Back to Journals » Neuropsychiatric Disease and Treatment » Volume 10
Frontotemporal lobar degeneration: current perspectives
Authors Riedl L, Mackenzie I, Förstl H, Kurz A, Diehl-Schmid J
Received 5 October 2013
Accepted for publication 2 December 2013
Published 13 February 2014 Volume 2014:10 Pages 297—310
DOI https://doi.org/10.2147/NDT.S38706
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Lina Riedl,1 Ian R Mackenzie,2 Hans Förstl,1 Alexander Kurz,1 Janine Diehl-Schmid1
1Center for Cognitive Disorders, Department of Psychiatry and Psychotherapy, Klinikum rechts der Isar, Technische Universität München, Munich, Germany; 2Department of Pathology and Laboratory Medicine, University of British Columbia, Vancouver, Canada
Abstract: The term frontotemporal lobar degeneration (FTLD) refers to a group of progressive brain diseases, which preferentially involve the frontal and temporal lobes. Depending on the primary site of atrophy, the clinical manifestation is dominated by behavior alterations or impairment of language. The onset of symptoms usually occurs before the age of 60 years, and the mean survival from diagnosis varies between 3 and 10 years. The prevalence is estimated at 15 per 100,000 in the population aged between 45 and 65 years, which is similar to the prevalence of Alzheimer's disease in this age group. There are two major clinical subtypes, behavioral-variant frontotemporal dementia and primary progressive aphasia. The neuropathology underlying the clinical syndromes is also heterogeneous. A common feature is the accumulation of certain neuronal proteins. Of these, the microtubule-associated protein tau (MAPT), the transactive response DNA-binding protein, and the fused in sarcoma protein are most important. Approximately 10% to 30% of FTLD shows an autosomal dominant pattern of inheritance, with mutations in the genes for MAPT, progranulin (GRN), and in the chromosome 9 open reading frame 72 (C9orf72) accounting for more than 80% of familial cases. Although significant advances have been made in recent years regarding diagnostic criteria, clinical assessment instruments, neuropsychological tests, cerebrospinal fluid biomarkers, and brain imaging techniques, the clinical diagnosis remains a challenge. To date, there is no specific pharmacological treatment for FTLD. Some evidence has been provided for serotonin reuptake inhibitors to reduce behavioral disturbances. No large-scale or high-quality studies have been conducted to determine the efficacy of non-pharmacological treatment approaches in FTLD. In view of the limited treatment options, caregiver education and support is currently the most important component of the clinical management.
Keywords: review, frontotemporal dementia, frontotemporal lobar degeneration
Introduction
The term frontotemporal lobar degeneration (FTLD) refers to a group of progressive brain diseases, which are heterogeneous with regard to etiology and neuropathology, but share atrophy of the frontal and/or temporal cortex as a morphological feature. The clinical syndromes of FTLD are characterized by progressive deterioration of either behavior or language. The first patient was described in 1892 by Arnold Pick, a neurologist in Prague,1 and FTLD was named Pick’s disease for more than a century. Today, this term is used only for a small subgroup of FTLD with specific histopathological features. The nomenclature of FTLD has remained inconsistent. In the present review, FTLD is used as an overarching term for a clinicopathological complex, which includes two clinical syndromes and three major underlying neuropathological subtypes. The clinical syndromes – behavioral-variant frontotemporal dementia (bvFTD) and primary progressive aphasia (PPA) – are distinguished by the early and predominant symptoms. The neuropathological subtypes are characterized by an abnormal accumulation of proteins, microtubule-associated protein tau (MAPT), transactive response DNA-binding protein with molecular weight 43 kDa (TDP-43), and fused in sarcoma protein (FUS).
Clinical subtypes
bvFTD
The most common clinical syndrome of FTLD is bvFTD. Atrophy predominantly of the mesiofrontal, orbitofrontal, and anterior insular cortex regions of the frontal lobes leads to progressive personality changes and behavioral disturbances.2–4
Since 1994, four sets of diagnostic criteria have been published,5–8 which reflect the evolving understanding of the disorder.9 The most recent criteria, proposed by The International bvFTD Criteria Consortium,7 define three levels of diagnostic certainty and refer to six behavioral and cognitive symptom clusters (Table 1).
 | Table 1 International consensus criteria for bvFTD |
According to these criteria, bvFTD is characterized by early decline in social behavior and personal conduct, as indicated by early disinhibition, apathy, loss of sympathy, perseverative and stereotyped behaviors, and hyperorality.
