Back to Journals » Journal of Inflammation Research » Volume 18
Fumarate Hydratase Restrains mtDNA Attenuates LPS-Induced Acute Lung Injury Through cGAS-STING Pathways
Authors Jiang Z, He R, Zhong Y, Liu B, He Z
Received 20 January 2025
Accepted for publication 16 April 2025
Published 21 April 2025 Volume 2025:18 Pages 5399—5413
DOI https://doi.org/10.2147/JIR.S518589
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ning Quan
Zewen Jiang,1,* Ruyuan He,2,* Yujian Zhong,1,* Bohao Liu,3 Ziqi He4
1Department of Orthopedic Surgery, Renmin Hospital of Wuhan University, Wuhan, People’s Republic of China; 2Department of Thoracic Surgery, Renmin Hospital of Wuhan University, Wuhan, People’s Republic of China; 3Department of Thoracic Surgery, First Hospital of Jilin University, Changchun, People’s Republic of China; 4Department of Urology, Renmin Hospital of Wuhan University, Wuhan, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Bohao Liu, Department of Thoracic Surgery, The First Hospital of Jilin University, 1 Xinmin Street, Chaoyang District, Changchun, 130021, Jilin Province, People’s Republic of China, Email [email protected] Ziqi He, Department of Urology, Renmin Hospital of Wuhan University, 99 Zhangzhidong Road, Wuhan, Hubei Province, 430060, People’s Republic of China, Email [email protected]
Background: The metabolic reprogramming of alveolar macrophages, particularly mitochondrial energy metabolism centered on the tricarboxylic acid (TCA) cycle, plays a pivotal role in acute lung injury (ALI). Fumarate hydratase (FH), a key enzyme catalyzing fumarate-to-malate conversion in the TCA cycle, is implicated in macrophage inflammatory responses, but its specific role in ALI remains unclear.
Methods: We employed FHIN1 to assess its regulatory effects in LPS-induced ALI models. Wildtype C57BL/6 mice were randomly divided into control group, FHIN1 group, LPS group and LPS+FHIN1 group. FHIN1 and RU.521 was used to explored the interaction of FH and cGAS-STING in THP-1 cells.
Results: LPS stimulation suppressed FH expression and induced fumarate accumulation in macrophages. Pharmacological FH inhibition exacerbated LPS-triggered inflammatory cytokine release, oxidative stress and aggravated lung injury in mice. Mechanistically, FH inhibition promoted mtDNA leakage, activating the cGAS-STING pathway to amplify inflammation. Blocking cGAS with RU.521 significantly attenuated FHIN1-driven inflammatory responses and mitigated lung injury exacerbation.
Conclusion: FH critically modulates ALI progression by restraining cGAS-STING-dependent inflammation. Targeting the FH-mtDNA-cGAS axis may offer therapeutic potential for ALI management.
Keywords: ALI, fumarate hydratase, mtDNA
Graphical Abstract:

Introduction
Acute lung injury (ALI) is a severe, life-threatening condition affecting populations worldwide, often progressing to acute respiratory distress syndrome (ARDS). Despite advancements in pharmacological treatment and ventilatory support for ALI and ARDS, approximately 40% of affected patients do not survive.1 ALI is commonly characterized by inflammatory cell infiltration and a substantial release of pro-inflammatory cytokines, leading to significant damage to the alveolar epithelium and vascular endothelium.2 This damage results in key clinical symptoms: pulmonary edema and refractory hypoxemia, contributing to the high mortality rate associated with ALI.3 Currently, there is no effective therapy to reduce mortality or improve outcomes in ALI/ARDS patients. Consequently, further investigation into the pathogenesis and the identification of critical signaling pathways in ALI/ARDS may provide novel targets for therapeutic intervention. During ALI, macrophages undergo significant phenotypic and functional alterations alongside notable shifts in cellular metabolism, a field of study known as immunometabolism or metabolic reprogramming.4,5
The Krebs cycle, often called the TCA cycle, serves as the ultimate shared route for the oxidation of carbohydrates, fats, and amino acids, while also generating precursors for various biomolecules, including non-essential amino acids, nucleotide bases, and porphyrin.6 Fumarate, a metabolite in the TCA cycle, has been extensively studied within cancer metabolism and is now gaining relevance in immunology.7,8 Fumarate hydratase (FH) functions as a key regulatory enzyme, catalyzing the conversion of fumarate to malate, A loss of FH activity leads to an accumulation of fumarate and disruption of the TCA cycle’s normal regulation. Knock out of FH in kidney and macrophage results in mitochondrial structure and function disorders, release of mtRNA and mtDNA and cytokine production.9,10 The cyclic GMP-AMP synthase (cGAS)-stimulator of interferon genes (STING) pathway is a pivotal component of the innate immune system, responsible for detecting cytosolic DNA. Upon recognition of double-stranded DNA in the cytoplasm, cGAS catalyzes the synthesis of cyclic GMP-AMP (cGAMP) and activates STING, which subsequently triggers downstream signaling cascades involving TANK-binding kinase 1 (TBK1) and interferon regulatory factor 3 (IRF3), culminating in the production of type I interferons and other pro-inflammatory cytokines. This response is crucial for mounting antiviral defenses and initiating adaptive immunity.11 We speculate that FH may mediate cGAS-STING activation by regulating mtDNA release. As a metabolic regulator of immunity, the function of FH in acute lung injury remains uncertain. This study aimed to explore the role of FH in LPS-induced ALI and its molecular mechanism.
