Back to Journals » International Journal of Nanomedicine » Volume 11
Fusion between fluid liposomes and intact bacteria: study of driving parameters and in vitro bactericidal efficacy
Authors Wang Z, Ma Y, Khalil H, Wang R, Lu T ![]() , Zhao W, Zhang Y, Chen J, Chen T
, Zhao W, Zhang Y, Chen J, Chen T
Received 11 October 2013
Accepted for publication 12 December 2013
Published 17 August 2016 Volume 2016:11 Pages 4025—4036
DOI https://doi.org/10.2147/IJN.S55807
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Carlos Rinaldi
Zhao Wang,1,2* Yufan Ma,1,3,4* Hayssam Khalil,1 Rutao Wang,1–3 Tingli Lu,1 Wen Zhao,1 Yang Zhang,3 Jamin Chen,1,2 Tao Chen,1–3
1Key Laboratory for Space Bioscience and Biotechnology, School of Life Sciences, Northwestern Polytechnical University, Xi'an, Shaanxi, 2Shaanxi Liposome Research Center, Xi'an, Shaanxi, 3Xi'an Libang Pharmaceuticals Co, Ltd, Xi'an, 4School of Medicine, Xi'an Jiaotong University, Xi'an, People's Republic of China
*These authors contributed equally to this work.
Background: Pseudomonas aeruginosa represents a good model of antibiotic resistance. These organisms have an outer membrane with a low level of permeability to drugs that is often combined with multidrug efflux pumps, enzymatic inactivation of the drug, or alteration of its molecular target. The acute and growing problem of antibiotic resistance of bacteria to conventional antibiotics made it imperative to develop new liposome formulations for antibiotics, and investigate the fusion between liposome and bacterium.
Methods: In this study, the factors involved in fluid liposome interaction with bacteria have been investigated. We also demonstrated a mechanism of fusion between liposomes
(1,2-dipalmitoyl-sn-glycero-3-phosphocholine [DPPC]/dimyristoylphosphatidylglycerol [DMPG] 9:1, mol/mol) in a fluid state, and intact bacterial cells, by lipid mixing assay.
Results: The observed fusion process is shown to be mainly dependent on several key factors. Perturbation of liposome fluidity by addition of cholesterol dramatically decreased the degree of fusion with P. aeruginosa from 44% to 5%. It was observed that fusion between fluid liposomes and bacteria and also the bactericidal activities were strongly dependent upon the properties of the bacteria themselves. The level of fusion detected when fluid liposomes were mixed with Escherichia coli (66%) or P. aeruginosa (44%) seems to be correlated to their outer membrane phosphatidylethanolamine (PE) phospholipids composition (91% and 71%, respectively). Divalent cations increased the degree of fusion in the sequence Fe2+ > Mg2+ > Ca2+ > Ba2+ whereas temperatures lower than the phase transition temperature of DPPC/DMPG (9:1) vesicles decreased their fusion capacity. Acidic as well as basic pHs conferred higher degrees of fusion (54% and 45%, respectively) when compared to neutral pH (35%).
Conclusion: Based on the results of this study, a possible mechanism involving cationic bridging between bacterial negatively charged lipopolysaccharide and fluid liposomes DMPG phospholipids was outlined. Furthermore, the fluid liposomal-encapsulated tobramycin was prepared, and the in vitro bactericidal effects were also investigated.
Keywords: fusion, lipid-mixing assay, lipid composition
Introduction
Pseudomonas aeruginosa is a remarkably adaptable pathogen. It has assumed an important role in the infections of patients with various impairments of host defenses. In cystic fibrosis, chronic pulmonary infections with P. aeruginosa and other related strains are considered the most important factors determining the prognosis of these patients.1,2 In fact, Pseudomonas causes rapid, extensive and fatal diseases in the compromised host and claims the highest crude mortality of any gram-negative causing bacteremia.3 P. aeruginosa represents a good model of antibiotic resistance. These organisms have an outer membrane with a low level of permeability to drugs that is often combined with multidrug efflux pumps, enzymatic inactivation of the drug, or alteration of its molecular target.4,5 The acute and growing problem of antibiotic resistance of Pseudomonas to conventional antibiotics made it imperative to develop new approaches to overcome these mechanisms.
To increase the bactericidal efficacy of antibiotics, different liposomal formulations for pulmonary administration were developed with the aim of promoting effective interactions between bacteria and encapsulated drugs, increasing the resident time of the encapsulated antibiotics in the lungs and reducing systemic drug absorption.6 Tobramycin encapsulated in a negatively charged liposomal formulation presenting a low gel to liquid-crystalline transition temperature (Tc) ≤37°C succeeded, for the first time, in eradicating mucoid P. aeruginosa in an animal model of chronic pulmonary infection.7 This fluid liposomal-encapsulated tobramycin was later shown to be effective against other bacterial strains.8 It was found in a previous study that this enhanced bactericidal activity is due to a potential mechanism of fusion between the fluid liposomes and the bacterial membranes.9 In the present study, we systematically evaluated major driving forces behind such a fusion process.
The fusion process between two vesicles occurs in three distinct steps. First, the vesicles have to adhere or aggregate; this approach is subject to a number of different interaction forces. These forces comprise: 1) electrostatic interactions; 2) attractive Van der Waals interactions; and 3) hydration forces. Second, a local perturbation of the packing of the lipids bilayer at the site of contact seems required, initiating the merging of the outer monolayers of the two bilayers membranes. Finally, the aqueous vesicle interiors have to coalesce with the concomitant mixing of the inner-monolayer lipids.
A number of different fusogens have now been established as agents to accelerate the process of fusion. They range in structure from charged organic compounds, such as lecithin, to inert biological substances, such as metal ions.10–12 The divalent cations that induce interaction between negatively charged phospholipid vesicles have been studied extensively.13–15
Negatively charged phospholipid vesicles do not naturally fuse or aggregate due to the long-range electrostatic repulsion. Divalent cations, by binding to negatively charged vesicles, reduce electrostatic repulsion, inducing aggregation shortly followed by fusion. This is due to structural changes in bilayer vesicles resulting from the disruption of the strong repulsive hydration forces that prevent hydrophobic interaction between phospholipid bilayers at short distances of separation.13–15 To understand further how these liposomes interact with bacteria, we investigated the factors involved in this process. In addition, the fluid liposomal-encapsulated tobramycin was later prepared, and the in vitro bactericidal efficacy of it to P. aeruginosa, Stenotrophomonas maltophilia, Burkholderia cepacia, Escherichia coli, and Staphylococcus aureus were also investigated.
