Back to Journals » Drug Design, Development and Therapy » Volume 11
Functional characterization of wild-type and 24 CYP2D6 allelic variants on gefitinib metabolism in vitro
Authors Fang P, Zheng X, He JY, Ge HL, Tang PF, Cai JP ![]() , Hu GX
, Hu GX
Received 3 February 2017
Accepted for publication 30 March 2017
Published 21 April 2017 Volume 2017:11 Pages 1283—1290
DOI https://doi.org/10.2147/DDDT.S133814
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Qiongyu Guo
Ping Fang,1 Xiang Zheng,1 Jiayang He,1 Honglei Ge,1 Pengfei Tang,1 Jianping Cai,2 Guoxin Hu1
1Department of Pharmacology, School of Pharmacy, Wenzhou Medical University, Wenzhou, Zhejiang, 2The Ministry of Health (MOH) Key Laboratory of Geriatrics, Beijing Hospital, National Center of Gerontology, Beijing, People’s Republic of China
Background: Cytochrome P450 2D6 (CYP2D6), a member of the CYP450 enzyme super family, is a polymorphic enzyme that metabolizes ~25% of therapeutic drugs. CYP2D6 exhibits significant genetic polymorphisms which might cause adverse effects and therapeutic failures of some drugs.
Objective: The purpose of this study was to evaluate the catalytic activities of 22 novel CYP2D6 alleles (CYP2D6*87, *88, *89, *90, *91, *92, *93, *94, *95, *96, *97, *98, R25Q, F164L, E215K, F219S, V327M, D336N, V342M, R344Q, R440C, R497C) on the metabolism of gefitinib in vitro.
Methods and results: CYP2D6 variants were incubated with 1–100 µM gefitinib for 60 min at 37°C and the reaction was terminated by cooling to -80°C immediately. Gefitinib and its metabolite O-desmethyl gefitinib were analyzed by an ultra-performance liquid chromatography-tandem mass spectrometry system. Compared to CYP2D6.1, most CYP2D6 variants exhibited significantly decreased relative clearance values (from 3.11% to 79.35%), whereas CYP2D6.92 and CYP2D6.96 displayed no detectable enzyme activity. Only CYP2D6.94 exhibited a markedly increased intrinsic clearance value, and eight variants (CYP2D6.88, CYP2D6.89, CYP2D6.91, CYP2D6.97, V342M, R344Q, F219S, and F164L) showed no significant difference. In addition, 23 CYP2D6 allelic isoforms exhibited substrate inhibition trend toward gefitinib.
Conclusion: As the first study of all the aforementioned alleles for gefitinib metabolism, these comprehensive data may help in the clinical assessment of the metabolism of gefitinib, and may also offer a reference for personalized treatment with gefitinib in clinical settings.
Keywords: CYP2D6, genetic polymorphism, drug metabolism, gefitinib, enzymatic activity, personalized treatment, NSCLC
Introduction
Lung cancer remains the most frequently diagnosed malignancy and the leading cause of cancer-related death worldwide.1–3 Approximately 80%–85% of all lung cancers are non-small cell lung cancer (NSCLC) with ~55% of these presenting with locally advanced or metastatic disease causing the 5-year survival rate to be as low as <5%.1,3 In recent years, the discovery of the epidermal growth factor receptor tyrosine kinase inhibitor (EGFR-TKI) brings a new hope for patients in the treatment of advanced NSCLC. EGFR is a known transmembrane receptor, which upon ligand binding, induces receptor dimerization and tyrosine autophosphorylation and leads to cell proliferation.4–6 Mutation in this gene is significantly associated with lung cancer.7 In previous studies, the EGFR is detected by immunohistochemistry on 40%–80% of NSCLC.8,9 Accordingly, promising molecular inhibitors targeting the activating mutants of the EGFR have been intensely pursued in the treatment of EGFR-mutant positive NSCLC.