Materials and Methods
Animals and Treatment
This study adhered to ethical standards for animal research, receiving approval from the Animal Care and Use Committee of Wuhan University. All procedures complied with the US National Institutes of Health’s Guidelines for the Care and Use of Laboratory Animals (NIH Publication No. 85–23). Mice were maintained under specific pathogen-free (SPF) conditions at the Wuhan University Animal Center, with carefully controlled humidity (45–55%), temperature (20–25 °C), and a standard 12-hour light/dark cycle. Male wild-type C57BL/6 mice, aged 8–10 weeks were sourced from the Institute of Laboratory Animal Science, Chinese Academy of Medical Sciences (Beijing, China). To establish an acute lung injury (ALI) model in vivo, mice were injected intraperitoneally with lipopolysaccharide (LPS) at a concentration of 10 mg/kg, dissolved in 50 μL of sterile saline, following previously described methods. After 12 hours, mice were euthanized via cervical dislocation. The left lung was inflated with 10% neutral buffered formalin to preserve alveolar architecture, while the right lung was promptly dissected and preserved in liquid nitrogen for subsequent analysis. FHIN1 (HY-100004) was resuspended in 10% DMSO followed by 90% cyclodextrin in PBS (20% w/v), FHIN1 (50 mg/kg) was intraperitoneally 1 h before injected with LPS. RU.521 (5 mg/kg, RU.521 dissolved in saline solution, MedChemExpress, USA) was intraperitoneally 2 h before injected with LPS.
Cell Culture and Reagents
Hominine monocytic THP-1 cells were acquired from the China Cell Line Bank (Beijing, China) and cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum. Differentiation into macrophages was induced by treating the cells with 100 ng/mL of Phorbol 12-myristate 13-acetate (PMA) for 12 hours.12 To downregulate FH expression, the differentiated cells were transfected with siRNA obtained from GenePharma Co. Ltd. (Shanghai, China). Following the manufacturer’s protocol, cells were prepared with Lipofectamine™ 3000 Transfection Reagent (Thermo Fisher Scientific, Waltham, MA, USA) before siRNA introduction. FHIN1 was applied at concentrations of 10 or 20 μM for 3 hours prior to LPS stimulation. RU.521 (10 µM) was applied 1 h before LPS stimulation.
Western Blotting
For SDS-PAGE, a 10–12% separating gel, a 5% stacking gel, precooled electrophoresis buffer, and a membrane transfer solution chilled to 4 °C were prepared. The PVDF membrane was activated by soaking in methanol, and proteins were transferred at a constant current of 250 mA for 90 minutes. Blocking was carried out by incubating the membrane with 5% powdered skim milk at 37 °C for 2 hours. Subsequently, the membrane was incubated overnight at 4 °C with primary antibodies diluted 1:1000 in antibody diluent. Following this, the membrane was washed three times with TBST and then exposed to secondary antibodies (1:4000 dilution in TBST) for 2 hours at room temperature. After another wash with TBST, the membrane was developed and analyzed. Primary antibodies used included anti-STING (19851-1-AP), anti-cGAS (26416-1-AP), anti-FH (A5688), anti-β-actin (AC050), and anti-IRF3 (T55779), anti-pIRF3 (TA2436) sourced from Abmart (Shanghai, China) and Proteintech (Wuhan, China). Secondary antibodies were procured from Proteintech as well.
Lung Injury Score
The severity of pulmonary injury was evaluated using an established semiquantitative scoring protocol.11 Four histopathological parameters were systematically analyzed: inflammatory infiltration (red arrows), edema (blue arrows), hemorrhage (black arrows), and alveolar septal thickening (green arrows). Each parameter was graded on a 0–4 scale according to standardized criteria: Inflammatory infiltration: Quantified by enumerating total inflammatory cells per ×100 magnification field, with 5–7 random fields assessed per histological section. Edema and hemorrhage: Graded based on alveolar involvement percentage:0 (absent), 1 (mild, <10% alveoli affected), 2 (moderate, 10–30% involvement), 3 (severe, 30–50% involvement), 4 (critical, >50% involvement). Alveolar septal thickening: Measured using photomicrographs captured at 400x magnification, with vertical measurements taken at maximal thickness points. Thickness categories were defined as: 0 (<15 μm), 1 (15–30 μm), 2 (30–45 μm), 3 (45–60 μm), 4 (>60 μm). Final composite scores were derived by averaging individual parameter scores across all evaluated fields, providing a comprehensive assessment of pulmonary injury severity through integrated analysis of these four pathological features.