Material and methods
Bacterial strains and mediums
P. aeruginosa ATCC® 25619™ purchased from American Type Culture Collection (Rockville, MD, USA) and E. coli K12 was purchased from Invivogen (Nucliber, Spain). All other strains – B. cepacia 1368, Streptococcus agalactiae 910121, S. maltophilia (C6R9), and S. aureus 91131 – are clinical isolates. For the experiments, overnight cultures in Mueller-Hinton broth (MH) (BD Laboratories, Franklin Lakes, NJ, USA) were prepared. The following day, 30 μL of these cultures were used to inoculate 20 mL of fresh MH under agitation at 37°C. Experiments were always carried out when the culture reached an optical density (OD660nm) of 0.6. MH broth was used because it has the particularity of lacking all known divalent cations (trace amounts).
Fluid liposomes
Fluid liposomes are liposomes composed of dipalmitoyl phosphatidylcholine (DPPC) and dimyristoyl phosphatidylglycerol (DMPG) (9:1, mol/mol) (Avanti Polar Lipids, Inc., Alabaster, AL, USA), which have an overall low gel liquid-crystalline Tc (<37°C). They were prepared by a hydration-extrusion method previously described.7–9 Appropriate amounts of lipid mixtures were dissolved with a solution of methanol/chloroform (1:2). The fluorescence-labeled lipid markers solution (1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-[7-nitro-2–1,3-benzoxadiazol-4-yl] and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-[lissamine Rhodamine B sulfonyl]; 0.5 mol% each of the total lipids) was added and the solvents were evaporated under nitrogen stream with a warm water bath until a homogeneous lipid film was produced. The resultant film was dried under vacuum overnight, hydrated with hepes buffered saline (25 mM HEPES and 150 mM NaCl, pH 7.5) by vigorous mixing, and followed by five times freeze-and-thaw (freeze with dry ice acetone and thaw in hot water at 65°C). The sample was then extruded through two stacked nuclepore polycarbonate filter with a pore size of 100 nm using an extrusion device (Lipex Biomembranes, Inc., Vancouver, BC, Canada) as previously described.7
Bacteria and liposome fusion
Liposomal fusion with bacteria was monitored by a lipid-mixing assay based on the extent of resonance energy transfer (RET) between the lipid headgroup-labeled probes, 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine Rhodamine B sulfonyl) (Rh-PE), and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(7-nitro-2–1,3-benzoxadiazol-4-yl) (NBD-PE), as described by Gerritsen et al.16 All fluorescence measurements were carried out with a Perkin-Elmer LS-50B fluorescence spectrophotometer. In a typical experiment, the NBD-PE/Rh-PE -labeled liposomes were prepared as described previously containing both NBD-PE and Rh-PE at 0.2 mol% each.9 The NBD/Rh-labeled vesicles (10 μg/mL final concentration) were mixed with 1.9 mL of bacteria (OD660 =0.6) and incubated at 37°C under constant stirring. RET efficiency was monitored by measuring time-drives of the reaction mixture fluorescence in quartz cuvettes. The fusion of NBD/Rh-labeled liposomes with bacteria resulted in probe dilution (lipid-mixing) and an increased distance between the NBD-PE and Rh-PE, thereby decreasing RET efficiency and decreasing Rh-fluorescence intensity.
Continuous monitoring of rhodamine fluorescence (590 nm) was done at an interval of 1 minute under steady-state excitation at 475 nm. The final fluorescence intensity (Fmax), which represents maximal fluorescent lipid probe dilution in each sample, was determined following the solubilization of vesicles with Triton X-100 detergent (0.2% volume). The percentage of fusion (or lipid dilution) was calculated using the following equation:
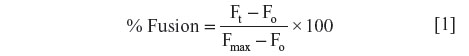
where Ft is the fluorescence intensity at each time point and Fo is the initial fluorescence intensity. Each experiment for the given conditions was repeated at least three times. Rhodamine fluorescence intensity was monitored in order to avoid any artifact caused by P. aeruginosa natural green fluorescence.
Effect of calcium concentration on fusion
In the MH (OD660 =0.6), 1.9 mL of bacteria was grown and supplemented with the proper amount of CaCl2 from a stock solution of 1 M to reach the desired concentration. Volume was adjusted to avoid any dilution artifacts. After 5 minutes of equilibration time, labeled liposomes were added to give a final concentration of 10 μg/mL as described in the Bacteria and liposome fusion section and measurements started immediately.
Effect of different divalent cations on liposomal fusion with intact bacteria
Stock solutions of each divalent cations in salt solutions (CaCl2, BaCl2, FeSO4, and MgCl2) were prepared at a concentration of 1 M. For each measurement, divalent cations were added to 1.9 mL of bacteria grown in MH (OD660 =0.6) to give a final concentration of 1 mM. Following 5 minutes of equilibration time, labeled liposomes were added (final concentration of 10 μg/mL) as described in the Bacteria and liposome fusion section and measurements started immediately.
Effect of pH on liposomal fusion
One point nine mL of bacteria grown in MH (OD660 =0.6) was taken and the medium pH adjusted to 5.4. Following 5 minutes of equilibration time, labeled liposomes were added as described in the Bacteria and liposome fusion section (final concentration of 10 μg/mL) and measurements started immediately.
Effect of temperature on liposomal fusion
Labeled liposomes were added at a final concentration of 10 μg/mL to 1.9 mL of bacteria grown in MH (OD660 =0.6). Fusion measurements were carried at three different temperatures (4°C, 22°C, and 37°C) with two gram-negative bacteria, P. aeruginosa ATCC® 29248™ and E. coli K12 (wild type) for 30 minutes.
Preparation of fluid liposomal-encapsulated tobramycin
This fluid liposomal-encapsulated tobramycin was prepared by a dehydration–rehydration vesicle method as previously described.7 The liposome preparation consisted of DPPC and DMPG in a molar ratio of 15:1, which corresponds to a global Tc of 29°C.6 Tobramycin in liposomal preparations was quantified by high performance liquid chromatography (HPLC) as previously described.7
Determination of liposome size, zeta-potential, and entrapment efficiency
Liposome size and zeta-potential were determined by Zetasizer Nano-ZS system (Malvern Instruments, Malvern, Worcestershire, UK). The emulsion samples were diluted 1:10 with purified water before measurement.
The entrapment efficiency (EE) of the developed liposome was determined by measuring the concentration of tobramycin in the dispersion phase. The emulsions were subject to Ultrafiltration tubes (Amicon Ultra-4, Ultracel-10 Membrane, 10 kDa; EMD Millipore Corp, Billerica, MA, USA) operated at 12,000 rpm for 0.5 hours. The aqueous phase was collected and the concentration of tobramycin in aqueous phase was estimated using HPLC. The EE was calculated according to the following equation:
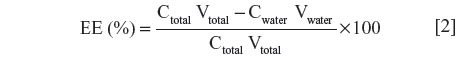
where C is the concentration of tobramycin and V is the volume of the developed liposome.
