Back to Journals » Drug Design, Development and Therapy » Volume 8
Functional analysis of tanshinone IIA that blocks the redox function of human apurinic/apyrimidinic endonuclease 1/redox factor-1
Authors Sui J, Li M, Qian C, Wang S, Cheng Y, Chen BP, Wang D
Received 14 July 2014
Accepted for publication 6 August 2014
Published 3 November 2014 Volume 2014:8 Pages 2147—2160
DOI https://doi.org/10.2147/DDDT.S71124
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Shu-Feng Zhou
Jiangdong Sui,1,2 Mengxia Li,1 Chengyuan Qian,1 Shufeng Wang,3 Yi Cheng,1 Benjamin PC Chen,2 Dong Wang1
1Cancer Center, Daping Hospital and Research Institute of Surgery, Third Military Medical University, Chongqing, People’s Republic of China; 2Division of Molecular Radiation Biology, Department of Radiation Oncology, University of Texas Southwestern Medical Center, Dallas, TX, USA; 3Institute of Immunology, PLA, College of Basic Medical Sciences, Third Military Medical University, Chongqing, People’s Republic of China
Abstract: Apurinic/apyrimidinic endonuclease 1/redox factor-1 (APE1/Ref-1) is a multifunctional protein possessing both DNA repair and redox regulatory activities. It has been shown that blocking redox function leads to genotoxic, antiangiogenic, cytostatic, and proapoptotic effects in cells. Therefore, the selective inhibitors against APE1’s redox function can be served as potential pharmaceutical candidates in cancer therapeutics. In the present study, we identified the biological specificity of the Chinese herbal compound tanshinone IIA (T2A) in blocking the redox function of APE1. Using dual polarization interferometry, the direct interaction between APE1 and T2A was observed with a KD value at subnanomolar level. In addition, we showed that T2A significantly compromised the growth of human cervical cancer and colon cancer cells. Furthermore, the growth-inhibitory or proapoptotic effect of T2A was diminished in APE1 knockdown or redox-deficient cells, suggesting that the cytostatic effect of T2A might be specifically through inhibiting the redox function of APE1. Finally, T2A pretreatment enhanced the cytotoxicity of ionizing radiation or other chemotherapeutic agents in human cervical cancer and colon cancer cell lines. The data presented herein suggest T2A as a promising bioactive inhibitor of APE1 redox activity.
Keywords: tanshinone IIA, APE1, redox activity, multifunctional protein
Introduction
The human apurinic/apyrimidinic endonuclease 1/redox factor-1 (APE1/Ref-1; hereafter referred to as APE1) is an essential enzyme in the DNA base excision repair (BER) pathway that plays a critical role in correcting single or short-patch base lesions under various genotoxic attacks.1 In addition, APE1 is involved in the redox modification activity of many redox-sensitive transcription factors (eg, nuclear factor-κB [NF-κB]; activator protein 1 [AP-1]; hypoxia-inducible transcription factor 1α [HIF-1α]; and others). Therefore, APE1 can influence stress response, DNA repair, and other cellular processes, including angiogenesis, inflammation, and cell survival.1 The distinct activities of APE1 are retained in two independent domains: the C-terminal domain is specific for its DNA repair function; and the N-terminal residue is required for redox regulation.1,2
Accumulating evidence reveals that APE1 supports the cell proliferation of numerous cancers, such as breast, cervical, ovarian, colorectal, nonsmall cell lung cancer, osteosarcoma, and hepatocellular carcinoma.3–9 Also, the elevated expression levels have been associated with accelerated angiogenesis, poor prognosis, chemoradiotherapy resistance, incomplete therapeutic responses, short relapse-free intervals, and short survival times.2 Furthermore, we previously showed that APE1 knockdown via Ad5/F35-mediated chimeric adenoviral small interfering RNA (siRNA) vector (Ad5/F35-siAPE1) potentiates cellular radiosensitivity in human osteosarcoma, colorectal cancer, and hepatocellular carcinoma cells, both in vitro and in the xenograft model.6,8,9 Therefore, the APE1 represents a favorable cancer therapeutic target in both preventive and curative treatment paradigms. Blocking the DNA repair function of APE1 by small molecule inhibitors potentiates the cytotoxicity of alkylating agents.
Theoretically, the inhibition of the APE1’s redox function might interfere with the transcription of a subset of human genome, which could further alter the stress response of cancer cells. Based on the analysis of downstream gene profiles of the transcription factors regulated by APE1, it strongly implied that the APE1 regulatory roles on the DNA repair capacity could be beyond its direct enzymatic activity in the BER pathway.10,11 The redox regulatory roles of APE1 on p53, AP-1, and HIF-1α affect the expression of a subset of DNA repair genes functioning not only in BER, but also in homologous recombination, mismatch repair, and global genome repair pathways.10,11 Therefore, the inhibition or alteration of the APE1 function, particularly the redox function, in cancer cells could potentially not only restrict tumor cell growth and angiogenesis, but it could also alleviate the resistance of cancer cells to chemoradiotherapy.
Hitherto, E3330 and its synthesized chemical analogues have been demonstrated to selectively inactivate the redox activity of APE1 and suppress proliferation, angiogenesis, and the migration of cancer cells in multiple studies.11 Moreover, the dietary agents, such as soy isoflavones (extracted in plants) and resveratrol (extracted in red wine), have been recently reported to impact the expression or activities of APE1, inferring the potential for natural regulation of APE1 functions.11 Interestingly, based on the high-throughput in silico screening from the traditional Chinese medicine compounds database, we have previously identified another dietary compound, tanshinone IIA (T2A) (C19H18O3), as an affinitive small molecule for APE1 protein. T2A is one of the main active components in danshen, the dried root of Salvia miltiorrhiza, which has been widely prescribed in traditional Chinese medicine practice for centuries in the treatment of cardiovascular and cerebrovascular diseases, with minimal side effects. In addition to its antioxidant12 and anti-inflammatory properties,13 T2A has been recently shown to possess growth inhibition effects on human cancer cells.14–18 However, the functional target and molecular mechanism of T2A remain elusive.
Therefore, the objectives of the present study were: 1) to validate the effectiveness of T2A in regulating APE1’s redox activity in vitro and the specific interaction between T2A and APE1; 2) to establish the APE1 redox residue as a functional target of T2A in inhibiting the growth of cervical cancer and colon cancer cells; and 3) to evaluate the synergistic cytotoxic effect of T2A combined with ionizing radiation (IR) and some other DNA-damaging agents on cervical cancer and colon cancer cells.
Materials and methods
Materials
T2A, E3330, CRT0044876, etoposide (Eto), mitomycin C (MMC), methyl methanesulfonate (MMS), hydroxyurea (HU), camptothecin (Cpt), doxycycline (DOX), dimethyl sulfoxide (DMSO), and 3-(4,5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). Cell culture media, fetal bovine serum, and trypsin were obtained from Thermo Scientific HyClone (Logan, UT, USA). The LightShift Chemiluminescence Electrophoretic Mobility-Shift Assay (EMSA) kit, SuperSignal West Pico chemiluminescence reagents, and horseradish peroxidase-conjugated antimouse or antirabbit IgG antibodies were purchased from Thermo Fisher Scientific, Waltham, MA, USA.