In the last decade, two small molecules, selective, orally bioavailable and reversible EGFR-TKIs gefitinib and erlotinib have been extensively developed in NSCLC.3 Gefitinib (Figure 1A) is the first EGFR-TKI, which has been approved as early as 2002 for the treatment of advanced NSCLC patients. In 2003, gefitinib received accelerated approval by the US Food and Drug Administration (FDA) as third-line treatment for advanced EGFR-mutated NSCLC patients. Then, in 2015 it was approved as the first-line treatment of patients with metastatic NSCLC harboring activating EGFR mutations by FDA.10–12 Gefitinib is metabolized by the hepatic cytochrome P450 (CYP450) enzyme CYP2D6, CYP3A4, and CYP3A5. And CYP2D6 is the enzyme primarily responsible for the formation of O-desmethyl gefitinib (Figure 1B).13 Previous studies indicate that among gefitinib metabolites, only O-desmethyl gefitinib can be identified in human plasma, whereas the plasma concentrations of other gefitinib metabolites, such as M537194 and M387783, which are metabolized by CYP3A4 are very low or even undetectable.2,13–16 There are several studies showing that patients with poor metabolizers (PMs) of CYP2D6 and inhibition of CYP3A4 may lead to prominent overdose of gefitinib, which may finally cause gefitinib-induced hepatotoxicity.13,16 Therefore, we believe that it is necessary to evaluate the effects of 24 CYP2D6 variants on the metabolism of gefitinib in vitro.
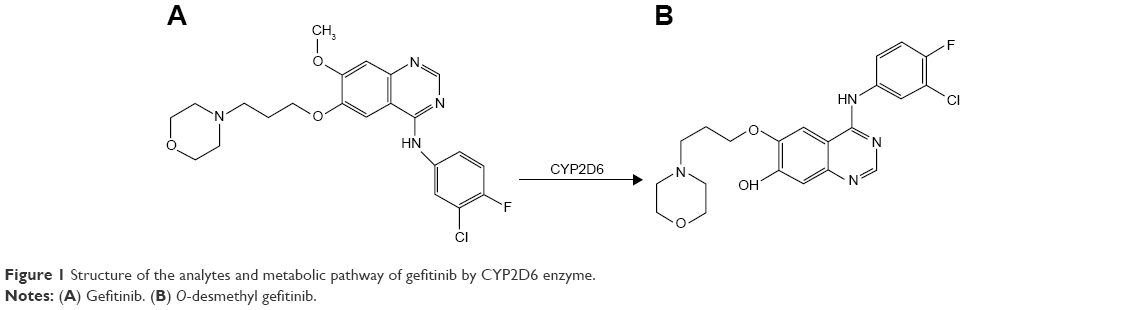 | Figure 1 Structure of the analytes and metabolic pathway of gefitinib by CYP2D6 enzyme. |
As a member of CYP450 enzyme superfamily, CYP450 2D6 plays a vital role in the metabolism of ~20%–30% of therapeutically important drugs, mainly including antipsychotic drugs, antiarrhythmic agents, tricyclic antidepressants, β-blockers, and opioids.17,18 Like other CYP450 enzymes, CYP2D6 exhibits significant genetic polymorphisms between individuals. To date, over 100 CYP2D6 alleles have been identified and named by the Human CYP Allele Nomenclature committee (http://www.cypalleles.ki.se/cyp2d6.htm). The genetic polymorphisms in CYP2D6 cause the tremendous variation in CYP2D6 enzyme activities. Thus, individuals can be classified into four phenotypes: PMs, intermediate metabolizers, extensive metabolizers (EMs), or ultrarapid metabolizers. Genetic polymorphisms in CYP2D6 have been shown to have clinical consequences, resulting in toxicity and/or altered efficacy of many drugs. Thus, the identification of the polymorphic allele, especially defective alleles, in different racial or ethnic populations, is of great importance to understand the differences in the clinical response to drugs.19
Previously, 22 new nonsynonymous mutation sites among the 2,129 people of the Chinese Han population were discovered, 12 of them were defined as the novel alleles (CYP2D6*87–*93, *94A, *94B, and *95–*98) by the Human CYP Allele Nomenclature committee.20 In our study, we systematically assessed the enzymatic characteristics of these 22 CYP2D6 allelic variants and 2 typical variants (CYP2D6.2 and CYP2D6.10) as well as the wild type CYP2D6.1 toward gefitinib. And we hope that our data can provide valuable information about CYP2D6 genetic polymorphisms and offer a reference to the personalized medication for CYP2D6-metabolized drugs.