Behavioral disinhibition is a frequent symptom of bvFTD. It has multiple appearances; eg, a lack of etiquette, with patients making offensive jokes, often with sexual reference, or exhibiting childish behavior. Patients may approach strangers in an aggressive manner or may touch them inappropriately. They may exhibit impulsive or risky behaviors such as gambling, or may fall for financial scams; eg, doorstep selling. In a recent study of pathologically confirmed bvFTD cases, 76% of patients showed behavioral disinhibition or impulsivity.7
Over 85% of patients in this study exhibited apathy and inertia as an early symptom.7 Inertia refers to a decreased action generating ability. Apathy corresponds to general passivity. Both result in a lack of motivation to pursue activities and hobbies, so patients may need assistance to initiate or continue those activities.
Human empathy relies on the ability to share emotions as well as the ability to understand other’s thoughts, desires, and feelings.10 The loss of sympathy in patients with bvFTD results in a cold and indifferent attitude. There is converging evidence from neuroimaging studies that the neurodegeneration in anterior regions of the medial frontal cortex is linked to the patients’ deficits in their ability to attribute mental states to one’s self and to others.11
Patients may perform repetitive, stereotyped actions such as scratching, lip smacking, or clapping hands. Some may repeat phrases or stories. Rituals involving fixed wandering routes, hoarding, or object counting are also observed. These inappropriate repetitive behaviors are linked to a dysfunction of a circuit involving the orbitofrontal and anterior cingulate cortices, the basal ganglia, and the thalamus.12
Altered food preferences with craving for sweets, or rigid preference of particular foods are frequently observed.13 Some patients show binge eating, sometimes even with negative health consequences (eg, diabetes mellitus). There has been discussion about the involvement of the orbitofrontal–insular–striatal brain network and the hypothalamus.14
While behavioral changes dominate the initial presentation of bvFTD, cognitive deficits appear as the disease progresses. The neuropsychological profile of bvFTD is characterized by executive deficits and by relative sparing of memory and visuospatial functions.
PPA
The language variants of FTLD, semantic variant PPA (svPPA; also referred to as semantic dementia or temporal variant frontotemporal dementia) and nonfluent variant PPA (nfvPPA; also referred to as progressive nonfluent aphasia) are summarized under the term PPA.
svPPA is caused by marked, often asymmetric (left > right) anterior and inferior temporal atrophy.15,16 As the disease progresses, posterior temporal regions are also involved, as well as the orbitofrontal lobe, insula and anterior cingulate, and homologous regions in the opposite hemisphere.17,18 Speech is fluent, and syntax, prosody, and motor speech are intact. However, patients gradually lose their semantic memory – the knowledge about words, objects, and concepts. Speech becomes empty and ultimately meaningless. Semantic paraphasias are frequent, and surface dyslexia and dysgraphia occur. In advanced disease stages, semantic knowledge is impaired beyond language, and the patients develop features of multimodal agnosia. During the course of svPPA many patients present behavioral disturbances similar to bvFTD.19 Personality changes are frequently among the first symptoms in patients who exhibit a marked atrophy of the right temporal lobe.20 In these patients, language deficits are less prominent than prosopagnosia and associative agnosia.21
nfvPPA is caused by a predominant left posterior fronto-insular atrophy. Patients with nfvPPA present with a nonfluent, effortful, halting speech that is dominated by obvious word-finding difficulties and agrammatism. Language production is simplified, phonematic paraphasias are frequent. Oral apraxia is observed in most patients, dysarthria occurs less frequently. In contrast to svPPA, object knowledge and single word comprehension are spared, while comprehension of syntactically complex sentences is impaired. In the late disease stages, the patients become mute.16,22
Recent diagnostic criteria suggest a third subtype of PPA that does not fit the criteria for svPPA or nfvPPA and has been termed logopenic variant of progressive aphasia (lvPPA).16 In lvPPA, atrophy is most prominent in the posterior cortical region with asymmetrical (left > right) temporo–parietal atrophy. Patients present phonological disorders as well as impaired word retrieval and sentence repetition. On the other hand, motor speech, grammar, and comprehension are relatively intact. A recently published study examined the clinicopathological correlations within the PPA variants.23 Fifty-two patients were included. From the 30 patients meeting the basic PPA criteria, all the svPPA patients (four) and 75% of the nfvPPA patients had frontotemporal lobar degeneration spectrum pathology. In the group of patients who met the logopenic variant PPA, the underlying pathology was heterogenous (46% Alzheimer’s disease [AD], 8% AD mixed with dementia with Lewy bodies, 23% frontotemporal lobar degeneration, and 23% others). It is obvious that this causes great difficulty in diagnosing an aphasic syndrome.