Cell lines
The human cervical cancer cell line (HeLa), human colon cancer cell line (HCT116), and human umbilical veins endothelial cells (HUVEC) were purchased from the American Type Culture Collection, Manassas, VA, USA, and cultured in Dulbecco’s Modified Eagle’s Medium supplemented with 10% volume/volume heat-inactivated fetal bovine serum and 100 U/mL penicillin and 100 μg/mL streptomycin in a humidified 5% CO2, 95% air incubator at 37°C. For inducible silencing of endogenous APE1, the HeLa cell clones were developed as described.19,20 The APE1 siRNA sequences were cloned into BglII and HindIII restriction sites of pTER vector, which present a DOX-responsive promoter to form the so-called pTER/APE1 vector. HeLa cells were transfected with pcDNA6/TR to generate stable tetracycline-repressor-expressing cell clones, which were further selected for the acquired resistance by incubation with 5 μg/mL blasticidin (Thermo Fisher Scientific, Waltham, MA, USA) for 14 days. Individual cell clones expressed the tetracycline-repressor at the highest levels and was, therefore, selected for transfection with the pTER/APE1 vector previously linearized with Bst 1107I (Thermo Fisher Scientific) and subjected to selection with Zeocin™ (200 ng/mL; Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA) for 14–21 days. For the generation of APE1 knockin cell lines, an APE1 expression vector was generated by cloning an EcoRI–BamHI fragment from pFLAG-CMV-5.1/APE1 (Sigma-Aldrich Co.) into p3XFLAG-CMV-14 vector (Sigma-Aldrich Co.).
To avoid the degradation of the ectopic APE1 messenger RNA (mRNA) acid by the specific siRNA sequence, two nucleotides of the APE1-cDNA coding sequence were mutated with site-directed mutagenesis kits (Agilent Technologies, Santa Clara, CA, USA), leaving the APE1 amino acid sequence unaffected. The site-directed mutagenesis kit was used to generate the APE1 C65S mutant, which was confirmed by DNA sequencing (MWG Biotech Ag, Ebersberg, Germany). Then, the APE1 siRNA clones were transfected with p3XFLAG-CMV empty vector, p3XFLAG-CMV/APE1 wild-type vector, and p3XFLAG-CMV/APE1 C65S mutant vector, previously digested with ScaI (Thermo Scientific).
Also, 48 hours after transfection, the cells were subjected to selection with Geneticin® (Thermo Fisher Scientific) for 14 days and selected for the acquired resistance. After DOX treatment at the final concentration of 1 μg/mL for 10 days, whole-cell lysates were analyzed for APE1 expression by Western blot.
EMSA
Experiments were accomplished, according to the user’s instruction manual of the Thermo Scientific LightShift Chemiluminescence EMSA kit with minor modifications. Briefly, the nuclear extracts were prepared by resuspending cell pellets in hypotonic lysis buffer (10 mM Tris-HCl; pH 7.5; 1.5 mM MgCl2; 5 mM KCl; protease and phosphatase inhibitors), followed by nuclear extraction buffer (50 mM Tris-HCl; pH 7.5; 0.5 M NaCl; 2 mM ethylenediaminetetraacetic acid [EDTA]; 10% sucrose; 10% glycerol; protease and phosphatase inhibitors). Subsequently, the nuclear extracts (4 μg) were treated with T2A at various concentrations for 30 minutes, then incubated with 3′-biotin-labeled and purified double-stranded oligonucleotide probes containing consensus sequences for NF-κB, AP-1, HIF-1α, and SP-1 binding sites (Invitrogen).
After incubation, the samples were separated on a 5% polyacrylamide gel at 100 V for 1 hour and then transferred to a Zeta-Probe GT nylon membrane (Bio-Rad Laboratories Inc., Hercules, CA, USA). The probes were detected by horseradish peroxidase-conjugated streptavidin (1:300), and the bands were visualized by enhanced chemiluminescence reagents provided with the kit. The resultant bands were quantified using the Quantity One imaging software (Bio-Rad Laboratories Inc.).
Western blot assay and antibodies
The cells were collected after T2A treatment and lysed in the lysis buffer (100 mM NaCl; 1 mM EDTA; 20 mM Tris-HCl (pH 8.0); 0.5% NP-40; and protease inhibitors), sonicated and cleared by centrifugation (10,000× g for 10 minutes). The cleared lysates (50 μg) were electrophoresed by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis for 1 hour at 165 V. Separated proteins were then transferred onto polyvinylidene difluoride membranes (Bio-Rad Laboratories Inc.) for 2 hours at 380 mA. After being blocked in Tris-buffered saline with Tween (TBST) (50 mM Tris-HCl; pH 7.5; 150 mM NaCl; and 0.1% (volume/volume) Tween 20) containing 5% (weight/volume) nonfat dry milk for 1 hour at room temperature, membranes were incubated with the specific primary antibodies overnight at 4°C. After five washes with TBST, the membranes were incubated for 1 hour at room temperature with the appropriate peroxidase-conjugated secondary antibodies. Then, the membranes were washed five times with TBST, and the blots were reacted with chemiluminescence reagents and revealed with Biomax-Light films (Eastman Kodak Company, Rochester, NY, USA). Band intensities were analyzed using the Gel Doc 2000 apparatus and software (Quantity One; Bio-Rad Laboratories Inc.). The primary antibodies and concentrations used were indicated as follows: anti-APE1 (1:5,000; Santa Cruz Biotechnology Inc., Dallas, TX, USA); anti-β-actin (1:5,000; Sigma-Aldrich Co.); and anticleaved poly adenosine diphosphate ribose polymerase (PARP) (1:1,000; Cell Signaling Technology, Inc., Danvers, MA, USA).
Radiotracer AP site incision assay
The purified human APE1 protein was obtained from inhouse purification following the protocol from the Dr David M Wilson Laboratory in National Institute on Aging, National Institutes of Health (Baltimore, MD, USA). This protein is tag free. We tested the protein activity by classical abasic site incision assay. The detailed protocol could be found in our previous publication.21 As for the redox activity, we did not test for purified protein due to the lack of sophisticated assay. The radiotracer assay was performed essentially as described.22 In brief, T2A and CRT0044876 were incubated at various concentrations with 500 pg purified human APE1 protein at room temperature for 30 minutes in 50 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, pH 7.5, 100 mM KCl, 1 mM MgCl2, and 1 mM dithiothreitol. At that time, 1 pmol 32P-radiolabeled DNA substrate (Midland Certified Reagent Co., Midland, TX, USA) was added. Incision reactions were then carried out immediately at 37°C for 10 minutes in a final volume of 10 μL. After the addition of an equal volume of stop buffer (0.05% bromophenol blue and xylene cyanol; 20 mM EDTA; 95% formamide), the radiolabeled substrate and product were separated on a standard polyacrylamide denaturing gel and quantified by phosphorimager analysis.
APE1 DNA-binding assay
Experiments were performed, as described previously, with slight modifications.23 Briefly, T2A and CRT0044876 were incubated at various concentrations with 30 ng purified human APE1 protein at room temperature for 30 minutes in 25 mM 3-(N-morpholino) propanesulfonic acid–KOH, pH 7.2, 100 mM KCl, 10% glycerol, 1 mM dithiothreitol, 50 μg/mL bovine serum albumin, and 4 mM EDTA. At that time, incubations were mixed with 0.1 pmol 32P-radiolabeled oligonucleotide (Midland Certified Reagent Co.) on ice for 5 minutes. Binding reactions were resolved in nondenaturing polyacrylamide gel (20 mM Tris-HCl; pH 7.5; 10 mM Na acetate; 0.5 mM EDTA; 8% acrylamide; 2.5% glycerol) in 20 mM Tris-HCl, pH 7.5, 10 mM Na acetate, pH 7.5, 0.5 mM EDTA. Electrophoresis was performed at 4°C for 2 hours at 120 V. The gel was dried and autoradiographed to identify the location of bound and unbound DNA.
Dual polarization interferometry
Dual polarization interferometry (DPI) analysis was performed using an AnaLight® Bio200 workstation (Farfield Scientific Limited, Crew, UK), with 0.1 mg/mL APE1 immobilized onto the unmodified chips. T2A, dissolved in the running buffer (20 mM HEPES [pH 7.4]; 100 mM NaCl; and 1 mM MgCl2), was bound to the immobilized APE1 at the indicated concentrations. Data collection and analysis were performed using the AnaLight® software suite and AnaLight® Explorer, respectively. The DPI instrument provides absolute measurements of the change in the thickness and density of the molecular protein layer in real time, which enabled a calculation of the mass of the immobilized APE1 as T2A was added. These measurements provide the ability to determine binding constants and stoichiometry of binding.