Materials and methods
Chemicals and reagents
Gefitinib and its metabolite O-desmethyl gefitinib were purchased from Shanghai Canspec Scientific Instruments Co., Ltd (Perfemiker, Shanghai, China), the reduced nicotinamide adenine dinucleotide phosphate (NADPH) and carbamazepine (internal standard, IS) were obtained from Sigma-Aldrich Company (St Louis, MO, USA). P450 Cytochrome b5 microsomes and recombinant human CYP2D6 microsomes were kind gifts from Beijing Hospital (Beijing, China).21 High performance liquid chromatography (HPLC) grade organic solvents and LC-MS grade acetonitrile were purchased from Merck (Darmstadt, Germany), formic acid of HPLC grade (FA, purity 99.9%) was purchased from J&K scientific Ltd. (Shanghai, China), and all other reagents used were of analytical grade.
Instrumentation
Samples were analyzed by ultra-performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS) using a Waters Acquity I-Class and a Waters Xevo TQD triple-quadrupole mass spectrometry (Waters Corp., Milford, MA, USA) with an electrospray ionization source. Instrument control and data acquisition were performed by Masslynx 4.1 software (Waters Corp.).
Conditions for enzymatic activity analysis
Recombinant microsomes expressing CYP2D6 variants generated in Spodoptera frugioerda 21 (Sf21) insect cells were obtained according to the previously reported methods.21 Western blotting was performed to determine protein expression levels and the content of microsomes was determined using a BCA Protein Assay Kit with BSA as a standard.21 The volume of incubation mixture is 200 μL, which consisted of microsomes (5 pmol CYP2D6.1 or 5 pmol other CYP2D6 mutants, respectively), purified cytochrome b5, gefitinib (1–100 μM) and 0.1 M Tris-HCl buffer (pH 7.4). The reaction was allowed to preincubate for 5 min in a Fisher shaking water bath. Then 1 mM NADPH was added to start the reaction at 37°C. And incubations proceeded at 37°C for 60 min. Reactions were terminated by cooling to −80°C immediately. Then 400 μL acetonitrile and 20 μL carbamazepine (internal standard, 1 μg/mL) were added to the incubation mixture, which was then vortexed for 2 min and centrifuged at 13,000 rpm at 4°C for 10 min. The supernatant was 1:1 diluted with water, and 2 μL of the mixture was injected into the UPLC-MS/MS system for analysis. Incubations were performed in triplicate and data presented as the mean ± standard deviation.
Chromatographic and mass spectrometric conditions
The chromatographic separation was carried out using an ACQUITY UPLC-MS/MS and performed on a Waters ACQUITY UPLC BEH C18 column (2.1×50 mm, 1.7 μm, Waters Corp.) at 40°C. The initial mobile phase consisted of solvent A (water containing 0.1% formic acid) and solvent B (acetonitrile) with gradient elution at a flow rate of 0.4 mL/min, injection volume 2 μL. A gradient elution program was as follows: 0–0.6 min (60%–10% A), 0.6–1.2 min (10% A), 1.2–1.8 min (10%–60% A). The total run time was 2.5 min. Under above conditions, O-desmethyl gefitinib and carbamazepine were well separated and their retention times were 0.35 and 0.77 min, respectively.
A Waters XEVO TQD MS was set to positive electrospray ionization in multiple reaction monitoring mode, with ionization conditions as follows: desolvation temperature 500°C, ionization source temperature 150°C, and capillary voltage 2,000 V. Nitrogen was served as desolvation gas with flow rate of 1,000 L/h. The multiple reaction monitoring transitions were m/z 447.2→128.1, m/z 433.1→128.1, and m/z 237.1→194.2 for gefitinib, O-desmethyl gefitinib, and carbamazepine, respectively. The collision energy for gefitinib, O-desmethyl gefitinib, and carbamazepine was set at 25, 25, and 20 V, respectively; and the cone voltage for each was set at 40 V.
Prior to the experiments, the UPLC-MS/MS method was validated. A seven-point standard curve in the range of 1–50 μg/mL was used to quantify O-desmethyl gefitinib, with a correlation coefficient r2>0.99. The limit of quantification, defined as a signal/noise ratio of 3, was 0.2 ng/mL for O-desmethyl gefitinib. The precision and accuracy of the method were assessed by determination of quality control samples at three concentrations. The relative standard deviations of intra-day and inter-day precisions were both below 10%; the accuracy was from 87.77% to 93.73%. These results demonstrated that the method was accurate and precise.