MDA and Cytokines Detection
The tissue or cells were lysed with lysis buffer, centrifuged at 12,000 g for 10 minutes, and the supernatant was collected. Protein concentrations were measured using BCA protein assay kit (P0010S, beyotime). The working curve was drawn using the standard sample and detected MDA level of sample according to the kit instructions (S0131M, beyotime). The MDA level was detected and calculated using a spectrophotometer, and statistical analysis was performed after normalization. The cytokines (IL-1β, TNF-α, IL-10 and IFN-β) were detected through ELISA Kit (PI301, PT512, PI528 and PI568 beyotime) according to instruction.
Hematoxylin & Eosin (H&E) Staining
The lung tissues underwent fixation, dehydration, and paraffin embedding before being sectioned into 5 μm slices using a microtome. Hematoxylin staining was applied to visualize the tissue, followed by differentiation with hydrochloric acid alcohol, bluing in tap water, and counterstaining with eosin. The stained sections were gradually dried through graded ethanol and xylene treatments and mounted using neutral gum. Finally, the sections were observed and photographed under a microscope at magnifications of 100× and 200×.
Assay of Lung Wet/Dry Ratio
After the complete removal of the right lung in mice, residual blood was carefully cleared, and the wet weight was immediately recorded. The lung tissues were subsequently dried in an oven at 75 °C for four days until a constant weight was achieved, enabling the determination of the dry weight and calculation of the lung wet-to-dry ratio.
Detection of Fumarate and FH Activity
Fumarate levels in lung tissue and THP-1 cells were determined using the Fumarate Assay Kit (Abcam, 102516) in accordance with the manufacturer’s instructions. Briefly, transfected cells were plated in 96-well plates, and supernatants were collected at intervals of 24, 48, 72, and 96 hours post-transfection. Fumarate concentrations were calculated using a standard curve generated from known fumarate concentrations. FH activity was assessed with the Fumarase Activity Assay Kit (Beyotime, S0520S), following the experimental procedures specified in the kit’s protocol.
Relative Quantitative RT-PCR
Total RNA was extracted from lung tissues or cells using the TRIzol method. TRIzol reagent was added to each sample, followed by RNA separation with chloroform, precipitation using isopropanol, and washing with 75% ethanol. The purified RNA was dissolved in 20 μL of DEPC-treated enzyme-free water and quantified with a NANODROP spectrophotometer. The RNA was then reverse-transcribed into cDNA following the instructions provided with the reverse transcription kit. mRNA expression levels were quantified using SYBR® Green Real-time PCR Master Mix (QPK-201, TOYOBO), with β-actin serving as the internal control. The 2−ΔΔCt method was used for statistical analysis. Primer sequences utilized in this study are listed in Supplementary Table S1.
Detection of mtDNA in the Cytoplasm
Mitochondrial DNA (mtDNA) was isolated using the Mitochondrial DNA Isolation Kit (Abcam, #ab65321). THP-1 cells were lysed and centrifuged at 800×g for 10 minutes at 4 °C to remove debris. The resulting supernatant was collected and subjected to further centrifugation at 10,000×g for 30 minutes at 4 °C to obtain the mitochondrial fraction. Differential centrifugation ensured that cytosolic mtDNA remained in the supernatant, while mitochondrial mtDNA was concentrated in the pellet. Cytosolic mtDNA levels were quantified via real-time PCR by measuring Cytochrome C oxidase subunit 1 (COX1) expression, normalized to 18S.
Detection of mtRNA in the Cytoplasm
THP-1 cells were scraped on ice into ice-cold PBS and pelleted at 300g for 5 min at 4 °C. Remove supernatant and resuspend the pellet in 400 μL extraction buffer (150 mm NaCl, 50 mm HEPES pH 7.4, and 25 μg mL−1 digitonin). The resuspended samples were vortexed thoroughly and centrifuged again (2,000g at 4 °C for 5 min). Using an AllPrep DNA/ RNA Mini kit (Qiagen) to isolate RNA in supernatant. To ensure that any genomic DNA contamination is removed, treat the RNA with DNase I. Use NanoDrop to quantify RNA concentration. qPCR was performed using primers specific for mitochondrial D-loop on cDNA to determine the presence of mtRNA in the cytosol. The housekeeping control gene ACTB was used to normalized and amplified in cDNA. Using a SYBR Green qPCR Master Mix (Thermo Fisher, US) according to the kit’s instructions. Amplify cDNA in a qPCR machine and use the ΔΔCt method for quantifying mtRNA relative expression levels.
Bioinformatic Analysis
Raw and processed data were obtained from the public database DYRAD (https://datadryad.org/dataset/doi:10.5061/dryad.6wwpzgn28). Differential expression analysis of RNA-seq datasets was performed using the DESeq2 software. Statistical significance between groups was assessed with the Wald test. Heatmaps were generated using the R programming language.
Statistical Analysis
The data are expressed as the mean ± standard deviation (mean ± SD). Statistical analysis was conducted using SPSS 22.0 software (SPSS Inc., Chicago, IL, USA), and figures were generated with GraphPad Prism 8.0. Group differences were analyzed using the t-test for two-group comparisons or one-way analysis of variance (ANOVA) with Bonferroni post hoc testing for multiple-group comparisons. A P-value below 0.05 was deemed statistically significant.