Docking simulation
The three-dimensional (3D) model of DNA-bound APE1 structure was constructed based on the X-ray crystallographic data for 1DE8 in the Protein Data Bank database. Docking simulation was performed using Discovery Studio 2.55 (Accelrys Inc., San Diego, CA, USA). The possible binding sites of N-terminal region of APE1 for T2A were identified by clicking the command “find binding site from receptor cavities” in the toolbars of “define and edit binding site.” Then, the CDOCKER in Discovery Studio 2.55 was used to perform the ligand and receptor docking procedure. Finally, 50 random conformations were generated, and the top ten hits were recorded. The top five hits were shown the similar conformation, the best conformation according to the CDOCKER energy and CDOCKER interaction energy were used to perform the energy minimization (parameters: minimization 1: method: Steepest Descent, Max step: 50,000, RMS Gradient: 0.1; minimization 2: method: Conjugate Gradient, Max step: 50,000, RMS Gradient: 0.0001) and dynamic simulation (parameters: 500,000 steps, 2 fs/step).
Cell growth assay
Cell growth was determined using the MTT colorimetric assay. Cells were plated in each well of 96-well plate and allowed to adhere overnight. Cells were then treated with T2A and/or either of other reagents at desired concentrations or DMSO as the vehicle and incubated for indicated time points. MTT reagent (0.5 mg/mL in PBS) was added and incubated for 4 hours at 37°C in 5% CO2, and absorption was determined with an ELISA plate reader at 490 nm. The experiments were independently performed at least three times, each in quintuplicates. The results were confirmed by direct cell counting using a hemocytometer.
Statistical analysis
Statistical analysis was performed using statistics software SPSS 18.0 (SPSS Inc., Chicago, IL, USA). Comparisons between means of two groups were evaluated by the analysis of variance (ANOVA). P<0.05 was considered statistically significant.
Results
Effects of T2A on APE1 redox activity and its protein expression
To determine whether T2A potentially regulates the redox function of APE1, we evaluated the effects of T2A on APE1-dependent regulation of NF-κB, AP-1, and HIF-1α, since the DNA-binding activities of these transcription factors are known to be regulated by the redox function of APE1. HeLa cells were stably transfected with DOX inducible short hairpin RNA (shRNA) to exert knockdown of APE1 expression. Based on this inducible APE1 shRNA knockdown cell line (APE1shRNA), both wild-type APE1 (APE1wt) and redox-deficient mutant APE1 (APE1C65S) were reconstituted. The Western blot was performed to verify the downregulated endogenous APE1 protein levels and to demonstrate similar expression of APE1wt and APE1C65S exogenous proteins (Figure S1). The EMSA was carried out to determine whether T2A alters DNA-binding activities of NF-κB, AP-1, and HIF-1α. In the absence of T2A, we observed that APE1 silencing or C65S mutation alone attenuated the DNA-binding of all three transcription factors, thus confirming the involvement of APE1 redox activity in regulating these transcription factors (Figure 1A–F).
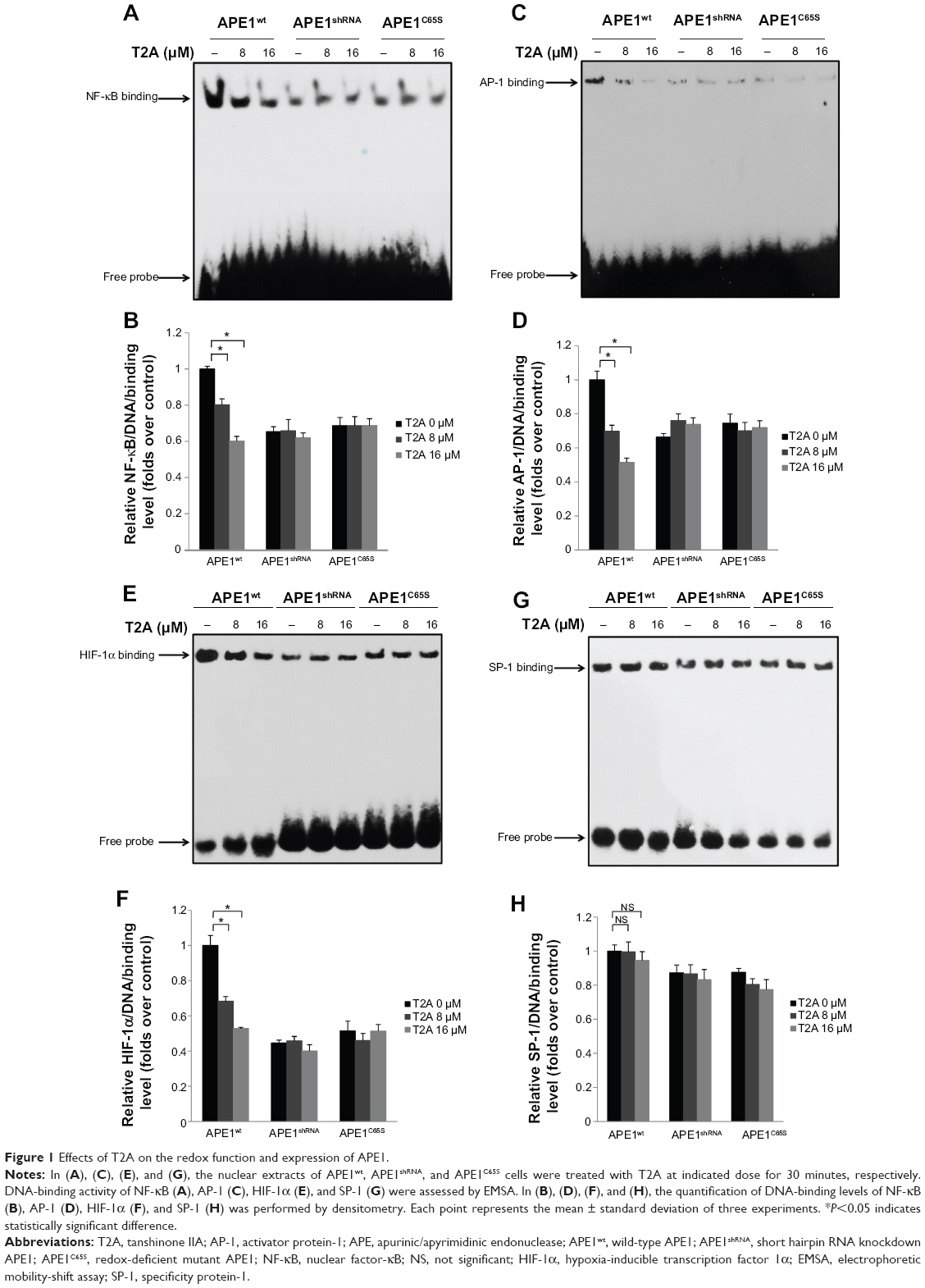 | Figure 1 Effects of T2A on the redox function and expression of APE1. |
In APE1-proficient cells (APE1wt), the addition of T2A attenuated the DNA-binding of NF-κB, AP-1, and HIF-1α in a dose-dependent manner; whereas, T2A failed to reduce DNA-binding activity of SP-1, as SP-1 is not the subject of APE1 redox regulation (Figure 1G and H). T2A also did not further suppress the attenuated DNA-binding of NF-κB, AP-1, and HIF-1α in both APE1shRNA and APE1C65S cells, confirming that T2A exerts its regulation specifically through the redox activity of APE1.
Since E3330 is considered to be a selective APE1 redox inhibitor, we then compared the redox-inhibiting effects between T2A and E3330. Interestingly, the half maximal inhibitory concentration (IC50) value for T2A (15.9 μM) was lower than that of E3330 (20.9 μM), by using NF-κB as the transcription factor target complex (Figure S2). Altogether, we provide compelling evidence that T2A appears to be a promising redox inhibitor of APE1.