Overlap of FTLD with corticobasal degeneration (CBD), progressive supranuclear palsy (PSP), and motor neuron disease
There is considerable clinical and histopathological overlap of FTLD with the atypical parkinsonian disorders CBD and PSP, and with motor neuron disease (Table 2). Patients with PSP show vertical gaze palsy and parkinsonian symptoms such as bradykinesia, axial rigidity, and postural instability, which poorly respond to levodopa treatment. Rapid eye movement sleep behavior disorder is observed in Parkinson’s disease, but not in PSP.24 Difficulties in swallowing and aspiration occur in the later disease stages. CBD is characterized by an insidious onset and slow disease progression, lack of response to levodopa therapy, akinetic rigidity, limb apraxia, and speech and language impairment.25 Myoclonus, asymmetric dystonia, alien limb phenomenon, frontal executive dysfunction, and visuospatial deficits are secondary criteria. Episodic memory is relatively preserved as hippocampi and temporal cortices are unaffected.26 Patients with PSP or CBD may develop behavioral disturbances similar to bvFTD or PPA. Vice versa, patients with bvFTD or nfvPPA may develop neurological symptoms which are characteristic for later-stage PSP or CBD.27
 | Table 2 Molecular classification of FTLD with genetic and clinical correlations |
Amyotrophic lateral sclerosis (ALS), as the most common form of motor neuron disease, presents with atrophic paresis, fasciculations, and muscular cramps as signs of involvement of the spinal motor neurons, and brisk reflexes, pyramidal signs, and increased muscular tonus indicating affection of cortical motor neurons. Weakness of the respiratory muscles and breathing insufficiency are the main cause of death. Up to 15% of patients with FTLD also show symptoms of ALS. About 5% of patients with ALS develop the full clinical phenotype of bvFTD, svPPA, or nfvPPA.28,29
Epidemiology
Only a few studies have attempted to determine the epidemiology of FTLD. It is difficult to obtain reliable estimates of prevalence and incidence because the concept and nomenclature of FTLD are changing and clinical diagnosis can be challenging. In the population as a whole, FTLD is relatively rare30 and has been included in the list of “orphan diseases” (Orpha number ORPHA282 of http://www.orpha.net). Among people below the age of 65 years, however, the prevalence of FTLD is comparable to that of AD. It has been estimated at 15 per 100,000 patients between 45 and 65 years of age.31 Population-based studies have shown that male and female persons are equally affected.30–32 The mean age of onset is approximately 58 years,32 but patients with an onset in their thirties have been described.33 Prognosis is poor, and FTLD leads to death within 3 to 10 years after diagnosis, with survival in svPPA being longer than in nfvPPA and bvFTD.34,35 Main causes of death are pneumonia, circulatory system failure, and cachexia.35
Diagnosis and differential diagnosis
Medical history and neuropsychology
The diagnostic latency between the symptom onset and the correct diagnosis ranges from 3 (nfvPPA) to 4 (bvFTD and svPPA) years,36 reflecting the challenge of diagnosis despite recent advances in the clinical characterization of FTLD.9 The majority of patients with bvFTD do not complain of any symptoms.37 Behavioral changes reported by relatives are often unspecific, and may suggest a psychiatric, rather than a neurodegenerative, disorder. Moreover, patients may perform within normal ranges on neuropsychological tests of memory and language, and structural imaging abnormalities may be subtle and confounded by age-dependent changes.
