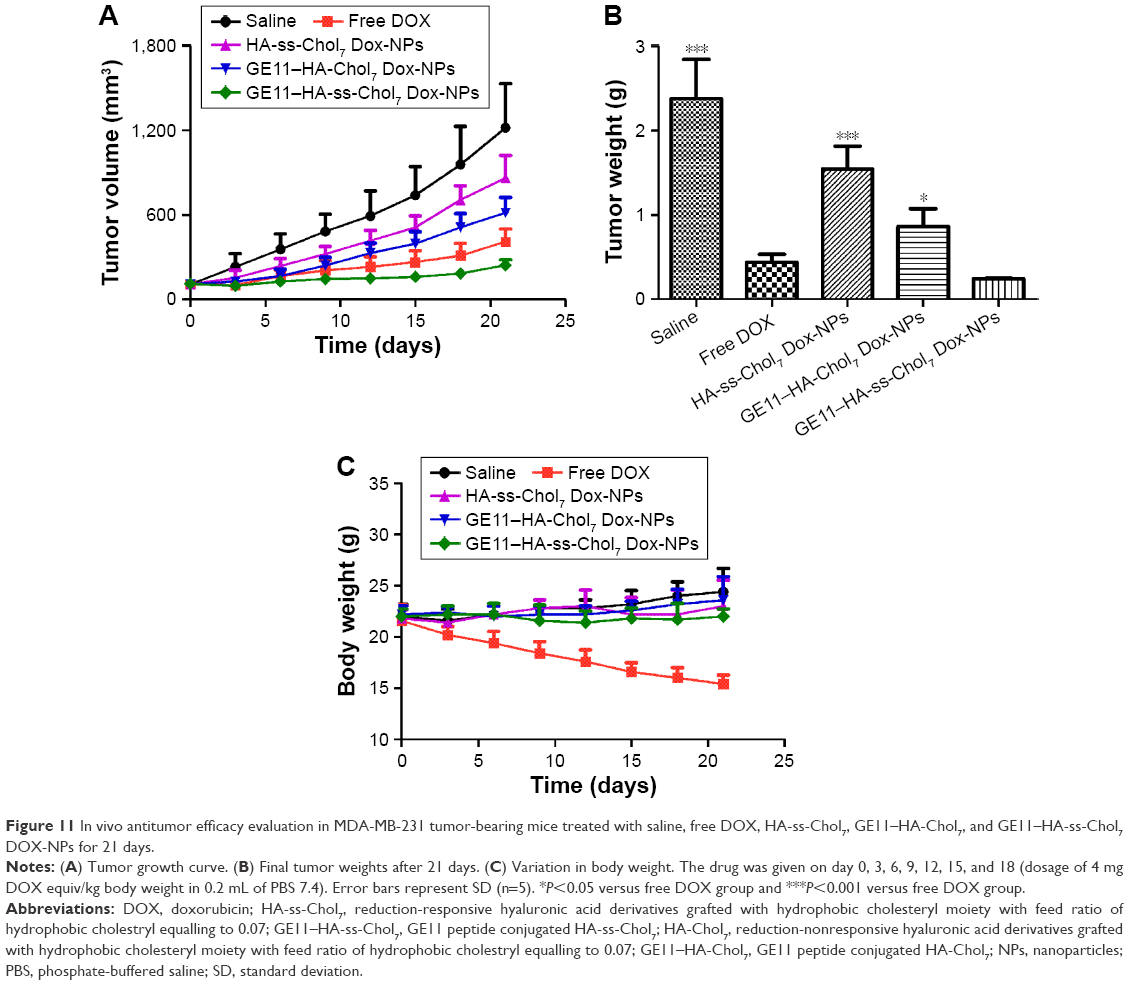 | Figure 11 In vivo antitumor efficacy evaluation in MDA-MB-231 tumor-bearing mice treated with saline, free DOX, HA-ss-Chol7, GE11–HA-Chol7, and GE11–HA-ss-Chol7 DOX-NPs for 21 days. |
Discussion
A well-designed anticancer drug delivery carrier should meet the following requirements: 1) it should be nontoxic and nonimmunogenic of the carrier itself; 2) maintenance of intact structure after dilution in the bloodstream, thereby limiting systemic side effects; 3) enhanced absorption of the drugs into the solid tumors either by passive or active targeting; and 4) acceleration of therapeutics’ burst release at the tumor site.46,47 Driven by the aims given earlier, the design of an “all-in-one” system that allows simultaneous tumor active targeting and stimuli-responsive drug release is of particular interest.
In this study, GE11-modified, HA-based NPs were successfully prepared by synthesizing amphiphilic conjugates of HA and cholesteryl chloroformate, connected by a disulfide bridge, and then modifying GE11 peptide on the surface. Such a vehicle was supposed to have the ability to stay intact in blood circulation, target the breast cancer cells by CD44 and EGFR, and accelerate intracellular drug release by redox-responsive disassembling of the whole structure.
