Back to Journals » Infection and Drug Resistance » Volume 12
Functional characteristics of CYP3A4 allelic variants on the metabolism of loperamide in vitro
Authors Lin QM ![]() , Li YH, Liu Q, Pang NH, Xu RA, Cai JP
, Li YH, Liu Q, Pang NH, Xu RA, Cai JP ![]() , Hu GX
, Hu GX ![]()
Received 9 May 2019
Accepted for publication 8 August 2019
Published 10 September 2019 Volume 2019:12 Pages 2809—2817
DOI https://doi.org/10.2147/IDR.S215129
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Suresh Antony
Qian-Meng Lin,1 Ying-Hui Li,1 Qian Liu,1 Ni-Hong Pang,1 Ren-Ai Xu,2 Jian-Ping Cai,3 Guo-Xin Hu1
1School of Pharmaceutical Sciences, Wenzhou Medical University, Wenzhou 325035, People’s Republic of China; 2Department of Pharmacy, The First Affiliated Hospital of Wenzhou Medical University, Wenzhou 325035, People’s Republic of China; 3The Ministry of Health (MOH) Key Laboratory of Geriatrics, Beijing Hospital, National Center of Gerontology, Beijing 100730, People’s Republic of China
Correspondence: Guo-Xin Hu
School of Pharmaceutical Sciences, Wenzhou Medical University, Chashan Street, Chashan Town, Wenzhou 325035, People’s Republic of China
Email [email protected]
Jian-Ping Cai
The Key Laboratory of Geriatrics, Beijing Hospital, Beijing Institute of Geriatrics, Ministry of Health, Dongdan Dahua Road, Beijing 100730, People’s Republic of China
Email [email protected]
Background: Cytochrome P450 3A4 (CYP3A4) appears to be genetically polymorphic, which in turn contributes to interindividual variability in response to therapeutic drugs. Loperamide, identified as a CYP3A4 substrate, is prone to misuse and abuse and has high risks of life-threatening cardiotoxicity.
Methods: Thus, this study is designed to evaluate the enzymatic characteristics of 29 CYP3A4 alleles toward loperamide in vitro, including the 7 novel CYP3A4 variants (*28–*34). The incubation system (containing CYP3A4 enzyme, cytochrome b5, 0.5–20 μM loperamide, potassium phosphate buffer and nicotinamide adenine dinucleotide phosphate) was subject to 40-mins incubation at 37°C and the concentrations of N-demethylated loperamide were quantified by UPLC-MS/MS.
Results: As a result, CYP3A4.6, .17, .20 and .30 showed extremely low activity or no activity and the rest of CYP3A4 variants presented varying degrees of decrements in catalytical activities when compared with CYP3A4.1.
Conclusion: As the first study to identify the properties of these CYP3A4 variants toward loperamide metabolism, our investigation may establish the genotype–phenotype relationship for loperamide, predict an individual’s capability in response to loperamide, and provide some guidance of clinical medication and treatment for loperamide.
Keywords: CYP3A4, genetic polymorphism, interindividual variability, loperamide, misuse and abuse, cardiotoxicity, personalized treatment
Introduction
Loperamide is routinely prescribed to treat acute and chronic diarrhea.1 In spite of being a μ-opioid receptor agonist, it is consider not having abuse potential, due to its poor oral bioavailability, extensive first-pass metabolism as well as inability of penetrating across the blood-brain barrier.1–5 In recent years, however, loperamide use has been on a steep rise, which is used for opioids substitution rather for diarrhea treatment.4,5 A growing number of clinical cases have been reported to disclose life-threatening cardiac events and death resulted from over-ingestion of loperamide.5–14 In June 2016, the FDA issued a safety communication aimed to warn about serious heart problems implicated in much higher than the therapeutic doses of loperamide.15 In Jan 2018, owing to the continually-increasing reports of loperamide-caused events, the FDA issued another communication to foster the safe use of loperamide further by limiting the packaging for loperamide.16
Loperamide is subjected to extensive first-pass metabolism in liver to form a principle metabolite, N-demethylated loperamide (DLOP) (Figure 1).17,18 Although DLOP has a lower cardiotoxicity relative to loperamide, unexpectedly, it can achieve a concentration systemically much higher than loperamide, leading DLOP to be an essential contributor to loperamide-associated cardiac events after loperamide.19 It is evidenced that cytochrome P450 3A4 (CYP3A4) and CYP2C8 play a critical role in this transformation.20 As the most abundant CYP450 enzyme in human liver, CYP3A4 is speculated to be more responsible for loperamide metabolism.20,21
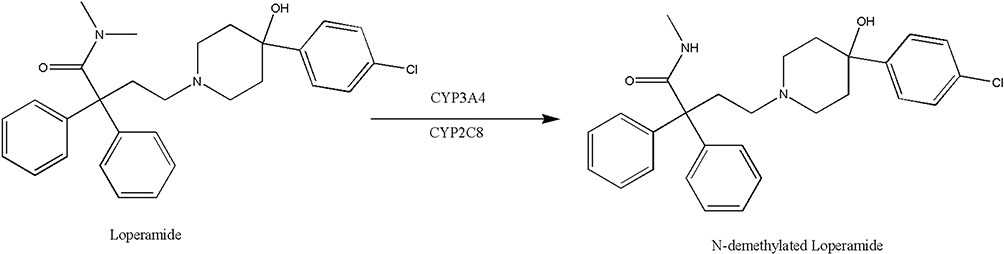 |
Figure 1 The transformation of loperamide to its main metabolite N-demethylated loperamide (DLOP). |
CYP3A4 has an extremely broad substrate spectrum, in charge of the oxidative metabolism of approximately 50% of clinically used drugs.22 Previous studies have confirmed that CYP3A4 appears genetically polymorphic, causing the wide interindividual variability in CYP3A4 enzymatic activity (up to 60-fold) and further causing the great difference of drugs metabolism, eventually which would result in undesirable side events or subtherapeutic effects.21–23 Taking loperamide, for example, the risk of loperamide-linked cardiotoxicity may be enhanced among poor metabolizers, especially who experience loperamide misuse or abuse. Therefore, it is imperative to research whether genetic variations of CYP3A4 would affect the enzymatic activities to predict the genotype–phenotype relationship for loperamide.