Statistical analysis
Michaelis–Menten curves (substrate vs velocity) and enzyme kinetic parameters were obtained by using GraphPad Prism 5 (GraphPad Software Inc., SanDiego, CA, USA). One-way analysis of variance (ANOVA) was used for intergroup comparison, and various variants were considered as factor whereas maximum initial velocity (Vmax), Michaelis constant (Km) or intrinsic clearance (Clint) values were deemed as dependent list. Statistical analysis was carried out with the Statistical Package for the Social Sciences (version 17.0; SPSS Inc., Chicago, IL, USA) by one-way ANOVA together with the post hoc Dunnet method, with *P<0.05 considered to be statistically significant.
Results
In our study, the catalytic activities of the wild-type CYP2D6.1 and 24 allelic variants were detected using gefitinib as substrate and intrinsic clearance (Vmax/Km) was used as evaluation criteria for each variant of CYP2D6 in vitro metabolic activity of gefitinib. The Michaelis–Menten curves and substrate inhibition plots for each of the CYP2D6 variants are shown in Figure 2, and the corresponding kinetic parameters (Vmax, Km, Ksi, intrinsic clearance, and relative clearance) are summarized in Table 1.
 | Figure 2 Michaelis–Menten curves of the enzymatic activity of the wild-type and 24 variants toward gefitinib O-demethylation. |
 | Table 1 Kinetic parameters for O-desmethyl gefitinib activities of recombinant wild-type and 24 mutant CYP2D6 proteins against gefitinib |
As shown in Table 1, among all these variants, CYP2D6.92 and CYP2D6.96 showed no enzymatic activity as no concentration of O-desmethyl gefitinib was detected. Except for CYP2D6.92 and CYP2D6.96, the remaining 22 variants displayed considerable differences in Km, Vmax, Ksi, or intrinsic clearance values compared with wild-type CYP2D6.1. Three allelic variants (CYP2D6.10, CYP2D6.93, and E215K) exhibited lower Vmax values and eight variants (CYP2D6.2, CYP2D6.10, CYP2D6.98, R25Q, E215K, D336N, R344Q, and R440C) had higher Km values than that of CYP2D6.1 (*P<0.05). According to the intrinsic clearance values when compared with the wild-type, only one variant (CYP2D6.94) had increased catalytic activity to 184.78%, whereas the remaining 13 variants (CYP2D6.2, CYP2D6.10, CYP2D6.87, CYP2D6.90, CYP2D6.93, CYP2D6.95, CYP2D6.98, R25Q, E215K, V327M, D336N, R440C, and R497C) had a significant decrease in enzyme activities compared with CYP2D6.1 (*P<0.05), for which the percentage varied from 3.11% to 69.42%. Especially, the intrinsic clearance values for variants CYP2D6.10, CYP2D6.93, and E215K were decreased to 3.11%, 3.68%, and 9.79% compared with CYP2D6.1, respectively. However, through the analysis of the kinetic parameters, there are eight variants (CYP2D6.88, CYP2D6.89, CYP2D6.91, CYP2D6.97, V342M, R344Q, F219S, and F164L) which showed no marked difference in intrinsic clearance values compared with wild type.
In addition, different from the previous studies,19,21–24 the metabolic ability toward gefitinib in vitro exhibited substrate inhibition trend, the inhibition constant of 25 tested CYP2D6 variants varied from nearly 5.9 to 158.1 μM.
Discussion
CYP2D6 plays a major role in the oxidation of ~20%–30% prescription drugs, and it is involved in the metabolism of gefitinib to its active metabolite, O-desmethyl gefitinib. According to the research, the formation of O-desmethyl gefitinib seems to be dependent on CYP2D6 activity. Therefore, polymorphisms in the CYP2D6 gene can greatly influence the rate of elimination of gefitinib. Individuals who are PMs of CYP2D6 have twofold higher plasma concentrations of gefitinib than EMs.14 Up to now, the enzymatic characteristics of CYP2D6 alleles toward gefitinib have not been well studied both in vivo and in vitro.