In general, an amphiphilic polymer can form a core/shell structure in water. The synthesized conjugate spontaneously self-assembles into NPs in aqueous solution, which is driven by the strong hydrophobic/hydrophilic interaction. It is believed that the physicochemical properties of NPs play an important role in their pharmaceutical application. It was demonstrated that the degree of modification of NPs with cholesteryl groups must range from 3% to 10% to ensure good solubility and effective cellular uptake.48 The PS of HA-ss-Chol NPs decreased with increase in the DS of cholesterol group, suggesting that higher hydrophobicity induces easier formation of NPs and more compact interaction in hydrophobic inner core. According to the previous reports, NPs with size approximately 100–200 nm were likely to exhibit long blood circulation effect by avoiding the recognition of reticuloendothelial system (RES) and accumulate in tumors by “leaky” vasculature through enhanced permeability and retention (EPR) effect.49,50 The size of the above-studied HA-based carriers was within the proper range of NPs. Zeta potential is a significant parameter to evaluate the stability of colloidal dispersion. The zeta potential of HA-based NPs was negatively charged (<−20 mV), which is attributed to the deprotonated carboxylic groups of HA backbone embracing the NPs’ surface. Such negatively charged surface permits long-term stability of NPs in the blood circulation, avoiding severe cytotoxicity and rapid clearance from the plasma compartment.51,52 DLS and TEM measurements revealed the influence of GE11 modification on NPs in size, zeta potential, and morphology, which was negligible despite the fact that the diameter increased slightly in DLS. Compared with the traditional low MW surfactants, the CAC value is much lower to guarantee the preservation of NPs’ stability and structural integrity under extensively dilute conditions in the systemic circulation before reaching the targeting site.
The results of redox-induced degradation assay of HA-ss-Chol7 NPs showed that HA-ss-Chol7 NPs under 10 μM DTT treatment maintained their initial structure in the reductive environment mimicking that of bloodstream. Conversely, when the HA-ss-Chol7 NPs were exposed to 20 mM DTT analogous to the tumor cell, the dramatic size swelling with a broadening of size distribution indicated rapid disassembly due to the cleavage of the disulfide linkages between the hydrophilic groups and the hydrophobic groups. Moreover, the steady size of HA-Chol7 NPs to 20 mM DTT was possibly on account of their nonresponsiveness to the reductive agent owing to the lack of disulfide bond in the NPs’ structure. The DLS results confirmed the redox-sensitive property of HA-ss-Chol7 NPs.
The self-assembled NPs formed bore a hydrophobic inner core that could serve as a desired container for hydrophobic drugs. The HA-based NPs could be obtained with a high DLC of 27.4%–33.6% in this work. The sizes of DOX-loaded NPs were smaller than their corresponding drug-free samples, indicating that the joining of hydrophobic drug DOX made the inner core more compact by the strong interaction between DOX and the hydrophobic region. Also, ionic interactions between DOX base and carboxylic groups of HA might increase the entrapment capability of the NPs.53 Zeta potential determination values (approximately −20 mV) suggested that the existence of ionic interactions between the carboxylic moieties of HA and the amine groups of DOX, and the addition of DOX did not reduce the NPs’ stabilities significantly.
From the in vitro release profile in different media, it was found that the drug release from designed redox-responsive HA-ss-Chol7 DOX-NPs depended on the concentration of DTT, and the release rate increased as the amount of DTT in the media increased. Similar fast release found at the initial 4 hours might be ascribed to the drug located in the hydrophilic shell or adhered to the surface of the NPs.54 Then the remaining DOX, probably loaded deep in the hydrophobic cores, presented a different release profile in different DTT concentrations. In the presence of 10 μM DTT, the less-efficient release during the following 44 hours suggested that the insoluble DOX was difficult to be released from the inner core as reported before,55 which met the demand that the integrity of vehicle was maintained well and the loss of drug minimized before reaching the target cells. The dramatic fast drug release that occurred when the HA-ss-Chol7 DOX-NPs were exposed to 10 or 20 mM DTT was probably attributed to the DTT-induced degradation of disulfide linkage, which is in line with the size swelling observed in the DLS assay. Therefore, exactly as the aim of designed, the reduction-sensitive NPs are likely to be stable in the bloodstream after intravenously administered, whereas release the encapsulated drug rapidly after accumulating in the tumor sites.
The cellular uptake study showed that both MDA-MB-231 and MCF-7 cells took in more dual-targeting GE11–HA-ss-Chol7 C6-NPs than single-targeting HA-ss-Chol7 C6-NPs. Moreover, the NPs’ internalization capability of MDA-MB-231 cells was stronger than that of MCF-7 cells. It has been demonstrated that the CD44 and EGFR are highly overexpressed in MDA-MB-231 cells compared with those in MCF-7 cells.39,40 We further proved that free HA and GE11 peptide suppressed the uptake of GE11–HA-ss-Chol7 C6-NPs, and the specificity of uptake was verified by competitive inhibitory studies. Therefore, it could be concluded that the cellular uptake of GE11–HA-ss-Chol7 C6-NPs involves the interaction with CD44 and EGFR in both cells, suggesting the existence of a dual-ligand tumor-targeting mechanism. Then, the uptake mechanism study indicated that internalization of the investigated NPs mainly involved the clathrin-mediated pathway and macropinocytosis.