Effects of T2A on APE1 endonuclease activity and APE1/AP site interaction
Human APE1 comprises above 95% of cellular AP endonuclease activity, which is essential for cell survival.24 We then investigated the effect of T2A on AP endonuclease activity by assaying the incision of the typical radiolabeled abasic site containing oligonucleotide substrate. As shown in Figure 2A and C, 10 μM T2A did not affect the incision activity of APE1 where this dose is sufficient to inhibit the redox activity of APE1 (Figure 1). Concurrently, 10 μM of T2A also had no impact on the AP site-binding activity of APE1 as expected (Figure 2B and D). Even at ten times higher concentration (100 μM), T2A displayed mild inhibition of APE1 endonuclease activity relative to the control (no inhibitor, DMSO control) reaction. In contrast, CRT0044876 at 10 μM readily abolished the AP site-binding activity of APE1, as well as its endonuclease activity.
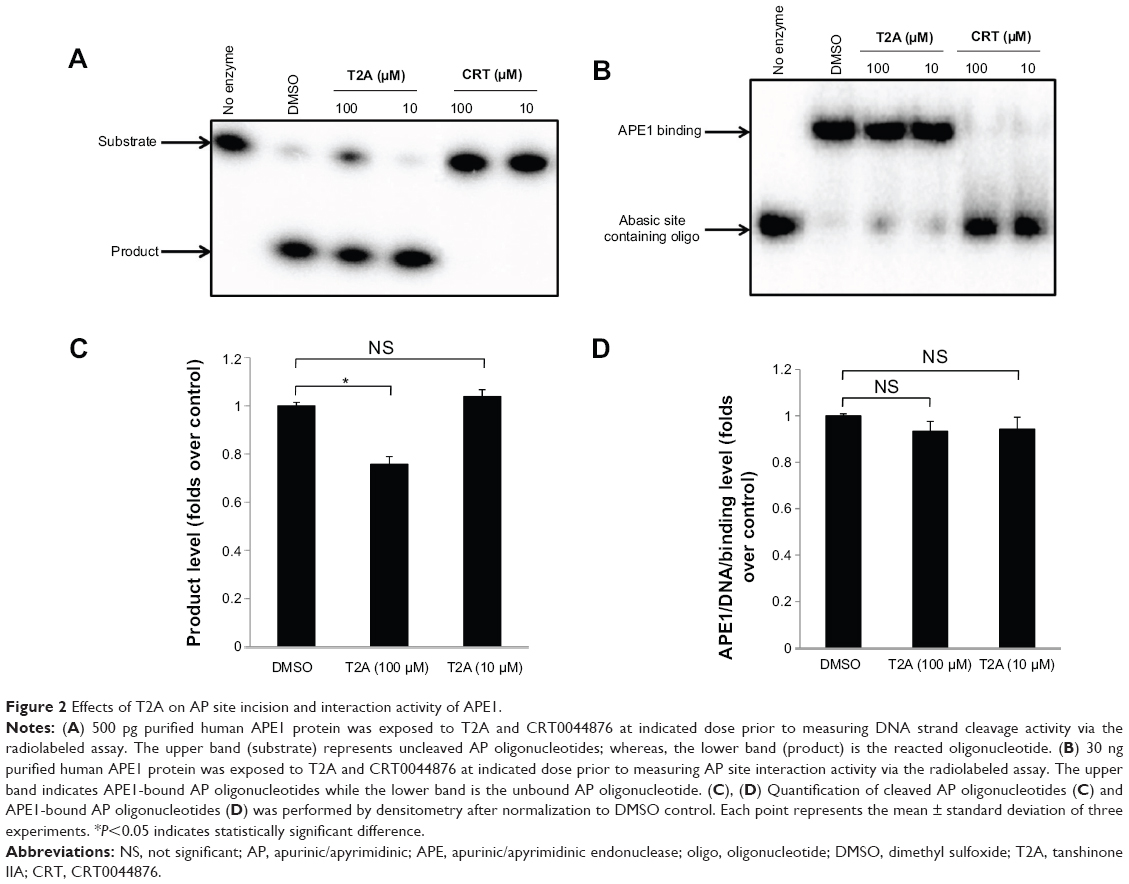 | Figure 2 Effects of T2A on AP site incision and interaction activity of APE1. |
Overall, these observations demonstrate the specificity of T2A on the redox activity of APE1 but not on its DNA repair activity.
Specific interaction between APE1 and T2A
The direct interaction as well as the binding affinity of T2A compound to APE1 protein was measured using DPI as mentioned in the “Materials and methods” section. The instrument, AnaLight® Bio200, provides a quantitative measurement of the change in the mass of immobilized APE1 protein in real time, as the different concentrations of T2A is added to the solution (Figure 3A). Additionally, the increased densities of the T2A–APE1 complexes were determined over time (Figure 3B), with the dissociation constant (KD) for the interaction estimated to be 0.88 nM (Figure 3C). These data strongly suggest that T2A specifically interacts with APE1 protein with a high affinity.
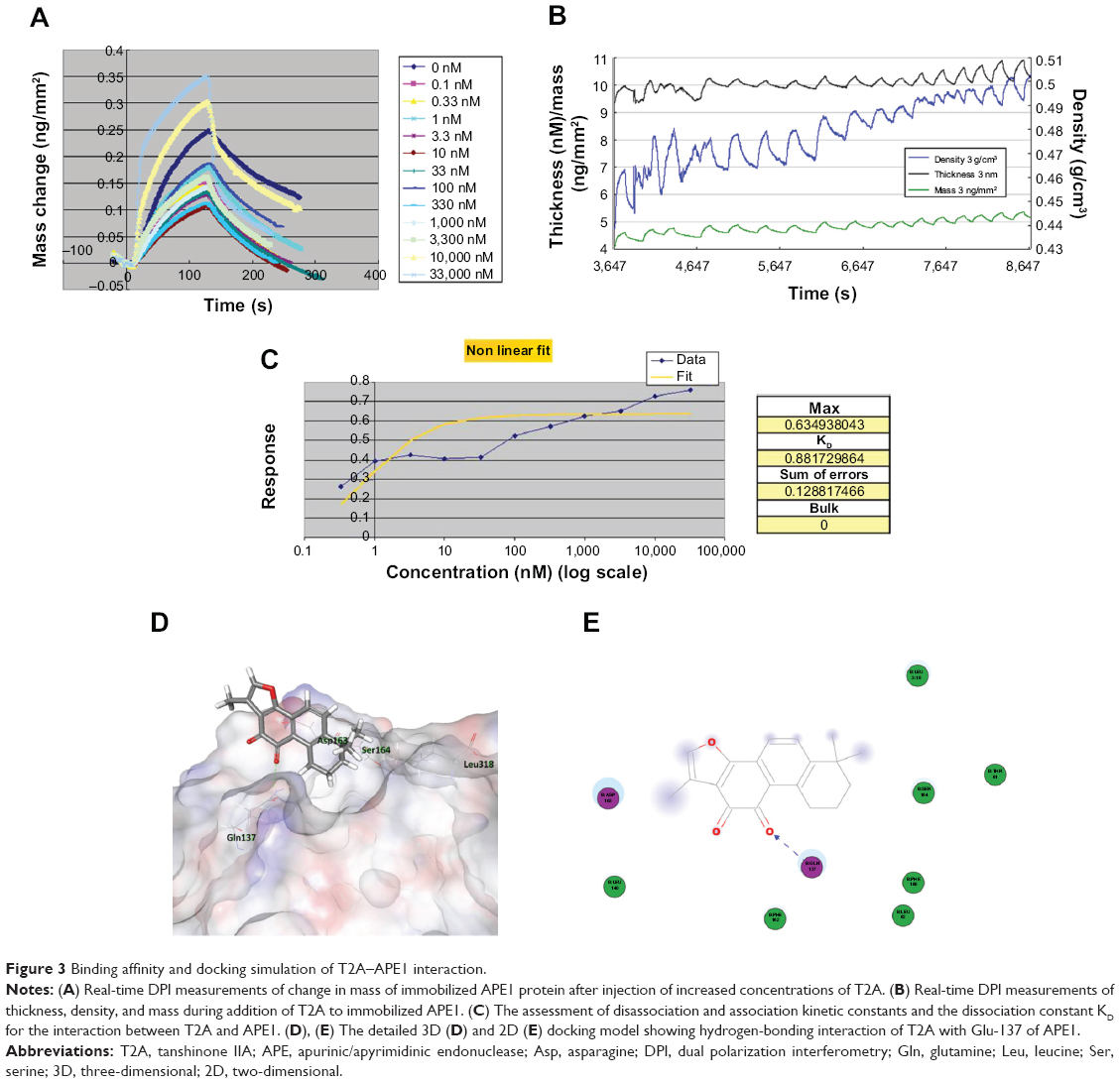 | Figure 3 Binding affinity and docking simulation of T2A–APE1 interaction. |
To further estimate the possible interaction interface between the two molecules, a computational molecular docking approach was employed. We adopted the crystal structure of APE1 (Protein Data Bank entry, 1DE8) as the APE1 protein modeling for the in silico docking and eight starting configurations of T2A molecule were applied. The docking results showed that T2A forms a hydrogen bond with the Glu-137 residue of APE1, and this interaction interface partly occupies the N-terminal domain of APE1, which has been previously recognized to be of importance of its redox activity (Figure 3D and E; Figure S3).