CYP2D6.2 and CYP2D6.10 tend to undergo the most study of the variants. In order to improve the credibility of our experiment, CYP2D6.2 and CYP2D6.10, two typical variants, served as the positive controls for the functional analyses. CYP2D6*10 is the most common allele in Asians, occurring in 42.86% of the Chinese Han population.20,25 Several functional analyses have been shown that CYP2D6.10 has higher Km, lower Vmax and behave with the least Vmax/Km values for dextromethorphan, bufuralol, nortriptyline, atomoxetine, and debrisoquine in vitro.21,25 CYP2D6*10 is generally considered to be an allele associated with the intermediate metabolizer phenotype. For typical defective allele CYP2D6*2, Cai et al found it exhibited lower intrinsic clearance value for dextromethorphan and bufuralol, ~43.85% and 40.41% of wild type, respectively.21 As a consequence, in our study the relative clearance of CYP2D6.2 (23.43% of CYP2D6.1) and CYP2D6.10 (3.11% of CYP2D6.1) were both presented significantly decreased. These results were consistent with previous work and indicate that it is appropriate to use our CYP expression and in vitro metabolism system for analyzing the catalytic activities of the other CYP2D6 variants.21,22,26,27
To better understand the enzymatic characteristics of CYP2D6 allelic isoforms and their effects on the metabolism of gefitinib in vitro, we functionally analyzed the enzymatic activity of 22 novel CYP2D6 alleles toward gefitinib using recombinant insect microsomes. We found that some of the variants showed similar trend about the enzyme activity as studies reported,21,27 but some caused increased or no change in enzyme activity, which is not in accordance with the results of previous research. It indicates that CYP2D6 not only exhibits significant genetic polymorphisms between individuals and among different races and ethnicities, but also showed marked differences on the metabolism of various clinical drugs. Therefore, it is of great necessity to undertake further research on the function of CYP2D6 alleles on the metabolism of gefitinib.
In our study, both CYP2D6.92 and CYP2D6.96 had undetectable enzyme activity in gefitinib metabolism, similar to previous reports.19,21,23,24 The reason for this may be because both of these variants were expressed as truncated proteins due to the premature termination in protein translation process, which led to the function loss of enzymatic activity. Particularly, only one variant CYP2D6.94, with a decrease Km and an increase Vmax, showed significant increased relative clearance compared with CYP2D6.1, suggesting higher catalytic activity than CYP2D6.1. On the contrary, as reported in other studies, CYP2D6.94 exhibited markedly different decreased catalytic activity toward propranolol (31.37%), nebivolol (58.23%), dextromethorphan (67.03%), venlafaxine (71.9%), and bufuralol (74.96%) in vitro.19,21,23,24 These data suggest that CYP2D6.94 may be reclassified as an ultrarapid metabolizer phenotype for gefitinib. Moreover, CYP2D6.88, CYP2D6.89, CYP2D6.91, CYP2D6.97, F164L, F219S, and V342M were found in Chinese people and exhibited similar metabolic activity of CYP2D6.1 for gefitinib. These results were significantly different from previous studies. Cai et al showed that CYP2D6.91, CYP2D6.97, F164L, F219S, and V342M exhibited markedly decreased catalytic activity toward bufuralol and dextromethorphan. In addition, CYP2D6.88 and CYP2D6.89 exhibited decreased intrinsic clearance for bufuralol as compared with wild type but no significant difference toward dextromethorphan.21 These results suggest that CYP2D6 had substrate-specific alterations in metabolic activity and that results obtained from one substrate cannot necessarily be extrapolated to other substrates. We speculate that the different affinity of enzyme and substrate might be responsible for this inconsistency.
It is important to point out that two allelic variants CYP2D6.93 and E215K exhibited a drastic decrease in enzymatic activity. CYP2D6.93 exhibited ~94.4% lower Vmax, 1.53-fold higher Km, and 96.32% lower intrinsic clearance values compared with wild type. As for this result, Dai et al speculated that the normal entrance of the CYP2D6 substrate might be blocked, resulting from the replacement of the hydrophilic Thr with the hydrophilic Pro at position 249, which would influence the spatial structure of the adjacent egress channel. E215K contains an amino acid switch from Glu to Lys at position 215, which is located in F-helix in the active site cavity of the CYP2D6 protein.27 That may be related with E215K’s extremely decreased intrinsic clearance values (9.79%) of CYP2D6.1 owing to high Km (5.67-fold). Our data suggests that CYP2D6.93 and E215K may have to be associated with a PM phenotype in vivo, with gefitinib as a substrate. Several studies have reported that patients with NSCLC who possessed PM phenotype have severe hepatotoxicity after treatment with gefitinib.16,28 Our work may serve as a reference for rational administration clinically, thereby can avoid adverse drug reactions such as gefitinib-induced hepatotoxicity.