In order to detect behavioral alterations, the diagnostic procedure should include a careful medical history, which needs to be obtained not only from the patient, but also from a close relative or friend. Standardized caregiver questionnaires (ie, the Frontal Behavioral Inventory38 or the Frontal Systems Behavioral Scale39) have been developed to assess bvFTD.
Detailed neuropsychological examination involving tests of memory, language, visuoconstruction, and executive tasks may identify cognitive deficits that are not apparent in everyday life. The cognitive profile might also help to discriminate FTLD from other causes of dementia, particularly from AD.
The cognitive profile in bvFTD is usually characterized by executive and generation deficits in the context of relatively preserved memory and visuospatial functions.7 However, unimpaired performance in neuropsychological tasks, even in measures of executive functions, does not exclude a diagnosis of bvFTD. Language deficits in PPA can be detected by careful analysis of spontaneous speech and by using standardized language tasks.
One of the most frequent misdiagnoses of bvFTD is major depression, which frequently is suggested by relatives who interpret the patients’ apathy, loss of interest, and social withdrawal as indicating a mood disorder. Therefore, a detailed psychiatric examination is also required – most patients with bvFTD do not complain at all about depressive symptoms such as sadness, self-doubts, or suicidal ideation. Further psychiatric diseases that sometimes mimic bvFTD are bipolar affective disorders or schizophrenia, as well as substance abuse.
Laboratory tests, neuroimaging, and cerebrospinal fluid analysis
Laboratory tests, neuroimaging, and cerebrospinal fluid analysis are needed to exclude vascular, infectious, inflammatory, neoplastic or paraneoplastic, and metabolic diseases, some of which may be amenable to treatment.
In bvFTD, structural imaging studies revealed grey matter atrophy in the frontal and temporal lobes, the anterior cingulate, anterior insula, and in subcortical structures.40,41 However, in early stages, changes may be very subtle. Visual rating scales are available to detect and quantify bvFTD-specific atrophy patterns (eg, Kipps et al42). Atrophy of the amygdala can help to discriminate between bvFTD and AD,43 while atrophy of the hippocampus44 does not differentiate between bvFTD and AD.43,45 In svPPA, marked anterior and inferior temporal atrophy is observed, whereas in nfvPPA, the size of the inferior frontal gyrus, the insula, and the premotor and supplementary motor area of the left hemisphere is reduced.46–48 Cerebral fluorodeoxyglucose positron emission tomography typically reveals glucose hypometabolism in the corresponding regions and may provide additional information in cases with borderline findings. Amyloid positron emission tomography imaging can be employed to exclude the presence of AD pathology in patients with FTLD.49,50
So far there are no cerebrospinal fluid biomarkers that can positively identify FTLD. Phospho-tau and ß-Amyloid appear to be helpful for discriminating FTLD from AD, with high cerebrospinal fluid p-tau and low ratios of Aβ(1-42)/Aβ(1-40) and Aβ(1-42)/Aβ(1-38) being specific for AD.51
Genetics of FTLD
Approximately 40% of FTLD patients have a history that is suggestive of familial transmission, but only 10%–30% of family pedigrees show an autosomal dominant inheritance pattern. To date, mutations in five genes have been identified which are displayed in Table 3. Mutations of chromosome 9 open reading frame 72 (C9orf72), microtubule-associated protein tau (MAPT), and progranulin (GRN) explain over 80% of cases in FTLD families with a strong autosomal dominant family history.