Effects of T2A on cell growth of human cervical cancer, colon cancer, and endothelial cell lines
To further evaluate the growth-inhibitory activity of T2A, human cervical cancer HeLa cells and colon cancer HCT116 cells were treated with increasing concentrations of T2A for various durations, and the cell viabilities were measured by MTT assay. As shown in Figure 4A, T2A significantly inhibited the proliferation of HeLa cells in dose- and time-dependent manners with IC50 are 29.6, 12.1, and 4.8 μM for 24-hour, 48-hour, and 72-hour treatment, respectively. Treatment with T2A doses ≤8 μM caused cytostatic effect and inhibition of HeLa cell proliferation; however, they did not significantly affect the viability of HeLa cells. On the other hand, treatment with T2A doses ≥16 μM caused not only cytostatic effect, but it also led to decreased viability (Figure 4A). Similarly, T2A suppressed proliferation of HCT116 cells at lower doses and became reduced viability of HCT116 cells at higher doses. The IC50 are 30.5, 8.9, and 4.5 μM for 24-hour, 48-hour, and 72-hour treatment, respectively (Figure 4B).
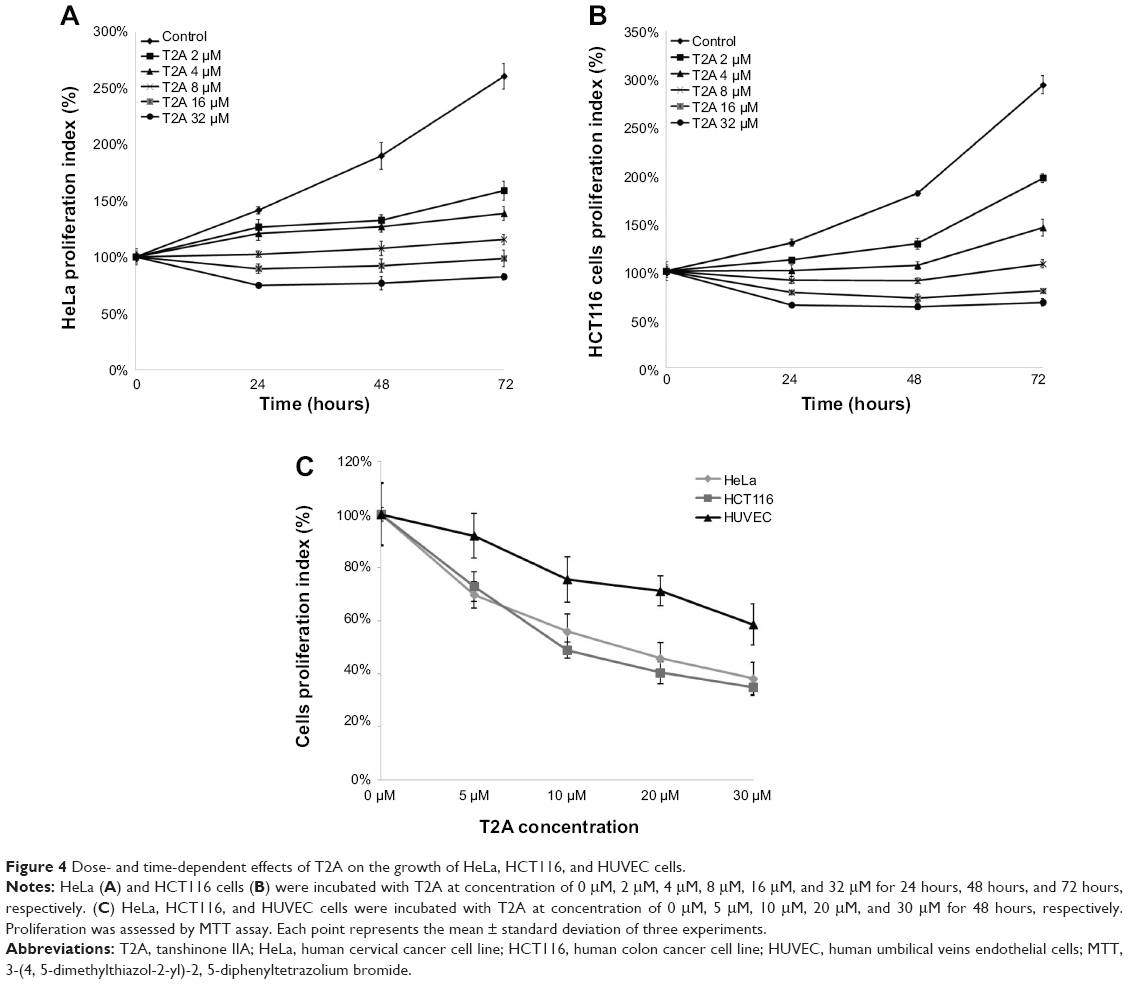 | Figure 4 Dose- and time-dependent effects of T2A on the growth of HeLa, HCT116, and HUVEC cells. |
To further investigate whether T2A was also cytostatic to noncancer cells, we conducted toxicity analysis in HUVEC. Figure 4C showed that T2A induced less cytostatic effect on HUVEC cells in comparison with HeLa and HCT116 cells and did not decrease the 50% viability of HUVEC, even at 30 μM after 48 hours. The selectivity indexes were 3.25 for HeLa and 4.38 for HCT116 cells. Collectively, these results implied that T2A is a potential antitumor agent with modest growth-inhibitory activity in cancer cells but limited effects on normal cells.