CLSM results showed that after incubation for 1 hour, both studied NPs did not release the small amount of DOX taken up. The different intracellular release behaviors between GE11–HA-ss-Chol7 and GE11–HA-Chol7 DOX-NPs during 4 hours incubation are most likely due to fast DOX release from redox-responsive GE11–HA-ss-Chol7 NPs. The uptake of DOX by the cell nuclei is crucial because DOX has to intercalate with DNA to induce cell death.56 The results indicate that the redox-sensitive NPs provide an effective strategy for rapid delivery of the drug into cytosol. Moreover, higher uptake efficiency was found in MDA-MB-231 cells than MCF-7 cells, which can be probably ascribed to the dual receptor-mediated endocytosis of NPs.
MTT assays of the blank NPs confirm that these HA-based NPs have good biocompatibility to be safely used as pharmaceutical vehicles. The cytotoxicity results suggest that GE11–HA-ss-Chol7 DOX-NPs showed the utmost cellular inhibition in MDA-MB-231 cells which showed high CD44 and EGFR expression. These desirable results may involve the combination of redox-responsive effect in the cytosol and dual receptor-mediated endocytosis, which are consistent with the cellular uptake and intracellular DOX distribution results. Although free DOX showed the highest inhibitory rate in MCF-7 cells, the in vivo use might be limited due to an undesirable pharmacokinetic behavior and nonspecific cytotoxicity that produce severe side effects.
In the in vivo imaging analysis study, compared with free DiR group and HA-ss-Chol7 DiR-NPs, the highest tumor-targeting efficiency was found in GE11–HA-ss-Chol7 DiR-NPs group, which might be ascribed to a combination of EPR effect and dual receptor-mediated uptake of NPs.57 In comparison with the free DiR control group, the continuous higher level of DiR detected in tumor sites implies that NPs effectively prolong the circulation time of the encapsulated drug, probably owing to the hydrophilic shell of the carrier.58 Therefore, it provides a powerful evidence that the designed GE11–HA-ss-Chol7 NPs are available for tumor-specific drug delivery.
In the in vivo tumor growth suppression assay, compared with any other group, the most outstanding antitumor efficiency was found in GE11–HA-ss-Chol7 DOX-NPs group, which is consistent with the in vitro studies. The predominant antitumor efficacy could be ascribed to the passive targeting by the EPR effect and the enhanced intracellular delivery by receptor-mediated endocytosis.59 Also, the reduction-triggered intracellular drug release strategy is an efficient way to improve the cancer therapy. Additionally, the distinct weight loss in free DOX group indicates its severe systemic side effect, as reported previously.60 The little difference in body weight between the saline group and the HA-based groups suggests the low in vivo toxicity of the NPs.
Conclusion
In this study, we have demonstrated that the novel redox-responsive degradable NPs based on GE11–HA-ss-Chol conjugates selectively target the breast tumor not only by EPR effect, but also through active dual-targeting endocytosis mechanism. Both in vitro and in vivo results confirmed their predominant effects in enhancing drug efficacy. Therefore, we can conclude that GE11–HA-ss-Chol NPs are supposed to be a promising and desirable vehicle for anticancer drugs like DOX to treat breast cancer.
Acknowledgments
This work was financially supported by the Program of the 333 High-level Personnel Training Project (number BRA2015392) of the Young Leaders in Science and Technology of Jiangsu Province. The authors express their gratitude to Jiangsu Province Public Technology Service Center of Nanodrug Preparation and Evaluation and Jiangsu Key Laboratory of Carcinogenesis and Intervention (China Pharmaceutical University) for assistance with the cell and animal experiments.
Disclosure
The authors report no conflicts of interest in this work.
References
Carvalho C, Santos RX, Cardoso S, et al. Doxorubicin: the good, the bad and the ugly effect. Curr Med Chem. 2009;16(25):3267–3285. | ||
Tacar O, Sriamornsak P, Dass CR. Doxorubicin: an update on anticancer molecular action, toxicity and novel drug delivery systems. J Pharm Pharmacol. 2013;65(2):157–170. | ||
Mura S, Nicolas J, Couvreur P. Stimuli-responsive nanocarriers for drug delivery. Nat Mater. 2013;12(11):991–1003. | ||
Samadi N, van Nostrum CF, Vermonden T, Amidi M, Hennink WE. Mechanistic studies on the degradation and protein release characteristics of poly (lactic-co-glycolic-co-hydroxymethylglycolic acid) nanospheres. Biomacromolecules. 2013;14(4):1044–1053. | ||
Locatelli E, Matteini P, Sasdelli F, et al. Surface chemistry and entrapment of magnesium nanoparticles into polymeric micelles: a highly biocompatible tool for photothermal therapy. Chem Commun. 2014;50(58):7783–7786. | ||
Purcell BP, Lobb D, Charati MB, et al. Injectable and bioresponsive hydrogels for on-demand matrix metalloproteinase inhibition. Nat Mater. 2014;13(6):653–661. | ||
Tong R, Tang L, Ma L, Tu CL, Baumgartner R, Cheng JJ. Smart chemistry in polymeric nanomedicine. Chem Soc Rev. 2014;43(20):6982–7012. | ||