Hitherto, there are 53 CYP3A4 variants that are identified and named by the Human CYP Allele Nomenclature Committee website (http://www.cypalleles.ki.se/cyp3a4.htm). In this study, we aim to investigate the catalytic activity of wild-type CYP3A4*1, 21 previously reported CYP3A4 variants and 7 novel variants discovered by Hu et al.21 on the metabolism of loperamide in vitro, which may assist prediction of an individual’s capability for responding to loperamide.
Materials and methods
Chemicals and materials
Loperamide, DLOP and midazolam (used as internal standard) were purchased from Shanghai Canspec Scientific & Technology Co., Ltd. (Shanghai, China) Acetonitrile and methanol of analytical grade were bought from Merck (Darmstadt, Germany); formic acid was from Sigma-Aldrich Co. (St. Louis, MO, USA). Ultra-pure water was produced with a Milli-Q reagent system (Millipore, Bedford, MA, USA). The reduced nicotinamide adenine dinucleotide phosphate (NADPH) was purchased from Roche Pharmaceutical Ltd (Basel, Switzerland). Recombinant human CYP3A4 enzymes and purified cytochrome b5 were kind gifts from Beijing Hospital (Beijing, China).24–26
Incubation conditions
The 200-μL incubation system involved 100 mM potassium phosphate buffer (pH 7.4), 1 pmol CYP3A4.1 or other CYP3A4 variants, 2 pmol cytochrome b5, 1 mM NADPH and 0.5–20 μM loperamide that was sequentially diluted by methanol. The mixture without NADPH went through 5-mins prewarm at 37°C, then with the help of 1 mM NADPH initiated a 40-mins reaction process, and finally was frozen at −80°C to terminate the reaction. 600 μL of Acetonitrile and 20 μL of midazolam (10 μg/mL) were added into the incubation system prior to 2-mins vortex and 10-mins centrifugation at 11,357×g, and the supernatant was diluted (1:20) with ultra-pure water for UPLC-MS/MS analysis. Collectively, all incubations were performed in triplicate and data were in the form of mean ± SD.
Equipment and operation conditions
DLOP and midazolam were determined by UPLC-MS/MS, which was equipped with an Acquity UPLC system (Waters Corp., Millipore, Bedford, MA, USA) and a Waters Xevo TQ-S Micro-triple quadrupole mass spectrometer with an electrospray ionization source (Waters Corp., Millipore, Bedford, MA, USA). Analytes were separated on BEH C18 Column (2.1 mm × 100 mm, 1.7 μm; Waters Corp., Millipore, Bedford, MA, USA) at 40°C. The mobile phase involved ACN (A) and 0.1% formic acid (B) with a gradient elution at 0.35 mL/min for 3.0 mins. The gradient condition was as follow: 70–15% B (0–1.4 mins), 15–70% B (1.4–2.6 mins), and 70% B (2.6–3.0 mins). Multiple reaction monitoring in a positive mode was selected for detecting analytes. The transitions were m/z 463.3→252.1 and m/z 326.1→291.1 for DLOP and midazolam, respectively. Under these circumstances, DLOP and midazolam were well separated and their retention times were 1.39 and 1.16 mins, respectively (Figure 2).