In vitro experimental design
In vitro sterilizing effects of free tobramycin, liposomes containing tobramycin, or phosphate buffered solution (PBS) alone were performed in the presence of sub-MIC (minimum inhibitory concentration) of free or encapsulated tobramycin. Bacteria were inoculated in 250 mL of Proteose Peptone Broth no 2 (BD) for 17 hours to obtain a log-phase cell density of 105 colony forming units (cfu)/mL by optical density at 660 nm. In order to evaluate the number of bacteria, culture dilutions were plated in triplicate.
After that, 1 mL of either free antibiotic, liposome-encapsulated tobramycin, or controls was added to 29 mL of the cultures described above. Tobramycin in liposomal preparations was first quantified by HPLC analysis after methanol extraction.17 A second HPLC analysis was performed after the final dilutions to compare free and encapsulated antibiotic preparations. P. aeruginosa, S. maltophilia, B. cepacia, E. coli, and S. aureus cultures received, respectively, 30, 1, 3, 0.5, and 1.5 mg/L of free tobramycin or liposomal-encapsulated tobramycin. These values were lower than the MICs for each bacterial strain. The cultures were then incubated on a shaker at 37°C for 24 hours. Samples of 1.5 mL were collected after 0, 6, and 24 hours of incubation. One milliliter was read at 660 nm and 0.5 mL was used immediately for serial dilutions in cold sterile PBS. Appropriate dilutions were plated and cultured in triplicate on Proteose Peptone no 2 agar. Then the number of cfu were determined after overnight incubation at 37°C in 5% CO2.
Data analysis
Bacterial counts were expressed as means ± six standard errors obtained from at least three plates per dilution. All values in the present study are reported as mean ± standard errors from at least three independent experiments. The significance of treatment effect was evaluated using SPSS 11.5 (SPSS Inc., Chicago, IL, USA), with significance for P<0.05. All treated groups were compared with a negative control group. One-way analysis of variance was used to test statistical differences for single group analysis, followed by Tukey’s multiple comparisons. Two-way analysis of variance was used for grouped analysis of statistical differences followed by posttests.
Results
Monitoring and quantifying fusion by lipid-mixing assay
Previously, our group demonstrated a fusion process between fluid liposomes and bacteria by several techniques: 1) negative staining; 2) fluorescence activated cell sorter; 3) immunoelectron microscopy; and 4) lipid-mixing.25 Therefore, in the present study, we investigated the driving parameters of liposome fusion with bacteria by the lipid-mixing assay with Rh-PE and NBD-PE as fluorescent probes. It has been reported that the lipid-mixing assay, in general, gives a more reliable measurement of membrane fusion since the fluorescence intensity changes are directly related to the distance between NBD and Rh, and not to vesicular aggregation.16–18 This assay has been widely used for study of membrane fusion.16–22 It represents a typical fluorescence emission scan profile of liposome-bacteria fusions monitored by a spectrofluorometer with a constant excitation at 475 nm (excitation peak of NBD) as shown in Figure 1A. As time progresses, fluorescence intensity of Rh (590 nm) decreases and NBD signal (520 nm) increases. The decrease in resonance energy transfer efficiency indicates that a significant fusion occurred when fluid liposomes were mixed with bacteria and incubated at 37°C.
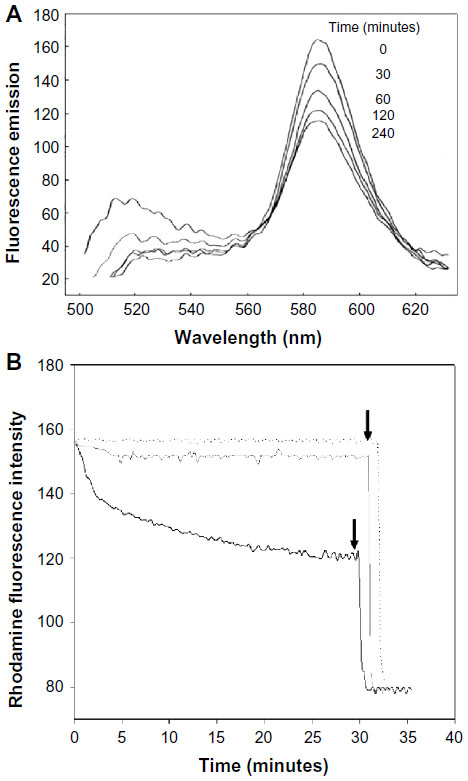 | Figure 1 Fusion of fluid liposomes with Pseudomonas aeruginosa 29248 monitored by lipid mixing assay. |
There is a time-drive representation of rhodamine fluorescence intensity decrease after liposomal fusion with P. aeruginosa shown in Figure 1B. A rapid accelerating phase can be observed in the first 5 minutes followed by a slower phase. Addition of Triton X-100 leads to a maximal dilution of the probes allowing for calculation of the extent of fusion using equation [1].
In control experiments, a mixture of NBD/Rh-labeled liposomes and non-labeled liposomes did not produce any decrease in Rh fluorescence; hence, these vesicles do not fuse together. Controls were also made by incubating vesicles made of DPPC/DMPG/cholesterol (Chol) (8:1:1 molar ratio) with intact bacteria under the same experimental conditions. This latter control, where only 10% of cholesterol is added to the phospholipids, clearly demonstrates that the decrease in Rh fluorescence is not due to lipid exchange.
Effect of calcium concentration on fusion
It has been reported that low calcium concentrations increased the interaction and fusion of vesicles from acidic phospholipid membranes with bacteria.22,23 To verify whether the interaction of liposomes with bacteria can be enhanced by calcium, increasing concentrations of calcium were added to the medium prior to fusion measurements.
Fusion of liposomes with intact cells of P. aeruginosa was greatly enhanced by increasing the concentration of calcium in the medium (Figure 2). Concentrations of calcium ranging from 1 mM to 7 mM were used in a medium deficient in calcium and magnesium (MH). Calcium mainly affects the rapid acceleration phase in the first 5 minutes of the reaction. Concentrations above 7 mM could not be used due to bacterial precipitation. Controls in the presence of calcium, where labeled liposomes were mixed with non-labeled ones, or DPPC/DMPG/Chol (8:1:1) vesicles were mixed with bacteria, showed no decrease or significantly lower decrease in RET (Figure 1B). DPPC/DMPG/Chol has roughly the same surface charge ratio as liposomes but is less fluid due to the addition of cholesterol. Fluorescence emissions of labeled liposomes were neither affected by calcium nor by the natural fluorescence of bacteria (data not shown).