Furthermore, the most meaningful difference between our study and previous reports19,21–24 is that gefitinib exhibited a substrate inhibition trend of metabolic ability by CYP2D6 in vitro. Twenty-three tested CYP2D6 allelic variants showed this substrate inhibition trend toward gefitinib and their Ksi values varied from ~6 to 158 μM. As reported in a study by Swaisland et al, suggested that gefitinib produced a little inhibition of CYP2D6 isoenzyme activity which resulted in a small increase in the metoprolol area under the curve and the maximum serum concentration.29 Hereby, we know that gefitinib was both an inhibitor and a substrate of CYP2D6. That might be the main reason for this substrate inhibition. In addition, we believe that it is still valuable to pay more consideration to the dose of CYP2D6 substrates which have a narrow therapeutic window when in the presence of gefitinib.
Conclusion
In summary, we systematically assessed the enzymatic activity of 24 CYP2D6 allelic isoforms on the metabolism of gefitinib. Our results indicate that most of these CYP2D6 variants exhibit significantly decreased enzymatic activities. In addition, it has been reported that inhibition of CYP3A4 leads to a shunt to a CYP2D6-dependent metabolic pathway. Therefore, we speculate that patients with these dysfunctional CYP2D6 alleles should reduce the dosage of gefitinib in order to avoid gefitinib-induced hepatotoxicity when functions of CYP3A4 are also inhibited. Moreover, further studies in vivo or in vitro on the effects of CYP2D6 and CYP3A4 toward gefitinib are required to make sense of the relation between CYPs and gefitinib clearance. As the first study of these alleles for gefitinib metabolism, it may help in guiding individualization and optimization in patient treatment.
Acknowledgments
This work was supported by the Ministry of Health, National Health and Family Planning Commission of the People’s Republic of China. The authors thank the members of the Beijing Institute of Geriatrics of the Ministry of Health for their advice and assistance.
Disclosure
The authors report no conflicts of interest in this work.
References
Cappuzzo F, Morabito A, Normanno N, et al. Efficacy and safety of rechallenge treatment with gefitinib in patients with advanced non-small cell lung cancer. Lung Cancer. 2016;99:31–37. | ||
Han SY, Zhao HY, Zhou N, Zhou F, Li PP. Marsdenia tenacissima extract inhibits gefitinib metabolism in vitro by interfering with human hepatic CYP3A4 and CYP2D6 enzymes. J Ethnopharmacol. 2014;151(1):210–217. | ||
Nurwidya F, Takahashi F, Takahashi K. Gefitinib in the treatment of nonsmall cell lung cancer with activating epidermal growth factor receptor mutation. J Nat Sci Biol Med. 2016;7(2):119–123. | ||
Bocharov EV, Lesovoy DM, Pavlov KV, Pustovalova YE, Bocharova OV, Arseniev AS. Alternative packing of EGFR transmembrane domain suggests that protein-lipid interactions underlie signal conduction across membrane. Biochim Biophys Acta. 2016;1858(6):1254–1261. | ||
Bremer E, Samplonius DF, van Genne L, et al. Simultaneous inhibition of epidermal growth factor receptor (EGFR) signaling and enhanced activation of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) receptor-mediated apoptosis induction by an scFv:sTRAIL fusion protein with specificity for human EGFR. J Biol Chem. 2005;280(11):10025–10033. | ||
Yu X, Sharma KD, Takahashi T, Iwamoto R, Mekada E. Ligand-independent dimer formation of epidermal growth factor receptor (EGFR) is a step separable from ligand-induced EGFR signaling. Mol Biol Cell. 2002;13(7):2547–2557. | ||
Zhang W, Stabile LP, Keohavong P, et al. Mutation and polymorphism in the EGFR-TK domain associated with lung cancer. J Thorac Oncol. 2006;1(7):635–647. | ||
Ettinger DS. Clinical implications of EGFR expression in the development and progression of solid tumors: focus on non-small cell lung cancer. Oncologist. 2006;11(4):358–373. | ||