Effect of APE1 silencing or its redox mutation on T2A action
APE1-knockdown and redox-deficient HeLa cells were subjected to T2A treatment for cell proliferation and cytostatic analysis to further determine the biological potential of T2A and its dependency on APE1. As shown in Figure 5, APE1 silencing and C65S mutation suppressed cell viability by 57% and 48%, respectively (P<0.01). T2A at 8 μM and 16 μM inhibited the viability of APE1wt cells by 50% and 56%, respectively. In contrast, T2A failed to cause significant further growth-inhibitory effect on the basis of APE1 deficient or redox mutated. Furthermore, we determined if the proapoptotic effects of T2A is also dependent on APE1 status. The results showed that T2A significantly increased the level of an important apoptotic marker, cleaved PARP (c-PARP) in APE1wt cells in a dose-dependent manner, but it induced much less increase of c-PARP in APE1 knockdown, and C65S mutated cells that was already elevated in both cell lines (Figure 5B and C; P<0.05). Taken together, these results suggest that the cytostatic effect of T2A on cervical cancer cells is mainly through apoptosis and, specifically, through its redox inhibition on the APE1 protein.
 | Figure 5 Effect of APE1 knockdown or its redox mutation on the T2A activities in cell growth. |
Effects of T2A on chemotherapeutic agents and ionizing radiation-induced cell killing in cervical cancer and colon cancer cell lines
In addition to the potent antitumor effect of T2A alone treatment, we evaluated if T2A, at relatively nontoxic concentrations, could potentiate tumor cells’ killing effect of some cancer therapeutic factors, including IR or other chemotherapeutic agents, such as Eto, MMC, MMS, HU, and Cpt. Based on MTT assay, both HeLa and HCT116 cells were pretreated with T2A concentrations of 2 μM and 4 μM and then exposed to each of the aforesaid agents, separately. Figure 6A and C demonstrated that T2A at both concentrations significantly potentiate the cytotoxicity of MMS (the inhibitory rate is elevated by 16.5%–48.7% in HeLa cells and by 20.5%–46.3% in HCT116 cells. P<0.01) and IR (the inhibitory rate is elevated by 32%–61.3% in HeLa cells and by 20.4%–51.8% in HCT116 cells. P<0.01). T2A at both concentrations moderately augmented the effect of Eto (the inhibitory rate is elevated by 5.5%–14.1% in HeLa cells and by 14.7%–23.9% in HCT116 cells P<0.05), MMC (the inhibitory rate is elevated by 10.5%–16.2% in HeLa cells and by 11.1%–17.9% in HCT116 cells P<0.05), and Cpt (the inhibitory rate is elevated by 7.5%–12.3% in HeLa cells and by 5.7%–11.7% in HCT116 cells P<0.05).
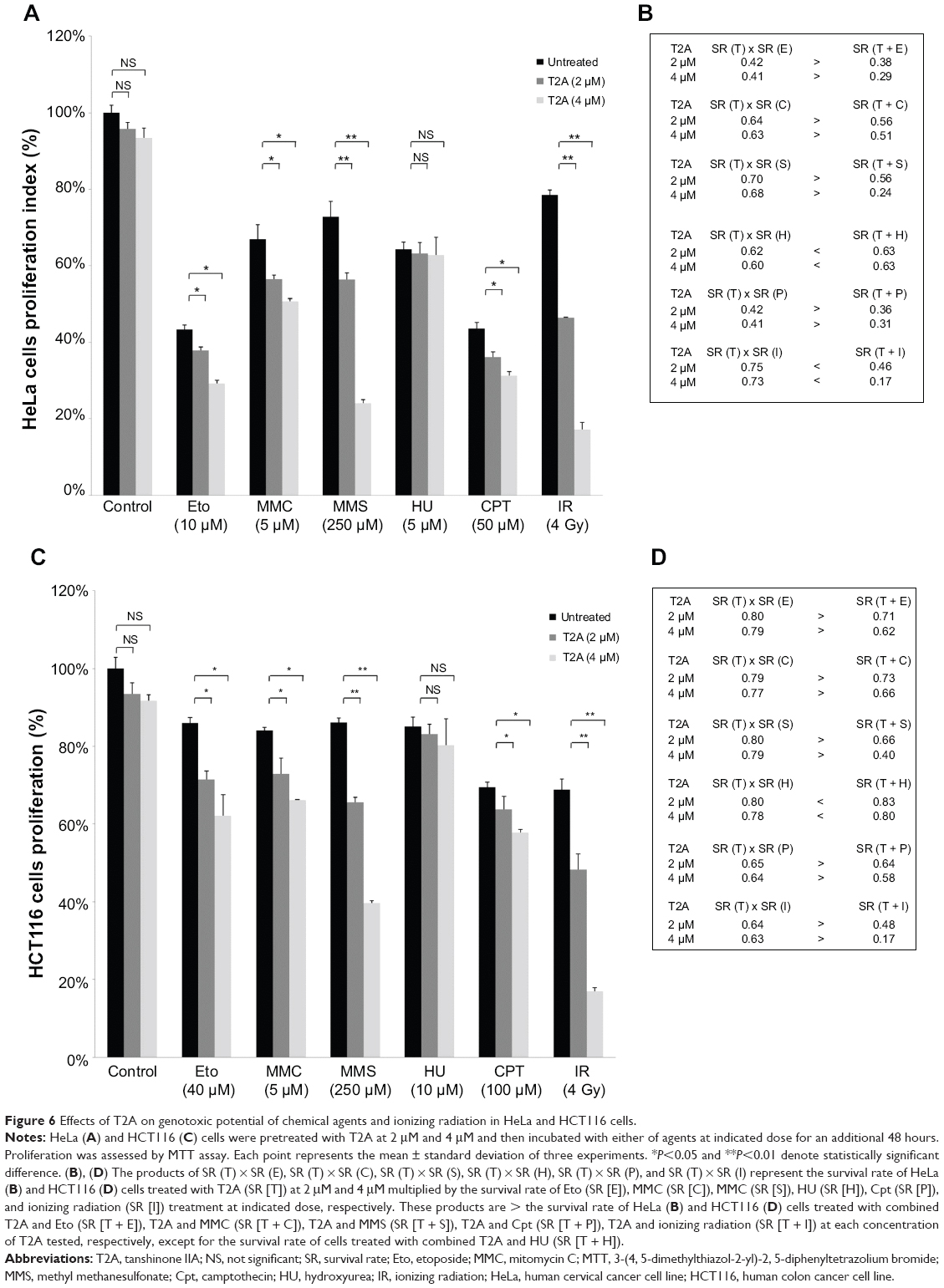 | Figure 6 Effects of T2A on genotoxic potential of chemical agents and ionizing radiation in HeLa and HCT116 cells. |
Our results also demonstrated that T2A has no sensitizing effects on HU-induced cytotoxicity (P>0.05). To address whether this effect is synergistic or additive, the survival rate of T2A treatment was multiplied by the survival rate of each agents, and their products were compared with the value of the survival rate of cells treated with combined T2A and the corresponding agents.25,26 These data showed that in both T2A concentrations tested, the products of the survival rate of each chemical treatment alone was slightly greater than that of the combined T2A and either of Eto, MMC, and Cpt treatment, and was obviously higher than that of T2A combined with either of MMS and IR treatment, except for that of T2A combined with HU, indicating that this effect could be synergistic, especially for MMS and IR treatment (Figure 6B and D).
Discussion
Inhibition of APE1 redox activity in promoting cancer cell death is a novel approach that has been relatively unexplored to date but displayed promising anticancer potential.11,27–30 The underlying mechanism likely is due to modulation in the transcriptome profile as the redox activity of APE1 is essential for several key transcription factors, including NF-κB, AP-1, HIF-1α, p53, and CREB, which have been implicated in tumor growth and progression, resistance to therapy, and tumor metastasis.1,2,10,11 Inhibition of APE1 redox activity would render these transcription factors unable to bind to DNA, thereby attenuating the expression of various proteins involved in DNA repair and stress responses.1,2,10,11 Our previous studies provided the first evidence that adenoviral delivery of anti-APE1 shRNA to inhibit APE1 expression is a viable anticancer strategy.6,8,9 Based on a pharmacophore match for docking-based virtual screening from the Traditional Chinese Medicine Library, we identified that T2A, one of the pharmacologically active components from S. miltiorrhiza, as a potential APE1 inhibitor.
The current study further examined whether T2A can function as a potent and druggable inhibitor of APE1 redox activity. Our result demonstrated that T2A blocked APE1’s ability to activate NF-κB, AP-1, and HIF-1α in a dose-dependent manner and enhanced tumor cell death as expected. These inhibitory effects of T2A were compromised in cells lacking APE1 or expressing the loss-of-redox function APE1C65S mutant, thus confirming that the redox activity of APE1 is indeed the functional target of T2A action. Our data also revealed that the in vitro redox IC50 of T2A is significantly lower than that of E3330, indicating that T2A has the potential to block APE1 redox function more efficiently than E3330 in a biologic context. Additionally, T2A exhibits the effect on neither APE1 protein expression (Figure S4) nor AP site-binding affinity/AP endonuclease function of APE1, further providing a supplemental explanation for T2A as APE1-specific redox inhibitor.