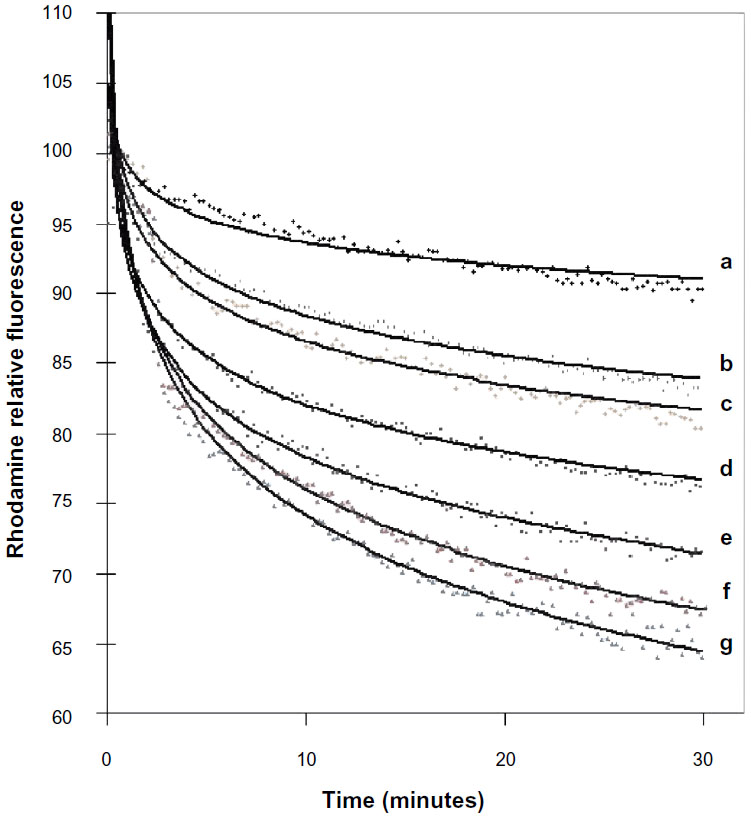 | Figure 2 Effect of calcium concentration on degree of fusion between liposomes and intact Pseudomonas aeruginosa 29248. |
Effect of various divalent cations on the degree of fusion
Figure 3 shows the degree of fusion calculated with equation [1] between liposomes and bacteria when the medium is supplemented with 1 mM of each of the appropriate cations. The highest degree of fusion is observed with iron and decreases in the sequence Fe2+ > Mg2+ > Ca2+ > Ba2+. The extent of fusion seems to be inversely proportional to the atomic radius of the ions, suggesting a possible steric crowding effect. The model for calcium-induced fusion between fluid liposomes and gram-negative bacterial membranes is shown in Figure 4. Upon addition of calcium or other divalent cations, the lipopolysaccharides (LPS) may be reoriented in such a conformation that their negatively charged polysaccharide chains are aggregated and flat, close to the bacterial membrane. This would result in the exposition of their negatively charged lipid A part. Calcium may then bridge the fluid liposomes and the bacteria causing aggregation, neutralizing the negative surface charges, dehydrating the head group of phosphatidylethanolamine (PE), and inducing local defect and fusion.
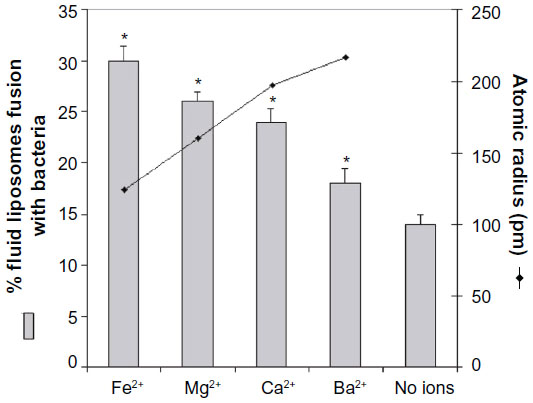 | Figure 3 Effect of divalent cations on percentage of fusion between liposomes and intact Pseudomonas aeruginosa ATCC 25619 correlated to atomic radius. |
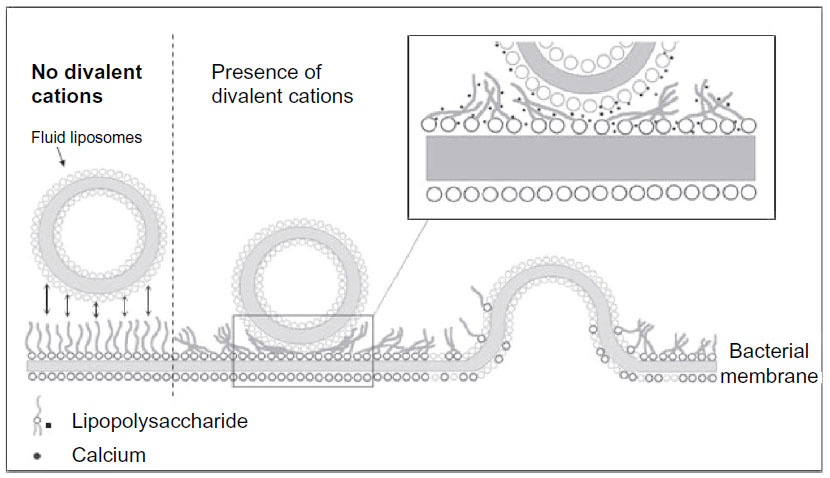 | Figure 4 Model for calcium-induced fusion between fluid liposomes and gram-negative bacterial membrane. |
Effect of pH on the degree of fusion
Previous studies have shown that pH could affect the degree of fusion of negatively charged liposomes.24 We therefore studied the effect of pH on the degree of fusion of liposomes and P. aeruginosa in a medium containing calcium. Figure 5 shows that a fusion level of almost 60% can be achieved at a low pH of 5.5 in the presence of 3 mM Ca2+. In fact, significant increase in the degree of fusion is observed at both low pH and high pH. All experimental groups were unaffected by the different pH values.
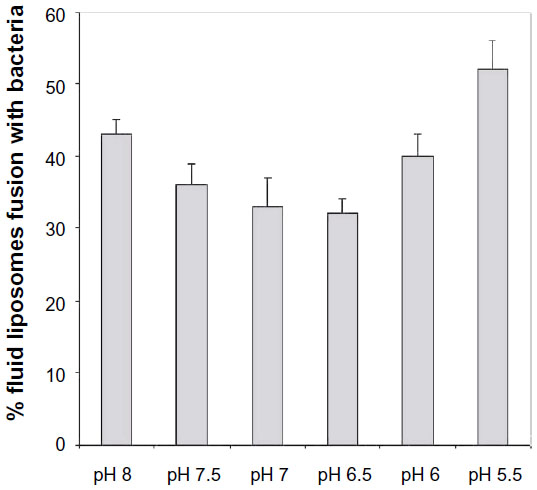 | Figure 5 Effect of pH on degree of fusion between fluid liposomes and Pseudomonas aeruginosa ATCC 25619 after 30 minutes at 37°C. |
Effect of temperature on fusion of fluid liposomes
The extent of fusion between liposomes with two different gram-negative bacteria, P. aeruginosa 25619 and E. coli K12 were measured at three different temperatures (Figure 6). At 4°C, no decrease in RET or fusion could be observed for both bacterial strains. The highest percentage of fusion was obtained at 37°C (near the Tc of fluid liposomes) whereas an intermediate level was measured at 22°C.