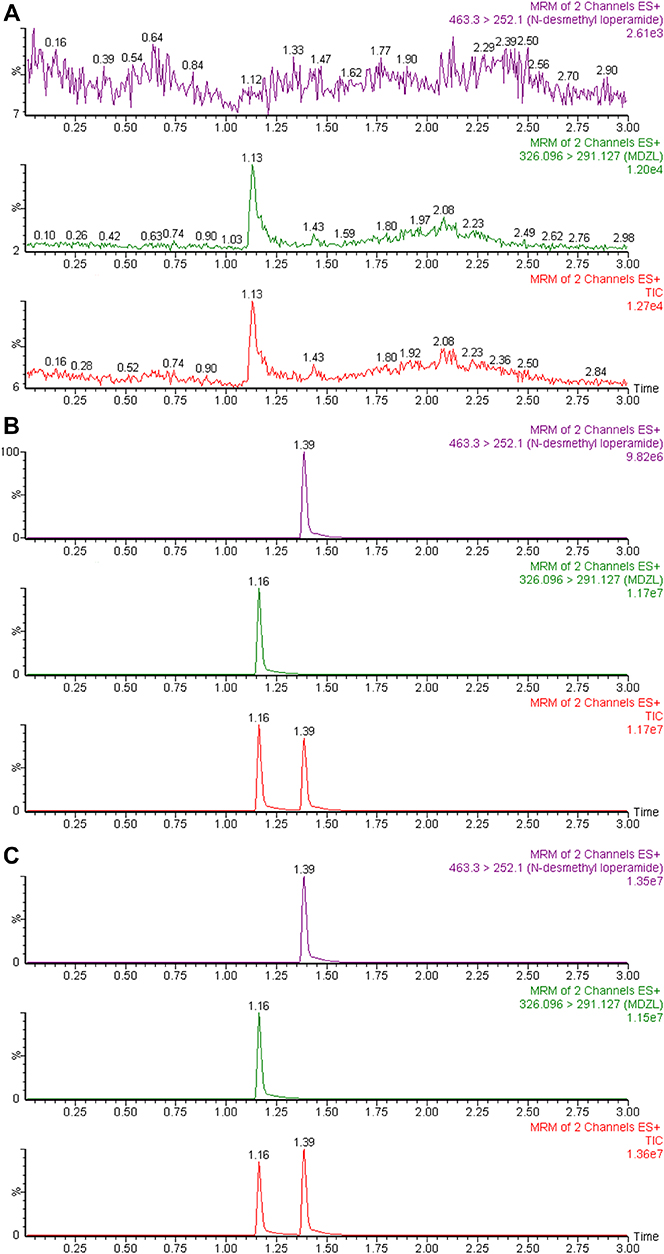 |
Figure 2 UPLC-MS/MS chromatographs of N-demethylated loperamide and midazolam (10 μg/mL midazolam) in the 200-μL incubation system: (A) without loperamide and midazolam; (B) with activity-abolished microsomes and spiked with 0.25 μM N-demethylated loperamide; (C) incubating with 20 μM loperamide and 1 pmol CYP3A4.1. Abbreviations: CYP3A4, cytochrome P450 3A4; MRM, multiple reaction monitoring. |
Statistical analysis
The kinetic parameters (Km and Vmax) were calculated via GraphPad Prism 5.0 (GraphPad Software Inc., San Diego, CA, USA) by Michaelis–Menten model to obtain intrinsic clearances (Clint) as the ratio of Vmax/Km and to depict Michaelis–Menten plots. All data were in the form of mean ± SD and subject to one-way ANOVA with Dunnett’s test by means of GraphPad Prism 5.0 to compare parameters of wild-type CYP3A4.1 with these of other variants. P-value below 0.05 means statistic difference.
Results
Michaelis–Menten kinetics of loperamide for CYP3A4.1 as well as other variants and their corresponding parameters are revealed in Table 1 and Figures 3 and 4. According to the alterations of values in Vmax, there are 3 situations: no evident changes between CYP3A4.1 and CYP3A4.3 and .5; significant increments in CYP3A4.7, .8, .11, .14, .16, and .18 compared with CYP3A4.1; remarkable decrements in the remaining variants. According to the alterations of values in Km, there are 3 situations: no considerable difference between CYP3A4.1 and CYP3A4.9, .10, .19, .24, .28, .31, and 0.34; moderate increments with P<0.05 in CYP3A4.4, .15, .29 and .32 relative to CYP3A4.1; serious increments with P<0.01 in the remaining variants. According to the comparison of values in Clint with CYP3A4.1, there are 2 situations: marginal decrease to 55.06–83.85% observed in CYP3A4.8, .16, .18 and .28; sharp collapse to 7.45–49.65% shown in the rest of CYP3A4 variants. Additionally, the concentrations of DLOP could not be detected for CYP3A4.6, .17, .20 and .30.