Results
FH Is Decreased After LPS Stimulation
To further elucidate the changes in fumarate and FH (fumarate hydratase) in the lungs of mice with sepsis-induced acute lung injury (ALI) and in macrophages following LPS stimulation, we established mouse and macrophage injury models induced by LPS. We observed that the levels of fumarate in the lung tissue of mice significantly increased after LPS stimulation (Figure 1A); however, this accumulation of fumarate was not caused by changes in FH enzyme activity (Figure 1B). This finding is consistent with previous studies indicating that LPS stimulation reprograms the TCA cycle in macrophages, leading to fumarate accumulation.9 In the lung tissues of ALI mice, we found that LPS stimulation significantly reduced the protein and mRNA expression of FH (Figure 1C–E). Furthermore, in the macrophage model, we observed that FH protein and mRNA levels also significantly decreased with prolonged LPS stimulation (Figure 1F–H), suggesting a pivotal role for FH in LPS-induced ALI.
 |
Figure 1 FH decreases after LPS stimulated in lung tissue of ALI. (A) Fumarate level of lung tissue (n=6); (B) The activity of FH in lung tissue (n=6); (C–E) The protein and mRNA level of FH in lung tissue (n=6); (F–H) FH protein and mRNA level at different time points after LPS stimulation in THP-1 cell (n=6). (*p < 0.05, **p < 0.01, ***p < 0.001). |
The Inhibition of FH Accelerates the LPS Induced ALI
To further investigate the role of FH in LPS-induced ALI, we used the pharmacological FH inhibitor FHIN1, which has been demonstrated in numerous studies to effectively inhibit FH enzyme activity.13,14 Mice were pretreated with FHIN1 via intraperitoneal injection prior to LPS stimulation. Our results showed that FHIN1 did not affect the protein or mRNA expression of FH (Figure 2A–C), but leading to a notable accumulation of fumarate in lung tissues (Figure 2D). Pretreatment with FHIN1 markedly enhanced the LPS-induced increase in inflammatory factors IL-1β, TNFα, IFN-β and decrease of IL-10 (Figure 2E–H), indicating that inhibition of FH might promote LPS-induced lung inflammation. The lung dry/wet ratio results also confirmed that lung edema in the FHIN1+LPS group was significantly more severe compared to the LPS group (Figure 2I). Additionally, MDA analysis revealed that FHIN1 exacerbated oxidative stress in the lung tissues of LPS-induced ALI mice (Figure 2J). Furthermore, HE staining of pathological sections indicated that, compared to the control group, LPS significantly aggravated lung injury and increased the lung injury score, and this trend was further exacerbated by FHIN1-mediated inhibition of FH enzyme activity (Figure 2K and L). These findings preliminarily demonstrate that pharmacological inhibition of FH significantly worsens LPS-induced acute lung injury.
 |
Figure 2 Inhibition of FH exacerbates LPS induced ALI. (A–C) FH protein and mRNA level in lung tissue after FHIN1 treatment (n=6); (D) Fumarate level in lung tissue after FHIN1 treatment (n=6); (E–H) The level of IL-1β, TNFα, IFN-β and IL-10 in lung tissue (n=6); (I) The wet/dry ratio of lung tissue (n=6); (J) Relative MDA level of lung tissue (n=6); (K) Typical H&E staining for lung tissues; (L) Lung injury score according to H&E staining (n=6). (*p < 0.05, **p < 0.01, ***p < 0.001). |
The Inhibition of FH Increase the Release of mtDNA in Macrophage
Alveolar macrophages play a crucial role in the development and progression of acute lung injury (ALI) and are among the most important immune cell types regulating ALI. Previous studies have shown that knocking down FH in macrophages affects mitochondrial integrity and leads to fumarate accumulation.10 To further explore the mechanisms by which FH inhibition exacerbates LPS-induced ALI, we analyzed TCA metabolite profiling data from the DYRAD database. Heatmap analysis revealed that FHIN1 treatment significantly increased the levels of fumarate, lactate, and argininosuccinate, while reducing the levels of acetyl-CoA, succinate, and citrate, indicating that FHIN1 induces TCA metabolic reprogramming and promotes fumarate accumulation (Figure 3A). Further in vitro experiments confirmed that treatment of PMA-differentiated THP-1 cells with FHIN1 significantly inhibited FH enzyme activity and increased fumarate levels (Figure 3B and C). Other findings showed that FHIN1 elevated LPS-induced IL-1β, TNFα and IFN-β levels (Figure 3D–F), as well as MDA and GSH levels (Figure 3G and H). The results of JC-1 immunofluorescence staining confirmed that FHIN1 treatment further disrupted the mitochondrial membrane potential (MMP) (Figure 3I and J). Consistent with previously published studies,10 we also found that FHIN1 treatment not only disrupted the TCA cycle but also promoted LPS-induced release of mtDNA and mtRNA (Figure 3K and L).