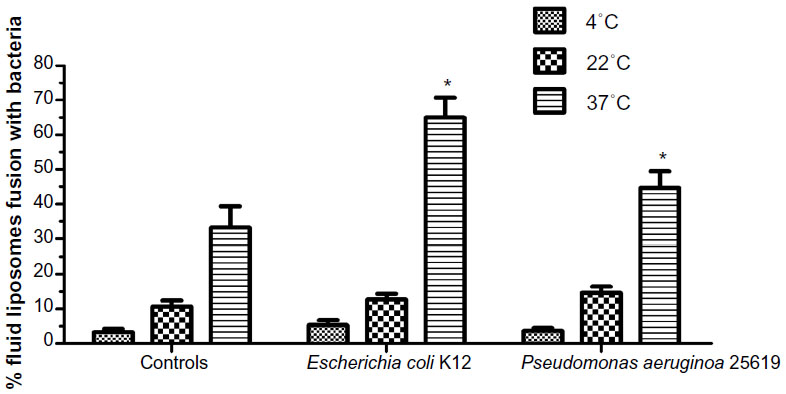 | Figure 6 Effect of temperature on the degree of fusion between fluid liposomes and two gram-negative strains, Escherichia coli K12 and Pseudomonas aeruginosa ATCC 25619, after 30 minutes at 37°C. |
Characterization of the developed liposomes
The mean diameter of all liposomes was <200 nm. Pd is a dimensionless measure of the broadness of size distribution calculated from distribution analysis, and the values of Pd were calculated for each peak as peak width/mean diameter, with values ranging from 0 to 1; the smaller the value, the more narrow distribution of the emulsion. The mean Pd of all liposomes was <0.2 and the results show that the lipid emulsions had good distribution.
ζ-potential is the key parameter of the determination of emulsion stability. Through it, it is possible to determine the size of the electrostatic repulsion between the particles, thus affecting the aggregation between the particles and the stability of the dispersion system. In our study, the ζ-potential of all the prepared formulations was around −20±5 mv, which proves the emulsions were stable.
We have previously demonstrated that fusion between fluid liposomes composed of DPPC/DMPG (9:1, mol/mol) and bacteria is not dependent on either vesicular size or lamellarity. Similar degrees of fusion were observed for liposomes with sizes ranging from 100 to 800 nm.25
The tobramycin loading efficiencies were >70% for all the prepared formulations, evaluated by HPLC analysis. The results indicated that tobramycin was suitable to be the material of liposome.
In vitro bactericidal efficacy of liposome capsulated versus free tobramycin
As indicated in “Material and methods”, sterilizing effects of bacteria exposed to free tobramycin and liposome-encapsulated tobramycin were evaluated by counting cfu on agar plates. Controls consisting of liposomes without tobramycin and liposomes combined with free tobramycin are not included in the figures because these results were similar with PBS and free tobramycin.
Six hours after treatment, a decrease in cfu/mL was observed for all the strains treated with liposome-encapsulated tobramycin while bacterial growth reached approximately 106–109 cfu/mL in PBS control cultures (Figure 7). Following the addition of liposome-encapsulated tobramycin, the growth of the P. aeruginosa and S. maltophilia groups was decreased dramatically compared to free tobramycin (Figure 7). The decrease was similar when B. cepacia, E. coli, and S. aureus were exposed to free tobramycin and liposome-encapsulated tobramycin. However, the cfu decreases with liposome-encapsulated tobramycin were probably 200, 160, and 120 times, respectively, compared with free antibiotic to B. cepacia, E. coli, and S. aureus (Figure 7).
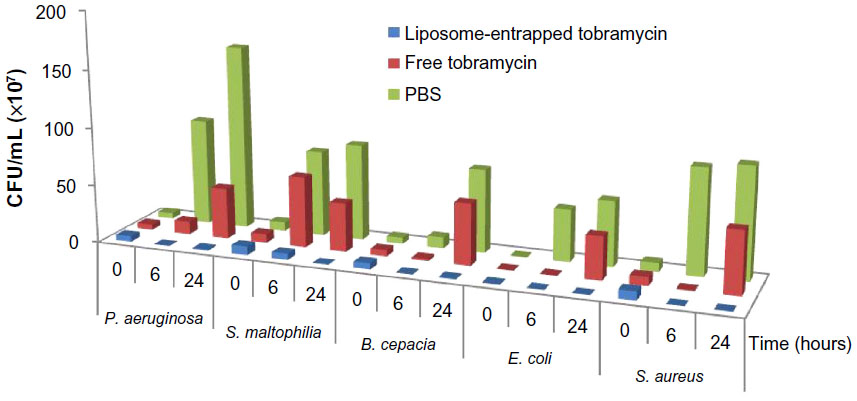 | Figure 7 Sterilizing effects of bacteria exposed to free tobramycin and liposome encapsulated tobramycin. |
Twenty-four hours after treatment, there was a significant increase in growth of P. aeruginosa, B. cepacia, E. coli, and S. aureus cultures when treated with free tobramycin. However, these bacteria were also inhibited when cultured with free liposome-encapsulated tobramycin (Figure 7).
Discussion
Membrane fusion between liposome–liposome and liposome–biological membrane has been extensively investigated, but few of these studies are related to prokaryotes.26–30 Liposomes have been successfully used in the delivery of bioactive proteins into flagellated E. coli envelope,31 and to transfect competent E. coli and Mycoplasma capricolum cells with encapsulated plasmid DNA.32,33 By using liposomes made of lipid extracts from the whole bacterial membranes of Salmonella minnesota RE595 (the composition and lipids were not identified), Tomlinson et al33 successfully incorporated phospholipid and proteins into gram-negative bacteria and confirmed that a fusion process was involved in this extensive phospholipid transfer. In this paper, we reported liposome-bacteria fusion with: 1) an artificial liposome of known lipid composition; and 2) intact bacterial cells (both gram-positive and gram-negative). Liposomes were developed in order to treat bacterial infections in cystic fibrosis with regard to the enhanced bactericidal activity of entrapped antibiotics released through their fusion with bacterial membranes. Considering this, it is desirable to understand liposome–bacteria fusion and further optimize this fusion potential of liposomes. We have previously demonstrated a fusion process between fluid liposomes and bacteria by several techniques: 1) negative staining; 2) fluorescence activated cell sorter; 3) immunoelectron microscopy; and 4) lipid-mixing.34 Although this mechanism of interaction has been demonstrated, the main factors that drive or control this fusion process are still not completely clear. We therefore used the lipid-mixing method in the present study to identify and systematically investigate such parameters. Several controls were added in our study to confirm our measurements. When DPPC/DMPG (9:1 molar ratio) vesicles where mixed with bacteria in the presence of calcium (5 mM), a significant decrease in Rh fluorescence intensity (590 nm) and an increase in NBD signal (520 nm) was observed when time progressed (Figure 1A). This decrease (Figure 1B) cannot be attributed to lipid exchange, light scattering, or aggregation because, when vesicles composed of DPPC/DMPG/Chol (8:1:1) are mixed with bacteria in the same conditions as previously described, a significantly lower decrease in Rh fluorescence intensity was observed. These vesicles possess roughly the same surface charges as DPPC/DMPG (9:1) vesicles, but are slightly more stable due to the addition of cholesterol.