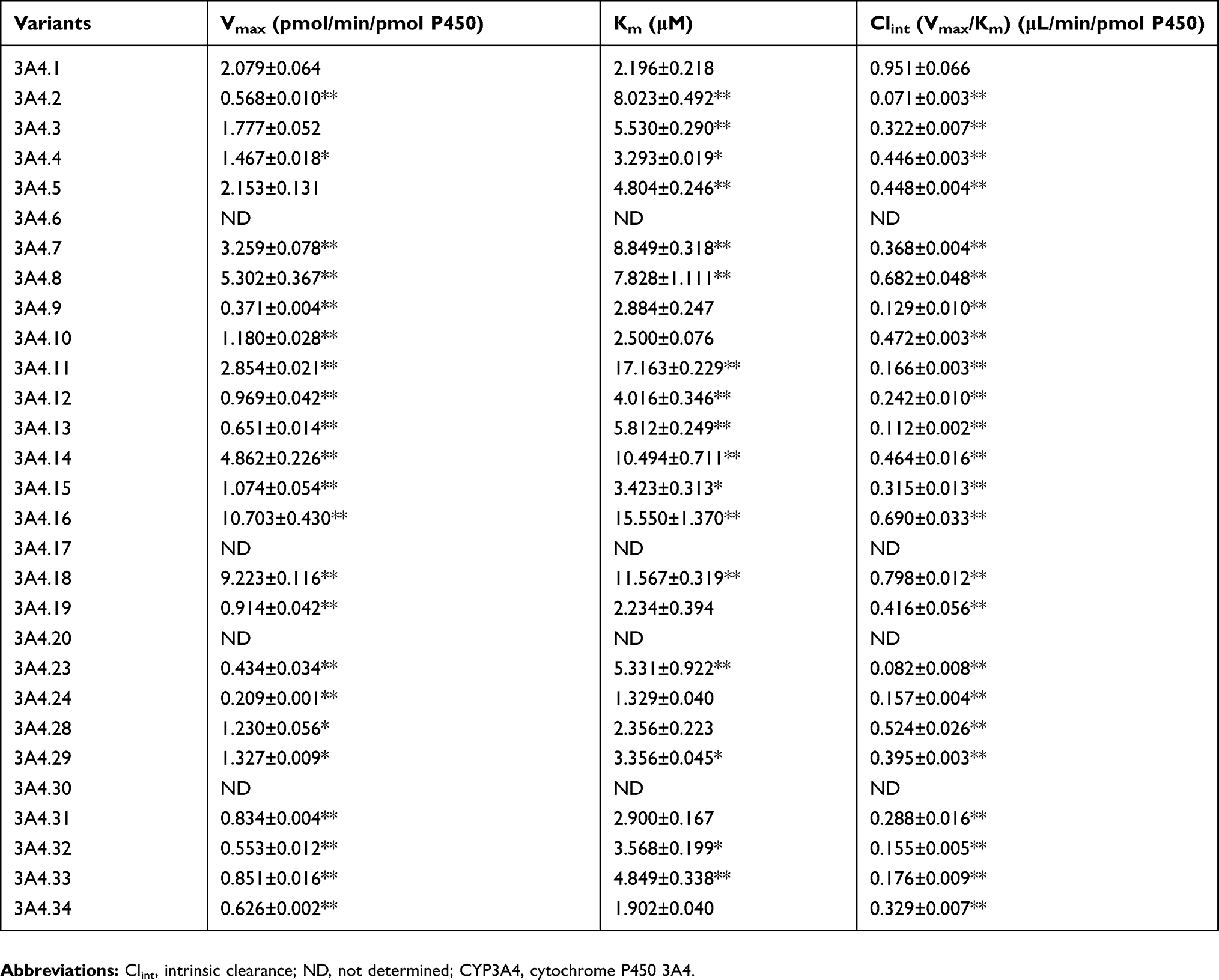 |
Table 1 Kinetic parameters for N-demethylated loperamide activity of CYP3A4.1 and other CYP3A4 variants on loperamide metabolism |
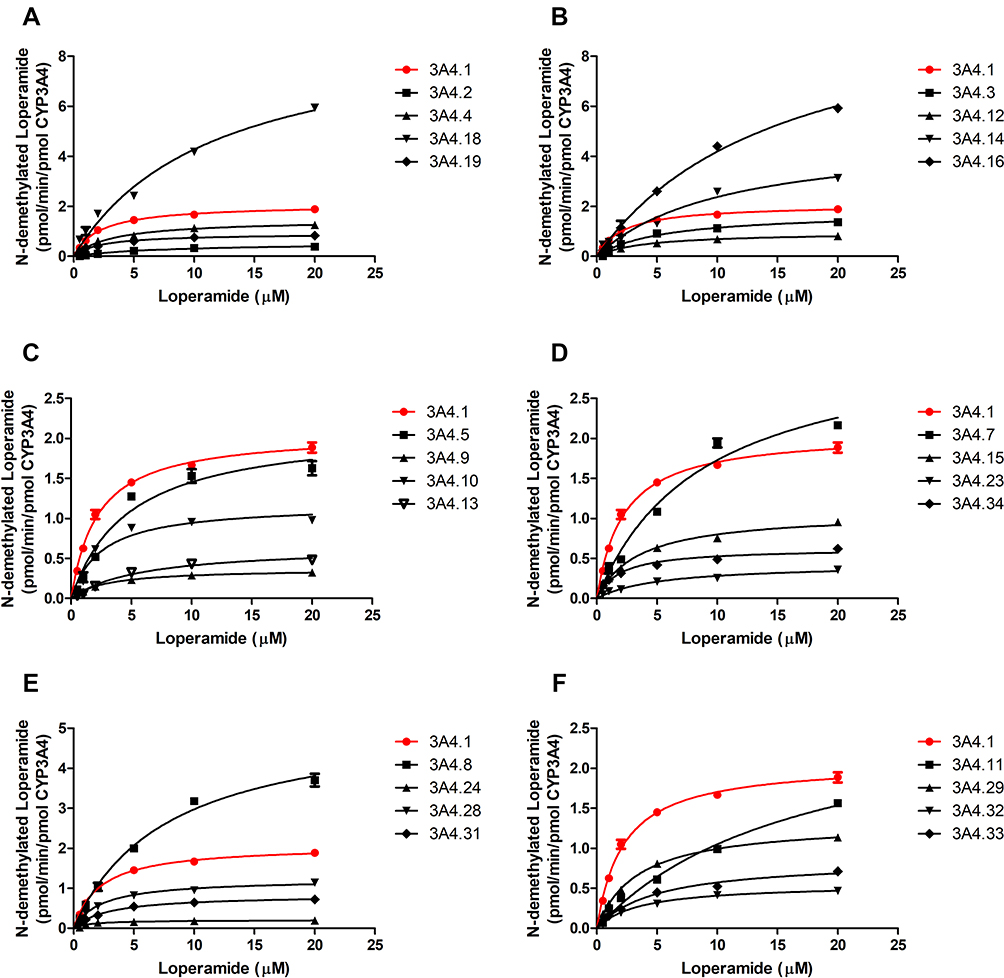 |
Figure 3 Michaelis–Menten curve of the enzymatic activities of the wild-type CYP3A4 and other CYP3A4 variants on loperamide metabolism. Data are presented as mean ± SD of 3 parallel experiments. The variants with designated allele names have been arranged into 6 groups (A–F).Abbreviation: CYP3A4, cytochrome P450 3A4. |
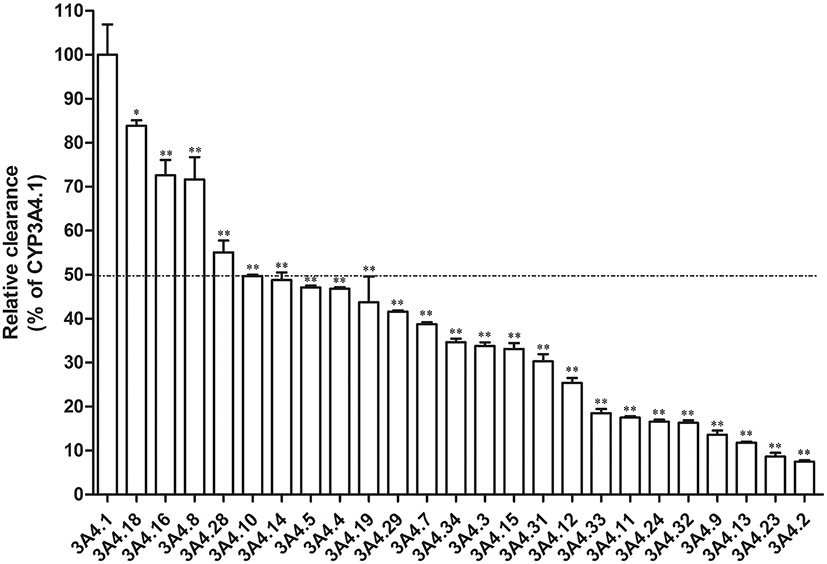 |
Figure 4 Relative clearance of CYP3A4 variants toward loperamide metabolism compared with the wild type, arranged in the order.Notes: Significant differences between the wild-type CYP3A4 and CYP3A4 variants analyzed by the mean of one-way ANOVA with Dunnett’s test, *P<0.05, **P<0.01. Abbreviation: CYP3A4, cytochrome P450 3A4. |
Overall, the reductions of Clint values could be caused by the changes in Vmax and Km into 4 situations: similar Vmax versus reduced Km (CYP3A4.3 and 0.5); reduced Vmax versus similar Km (CYP3A4.9, 0.10, 0.19, 0.24, 0.28, 0.31 and 0.34); reduced Vmax versus enhanced Km (CYP3A4.2, 0.4, 0.12, 0.13, 0.15, 0.23, 0.29, 0.32 and 0.33); increased Vmax versus greater-surged Km (the rest of CYP3A4 variants).
Discussion