 | Table 3 FTLD – genes |
MAPT
Up to 40 different mutations have been found in MAPT, accounting for 50% of familial cases of FTLD. These cases show a considerable variability of the clinical presentation.52,53 Mutations of MAPT lead to impaired microtubule assembly, impaired axonal transport, and promote pathological tau filament aggregation.54 Very early age at onset (<50 years), parkinsonism, and oculomotor dysfunction are suggestive of MAPT mutations.55
GRN
Mutations in the GRN gene account for 3% to 26% of familial cases.56,57 GRN is a growth factor that is expressed by many cell types, including neurons. The role of GRN for neuronal survival and function is still unclear/under debate. Null mutations cause disease via haploinsufficiency. As a result, reduced levels of GRN protein are observed in bodily fluids. The corticobasal syndrome and nfvPPA were related to GRN mutations.58
C9orf72
Only recently, an abnormal expansion of a hexanucleotide repeat in the C9orf72 gene was found to be a relatively common genetic cause of FTLD, accounting for 20% of familial cases.59,60 So far, the function of C9orf72 is unknown. Fewer than 20 GGGGCC hexanucleotide repeats in the intronic region of the C9orf72 gene are regarded as normal, the lower limit of the pathogenic range is considered as 65 repeats.61 Patients with C9orf72 expansions appear to have a family history of ALS and FTLD. Psychotic symptoms occur in up to 38% of patients with C9orf72 mutations.62
Valosin-containing protein (VCP) and charged multivesicular body protein 2B (CHMP2B)
Mutations in two other genes, VCP63 and CHMP2B,64 leading to TDP-43-positive FTLD, account for a minority of familial cases.57
Genetic counseling
Genetic testing of symptomatic persons is of high importance to confirm the diagnosis if one or more relatives are affected. With the advent of disease-modifying drugs tailored to the different neuropathological subtypes, results of genetic testing will probably influence therapeutic decisions. Frequently, unaffected relatives seek advice with regard to predictive genetic testing. Formal counseling by a geneticist or genetic counselor is recommended. While practice guidelines exist for numerous diseases, including AD,65 international guidelines for genetic counseling in FTLD have not yet been developed.
Neuropathology of FTLD
Considering the variability in clinical features and molecular genetics, it is not surprising that the neuropathology associated with FTLD is heterogeneous as well.66 A consistent feature is the relatively selective degeneration of the frontal and temporal cerebral lobes. In addition, most cases of FTLD have abnormal intracellular accumulation of some disease-specific protein, so it has become popular to classify FTLD into broad categories, based on the molecular defect thought to be most characteristic (Table 2).67
Until recently, the best studied FTLD subgroups were those conditions characterized by the accumulation of hyperphosphorylated tau protein in neurons and glia (FTLD-tau). These cases represent ~40% of FTLD and include those with the neuropathology of Pick’s disease, PSP, CBD, and cases of familial FTLD caused by mutations in MAPT. Although there is significant overlap in the morphology, and cellular and anatomical distribution of tau-positive pathology among the FTLD-tau subtypes, each condition is characterized by some specific type of inclusion that allows pathological diagnosis; Pick bodies in Pick’s disease, tufted astrocytes and numerous neurofibrillary tangles in subcortical nuclei in PSP, and astrocytic plaques and abundant thread pathology in CBD. In addition to these morphological differences, the biochemical form of tau that accumulates in the inclusions varies among the different conditions, with Pick bodies composed primarily of tau isoforms with three microtubule binding domains (3-repeat, 3R tau), while the inclusions of PSP and CBD contain 4R tau.
The majority of cases of tau-negative FTLD are characterized by neuronal cytoplasmic inclusions and dystrophic neurites in the superficial layers of the frontotemporal neocortex, and dentate granule cells of the hippocampus, that were originally recognized with ubiquitin immunohistochemistry (FTLD-U). In 2006, TDP-43 was identified as the ubiquitinated pathological protein in most cases of FTLD-U (subsequently renamed FTLD-TDP), as well as the vast majority of ALS.68 This discovery provided strong evidence that FTLD and ALS are closely related conditions with overlapping molecular pathogenesis. The pathological form of TDP-43 that accumulates in FTLD and ALS is composed of abnormal C-terminal fragments that are ubiquitinated and hyperphosphorylated. Different patterns of FTLD-TDP are now recognized, based on the cortical distribution and relative abundance of cytoplasmic inclusions versus neurites, with each having fairly specific clinical and genetic correlations (Table 2).69 In most series, FTLD-TDP represents the largest molecular FTLD subgroup, present in about half of all FTLD cases.