With few exceptions, all membrane fusion processes are regulated by three parameters: the nature of attacking vesicles, the property of the recipient membrane, and the potential fusogens that facilitate or regulate the fusion process. We will discuss these three parts in regard to the fusion between fluid liposomes and bacterial membranes in the following sections.
Nature of attacking vesicles
Liposomes’ fluid state has been shown to be an essential prerequisite to enhance the bactericidal activity of encapsulated antibiotics both in vivo and in vitro.18 Disaturated phosphatidylcholine/DMPG vesicles containing tobramycin were shown to lack the bactericidal enhancement capacity conferred by DPPC/DMPG vesicles. This requirement of fluidity was further confirmed in the present study by the lipid-mixing assay. Figure 6 shows that the percentage of fusion, between two distinct gram-negative bacteria and liposomes, is temperature-dependent. The rate of fusion decreases dramatically when temperatures are lower than the Tc of fluid liposomes (around 35°C). When cholesterol is added to liposomes, at concentrations as low as 10%, the degree of fusion is dramatically decreased (Figure 1B).
Property of recipient membrane
However, regardless of the fluidity of the liposomes, vesicles composed of DPPC/DMPG are not fusogenic by themselves. They can be fused with other actively fusogenic vesicles. Such nonfusogenic properties of DPPC vesicles are well documented by Ma et al.25 This same study found that vesicles composed of DPPC with or without negative lipids (up to 50%) are unable to fuse even if calcium is added. In our study, a similar observation was obtained by a lipid-mixing assay with a mixture of fluorescent-labeled and unlabeled liposomes as shown in Figure 1B. Upon mixing fluid liposomes with intact bacteria, a dramatic fusion was observed. Since it seems that the main driving force of such liposome–bacteria fusion is not due to the DPPC vesicles, it must come from the bacteria. It was later confirmed that the fusion between liposomes and intact bacteria was certainly dependent upon the nature of the bacterial membrane (Table 1). We suggest that the main driving force for the liposome–bacterial fusion is due to the nature of bacterial membranes. As indicated in Table 1, fluid liposomes fused with both gram-positive and gram-negative bacteria. In general, for the strains studied in our experiments, gram-negative bacteria gave a higher degree of fusion than gram-positive bacteria. It is well known that two of the most significant differences between gram-positive and gram-negative bacteria are an outer-leaflet layer of LPS in the outer membrane and a high amount of PE in the membranes of gram-negative bacteria. It is believed that the mechanisms involved in liposome–bacterial fusion may be different for gram-negative and gram-positive bacteria.
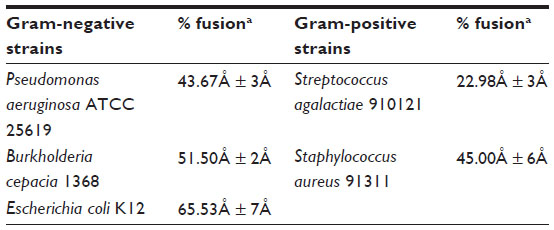 | Table 1 Comparison of the percentage of fusion between fluid liposomes and gram-positive bacteria |
In gram-negative bacteria, the LPS form protruding chains of polysaccharides, which are highly negatively charged, around the bacteria. In order to fuse with gram-negative bacteria, liposomes must therefore overcome this natural barrier. Only a few studies have been published on the interaction of liposomes with prokaryotic cells.32,33 Our result undoubtedly seems to correlate with these groups and, therefore, a possible mechanism to the fusion process between fluid liposomes and gram-negative bacteria was explained. Due to phosphatidylglycerol (PG) and LPS, liposomes have a negative charge. Long-range electrostatic repulsion usually prevents negatively charged vesicles from aggregation and fusion. However, aggregation can occur upon addition of calcium by bridging liposomes with bacteria. This would lead to neutralization of negative surface charges and dehydration of phospholipid headgroups, which would induce a local defect in membranes and fusion. Vesicle fusion induced by divalent cations requires that the lipids of the interacting membranes are in a fluid state (T≥Tc). This has been previously reported by different groups in the case of artificial negatively-charged vesicles.17,21,23 It is suggested that, under these conditions, membranes become transiently susceptible to fusion as a result of changes in molecular packing and creation of new phase boundaries induced by Ca2+. This requirement would further explain the importance of fluidity for fluid liposomes in order to achieve bactericidal enhancement and fusion. Following this stage of altered membranes, LPS are displaced by the divalent cations and fusion can occur with the inner membrane, which is mainly composed of PE, a fusogenic phospholipid. It is interesting to note that there is a correlation between the degree of liposome–bacterium fusion and the percentage of PE in the membrane. The inner layer of the outer membrane of P. aeruginosa, B. cepacia, and E. coli contains, respectively, 71%, 87%, and 91% PE phospholipids.35 The outer layer of gram-negative bacteria being composed predominantly of LPS indicated that the LPS can be fusion inducers. This would most likely be one of the reasons for the higher degree of fusion observed in gram-negative bacteria when compared to gram-positive bacteria, although the observed fusion difference may also be possible due to lack of PE in the membrane of the two gram-positive bacterial strains used for this study.
In the case of gram-positive bacteria, a higher degree of the liposome–bacteria fusion was also observed and correlated well to the membrane PG content. Forty-five percent fusion between liposomes and S. aureus (with 57% PG content) was observed while only 23% fusion for S. agalactiae (23% PG) was observed.36 Nevertheless, our suggested mechanism cannot fully explain the extent of fusion observed with gram-positive bacteria (Table 1), which lacks PE and LPS. An overall highly negatively-charged surface resulted from the presence of a higher percentage of PG in gram-positive bacterial membranes. Recently, Nikolaus et al27 observed membrane fusion and lamellar-to-inverted-hexagonal phase transition between negatively-charged vesicles composed of phospholipids similar to PG when calcium was added. Further studies with artificial liposomes composed of a high content of PG and gram-positive bacterial membranes are needed in order to identify the main inducers and understand their involvement.