The huge interindividual variations in drug metabolism may attribute to the collective effect of genetic polymorphisms, regulation of gene expression as well as interactions with therapeutic agents or environmental chemicals.27 Genetic polymorphisms, one of the influential factors, account for approximately 90% of variances of CYP3A4 enzymatic activities, indicating the great contribution of CYP3A4 genetic polymorphism to the tremendous interindividual variability in response to drugs such as loperamide (a CYP3A4 substrate).20–22,28 There are no related reports about enzymatic properties of CYP3A4 variants toward loperamide. In addition to the life-threatening cardiotoxicity of loperamide and the epidemic of loperamide misuse and abuse, it is of clinical value to identify the properties to establish the genotype–phenotype relationship to forecast the probability of response or severe toxicity to loperamide.5,13,29
A previous study has reported that CYP3A4, CYP2C8, CYP2D6, and CYP2B6 could catalyze loperamide N-demethylation in human liver microsomes.20 And among them, CYP3A4 and CYP2C8 played more important roles (53% and 38%, respectively) in loperamide N-demethylation.20 CYP3A4 is the most abundantly expressed enzyme in human liver (accounting for on average approximately 30% of the microsomal P450 pool) and CYP2C8 only accounts for 4.7%.30 Besides, loperamide exposure was rather increased in the presence of itraconazole (a CYP3A4 inhibitor) with P<0.001 than in the presence of gemfibrozil (a CYP2C8 inhibitor) with P<0.05.2 All findings imply that CYP3A4 makes greater contribution to loperamide N-demethylation. Therefore, we primarily investigate the effect of CYP3A4 genetic polymorphisms on the metabolism of loperamide.