An important recent discovery has been the identification of abnormal expansion of a GGGGCC hexanucleotide repeat in a noncoding region of the C9orf72 gene as the most common genetic cause of both FTLD and ALS.70 The neuropathology of these cases is a combination of FTLD-TDP and classical ALS with TDP-positive inclusions. In addition, ubiquitin-positive, TDP-negative neuronal inclusions in the neocortex, hippocampus, and cerebellum are a consistent and unique pathological feature of cases with the C9orf72 mutation. It has recently been shown that this TDP-negative pathology is the result of unconventional translation of the expanded GGGGCC repeat. Sense (and possibly antisense) translation in the three alternate reading frames results in the generation of several different peptides, each composed of repeating units of two amino acids (ie, glycine–alanine, glycine–proline, glycine–arginine).71,72 Novel antibodies against these various dipeptide repeats are proving to be a highly sensitive and specific tool for demonstrating this unique pathology. The relationship between the dipeptide repeats and TDP-43 mismetabolism and their relative roles in the pathogenesis of disease in cases with the C9orf72 mutation is currently an important area of investigation.
In 10%–20% of cases originally classified as FTLD-U (5%–10% of all FTLD), the cellular inclusions do not stain for either tau or TDP-43. The term “atypical” FTLD-U recognizes that these cases have a consistent and unique phenotype (sporadic, with very early onset severe psychobehavioral abnormalities in the absence of language or motor features), as well as novel pathological features (TDP-negative inclusions that include unusual vermiform neuronal intranuclear inclusions).33 Following the discovery of mutations in FUS as a cause of ALS, it was recognized that most of the remaining tau/TDP-negative FTLD subtypes were characterized by inclusions that are immunoreactive for FUS as well as the other members of the FET family of proteins (Ewing sarcoma protein and TAF-15).73 In addition to atypical FTLD-U, this FTLD-FUS group also includes two rare conditions in which FTLD usually coexists with pyramidal and/or extrapyramidal motor dysfunction; basophilic inclusion body disease and neuronal intermediate filament inclusion disease. As with the FTLD-tau subtypes, each of the conditions in the FTLD-FUS group have overlapping but distinct neuropathological features.
There remain very rare FTLD cases, such as the Danish family with autosomal dominant FTLD caused by a mutation in the CHMP2B gene, in which the pathological inclusions are only demonstrated with nonspecific markers of the ubiquitin proteasome system, and possibly some cases in which no inclusions can be detected. However, recent advances now make it possible to assign the vast majority of FTLD cases to one of three major molecular subgroups (FTLD-tau, FTLD-TDP, or FTLD-FUS).
Treatment
Pharmacological treatment
Compared with AD, the cholinergic system is relatively intact in FTLD.74,75 On the other hand there is strong evidence for abnormalities in the serotonergic system of FTLD patients, with a decrease in 5-HT1A and 5-HT2A receptors and neuronal loss in the raphe nuclei.75 Furthermore, a disruption of the dopaminergic system has been demonstrated,76 including low levels of dopamine metabolites and reduced presynaptic dopamine receptors in the putamen and caudate of FTLD patients.77