Potential fusogens
Besides the involvement of liposomes’ fluidity and bacterial membrane characteristics, other fusogens or environmental factors such as divalent cations, pH, and temperature are known to affect membrane fusion. It is well known that calcium induces membrane fusion in several model liposomal vesicles. We demonstrated in this study that liposome–bacteria fusion is directly proportional to the amount of calcium in the medium (Figure 2). It is thought that calcium neutralizes the negative surface charges of membranes resulting in dehydration of cell surfaces.37 It has been suggested that hexagonal II phase lipids are important for membrane fusion and that calcium is able to trigger a bilayer-hexagonal II phase transition of some phospholipids.38 PE, the major phospholipid of gram-negative bacteria, can also adopt both bilayer and hexagonal II arrangements. This would suggest that fusion between liposomes and gram-negative bacteria is dependent on the presence of phospholipid in the outer leaflet of the outer membrane, although it is thought that the outer membrane outer leaflet of gram-negative bacteria is completely deficient in phospholipids.39 This may in fact be due to the effect of calcium, which has been reported to induce a loss of outer-membrane proteins directly leading to redistribution of lipids and increased levels of phospholipids in the outer membrane.40
Two different mechanisms of calcium-induced fusion have been reported in the literature. Fusion can be initialized by a calcium triggered enzymatic reaction41 or simply as a bridge that induces aggregation and destabilization.37 Presently, all of the evidence suggests that calcium with its divalent cationic charges plays a major role as a bridge rather than as an activator of phospholipase activity in liposome–bacteria fusion. Firstly, calcium-induced enzymatic reaction is ion-specific, but we observed similar fusion increases for other divalent cations as indicated in Figure 3. This is also supported by the fact that only liposomes in the fluid state are able to fuse with bacterial membranes. Secondly, fusion is triggered by different divalent cations, which increased in the sequence of Fe2+ > Mg2+ > Ca2+ > Ba2+. This difference between the different cations could be due to their different capacity to cause aggregation and fusion. The degree of aggregation between liposomes and bacteria would increase according to two parameters: first, each cation has an electronegative affinity to phosphatidylcholine decreasing in the sequence Mg2+ > Ca2+ > Ba2+ as demonstrated by Coughlin et al;34 and second, fusion could be dependent on the atomic radius of the ions. Figure 3 shows that the extent of fusion is inversely proportional to the atomic radius. The smaller the ions, the closer liposomes interact with the bacterial membrane and the higher the fusion between them. The present study demonstrates the importance of cations in liposome–bacterium fusion.
The pH of the reaction solution has also influenced liposome–bacteria fusion as shown in Figure 5. We were surprised to find that acidic pH and basic pH induced, respectively, 19% and 10% more fusion than neutral pH. One hypothesis is provided by Coughlin et al,34 who reported that the lamellar structure of LPS is stabilized at neutral pH both by ionic interactions within the LPS and by divalent cation bridges. Hydrogen bonds would be lost at basic pH resulting in an increase of hydration and an increase of negative charge of the headgroups. This would cause the formation of micellar structure or unstable outer membranes. In the acidic range, there would be reduction of the repulsion by charges and hydration due to an increase in hydrogen bonds. This would lead to insoluble complexes and would also be responsible for an unstable membrane. These altered membranes will therefore be more susceptible to easy fusion with liposomes. While no studies on the condition of liposomes at these pHs were done, one can easily apply the same reasoning to the vesicles. DMPG, which is negatively charged, could react in the same manner as LPS and cause destabilization of the membranes.
Conclusion
In summary, the nature of the bacterial membrane is identified as the most important driving force in liposome–bacteria fusion. However, the fluidity of liposomes, divalent cations, pH, and temperature all have important effects on the rate and extent of fusion. Based on the results of this study, a possible mechanism was outlined. The study suggests that a cationic-induced aggregation is the initial step in the fusion process. In addition, the fluid liposomal-encapsulated tobramycin was prepared, and the in vitro bactericidal efficacy of it to P. aeruginosa, S. maltophilia, B. cepacia, E. coli, and S. aureus was also investigated. The bactericidal effect occurred more quickly when bacteria were cultured with liposomal-encapsulated tobramycin (Figure 7). This cannot be explained as only a result of prolonged residence time of liposome-encapsulated tobramycin and the resulting release of entrapped antibiotic at the bacterial site, but the enhanced fusion process between liposomes and bacteria may explain the bactericidal effect.
Acknowledgments
This paper was supported by the Xi’an High-tech Zone of innovative drug incubation and industrialization transformed integrated platform project (Number 2011ZX09401-308), and the Shaanxi Provincial Natural Science Foundation of China (Number 2009JQ2013).
Disclosure
The authors report no conflicts of interest in this work.
References
Ren CL, Konstan MW, Yegin A, et al. Multiple antibiotic-resistant Pseudomonas aeruginosa and lung function decline in patients with cystic fibrosis. J Cyst Fibros. 2012;11:293–299. | |
Hodson ME, Simmonds NJ, Warwick WJ, et al. An international/multicentre report on patients with cystic fibrosis (CF) over the age of 40 years. J Cyst Fibros. 2008;7:537–542. | |
Gellen-Dautremer J, Bert F, Panhard X, Fantin B, Lefort A. Physicians fail to consider Pseudomonas aeruginosa as a potential pathogen in medicine patients with bacteremia. J Infect. 2011;63:99–101. | |
Kerr KG, Snelling AM. Pseudomonas aeruginosa: a formidable and ever-present adversary. J Hosp Infect. 2009;73:338–344. | |
Beaulac C, Clement-Major S, Hawari J, Lagace J. In vitro kinetics of drug release and pulmonary retention of microencapsulated antibiotics in liposomal formulations in relation to lipid composition. J Microencapsul. 1997;14:335–348. | |