In this study, wild-type CYP3A4.1 was set as the control group while CYP3A4.6, .20 and .30 served as negative controls for the functional validation so as to make certain of the reliability and accuracy of this study. Previous experiments confirmed that CYP3A4.6, .20 and .30 were devoid of catalytical function, in agreement with the results in our study, as CYP3A4.6, .20 and .30 carried premature stop codons and then yielded truncated proteins.21,24,31,32 Apart from CYP3A4.6, .20 and .30, CYP3A4.17 also exhibited extremely weak enzymatic activity without detectable concentrations of DLOP, consistent with the results in lidocaine, ibrutinib, amiodarone, testosterone and chlorpyrifos metabolism.24,25,33,34 These consistencies indicate that the outcomes in this work are reliable and valid for functional analysis of CYP3A4 variants. Therefore, patients carrying these four variants could be classified as poor metabolizers for loperamide and should be paid more attention in order to avoid loperamide-caused toxicity.
In order to comprehensively expand the understanding of the effects of CYP3A4 genetic polymorphisms on the metabolism of loperamide, we analyzed the remaining CYP3A4 variants in detail. CYP3A4.4, with an allele frequency of 2.4% in Chinese subjects, was point mutation 352A>G in exon 5, which led to the amino acid exchange from Ile to Val in 118 site.35,36 It showed different catalytic activity with CYP3A4.1 and this difference might attribute to Ile118Val that may have an effect upon the substrate binding.36
CYP3A4.18, fairly common in Asians, was previously reported having an increment in turnover numbers for testosterone and chlorpyrifos.21,34 In this study, a similar result was acquired where CYP3A4.18 was associated with ~4.4-fold higher Vmax value and ~5.3-fold higher Km value, resulting in moderately lower catalytical activity.
For CYP3A4.28, .29, .31, .32, .33 and .34, six novel variants detected by Hu et al21 were collectively demonstrated with decreased intrinsic clearance rate averaged from 16.31% to 55.06% when compared with CYP3A4.1. Specifically, Hu et al predicted that CYP3A4.31 (H324Q), and .32 (I335T) likely have a damaging effect on enzymatic functions, even though H324Q and I335T were not implicated in the active site for drug-substrate binding.21,37,38
For CYP3A4.3 and .8, we speculated that the genetic mutations in CYP3A4 might have an impact on heme incorporation, which in turn altered the enzymatic functions. CYP3A4.3, had a nucleotide exchange of 1334T>C in exon 12, which encoded the amino acid from Met445 to Thr in the conserved heme-binding region.39 Consequently, CYP3A4.3 presented a massive reduction in Clint value (33.8±0.78% of wild type) with similar Vmax value and increased Km, relative to CYP3A4.1. When compared with CYP3A4.1, CYP3A4.8 exhibited lower Clint value (71.67±5.04% of wild type) with ~2.5-fold higher Vmax value and ~3.6-fold higher Km value. It may result from the substitution of Arg130 with Gln disturbing heme incorporation, in which Arg130 played a pivotal role vital for heme binding.40,41
For CYP3A4.11 and .13, we speculated that the genetic mutations in CYP3A4 might alter stability and conformations of proteins, which in turn affected the functions. CYP3A4.11 involves a C1088T point mutation where Thr363 is replaced by Met.42 As previously reported, CYP3A4.11 appeared unstable, probably due to the introduction of the rather large Met at the location of residue 363 disrupting the tertiary structure of the protein in this region.40 Thus, Clint value of CYP3A4.11 was about 5.7-fold lower than that of CYP3A4.1. Likewise, the reduction of CYP3A4.13 enzymatic activity may be caused by destabilization of the protein via perturbing the orderly framework of the structure, in which an amino acid Leu took the place of Pro416.40
Interestingly, there were substrate-selective differences of kinetic values observed in CYP3A4.2 that was discovered in white subjects with a frequency of 2.7% and was absent in Chinese or black subjects.39 This variant involved an amino acid substitution of Ser222Pro that could lead to a significant change in the three-dimensional structure of the enzyme since Pro is a known helix breaker.39 In comparison with CYP3A4.1, CYP3A4.2 showed a sharp decrement in the intrinsic clearance rate toward loperamide, and this similar decrement was also found in nifedipine, lidocaine and ibrutinib metabolism.24,33,39 In contrast, CYP3A4.2 exhibited an increased intrinsic clearance rate toward amiodarone and showed little or no alteration toward testosterone.25,39 This inconsistency might attribute to the different specificities for these substrates, suggesting that the results from loperamide could not be analogized to other substrates.
Conclusion
This is the first study performed to elucidate the enzymatic characteristics of 29 CYP3A4 alleles toward the metabolism of loperamide in vitro. Our results find that most of the variants manifest extremely lower enzymatic activities toward loperamide than the wild type, which means patients with these defective alleles may have to pay attention to the dosage when ingesting loperamide. Although the allelic frequencies of these variants are relatively low, considering the large population base in the world as well as the non-standard use and the consequent cardiotoxicity of loperamide, this study can be granted some clinical value in establishing the genotype–phenotype relationship for loperamide, predicting an individual’s capability in response to loperamide, and providing some guidance of clinical medication and treatment for loperamide. Additionally, with the existence of substrate specificity, further researches are warranted to investigate the functional impacts of CYP3A4 genetic polymorphisms on the metabolism of a more various and wider range of CYP3A4 substrates, especially those with high risk of adverse effects or narrow therapeutic window.