Given the relative preservation of cholinergic neurons in brains of patients with FTLD, there is no reason to expect a benefit from cholinesterase inhibition as in AD. Nonetheless, a number of small trials have evaluated cholinesterase inhibitors in FTLD.115–118 These studies are summarized in Table 4. In summary, cholinesterase inhibitors do not improve cognition or behavior in FTLD. This also applies to memantine, an N-methyl-D-aspartate-type glutamate receptor antagonist that is approved for the treatment of AD. Several recent studies failed to demonstrate a benefit of this compound in FTLD (Table 4).119–122 Nonetheless, about 30% to 40% of patients with FTLD currently receive treatment with cholinesterase inhibitors or memantine.78,79
 | Table 4 Clinical drug trials in FTLD |
The effect of antidepressants, particularly of selective serotonin reuptake inhibitors on neuropsychiatric symptoms in FTLD has been studied in several trials (Table 5).85,123–128 There is some evidence that the serotonergic antidepressants have potential for reducing behavioral disturbances, in particular disinhibition, apathy, repetitive behaviors, sexually inappropriate behaviors, and hyperorality.75,80 The British Association for Psychopharmacology has recently assigned a B rating for the treatment of behavioral disturbances in FTLD with selective serotonin reuptake inhibitors, indicating good overall clinical evidence.81
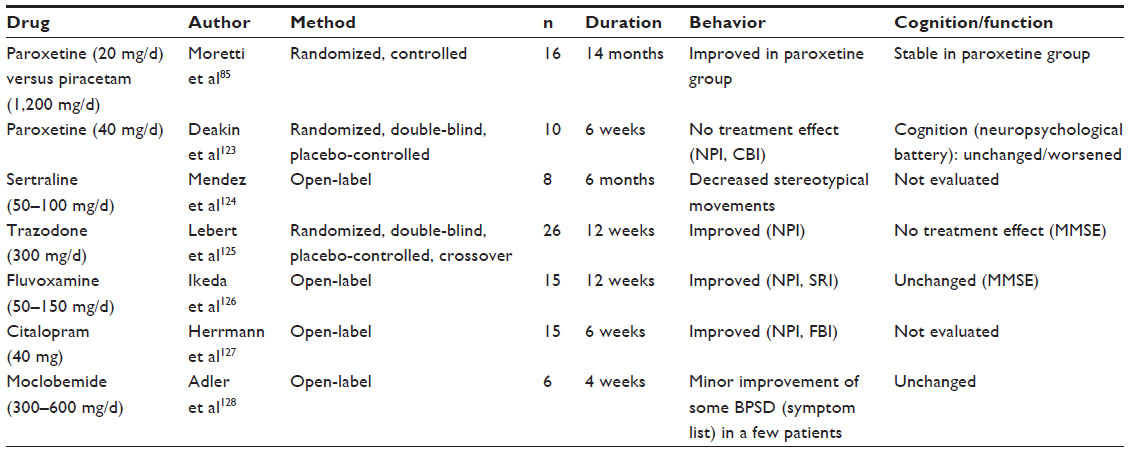 | Table 5 Serotonergic antidepressants |
A handful of small studies have been performed in patients with FTLD using dopaminergic agents (Table 6).119–121 Positive effects on behavior have been reported, but the results have not been replicated in larger studies.
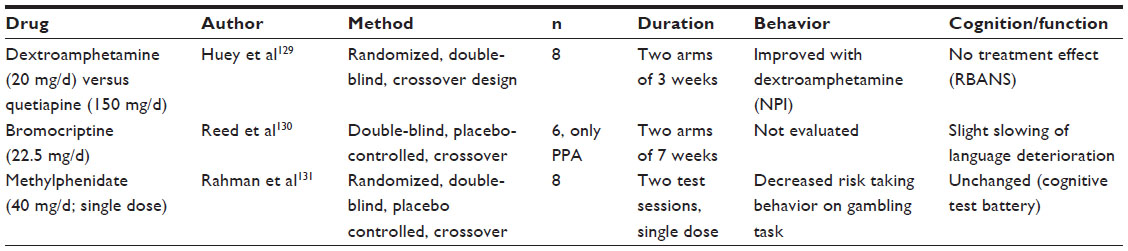 | Table 6 Dopaminergic drugs |
Antipsychotics are commonly prescribed to treat disinhibition, agitation, and psychosis in dementia regardless of etiology.82 No controlled studies are available on antipsychotics in FTLD. Positive effects of risperidone, olanzapine, and aripiprazole have been described.83–85 However, it is important to keep in mind that patients with FTLD may be susceptible to extrapyramidal side effects of antipsychotics86 and that the use of atypical antipsychotics for the treatment of behavioral disorders in elderly patients with dementia is associated with increased mortality.87
Non-pharmacological interventions
As long as effective pharmacological therapies for symptom management and disease-modification are not available, it is important that a multidisciplinary team identifies non-pharmacological interventions that support the patients’ engagement in activities and ensures an optimal quality of life.88 Treatment strategies should be individually tailored because symptom pattern, rate of decline, needs, and available resources are highly variable. Where necessary, specific disciplines (neurologists, psychiatrists, neuropsychologists, social workers, speech–language pathologists, occupational therapists, physiotherapists, neurorehabilitation professionals) should be consulted.88 It is essential to assess and monitor the patient’s responses to stimuli, to structure and modify the living environment, to take appropriate measures regarding safety, and to help caregivers develop strategies for managing behavioral disturbances. These tasks are very often the responsibilities of advanced practice nurses.89–91
Large non-pharmacological intervention-studies with the aim to improve or maintain behavior, cognition, or language in people with FTLD have not yet been performed. In an uncontrolled study, the efficacy of a 4-week inpatient treatment program for patients with FTLD or AD and their caregivers was evaluated.92 The intervention consisted of a multimodal rehabilitation for patients in combination with an educational program for caregivers. The pre–post comparison showed that the treatment program was associated with an improvement of depression and other psychopathological symptoms in patients with FTLD and with reduced burden on their caregivers.