Beaulac C, Clement-Major S, Hawari J, Lagace J. Eradication of mucoid Pseudomonas aeruginosa with fluid liposome-encapsulated tobramycin in an animal model of chronic pulmonary infection. Antimicrob Agents Chemother. 1996;40:665–669. | |
Beaulac C, Sachetelli S, Lagace J. In-vitro bactericidal efficacy of sub-MIC concentrations of liposomes-encapsulated antibiotic against Gram-negative and Gram-positive bacteria. J Antimicrob Chemother. 1998;41:35–41. | |
Sachetelli S, Khalil H, Chen T, Beaulac C, Sénéchal S, Lagacé J. Demonstration of a fusion mechanism between a fluid bactericidal liposomal formulation and bacterial cells. Biochim Biophys Acta. 2000;1463:254–266. | |
Miller C, Racker E. Ca+-induced fusion of fragmented sarcoplasmic reticulum with artificial planar bilayers. J Membr Biol. 1976;30:283–300. | |
Oteiza PI, Verstraeten SV. Interactions of al and related metals with membrane phospholipids: consequences on membrane physical properties. In: Liu AL, editor. Advances in Planar Lipid Bilayers and Liposomes. Vol 4. London: Academic Press; 2006:79–106. | |
Poole AR, Howell JI, Lucy JA. Lysolecithin and cell fusion. Nature. 1970;227:810–814. | |
Wilschut J, Duzgunes N, Papahadjopoulos D. Calcium/magnesium specificity in membrane fusion: kinetics of aggregation and fusion of phosphatidylserine vesicles and the role of bilayer curvature. Biochem. 1981;20:3126–3133. | |
Kim C, Spano J, Park EK, Wi S. Evidence of pores and thinned lipid bilayers induced in oriented lipid membranes interacting with the antimicrobial peptides, magainin-2 and aurein-3.3. Biochim Biophys Acta. 2009;1788:1482–1496. | |
Tarahovsky YS, Yagolnik EA, Muzafarov EN, Abdrasilov BS, Kim YA. Calcium-dependent aggregation and fusion of phosphatidylcholine liposomes induced by complexes of flavonoids with divalent iron. Biochim Biophys Acta. 2012;1818:695–702. | |
Uster PS. In situ resonance energy transfer microscopy: Monitoring membrane fusion in living cells. Method Enzymol. 1993;221:239–246. | |
Gerritsen WJ, Verkleij AJ, Van Deenen LL. The lateral distribution of intramembrane particles in the erythrocyte membrane and recombinant vesicles. Biochim Biophys Acta. 1979;555:26–41. | |
Sugiki T, Takahashi H, Nagasu M, Hanada K, Shimada I. Real-time assay method of lipid extraction activity. Anal Biochem. 2010;399:162–167. | |
Chen T, Choi LS, Einstein S, Klippenstein MA, Scherrer P, Cullis PR. Proton-induced permeability and fusion of large unilamellar vesicles by covalently conjugated poly(2-ethylacrylic acid). J. Liposome Res. 1999;9:387–405. | |
Trikash I, Gumenyuk V, Lishko V. The fusion of synaptic vesicle membranes studied by lipid mixing: the R18 fluorescence assay validity. Chem Phys Lipids. 2010;163:778–786. | |
Nir S, Bentz J, Wilshut J, Düzgünes N. Binding of monovalent cations to phosphatidylserine and modulation of Ca2+- and Mg2+-induced vesicle fusion. Biochim Biophys Acta. 1983;735:160–172. | |
Belenky P, Bogan KL, Brenner C. NAD+ metabolism in health and disease. Trends Biochem Sci. 2007;32:12–19. | |
Papahadjopoulos D, Vail WJ, Pangborn WA, Poste G. Studies on membrane fusion. I. Interactions of pure phospholipid membranes and the effect of myristic acid, lysolecithin, proteins and dimethylsulfoxide. Biochim Biophys Acta. 1976;448:245–264. | |
Ortiz A, Killian JA, Verkleij AJ, Wilschut J. Membrane fusion and the lamellar-to-inverted-hexagonal phase transition in cardiolipin vesicle systems induced by divalent cations. Biophys J. 1999;77:2003–2014. | |
Sanchez-Migallon MP, Aranda FJ, Gomez-Fernandez JC. The dissimilar effect of diacylglycerols on Ca(2+)-induced phosphatidylserine vesicle fusion. Biophys J. 1995;68:558–566. | |
Ma Y, Wang Z, Zhao W, et al. Enhanced bactericidal potency of nanoliposomes by modification of the fusion activity between liposomes and bacterium. Int J Nanomedicine. 2013;8:2351–2360. | |
Rogasevskaia TP, Churchward MA, Coorssen JR. Anionic lipids in Ca2+-triggered fusion. Cell Calcium. 2012;52:259–269. | |
Nikolaus J, Warner JM, O’Shaughnessy B, Herrmann A. The pathway to membrane fusion through hemifusion. Curr Top Membr. 2011;68:1–32. | |
Sahay G, Alakhova DY, Kabanov AV. Endocytosis of nanomedicines. J Control Release. 2010;145:182–195. | |
Komatsu H, Okada S. Ethanol-induced aggregation and fusion of small phosphatidylcholine liposome: participation of interdigitated membrane formation in their processes. Biochim Biophys Acta. 1995;1235:270–280. | |
Lorizate M, Huarte N, Sáez-Cirión A, Nieva JL. Interfacial pre-transmembrane domains in viral proteins promoting membrane fusion and fission. Biochim Biophys Acta. 2008;1778:1624–1639. | |
Nicolau C, Rottem S. Expression of a beta-lactamase activity in Mycoplasma capricolum transfected with the liposome-encapsulated E.coli pBR 322 plasmid. Biochem Biophys Res Commun. 1982;108:982–986. | |
Zhang XX, McIntosh TJ, Grinstaff MW. Functional lipids and lipoplexes for improved gene delivery. Biochimie. 2012;94:42–58. | |
Tomlinson S, Taylor PW, Luzio JP. Transfer of phospholipid and protein into the envelope of gram-negative bacteria by liposome fusion. Biochemistry. 1989;28:8303–8311. | |
Coughlin RT, Peterson AA, Haug A, Pownall HJ, McGroarty EJ. A pH titration study on the ionic bridging within lipopolysaccharide aggregates. Biochim Biophys Acta. 1985;821:404–412. | |
Cook PL, Vanderhill JL, Cook AE, Van Norstrand DW, Gordon MT, Harper PE. Light scattering measurement and Avrami analysis of the lamellar to inverse hexagonal phase transition kinetics of the lipid DEPE. Chem Phys Lipids. 2012;165:270–276. | |
Siegel DP, Epand RM. The mechanism of lamellar-to-inverted hexagonal phase transitions in phosphatidylethanolamine: implications for membrane fusion mechanisms. Biophys J. 1997;73:3089–3111. | |
Evans CT, Ratledge C, Gilbert SC. A rapid screening method for lipid-accumulating yeast using a replica-printing technique. J Microbiol Meth. 1985;4:203–210. | |
Lugtenberg B, Van Alphen L. Molecular architecture and functioning of the outer membrane of Escherichia coli and other gram-negative bacteria. Biochim Biophys Acta. 1983;737:51–115. | |
Stan-Lotter H, Sanderson KE. Interactions of cations with membrane fractions of smooth and rough strains of Salmonella typhimurium and other Gram-negative bacteria. J Bacteriol. 1981;146:542–551. | |
Park JB, Lee TH, Kim H. Fusion of phospholipid vesicles induced by phospholipase D in the presence of calcium ion. Biochem Int. 1992;27:417–422. | |
Karabaliev M. Effects of divalent cations on the formation and structure of solid supported lipid films. Bioelectrochemistry. 2007;71:54–59. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.