Baldi G, Ravagli C, Mazzantini F, et al. In vivo anticancer evaluation of the hyperthermic efficacy of anti-human epidermal growth factor receptor-targeted PEG-based nanocarrier containing magnetic nanoparticles. Int J Nanomedicine. 2014;9:3037–3056. | ||
Nehoff H, Parayath NN, Domanovitch L, Taurin S, Greish K. Nanomedicine for drug targeting: strategies beyond the enhanced permeability and retention effect. Int J Nanomed. 2014;9:2539–2555. | ||
Han Y, Zhang Y, Li D, Chen Y, Sun J, Kong F. Transferrin-modified nanostructured lipid carriers as multifunctional nanomedicine for codelivery of DNA and doxorubicin. Int J Nanomedicine. 2014;9:4107–4115. | ||
Accardo A, Aloj L, Aurilio M, Morelli G, Tesauro D. Receptor binding peptides for target-selective delivery of nanoparticles encapsulated drugs. Int J Nanomedicine. 2014;9:1537–1557. | ||
Quang Nam B, Li Y, Jang M-S, Dai Phu H, Lee JH, Lee DS. Redox- and pH-sensitive polymeric micelles based on poly(beta-amino ester)-grafted disulfide methylene oxide poly(ethylene glycol) for anticancer drug delivery. Macromolecules. 2015;48(12):4046–4054. | ||
Cun X, Ruan S, Chen J, et al. A dual strategy to improve the penetration and treatment of breast cancer by combining shrinking nanoparticles with collagen depletion by losartan. Acta Biomater. 2016;31:186–196. | ||
Ma Y, Nolte RJM, Cornelissen JJLM. Virus-based nanocarriers for drug delivery. Adv Drug Deliv Rev. 2012;64(9):811–825. | ||
Wu LL, Zou Y, Deng C, Cheng R, Meng FH, Zhong ZY. Intracellular release of doxorubicin from core-crosslinked polypeptide micelles triggered by both pH and reduction conditions. Biomaterials. 2013;34(21):5262–5272. | ||
Ci T, Li T, Chang G, Yu L, Ding J. Simply mixing with poly(ethylene glycol) enhances the fraction of the active chemical form of antitumor drugs of camptothecin family. J Control Release. 2013;169(3):329–335. | ||
Ci T, Chen L, Yu L, Ding J. Tumor regression achieved by encapsulating a moderately soluble drug into a polymeric thermogel. Sci Rep. 2014;4:5473. | ||
Yang R, An Y, Miao F, Li M, Liu P, Tang Q. Preparation of folic acid-conjugated, doxorubicin-loaded, magnetic bovine serum albumin nanospheres and their antitumor effects in vitro and in vivo. Int J Nanomedicine. 2014;9:4231–4243. | ||
Cheng L, Huang F-Z, Cheng L-F, et al. GE11-modified liposomes for non-small cell lung cancer targeting: preparation, ex vitro and in vivo evaluation. Int J Nanomedicine. 2014;9:921–935. | ||
Cabeza L, Ortiz R, Arias JL, et al. Enhanced antitumor activity of doxorubicin in breast cancer through the use of poly(butylcyanoacrylate) nanoparticles. Int J Nanomed. 2015;10:1291–1306. | ||
Liu Z, Jiao Y, Wang Y, Zhou C, Zhang Z. Polysaccharides-based nanoparticles as drug delivery systems. Adv Drug Deliv Rev. 2008;60(15):1650–1662. | ||
Ganesh S, Iyer AK, Morrissey DV, Amiji MM. Hyaluronic acid based self-assembling nanosystems for CD44 target mediated siRNA delivery to solid tumors. Biomaterials. 2013;34(13):3489–3502. | ||
Kogan G, Soltes L, Stern R, Gemeiner P. Hyaluronic acid: a natural biopolymer with a broad range of biomedical and industrial applications. Biotechnol Lett. 2007;29(1):17–25. | ||
Oh EJ, Park K, Kim KS, et al. Target specific and long-acting delivery of protein, peptide, and nucleotide therapeutics using hyaluronic acid derivatives. J Control Release. 2010;141(1):2–12. | ||
Herrlich P, Sleeman J, Wainwright D, et al. How tumor cells make use of CD44. Cell Adhes Commun. 1998;6(2–3):141–147. | ||
Stern R. Hyaluronidases in cancer biology. Semin Cancer Biol. 2008;18(4):275–280. | ||
Mezghrani O, Tang Y, Ke X, et al. Hepatocellular carcinoma dually-targeted nanoparticles for reduction triggered intracellular delivery of doxorubicin. Int J Pharm. 2015;478(2):553–568. | ||
Sharma G, Modgil A, Sun C, Singh J. Grafting of cell-penetrating peptide to receptor-targeted liposomes improves their transfection efficiency and transport across blood-brain barrier model. J Pharm Sci. 2012;101(7):2468–2478. | ||
Sawant RR, Torchilin VP. Challenges in development of targeted liposomal therapeutics. AAPS J. 2012;14(2):303–315. | ||
Huang S, Li C, Armstrong EA, et al. Dual targeting of EGFR and HER3 with MEHD7945A overcomes acquired resistance to EGFR inhibitors and radiation. Cancer Res. 2013;73(2):824–833. | ||
Salomon DS, Brandt R, Ciardiello F, Normanno N. Epidermal growth factor-related peptides and their receptors in human malignancies. Crit Rev Oncol/Hematol. 1995;19(3):183–232. | ||
Li Z, Zhao R, Wu X, et al. Identification and characterization of a novel peptide ligand of epidermal growth factor receptor for targeted delivery of therapeutics. Faseb J. 2005;19(14):1978–1985. | ||
Cheng R, Feng F, Meng F, Deng C, Feijen J, Zhong Z. Glutathione-responsive nano-vehicles as a promising platform for targeted intracellular drug and gene delivery. J Control Release. 2011;152(1):2–12. | ||
Meng FH, Hennink WE, Zhong Z. Reduction-sensitive polymers and bioconjugates for biomedical applications. Biomaterials. 2009;30(12):2180–2198. | ||