Kim CH, Kim SH, Park SY, Yoo J, Kim SK, Kim HK. Identification of EGFR Mutations by Immunohistochemistry with EGFR Mutation-Specific Antibodies in Biopsy and Resection Specimens from Pulmonary Adenocarcinoma. Cancer Res Treat. 2015;47(4):653–660. | ||
Hsiue EH, Lee JH, Lin CC, Yang JC. Safety of gefitinib in non-small cell lung cancer treatment. Expert Opin Drug Saf. 2016;15(7):993–1000. | ||
Cohen MH, Williams GA, Sridhara R, et al. United States Food and Drug Administration Drug Approval summary: Gefitinib (ZD1839; Iressa) tablets. Clin Cancer Res. 2004;10(4):1212–1218. | ||
Prommer E. Gefitinib: a new agent in palliative care. Am J Hosp Palliati Care. 2004;21(3):222–227. | ||
McKillop D, McCormick AD, Millar A, Miles GS, Phillips PJ, Hutchison M. Cytochrome P450-dependent metabolism of gefitinib. Xenobiotica. 2005;35(1):39–50. | ||
Kobayashi H, Sato K, Niioka T, et al. Effects of polymorphisms in CYP2D6 and ABC transporters and side effects induced by gefitinib on the pharmacokinetics of the gefitinib metabolite, O-desmethyl gefitinib. Med Oncol. 2016;33(6):57. | ||
McKillop D, Guy SP, Spence MP, et al. Minimal contribution of desmethyl-gefitinib, the major human plasma metabolite of gefitinib, to epidermal growth factor receptor (EGFR)-mediated tumour growth inhibition. Xenobiotica. 2006;36(1):29–39. | ||
Takimoto T, Kijima T, Otani Y, et al. Polymorphisms of CYP2D6 gene and gefitinib-induced hepatotoxicity. Clin Lung Cancer. 2013;14(5):502–507. | ||
Zanger UM, Raimundo S, Eichelbaum M. Cytochrome P450 2D6: overview and update on pharmacology, genetics, biochemistry. Naunyn Schmiedebergs Arch Pharmacol. 2004;369(1):23–37. | ||
Zhou SF. Polymorphism of human cytochrome P450 2D6 and its clinical significance: part I. Clin Pharmacokinet. 2009;48(11):689–723. | ||
Liang B, Zhan Y, Huang X, et al. Effect of 22 novel cytochrome P450 2D6 (CYP2D6) variants found in the Chinese Population on Hemangeol metabolism in vitro. Eur J Drug Metab Pharmacokinet. 2016;41(6):759–765. | ||
Qian JC, Xu XM, Hu GX, et al. Genetic variations of human CYP2D6 in the Chinese Han population. Pharmacogenomics. 2013;14(14):1731–1743. | ||
Cai J, Dai DP, Geng PW, et al. Effects of 22 novel CYP2D6 variants found in the Chinese Population on the Bufuralol and Dextromethorphan metabolisms in vitro. Basic Clin Pharmacol Toxicol. 2016;118(3):190–199. | ||
Liang B, Zhan Y, Wang Y, et al. Effect of 24 cytochrome P450 2D6 variants found in the Chinese Population on Atomoxetine metabolism in vitro. Pharmacology. 2016;97(1–2):78–83. | ||
Zhan YY, Liang BQ, Wang H, et al. Effect of CYP2D6 variants on venlafaxine metabolism in vitro. Xenobiotica. 2016;46(5):424–429. | ||
Hu X, Lan T, Dai D, et al. Evaluation of 24 CYP2D6 variants on the metabolism of nebivolol in vitro. Drug Metab Dispos. 2016;44(11):1828–1831. | ||
Byeon JY, Kim YH, Na HS, et al. Effects of the CYP2D6*10 allele on the pharmacokinetics of atomoxetine and its metabolites. Arch Pharm Res. 2015;38(11):2083–2091. | ||
Shen H, He MM, Liu H, et al. Comparative metabolic capabilities and inhibitory profiles of CYP2D6.1, CYP2D6.10, and CYP2D6.17. Drug Metab Dispos. 2007;35(8):1292–1300. | ||
Dai DP, Geng PW, Wang SH, et al. In vitro functional assessment of 22 newly identified CYP2D6 allelic variants in the Chinese population. Basic Clin Pharmacol Toxicol. 2015;117(1):39–43. | ||
Kijima T, Shimizu T, Nonen S, et al. Safe and successful treatment with erlotinib after gefitinib-induced hepatotoxicity: difference in metabolism as a possible mechanism. J Clin Oncol. 2011;29(19):e588–e590. | ||
Swaisland HC, Ranson M, Smith RP, et al. Pharmacokinetic drug interactions of gefitinib with rifampicin, itraconazole and metoprolol. Clin Pharmacokinet. 2005;44(10):1067–1081. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.