 |
Figure 3 Inhibition of FH promote the LPS induced mitochondrial damage and mtDNA release in macrophages. (A) The heatmap of TCA metabolite profiling after FHIN1 treatment in macrophage (n=6); (B) Fumarate level after FHIN1 treatment in THP-1 (n=6); (C) The activity of FH in THP-1 (n=6); (D–F) The level of IL-1β, TNFα and IFN-β in THP-1 (n=6); (G) Relative MDA level in THP-1 (n=6); (H) The level of GSH in THP-1 (n=6); (I and J) Typical JC-1 immunofluorescence staining and statistical results in THP-1; (K and L) The level of mtDNA and mtRNA in THP-1 (n=6). (*p < 0.05, **p < 0.01, ***p < 0.001). |
The Inhibition of FH Activate cGAS-STING Pathways Through mtDNA in Cytoplasm
To further investigate how FH inhibition-induced mitochondrial damage and mtDNA release exacerbate sepsis-induced acute lung injury (ALI), we conducted KEGG pathway enrichment analysis on LPS-stimulated macrophages treated with FHIN1. The results showed that Cytosolic DNA−sensing pathway was significantly upregulated in the FHIN1 inhibition group (Figure 4A). As we all known, release of mtDNA into the cytosol has emerged as a prominent trigger of cGAS–STING pathway activation in multiple contexts. To further confirm the changes in the cGAS-STING pathway after FH inhibition, we examined the protein and mRNA levels of key downstream transcription factor IRF3 and its target genes in the cGAS-STING pathway using Western blot and mRNA assays. Our results indicated that in THP-1 cells, LPS significantly upregulated the protein levels of cGAS, STING, and p-IRF3 (Figure 4B and C), and increased the mRNA levels of IRF3 target genes Areg, Ifnb1, and Mx1 (Figure 4D–F). Although FHIN1 treatment alone did not upregulate cGAS or its downstream proteins, the combination of FHIN1 and LPS further increased p-IRF3 levels and enhanced the transcription of downstream target genes, confirming that FH inhibition significantly exacerbates LPS-induced activation of the cGAS-STING pathway.
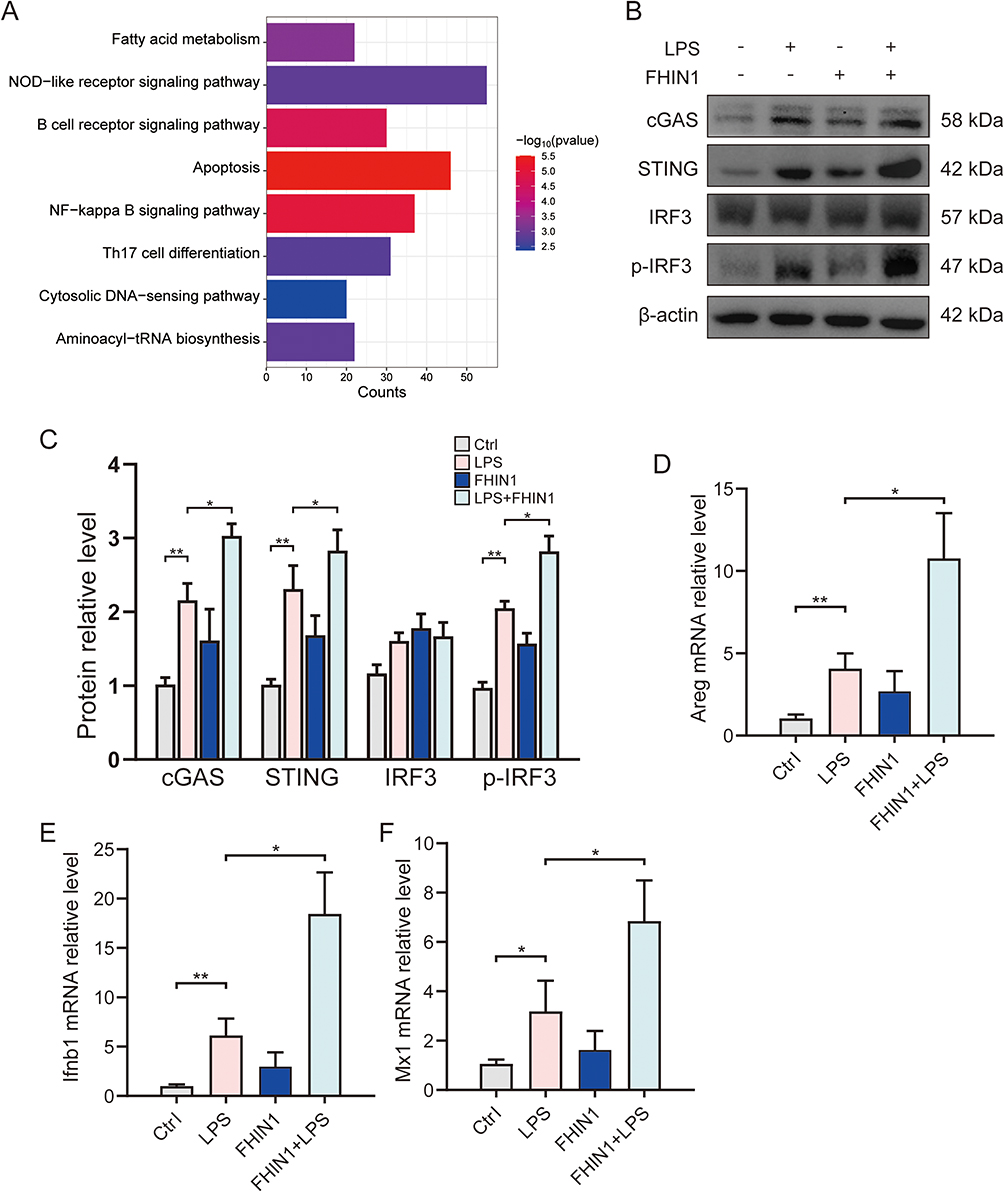 |
Figure 4 Inhibition of FH activates the cGAS-STING pathways in ALI. (A) KEGG enrichment between LPS and LPS+FHIN1 group; (B and C) Western blots for cGAS, STING, IRF3 and p-IRF3 in lung tissue (n=6); (D–F) Relative mRNA level of Areg, Ifnb1 and Mx1 in lung tissue (n=6). (*p < 0.05, **p < 0.01). |
Knockdown of cGAS Can Alleviate cGAS-STING Activation and Inflammatory Responses Induced by FH Inhibition