Huo M, Yuan J, Tao L, Wei Y. Redox-responsive polymers for drug delivery: from molecular design to applications. Polym Chem. 2014;5(5):1519–1528. | ||
Li D, Tang X, Pulli B, et al. Theranostic nanoparticles based on bioreducible polyethylenimine-coated iron oxide for reduction-responsive gene delivery and magnetic resonance imaging. Int J Nanomedicine. 2014;9:3347–3361. | ||
Liu X, Wang J, Xu W, et al. Glutathione-degradable drug-loaded nanogel effectively and securely suppresses hepatoma in mouse model. Int J Nanomedicine. 2015;10:6587–6602. | ||
Yao C, Tai Z, Wang X, et al. Reduction-responsive cross-linked stearyl peptide for effective delivery of plasmid DNA. Int J Nanomedicine. 2015;10:3403–3416. | ||
Sandoval MA, Sloat BR, Lansakara-P DSP, et al. EGFR-targeted stearoyl gemcitabine nanoparticles show enhanced anti-tumor activity. J Control Release. 2012;157(2):287–296. | ||
Huang JB, Zhang H, Yu Y, et al. Biodegradable self-assembled nanoparticles of poly (D,L-lactide-co-glycolide)/hyaluronic acid block copolymers for target delivery of docetaxel to breast cancer. Biomaterials. 2014;35(1):550–566. | ||
Alferiev IS, Connolly JM, Levy RJ. A novel mercapto-bisphosphonate as an efficient anticalcification agent for bioprosthetic tissues. J Organomet Chem. 2005;690(10):2543–2547. | ||
Xue M, Gao D, Chen XL, Liu KQ, Fang Y. New dimeric cholesteryl-based A(LS)(2) gelators with remarkable gelling abilities: organogel formation at room temperature. J Colloid Interf Sci. 2011;361(2):556–564. | ||
Ellman GL. A colorimetric method for determining low concentrations of mercaptans. Arch Biochem Biophys. 1958;74(2):443–450. | ||
Wu G, Fang YZ, Yang S, Lupton JR, Turner ND. Glutathione metabolism and its implications for health. J Nutr. 2004;134(3):489–492. | ||
Kuppusamy P, Li H, Ilangovan G, et al. Noninvasive imaging of tumor redox status and its modification by tissue glutathione levels. Cancer Res. 2002;62(1):307–312. | ||
Biswas S, Torchilin VP. Nanopreparations for organelle-specific delivery in cancer. Adv Drug Deliv Rev. 2014;66:26–41. | ||
Bertrand N, Wu J, Xu X, Kamaly N, Farokhzad OC. Cancer nanotechnology: the impact of passive and active targeting in the era of modern cancer biology. Adv Drug Deliv Rev. 2014;66:2–25. | ||
Wei X, Senanayake TH, Warren G, Vinogradov SV. Hyaluronic acid-based nanogel-drug conjugates with enhanced anticancer activity designed for the targeting of CD44-positive and drug-resistant tumors. Bioconjugate Chem. 2013;24(4):658–668. | ||
Torchilin V. Tumor delivery of macromolecular drugs based on the EPR effect. Adv Drug Deliv Rev. 2011;63(3):131–135. | ||
Wang J, Yao K, Wang C, Tang C, Jiang X. Synthesis and drug delivery of novel amphiphilic block copolymers containing hydrophobic dehydroabietic moiety. J Mat Chem B. 2013;1(17):2324–2332. | ||
Fischer D, Li YX, Ahlemeyer B, Krieglstein J, Kissel T. In vitro cytotoxicity testing of polycations: influence of polymer structure on cell viability and hemolysis. Biomaterials. 2003;24(7):1121–1131. | ||
Ma SF, Nishikawa M, Katsumi H, Yamashita F, Hashida M. Liver targeting of catalase by cationization for prevention of acute liver failure in mice. J Control Release. 2006;110(2):273–282. | ||
Kim JO, Kabanov AV, Bronich TK. Polymer micelles with cross-linked polyanion core for delivery of a cationic drug doxorubicin. J Control Release. 2009;138(3):197–204. | ||
Sun H, Guo B, Cheng R, Meng F, Liu H, Zhong Z. Biodegradable micelles with sheddable poly (ethylene glycol) shells for triggered intracellular release of doxorubicin. Biomaterials. 2009;30(31):6358–6366. | ||
Chan JM, Zhang LF, Yuet KP, et al. PLGA-lecithin-PEG core-shell nanoparticles for controlled drug delivery. Biomaterials. 2009;30(8):1627–1634. | ||
Gewirtz D. A critical evaluation of the mechanisms of action proposed for the antitumor effects of the anthracycline antibiotics adriamycin and daunorubicin. Biochem Pharmacol. 1999;57(7):727–741. | ||
Choi KY, Chung H, Min KH, et al. Self-assembled hyaluronic acid nanoparticles for active tumor targeting. Biomaterials. 2010;31(1):106–114. | ||
Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nanocarriers as an emerging platform for cancer therapy. Nat Nanotechnol. 2007;2(12):751–760. | ||
Maeda H, Nakamura H, Fang J. The EPR effect for macromolecular drug delivery to solid tumors: improvement of tumor uptake, lowering of systemic toxicity, and distinct tumor imaging in vivo. Adv Drug Deliv Rev. 2013;65(1):71–79. | ||
Octavia Y, Tocchetti CG, Gabrielson KL, Janssens S, Crijns HJ, Moens AL. Doxorubicin-induced cardiomyopathy: from molecular mechanisms to therapeutic strategies. J Mol Cell Cardiol. 2012;52(6):1213–1225. |
Supplementary materials
Synthesis procedure of insensitive conjugates of GE11–HA-Chol
The synthesis of GE11–hyaluronic acid (HA)-Chol conjugate consists of three steps: synthesis of reduction-insensitive linker of cholesteryl–ethylenediamine (Chol–eda), modification of HA with Chol–eda, and modification HA-Chol with GE11 peptide. The details have been provided in the “Materials and methods” section.