Acknowledgments
The authors thank the members of the Beijing Institute of Geriatrics of the Ministry of Health for their advice and assistance. This study was supported by the Ministry of Science and Technology of the People's Republic of China (No.2017ZX09304026).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Sheng J, Tran PN, Li Z, et al. Characterization of loperamide-mediated block of hERG channels at physiological temperature and its proarrhythmia propensity. J Pharmacol Toxicol Methods. 2017;88:109–122. doi:10.1016/j.vascn.2017.08.006
2. Niemi M, Tornio A, Pasanen MK, Fredrikson H, Neuvonen PJ, Backman JT. Itraconazole, gemfibrozil and their combination markedly raise the plasma concentrations of loperamide. Eur J Clin Pharmacol. 2006;62:463–472. doi:10.1007/s00228-006-0133-z
3. Mukwaya G, MacGregor T, Hoelscher D, et al. Interaction of ritonavir-boosted tipranavir with loperamide does not result in loperamide-associated neurologic side effects in healthy volunteers. Antimicrob Agents Chemother. 2005;49:4903–4910. doi:10.1128/AAC.49.12.4903-4910.2005
4. Jaffe JH, Kanzler M, Green J. Abuse potential of loperamide. Clin Pharmacol Ther. 1980;28:812–819. doi:10.1038/clpt.1980.239
5. Miller H, Panahi L, Tapia D, Tran A, Bowman JD. Loperamide misuse and abuse. J Am Pharm Assoc. 2017;57:S45–S50. doi:10.1016/j.japh.2016.12.079
6. Riaz IB, Khan MS, Kamal MU, et al. Cardiac dysrhythmias associated with substitutive use of loperamide: a systematic review. Am J Ther. 2017;26:170–182. doi:10.1097/MJT.0000000000000585
7. Katz KD, Cannon RD, Cook MD, et al. Loperamide-induced torsades de pointes: a case series. J Emerg Med. 2017;53:339–344. doi:10.1016/j.jemermed.2017.04.027
8. Mowry JB, Spyker DA, Brooks DE, McMillan N, Schauben JL. 2014 Annual Report of the American Association of Poison Control Centers’ National Poison Data System (NPDS): 32nd annual report. Clin Toxicol (Phila). 2015;53:962–1147. doi:10.3109/15563650.2015.1102927
9. Eggleston W, Clark KH, Marraffa JM. Loperamide abuse associated with cardiac dysrhythmia and death. Ann Emerg Med. 2017;69:83–86. doi:10.1016/j.annemergmed.2016.03.047
10. Stefek B, Wolfe LT, Cohen M. Brugada syndrome associated with adolescent loperamide abuse. Pediatrics. 2018;142:e20181423. doi:10.1542/peds.2018-1423
11. Ruan X, Luo JJ, Kaye AD. Loperamide-related deaths in North Carolina. Med Leg J. 2017;85:55–56. doi:10.1177/0025817216672116
12. Larsen TR, McMunn J, Ahmad H, AlMahameed ST. Ventricular tachycardia triggered by loperamide and famotidine abuse. Drug Saf Case Rep. 2018;5:11. doi:10.1007/s40800-018-0077-0
13. Wu PE, Juurlink DN. Clinical review: loperamide toxicity. Ann Emerg Med. 2017;70:245–252. doi:10.1016/j.annemergmed.2017.04.008
14. Dierksen J, Gonsoulin M, Walterscheid JP. Poor Man’s methadone: a case report of loperamide toxicity. Am J Forensic Med Pathol. 2015;36:268–270. doi:10.1097/PAF.0000000000000201
15. U.S. Food and Drug Administration. FDA drug safety communication: FDA warns about serious heart problems with high doses of the antidiarrheal medicine loperamide (Imodium), including from abuse and misuse. Available at: https://www.fda.gov/drugs/drug-safety-and-availability/fda-drug-safety-communication-fda-warns-about-serious-heart-problems-high-doses-antidiarrheal. Accessed June, 2016.
16. U.S. Food and DrugAdministration. FDA drug safety communication: FDA limits packaging for anti-diarrhea medicine loperamide (Imodium) to encourage safe use. Available at: https://www.fda.gov/drugs/drug-safety-and-availability/fda-drug-safety-communication-fda-limits-packaging-anti-diarrhea-medicine-loperamide-imodium. Accessed January, 2018.