To further confirm whether cGAS-STING is the primary regulatory mechanism by which pharmacological inhibition of FH exacerbates ALI, we used siRNA to knock down cGAS in THP-1 cells. The results showed that after cGAS knockdown, LPS stimulation significantly reduced the expression of STING and downstream p-IRF3 (Figure 5A). Moreover, cGAS knockdown significantly inhibited FHIN1-induced activation of the cGAS-STING pathway, with a notable reduction in p-IRF3 protein levels and the mRNA levels of its target genes Areg and Irnb1 (Figure 5A–D). MDA results also indicated that cGAS knockdown significantly alleviated the accumulation of oxidative stress caused by FHIN1 (Figure 5E). Additionally, ELISA results for cellular inflammatory factors confirmed that cGAS knockdown alleviated the increases of IL-1β and TNF-α induced by FHIN1+LPS, while significantly elevating IL-10 levels (Figure 5F). These findings demonstrate that cGAS knockdown significantly reduces cGAS-STING pathway activation and cellular inflammation levels induced by FH inhibition, confirming that the macrophage damage exacerbated by pharmacological FH inhibition via FHIN1 is cGAS-STING dependent.
 |
Figure 5 FH inhibit the inflammation through cGAS-dependent pathway in macrophage. (A and B) Western blots for cGAS, STING, IRF3 and p-IRF3 (n=6); (C and D) Relative mRNA level of Areg and Ifnb1 (n=6); (E) Relative MDA level in macrophage (n=6); (F) The level of TNFα, IL-1β and IL-10 in lung tissue (n=6). (*p < 0.05, **p < 0.01, ***p < 0.001). |
The cGAS Inhibitor RU.521 Can Alleviate the Exacerbation of ALI Caused by FH Inhibition
We confirmed that FH inhibition primarily exacerbates the inflammatory response of alveolar macrophages by affecting mtDNA release and activating the cGAS-STING pathway in vitro. However, it remains unclear whether oral administration of the cGAS inhibitor RU.521, a widely used selective inhibitor of cGAS activity15 can alleviate lung injury exacerbated by FH inhibition in vivo. Therefore, we administered RU.521 orally to mice prior to FHIN1 and LPS pretreatment. The results showed that RU.521 significantly reduced the levels of inflammatory factors IL-1β and TNF-α induced by FHIN1 (Figure 6A and B), decreased MDA levels (Figure 6C), and improved lung edema in mice compared to the FHIN1+LPS group. Histopathological analysis with HE staining and lung injury scoring confirmed that RU.521 pretreatment protected mice from exacerbated lung injury caused by FH inhibition (Figure 6D–F). Collectively, these findings demonstrate that pharmacological inhibition of FH exacerbates the inflammatory response of alveolar macrophages and promotes ALI through mtDNA release and activation of the cGAS-STING pathway.