1H-nuclear magnetic resonance (1H-NMR) of cholesteryl–cystamine (Chol–cys) and Chol–eda linkers
1H-NMR of the raw material of cholesteryl chloroformate is shown in Figure S1A with characteristic peaks of δ (ppm) 5.36 (s, 1H, alkenyl, a), 4.48 (m, 1H, oxycyclohexyl, b), and 0.66–2.33 (m, 43H, cholesteryl protons).
The formation of Chol–cys was confirmed with characteristic peaks of δ (ppm) 5.36 (s, 1H, alkenyl, a), 5.05 (s, 1H, CONH, c), 4.48 (m, 1H, oxycyclohexyl, b), 3.48 (m, 2H, CH2NH, d), 3.00 (m, 2H, CH2N(H2), e), 2.76–2.79 (m, 4H, 2CH2S, f), 0.66–2.33 (m, 45H, cholesteryl protons, and NH2), as shown in Figure S1B.
The formation of Chol–cys was confirmed with characteristic peaks of δ (ppm) 5.37 (s, 1H, alkenyl, a), 4.99 (s, 1H, CONH, g), 4.50 (m, 1H, oxycyclohexyl, b), 3.21–3.23 (m, 2H, N(H2)CH2, i), 2.82–2.84 (m, 2H, CONCH2, h), 0.68–2.38 (m, 45H, cholesteryl protons, and NH2), as shown in Figure S1C.
High-performance liquid chromatography assessment of GE11 peptide conjugation efficiency
GE11 peptide conjugation efficiency was evaluated with high-performance liquid chromatography utilizing the ultraviolet absorption of GE11 peptide at 220 nm, while at the same wavelength, the polymer conjugate of HA-ss-Chol had no absorption. The mobile phase was as follows: acetonitrile (solution A) and ultrapure water (solution B), both containing 0.1% trifluoroacetic acid; gradient elution mode: 0–20 minutes, 25%–50% A and 20.1–30 minutes, 50%–25% A. The retention time of GE11 peptide was 8.2 minutes. The conjugation efficiency was calculated using the formula, Cpeptide, before/Cpeptide, after, where Cpeptide, before and Cpeptide, after denote the peptide concentration before and after the conjugation reaction, respectively.
Evaluation of cellular uptake pathways of GE11–HA-ss-Chol7 nanoparticles (NPs)
Different endocytic inhibitors were adopted to evaluate the cellular uptake pathways of GE11–HA-ss-Chol7 NPs, as shown in Figure S4, in which nystatin was regarded as the caveolae-mediated endocytosis inhibitor, chloropromazine (CPZ) as the clathrin-mediated endocytosis pathway inhibitor, and amiloride as the macropinocytosis pathway inhibitor. The relative uptake efficiency was calculated as the ratio between the fluorescence intensities of groups treated with endocytic inhibitors and the control group (cells only treated with NPs).
In vitro cytotoxicity assay of blank NPs
The cytotoxicity of blank NPs was evaluated by MTT assay. As shown in Figure S5, after incubating for 24 hours with a medium containing GE11–HA-ss-Chol7, GE11–HA-Chol7, and HA-ss-Chol7 NPs, the cell viability was above 90% in all groups, even for the groups with the dosage 500 μg/mL, indicating the low cytotoxicity and good cell compatibility of the drug carriers studied.
Target abilities evaluated by in vivo imaging analysis
The target abilities of NPs were analyzed semi-quantitatively by relative tissue exposure (Re), tumor-targeting efficiency, and relative tumor-targeting efficiency (RTe) through in vivo imaging analysis. The calculation equations were as follows:
|
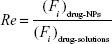
|
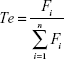
|
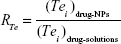
where Fi refers to the fluorescence intensity in the targeted tissue or organ.