17. Miyazaki H, Nambu K, Matsunaga Y, Hashimoto M. Disposition and metabolism of [14C]loperamide in rats. Eur J Drug Metab Pharmacokinet. 1979;4:199–206. doi:10.1007/BF03189427
18. Yoshida K, Nambu K, Arakawa S, Miyazaki H, Hashimoto M. Metabolites of loperamide in rats. Biomed Mass Spectrom. 1979;6:253–259. doi:10.1002/bms.1200060606
19. Vaz RJ, Kang J, Luo Y, Rampe D. Molecular determinants of loperamide and N-desmethyl loperamide binding in the hERG cardiac K(+) channel. Bioorg Med Chem Lett. 2017;28:446–451. doi:10.1016/j.bmcl.2017.12.020
20. Kim KA, Chung J, Jung DH, Park JY. Identification of cytochrome P450 isoforms involved in the metabolism of loperamide in human liver microsomes. Eur J Clin Pharmacol. 2004;60:575–581. doi:10.1007/s00228-004-0815-3
21. Hu GX, Dai DP, Wang H, et al. Systematic screening for CYP3A4 genetic polymorphisms in a Han Chinese population. Pharmacogenomics. 2017;18:369–379. doi:10.2217/pgs-2016-0179
22. Zhou Q, Yu X, Shu C, et al. Analysis of CYP3A4 genetic polymorphisms in Han Chinese. J Hum Genet. 2011;56:415–422. doi:10.1038/jhg.2011.30
23. Nicolas JM, Espie P, Molimard M. Gender and interindividual variability in pharmacokinetics. Drug Metab Rev. 2009;41:408–421. doi:10.1080/10837450902891485
24. Fang P, Tang PF, Xu RA, et al. Functional assessment of CYP3A4 allelic variants on lidocaine metabolism in vitro. Drug Des Devel Ther. 2017;11:3503–3510. doi:10.2147/DDDT.S152366
25. Yang CC, Zheng X, Liu TH, et al. Functional characterization of 21 CYP3A4 variants on amiodarone metabolism in vitro. Xenobiotica. 2019;49:120–126. doi:10.1080/00498254.2017.1414971
26. Zhou X-Y, Hu X-X, Wang C-C, et al. Enzymatic activities of CYP3A4 allelic variants on quinine 3-hydroxylation in vitro. Front Pharmacol. 2019;10. doi:10.3389/fphar.2019.00591
27. Wilkinson GR. Genetic variability in cytochrome P450 3A5 and in vivo cytochrome P450 3A activity: some answers but still questions. Clin Pharmacol Ther. 2004;76:99–103. doi:10.1016/j.clpt.2004.04.005
28. Ozdemir V, Kalow W, Tang BK, et al. Evaluation of the genetic component of variability in CYP3A4 activity: a repeated drug administration method. Pharmacogenetics. 2000;10:373–388. doi:10.1097/00008571-200007000-00001
29. Klein MG, Haigney MCP, Mehler PS, Fatima N, Flagg TP, Krantz MJ. Potent inhibition of hERG channels by the over-the-counter antidiarrheal agent loperamide. JACC Clin Electrophysiol. 2016;2:784–789. doi:10.1016/j.jacep.2016.07.008
30. Zanger UM, Schwab M. Cytochrome P450 enzymes in drug metabolism: regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol Ther. 2013;138:103–141. doi:10.1016/j.pharmthera.2012.12.007
31. Westlind-Johnsson A, Hermann R, Huennemeyer A, et al. Identification and characterization of CYP3A4*20, a novel rare CYP3A4 allele without functional activity. Clin Pharmacol Ther. 2006;79:339–349. doi:10.1016/j.clpt.2005.11.015
32. Apellaniz-Ruiz M, Inglada-Perez L, Naranjo ME, et al. High frequency and founder effect of the CYP3A4*20 loss-of-function allele in the Spanish population classifies CYP3A4 as a polymorphic enzyme. Pharmacogenomics J. 2015;15:288–292. doi:10.1038/tpj.2014.67
33. Xu RA, Wen J, Tang P, et al. Functional characterization of 22 CYP3A4 protein variants to metabolize ibrutinib in vitro. Basic Clin Pharmacol Toxicol. 2018;122:383–387. doi:10.1111/bcpt.12934
34. Dai D, Tang J, Rose R, et al. Identification of variants of CYP3A4 and characterization of their abilities to metabolize testosterone and chlorpyrifos. J Pharmacol Exp Ther. 2001;299:825–831. doi:10.1111/bcpt.12934
35. Liu CH, Peck K, Huang JD, et al. Screening CYP3A single nucleotide polymorphisms in a Han Chinese population with a genotyping chip. Pharmacogenomics. 2005;6:731–747. doi:10.2217/14622416.6.7.731
36. Hsieh KP, Lin YY, Cheng CL, et al. Novel mutations of CYP3A4 in Chinese. Drug Metab Dispos. 2001;29:268–273. doi:10.1016/S1359-6446(00)01678-0
37. Yano JK, Wester MR, Schoch GA, Griffin KJ, Stout CD, Johnson EF. The structure of human microsomal cytochrome P450 3A4 determined by X-ray crystallography to 2.05-A resolution. J Biol Chem. 2004;279:38091–38094. doi:10.1074/jbc.C400293200
38. Williams PA, Jose C, Dijana Matak V, et al. Crystal structures of human cytochrome P450 3A4 bound to metyrapone and progesterone. Science. 2004;305:683–686. doi:10.1126/science.1099736
39. Sata F, Sapone A, Elizondo G, et al. CYP3A4 allelic variants with amino acid substitutions in exons 7 and 12: evidence for an allelic variant with altered catalytic activity. Clin Pharmacol Ther. 2000;67:48–56. doi:10.1067/mcp.2000.104391
40. Eiselt R, Domanski TL, Zibat A, et al. Identification and functional characterization of eight CYP3A4 protein variants. Pharmacogenetics. 2001;11:447–458. doi:10.1097/00008571-200107000-00008
41. Hasemann CA, Kurumbail RG, Boddupalli SS, Peterson JA, Deisenhofer J. Structure and function of cytochromes P450: acomparative analysis of three crystal structures. Structure. 1995;2:41–62. doi:10.1016/S0969-2126(01)00134-4
42. Werk AN, Cascorbi I. Functional gene variants of CYP3A4. Clin Pharmacol Ther. 2014;96:340–348. doi:10.1038/clpt.2014.129
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.