E3330 has been reported to specifically bind to APE1 with the kinetic constant value of 1.6 nM.31 Additionally, Naidu et al have shown that a potent APE1 inhibitor hycanthone was 10 nM KD values (affinity constants) for APE1.32 Using the DPI approach, we observed that the APE1 mass change is taking place upon addition of T2A and the estimated KD was 0.88 nM, indicating that T2A has much higher binding affinity as compared to E3330 or hycanthone. Insight into how T2A specifically interacts with APE1 was gained from molecular docking simulations performed with the 3D structure of hydrogen-bonding interaction of T2A with Glu-137 residue of APE1. Since the APE1 redox-active site, Cys-65 residue (together with Cys-93), appears to be buried in the 3D model structure of the protein, the redox regulation may implicate the occurrence of unfolding of APE1 to allow interaction with transcription factors.2 However, due to the occupation of N-terminal pocket of APE1, T2A may further prevent conformational change of APE1.
Intrinsic growth-inhibitory activity of T2A was demonstrated in human cervical cancer and colon cancer cell lines. In accord with the previous studies by others,15,16 T2A inhibited the proliferation of HeLa and HCT116 cells in both dose- and time-dependent manners. Interestingly, T2A was relatively nontoxic to HUVEC cells, implying the selectivity of T2A to cancer cells. This is probably due to the elevation of intrinsic oxidative stress in most cancer cells. Furthermore, we, at the first time, demonstrated that APE1 knockdown or redox function deficiency alleviates the tumor cell apoptosis induced by T2A. This provides a strong evidence for therapeutic targeting the redox residue of APE1 using T2A or more potent small molecule inhibitors in cancer treatment.
APE1 is highly expressed in several human tumors, such as cervical cancer, ovarian cancer, osteosarcoma, and colorectal cancer. The vector-based APE1 shRNA enhanced the chemosensitivity of multiple myeloma to melphalan33 and radiosensitivity of human osteosarcoma, colorectal cancer, and hepatocellular carcinoma.6,8,9 However, it is unclear which of APE1’s functions may contribute more to this effect. In this study, T2A combined with either of DNA-damaging agents, including Eto, MMC, MMS, Cpt, and IR, decreased the survival of HeLa and HCT116 cells compared to cells solely treated with either of reagents. Especially, T2A potentiated the cytotoxicity of MMS and IR, but not that of HU. Our observations implied that T2A caused the increased sensitivity to DNA-damaging agents, presumably due to decreasing the amount of APE1 available to reduce NF-κB, AP-1, HIF-1α, or all of them by which the expression of key genes within DNA repair pathways was downregulated.
In conclusion, the current study presented indicates the specific interaction between T2A and APE1 and the biologic relevance of T2A that has anti-APE1 redox activity, which can be utilized as a novel tool to separate the two functions of APE1 for further study on the selective contribution of the redox and the DNA repair signaling to maintenance of cellular homeostasis in cancer. Our results also provided for the first time, to the best of our knowledge, the evidence to reveal that T2A has potent antitumor activity in part via inhibition of redox function of APE1. Furthermore, we found that inhibition of APE1 redox activity by T2A may be partially involved in the mechanism by which T2A enhances sensitivity to DNA-damaging agents and leads to greater cell killing in HeLa and HCT116 cells. Therefore, we believe that T2A, a widely used drug with a safe dietary compound, may serve as a complementary approach to enhance the therapeutic efficacy of DNA-damaging agents, which seems to be very promising for designing novel clinical strategies for optimizing the treatment outcome of cancer patients.
Acknowledgments
The authors are grateful to Professor Gianluca Tell for generously providing the APE1 knockdown and reconstituted cell lines; Dr David M Wilson III for purified human APE1 protein; Yanping Cun and Zengfu Shang for helpful discussions; Nan Dai and Yufen Lin for generously sharing reagents; and Guillermo Palchik for critical review of the manuscript. This work was supported by the National Natural Science Foundation of China (81171904) to Dr Dong Wang and by the National Institutes of Health (CA166677) to Professor Benjamin PC Chen.
Disclosure
The authors declare no conflict of interest in this work.
References
Bhakat KK, Mantha AK, Mitra S. Transcriptional regulatory functions of mammalian AP-endonuclease (APE1/Ref-1), an essential multifunctional protein. Antioxid Redox Signal. 2009;11(3):621–638. | ||
Tell G, Fantini D, Quadrifoglio F. Understanding different functions of mammalian AP endonuclease (APE1) as a promising tool for cancer treatment. Cell Mol Life Sci. 2010;67(21):3589–3608. | ||
Kakolyris S, Kaklamanis L, Engels K, et al. Human AP endonuclease 1 (HAP1) protein expression in breast cancer correlates with lymph node status and angiogenesis. Br J Cancer. 1998;77(7):1169–1173. | ||
Schindl M, Oberhuber G, Pichlbauer EG, Obermair A, Birner P, Kelley MR. DNA repair-redox enzyme apurinic endonuclease in cervical cancer: evaluation of redox control of HIF-1alpha and prognostic significance. Int J Oncol. 2001;19(4):799–802. | ||
Tanner B, Grimme S, Schiffer I, et al. Nuclear expression of apurinic/apyrimidinic endonuclease increases with progression of ovarian carcinomas. Gynecol Oncol. 2004;92(2):568–577. | ||
Wang D, Luo M, Kelley MR. Human apurinic endonuclease 1 (APE1) expression and prognostic significance in osteosarcoma: enhanced sensitivity of osteosarcoma to DNA damaging agents using silencing RNA APE1 expression inhibition. Mol Cancer Ther. 2004;3(6):679–686. | ||