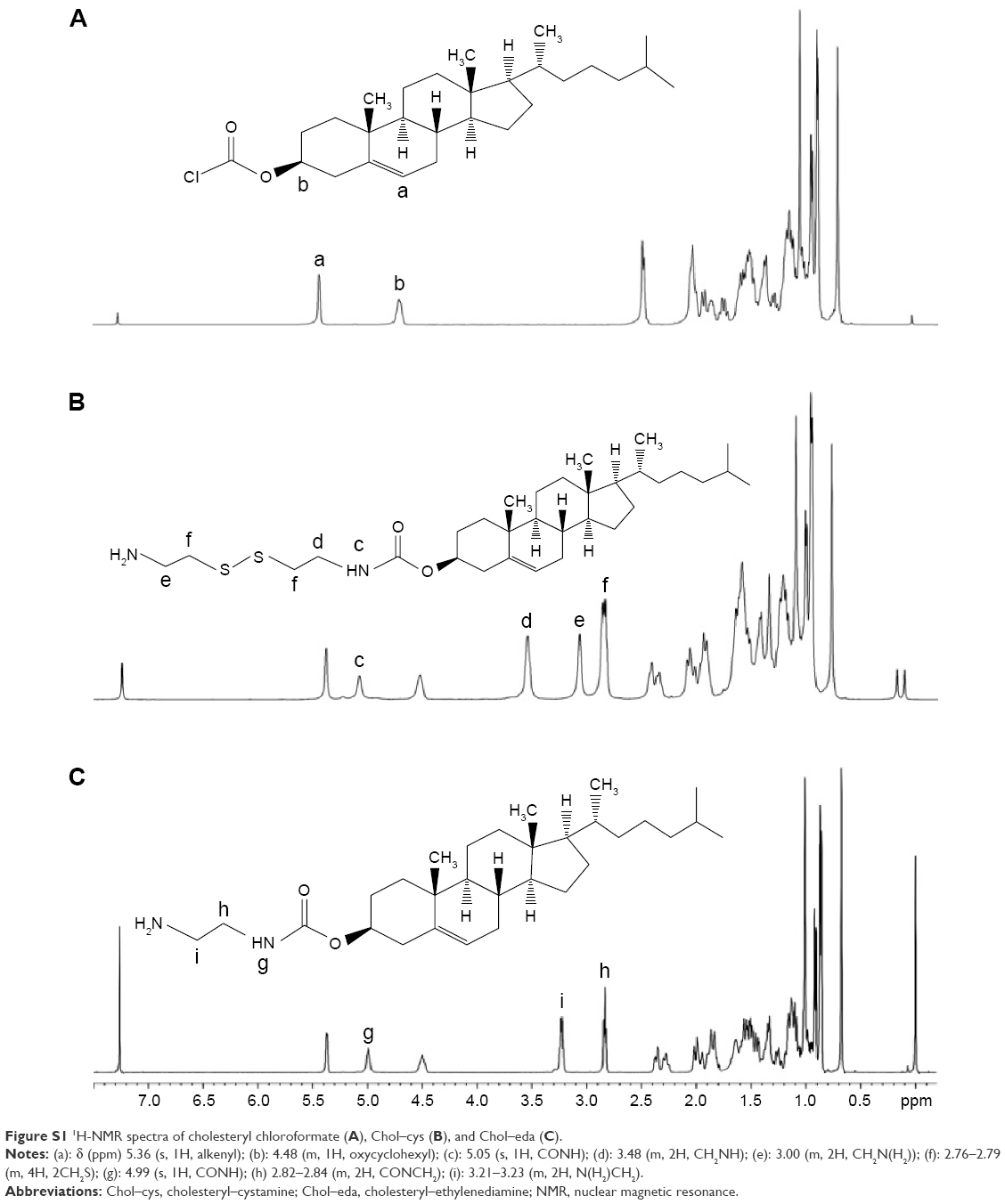 | Figure S1 1H-NMR spectra of cholesteryl chloroformate (A), Chol–cys (B), and Chol–eda (C). |
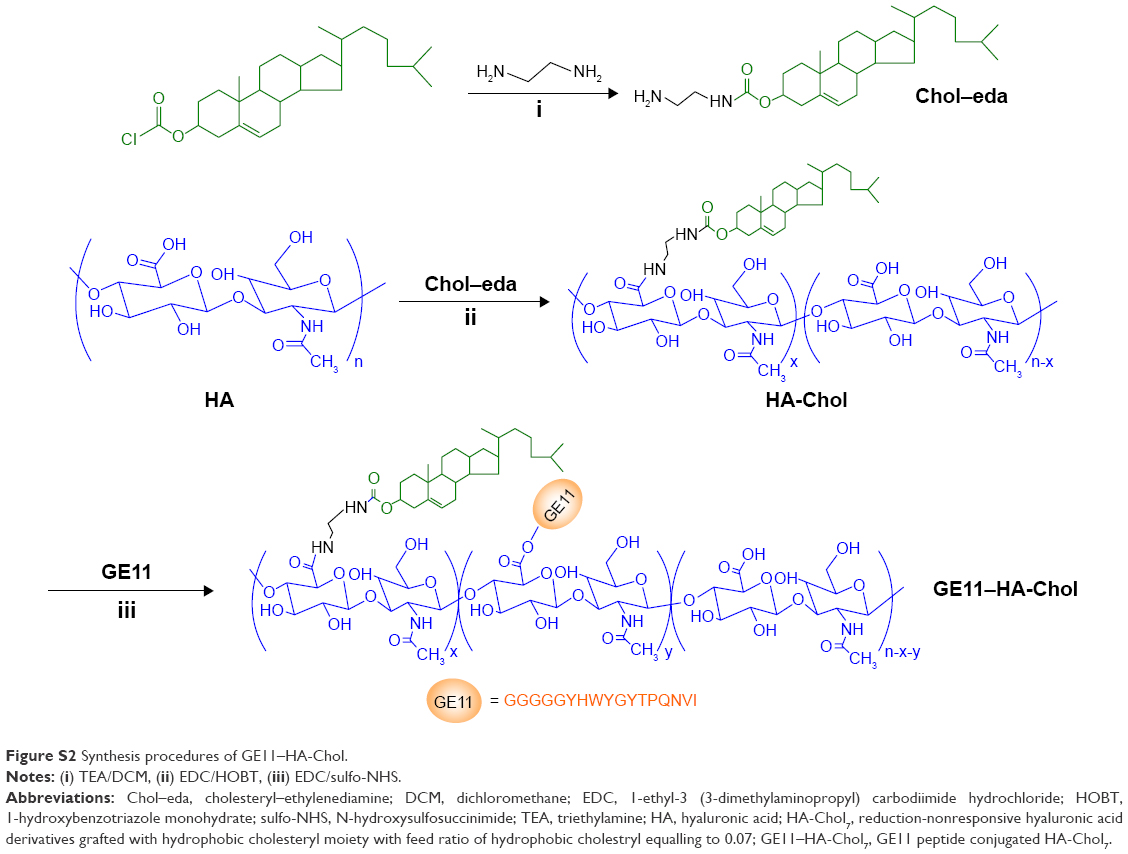 | Figure S2 Synthesis procedures of GE11–HA-Chol. |
 | Figure S3 HPLC assessment of GE11 peptide conjugated to the distal end of HA-ss-Chol by monitoring absorbance at 220 nm. |
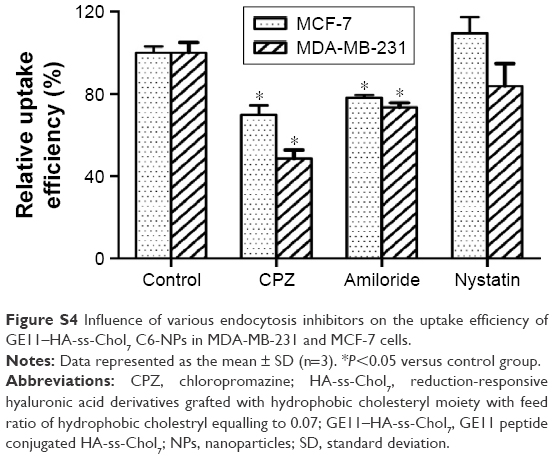 | Figure S4 Influence of various endocytosis inhibitors