 |
Figure 6 RU.521 can alleviate the exacerbation of ALI caused by FH inhibition. (A and B) The level of TNFα and IL-1β in lung tissue (n=6); (C) Relative MDA level in lung tissue (n=6); (D) Typical H&E staining for lung tissues; (E) Lung injury score according to H&E staining (n=6); (F) The wet/dry ratio of lung tissue (n=6). (*p < 0.05, **p < 0.01). |
Discussion
In this study, we confirmed the pivotal role of FH in sepsis-induced acute lung injury (ALI). Our findings indicate that pharmacological inhibition of FH markedly worsens LPS-induced lung damage and macrophage injury responses in mice. Metabolomics and RNA-seq analyses revealed that FH inhibition reprograms macrophage TCA metabolism, resulting in the accumulation of fumarate, lactate, and argininosuccinate, coupled with a decrease in acetyl-CoA, succinate, and citrate. Moreover, FH inhibition impairs mitochondrial function, promotes the release of mtDNA, and activates the cGAS-STING pathway, thereby amplifying inflammatory responses and exacerbating lung injury. Notably, pharmacological or genetic inhibition of cGAS effectively mitigates FH inhibition-induced lung injury. Fumarate, a key TCA cycle metabolite, has been extensively studied in cancer metabolism and is now gaining increased recognition in immunological research. Cancer studies have predominantly examined tumors with FH deficiency or mutations. As FH catalyzes the conversion of fumarate to malate, its loss leads to fumarate accumulation and TCA cycle dysregulation. This metabolic disruption activates multiple pathways that contribute to cancer initiation and progression.16 Consistent with recent studies,10 our findings confirm that FH inhibition triggers fumarate accumulation and disrupts mitochondrial metabolism and function. This accumulation is not attributed solely to reduced FH enzymatic activity; the upregulation of argininosuccinate synthase (ASS1) likely contributes as well.9 Evidence for this includes the observation that pharmacological inhibition of the pathway with the GOT2 inhibitor aminooxyacetic acid, combined with the genetic knockdown of argininosuccinate lyase, successfully prevented fumarate accumulation following LPS exposure.17
Furthermore, FH inhibition significantly increased IL-1β and TNF-α expression while reducing IL-10 levels compared to LPS stimulation alone. We propose that fumarate accumulation partially drives these alterations in inflammatory factors, though it is not the primary cause of worsened lung injury. Fumarate plays diverse roles in immune regulation. For example, dimethyl fumarate (DMF), a fumarate derivative, is a widely used immunomodulatory drug for treating Multiple Sclerosis (MS).18 Available research suggests that DMF targets several cellular proteins through which it manifests its anti-inflammatory and antitumoral activities. Interestingly, while accumulation of endogenous fumarate promotes inflammatory factor secretion, its derivative DMF exhibits anti-inflammatory properties.19,20 This paradox may stem from differences between endogenous fumarate and exogenously administered DMF, as well as variations in their cytoplasmic concentrations.
The primary mechanism by which FH inhibition exacerbates lung injury appears to be caused by macrophage metabolic reprogramming. Published results have shown that while both exogenous DMF supplementation and FH inhibition result in fumarate accumulation, their effects on mitochondrial function and immune responses differ significantly. For instance, FH inhibition (via FHIN1) reduces ATP/ADP, ATP/AMP, and phosphocreatine/creatine ratios, highlighting FH’s essential role in maintaining mitochondrial bioenergetics. Unlike DMF, FHIN1 impairs basal respiration, ATP production, and maximal respiration, as measured by oxygen consumption rates.9
In addition, FH inhibition alters not only fumarate levels but also other TCA metabolites, resulting in changes distinct from those caused by DMF treatment. We hypothesize that mitochondrial dysfunction, coupled with increased release of mtDNA and mtRNA, primarily drives the worsening of lung injury following FH inhibition. To investigate this further, we explored the downstream immune-regulatory effects of mtDNA. RNA-seq analysis revealed that the cytosolic DNA−sensing pathway was activated after FHIN1 treatment. While previous studies demonstrated that increased mtRNA due to FH inhibition activates RNA sensors such as TLR7, RIG-I, and MDA5, leading to elevated interferon-β production,9 the downstream mechanisms of mtDNA remain poorly understood. As a classical mtDNA sensor, cGAS-STING plays a pivotal role in this process. In the context of acute lung injury (ALI), the cGAS-STING pathway plays a complex role. While appropriate activation of this pathway is essential for host defense against pathogens, its dysregulation can contribute to excessive inflammation and tissue damage. For instance, studies have shown that overactivation of the cGAS-STING pathway due to self-DNA release can drive inflammatory responses in lung tissues, exacerbating conditions such as ALI. Conversely, certain interventions targeting this pathway have demonstrated therapeutic potential.21 Our previous studies have confirmed that cGAS-STING promotes ALI by regulating NLRP3 inflammasome activation and macrophage pyroptosis.22 Our findings show that cGAS-STING-dependent regulation is crucial in macrophage inflammatory responses following FH inhibition. Importantly, cGAS inhibitors significantly reduced the exacerbation of ALI induced by FHIN1.
This study also has some limitations. First, it primarily relied on the pharmacological inhibitor FHIN1 to suppress FH activity, without employing macrophage-specific FH knockout mice for further animal model validation. Consequently, the regulatory functions of FH related to its structural properties, rather than enzymatic activity, were not considered. Additionally, the mechanisms through which TCA metabolic reprogramming induced by FH inhibition leads to changes in mitochondrial structure and function were not comprehensively explored, highlighting the need for further investigation.
Data Sharing Statement
Data sharing not applicable to this article as no datasets were generated during the current study.
Ethics Approval and Consent to Participate
This study adhered to ethical standards for animal research, receiving approval from the Animal Care and Use Committee of Wuhan University.
Consent for Publication
Ethical approval for animal experiments was obtained from Animal Care and Use Committee of Wuhan University.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; All authors have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by grants from the National Natural Science Foundation of China (No. 82300103), the Fundamental Research Funds for the Central Universities (2042023kf0011).
Disclosure
The authors declare that they have no competing interests.