Yoo DG, Song YJ, Cho EJ, et al. Alteration of APE1/ref-1 expression in non-small cell lung cancer: the implications of impaired extracellular superoxide dismutase and catalase antioxidant systems. Lung Cancer. 2008;60(2):277–284. | ||
Xiang DB, Chen ZT, Wang D, et al. Chimeric adenoviral vector Ad5/F35-mediated APE1 siRNA enhances sensitivity of human colorectal cancer cells to radiotherapy in vitro and in vivo. Cancer Gene Ther. 2008;15(10):625–635. | ||
Cun Y, Dai N, Xiong C, et al. Silencing of APE1 enhances sensitivity of human hepatocellular carcinoma cells to radiotherapy in vitro and in a xenograft model. PLoS One. 2013;8(2):e55313. | ||
Luo M, He H, Kelley MR, Georgiadis MM. Redox regulation of DNA repair: implications for human health and cancer therapeutic development. Antioxid Redox Signal. 2010;12(11):1247–1269. | ||
Kelley MR, Georgiadis MM, Fishel ML. APE1/Ref-1 role in redox signaling: translational applications of targeting the redox function of the DNA repair/redox protein APE1/Ref-1. Curr Mol Pharmacol. 2012;5(1):36–53. | ||
Lin R, Wang WR, Liu JT, Yang GD, Han CJ. Protective effect of tanshinone IIA on human umbilical vein endothelial cell injured by hydrogen peroxide and its mechanism. J Ethnopharmacol. 2006;108(2):217–222. | ||
Li W, Li J, Ashok M, et al. A cardiovascular drug rescues mice from lethal sepsis by selectively attenuating a late-acting proinflammatory mediator, high mobility group box 1. J Immunol. 2007;178(6):3856–3864. | ||
Wang J, Wang X, Jiang S, et al. Growth inhibition and induction of apoptosis and differentiation of tanshinone IIA in human glioma cells. J Neurooncol. 2007;82(1):11–21. | ||
Zhou L, Chan WK, Xu N, et al. Tanshinone IIA, an isolated compound from Salvia miltiorrhiza Bunge, induces apoptosis in HeLa cells through mitotic arrest. Life Sci. 2008;83(11–12):394–403. | ||
Su CC, Chen GW, Kang JC, Chan MH. Growth inhibition and apoptosis induction by tanshinone IIA in human colon adenocarcinoma cells. Planta Med. 2008;74(11):1357–1362. | ||
Su CC, Lin YH. Tanshinone IIA inhibits human breast cancer cells through increased Bax to Bcl-xL ratios. Int J Mol Med. 2008;22(3):357–361. | ||
Chiu TL, Su CC. Tanshinone IIA induces apoptosis in human lung cancer A549 cells through the induction of reactive oxygen species and decreasing the mitochondrial membrane potential. Int J Mol Med. 2010;25(2):231–236. | ||
Vascotto C, Fantini D, Romanello M, et al. APE1/Ref-1 interacts with NPM1 within nucleoli and plays a role in the rRNA quality control process. Mol Cell Biol. 2009;29(7):1834–1854. | ||
Li M, Vascotto C, Xu S, et al. Human AP endonuclease/redox factor APE1/ref-1 modulates mitochondrial function after oxidative stress by regulating the transcriptional activity of NRF1. Free Radic Biol Med. 2012;53(2):237–248. | ||
Li M, Völker J, Breslauer KJ, Wilson DM 3rd. APE1 incision activity at abasic sites in tandem repeat sequences. J Mol Biol. 2014;426(11):2183–2198. | ||
Wilson DM 3rd, Takeshita M, Grollman AP, Demple B. Incision activity of human apurinic endonuclease (Ape) at abasic site analogs in DNA. J Biol Chem. 1995;270(27):16002–16007. | ||
Wilson DM 3rd, Takeshita M, Demple B. Abasic site binding by the human apurinic endonuclease, Ape, and determination of the DNA contact sites. Nucleic Acids Res. 1997;25(5):933–939. | ||
Chen DS, Herman T, Demple B. Two distinct human DNA diesterases that hydrolyze 3′-blocking deoxyribose fragments from oxidized DNA. Nucleic Acids Res. 1991;19(21):5907–5914. | ||
Steel GG, Peckham MJ. Exploitable mechanisms in combined radiotherapy-chemotherapy: the concept of additivity. Int J Radiat Oncol Biol Phys. 1979;5(1):85–91. | ||
Singh-Gupta V, Joiner MC, Runyan L, et al. Soy isoflavones augment radiation effect by inhibiting APE1/Ref-1 DNA repair activity in non-small cell lung cancer. J Thorac Oncol. 2011;6(4):688–698. | ||
Luo M, Delaplane S, Jiang A, et al. Role of the multifunctional DNA repair and redox signaling protein Ape1/Ref-1 in cancer and endothelial cells: small-molecule inhibition of the redox function of Ape1. Antioxid Redox Signal. 2008;10(11):1853–1867. | ||
Zou GM, Maitra A. Small-molecule inhibitor of the AP endonuclease 1/REF-1 E3330 inhibits pancreatic cancer cell growth and migration. Mol Cancer Ther. 2008;7(7):2012–2021. | ||
Zou GM, Karikari C, Kabe Y, Handa H, Anders RA, Maitra A. The Ape-1/Ref-1 redox antagonist E3330 inhibits the growth of tumor endothelium and endothelial progenitor cells: therapeutic implications in tumor angiogenesis. J Cell Physiol. 2009;219(1):209–218. | ||
Li M, Wilson DM 3rd. Human apurinic/apyrimidinic endonuclease 1. Antioxid Redox Signal. 2014;20(4):678–707. | ||
Shimizu N, Sugimoto K, Tang J, et al. High-performance affinity beads for identifying drug receptors. Nat Biotechnol. 2000;18(8):877–881. | ||
Naidu MD, Agarwal R, Pena LA, et al. Lucanthone and its derivative hycanthone inhibit apurinic endonuclease-1 (APE1) by direct protein binding. PLoS One. 2011;6(9):e23679. | ||
Yang ZZ, Chen XH, Wang D. Experimental study enhancing the chemosensitivity of multiple myeloma to melphalan by using a tissue-specific APE1-silencing RNA expression vector. Clin Lymphoma Myeloma. 2007;7(4):296–304. |
Supplementary materials
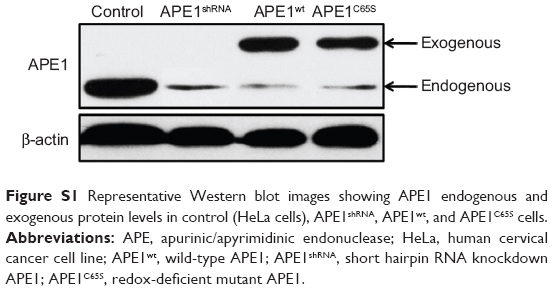 | Figure S1 Representative Western blot images showing APE1 endogenous and exogenous protein levels in control (HeLa cells), APE1shRNA, APE1wt, and APE1C65S cells. |
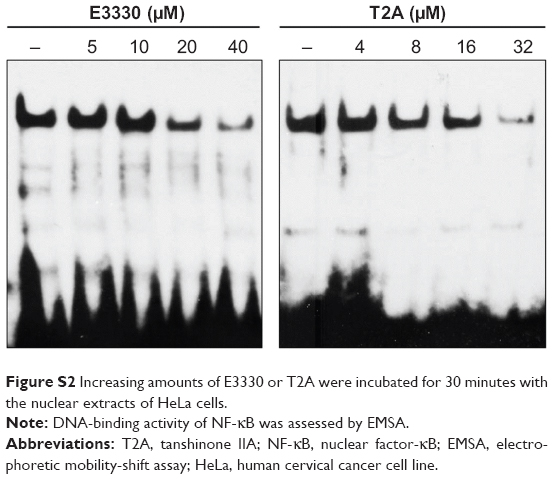 | Figure S2 Increasing amounts of E3330 or T2A were incubated for 30 minutes with the nuclear extracts of HeLa cells. |
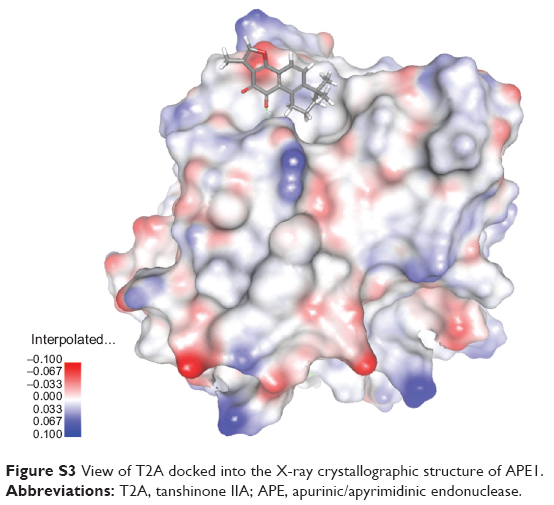 | Figure S3 View of T2A docked into the X-ray crystallographic structure of APE1. |
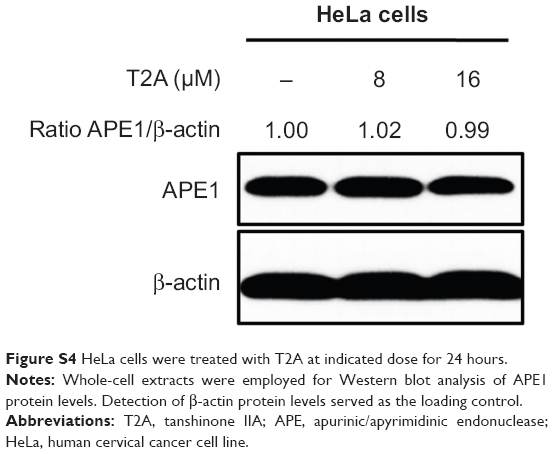 | Figure S4 HeLa cells were treated with T2A at indicated dose for 24 hours. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.