on the uptake efficiency of GE11–HA-ss-Chol7 C6-NPs in MDA-MB-231 and MCF-7 cells. |
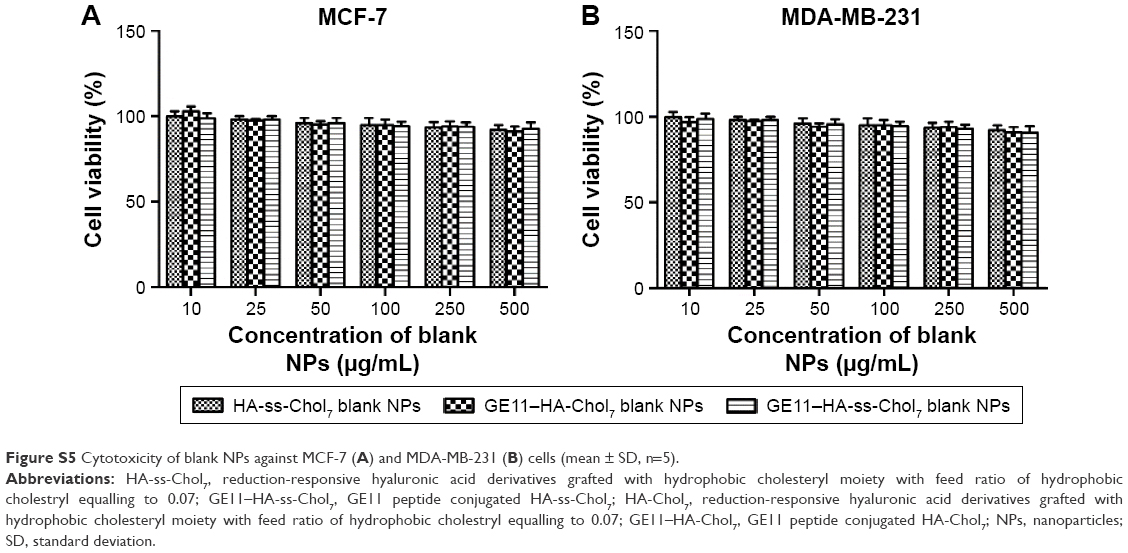 | Figure S5 Cytotoxicity of blank NPs against MCF-7 (A) and MDA-MB-231 (B) cells (mean ± SD, n=5). |
 | Table S1 Relative tissue exposures of DiR-NPs compared with DiR solution |
 | Table S2 Tumor-targeting efficiency and relative tumor-targeting efficiency of DiR-NPs |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.