References
1. Rubenfeld GD, Caldwell E, Peabody E, et al. Incidence and outcomes of acute lung injury. N Engl J Med. 2005;353:1685–1693. doi:10.1056/NEJMoa050333
2. Butt Y, Kurdowska A, Allen TC. Acute lung injury: a clinical and molecular review. Arch Pathol Lab Med. 2016;140:345–350. doi:10.5858/arpa.2015-0519-RA
3. Kumar V. Pulmonary innate immune response determines the outcome of inflammation during pneumonia and sepsis-associated acute lung injury. Front Immunol. 2020;11:1722. doi:10.3389/fimmu.2020.01722
4. Johnston LK, Rims CR, Gill SE, McGuire JK, Manicone AM. Pulmonary macrophage subpopulations in the induction and resolution of acute lung injury. Am J Respir Cell mol Biol. 2012;47:417–426. doi:10.1165/rcmb.2012-0090OC
5. Liu Y-C, Zou X-B, Chai Y-F, Yao Y-M. Macrophage polarization in inflammatory diseases. Int J Biol Sci. 2014;10:520–529. doi:10.7150/ijbs.8879
6. Martínez-Reyes I, Chandel NS. Mitochondrial TCA cycle metabolites control physiology and disease. Nat Commun. 2020;11:102. doi:10.1038/s41467-019-13668-3
7. Ge X, Li M, Yin J, et al. Fumarate inhibits PTEN to promote tumorigenesis and therapeutic resistance of type2 papillary renal cell carcinoma. Molecular Cell. 2022;82:1249–1260.e7. doi:10.1016/j.molcel.2022.01.029
8. Schmidt LS, Linehan WM. Hereditary leiomyomatosis and renal cell carcinoma. IJNRD. 2014;7:253–260. doi:10.2147/IJNRD.S42097
9. Hooftman A, Peace CG, Ryan DG, et al. Macrophage fumarate hydratase restrains mtRNA-mediated interferon production. Nature. 2023;615:490–498. doi:10.1038/s41586-023-05720-6
10. Zecchini V, Paupe V, Herranz-Montoya I, et al. Fumarate induces vesicular release of mtDNA to drive innate immunity. Nature. 2023;615:499–506. doi:10.1038/s41586-023-05770-w
11. Chen Q, Sun L, Chen ZJ. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat Immunol. 2016;17:1142–1149. doi:10.1038/ni.3558
12. He R, Liu B, Xiong R, et al. Itaconate inhibits ferroptosis of macrophage via Nrf2 pathways against sepsis-induced acute lung injury. Cell Death Discov. 2022;8:43. doi:10.1038/s41420-021-00807-3
13. Takeuchi T, Schumacker PT, Kozmin SA. Identification of fumarate hydratase inhibitors with nutrient-dependent cytotoxicity. J Am Chem Soc. 2015;137:564–567. doi:10.1021/ja5101257
14. Cheng J, Liu Y, Yan J, et al. Fumarate suppresses B-cell activation and function through direct inactivation of LYN. Nat Chem Biol. 2022;18:954–962. doi:10.1038/s41589-022-01052-0
15. Vincent J, Adura C, Gao P, et al. Small molecule inhibition of cGAS reduces interferon expression in primary macrophages from autoimmune mice. Nat Commun. 2017;8:750. doi:10.1038/s41467-017-00833-9
16. Isaacs JS, Yun JJ, Mole DR, et al. HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: novel role of fumarate in regulation of HIF stability. Cancer Cell. 2005;8:143–153. doi:10.1016/j.ccr.2005.06.017
17. Peace CG, O’Carroll SM, O’Neill LAJ. Fumarate hydratase as a metabolic regulator of immunity. Trends Cell Biol. 2024;34:442–450. doi:10.1016/j.tcb.2023.10.005
18. Okuda DT, Kantarci O, Lebrun-Frénay C, et al. Dimethyl fumarate delays multiple sclerosis in radiologically isolated syndrome. Ann Neurol. 2023;93:604–614. doi:10.1002/ana.26555
19. Cattani-Cavalieri I, da Maia Valença H, Moraes JA, et al. Dimethyl fumarate attenuates lung inflammation and oxidative stress induced by chronic exposure to diesel exhaust particles in mice. Int J mol Sci. 2020;21:9658. doi:10.3390/ijms21249658
20. Ryan TAJ, Hooftman A, Rehill AM, et al. Dimethyl fumarate and 4-octyl itaconate are anticoagulants that suppress tissue factor in macrophages via inhibition of Type I Interferon. Nat Commun. 2023;14:3513. doi:10.1038/s41467-023-39174-1
21. Ma R, Ortiz Serrano TP, Davis J, Prigge AD, Ridge KM. The cGAS-STING pathway: the role of self-DNA sensing in inflammatory lung disease. FASEB J. 2020;34:13156–13170. doi:10.1096/fj.202001607R
22. Ning L, Wei W, Wenyang J, Rui X, Qing G. Cytosolic DNA‐STING‐NLRP3 axis is involved in murine acute lung injury induced by lipopolysaccharide. Clin Transl Med. 2020;10:e228. doi:10.1002/ctm2.228
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.