A few single-subject studies have investigated the efficacy of speech therapy on word-finding ability in patients with PPA and demonstrated minor improvements of naming immediately following treatment completion.93–95 However, there is little evidence for sustainability and generalizability of these learning effects. To date, there is general consensus that the goal of speech therapy in PPA is not to regain lost language, but to maximize communication skills as long as possible. Interventions may include the development of skills for facilitating communication and teaching the use of communication tools.96
Caregiver support
Given the lack of pharmacological or non-pharmacological treatments with proven efficacy, caregiver support is the mainstay of the management of FTLD.
Several studies have consistently shown that FTLD caregivers are heavily stressed and burdened as a result of their caregiving role. Family caregivers of patients with FTLD report higher levels of psychological distress and subjective burden than caregivers of patients with AD or other dementias.97–99 Specific problems of FTLD caregivers include delayed diagnosis, uncertainty of diagnosis, young age of patients, behavioral problems, depression, lack of information, constraints regarding self-care, and lack of access to appropriate care facilities.100 Most health and social care services do not meet the needs of younger patients with dementia and their caregivers because they are tailored to older adults and to patients with AD.101
A recent survey of FTLD caregiver needs showed that the most important needs and requests of the caregivers are detailed information about the disease, psychosocial support provided by experienced staff, appropriate financial support, and better education of health and social care professionals about the disease.102 Counseling of patients and caregivers, either in individual or group format, should include detailed information about the disease and the worsening of symptoms that may be expected, recommendations for the management of challenging behaviors, relevant caring issues, as well as legal and financial advice. Suggestions for caregivers’ own well-being should also be offered. Caregivers should be motivated to participate in FTLD caregiver support groups or self-help groups, if available. Information about FTLD can also be found on the internet, for example on the webpage of the Association for FTLD (http://www.theaftd.org). In Canada, a regular biannual internet-based videoconferencing support group for FTLD spouses has been set up.103
Future directions
In recent years, the accuracy of the clinical diagnosis of FTLD has been improved by detailed clinical descriptions, enlarged knowledge about the performance of the patients in neuropsychological tests, development of novel FTLD-specific assessment instruments including interviews and clinical rating scales, refined imaging techniques, and better understanding of cerebrospinal fluid biomarker changes, as well as revised diagnostic criteria. However, in the absence of specific and accurate biomarkers, diagnosis and differential diagnosis of FTLD is still challenging. There is a need for diagnostic interviews that enable a reliable assessment of behavioral disturbances and personality changes. Tests of social cognition and theory of mind need to be developed. The early clinical stages of bvFTD, svPPA, and nfvPPA that correspond to “mild cognitive impairment” in AD should be defined as part of a strategy aiming at timely diagnosis and treatment.
Recently, remarkable progress has been made regarding the understanding of the genetic causes, molecular basis, and neuropathological features of FTLD. Most common gene alterations that cause FTLD have been discovered and the major pathological proteins have been identified. Nonetheless, there are plenty of future challenges. Some mutations underlying the proteinopathies await identification. Understanding the factors that modify the clinical expression of mutations will bring novel insights for the development of specific drug treatment. The contribution of susceptibility genes has yet to be explored.
A major goal of research is the identification of laboratory or imaging biomarkers that reliably identify pathological subtypes of FTLD: FTLD cases that are associated with tau and TDP-43 pathology represent potential targets for future therapies that aim at slowing or halting disease progression. Recently, an international multicenter Phase 3, 52-week study of methylene blue in bvFTD has been initiated (NCT01626378).104 Methylene blue is a tau aggregation inhibitor that also appears to protect against TDP-43 toxicity in animal models.105 This first clinical trial in bvFTD will most probably unveil potential difficulties with studies in bvFTD: the lack of appropriate outcome parameters; problems with recruitment; diminished patient compliance; behavioral disturbances that hamper the examinations, interviews, and neuroimaging; and concomitant psychiatric medication in high doses. A number of tau-based disease-modifying therapies have already been investigated in clinical trials:106 the glycogen synthase kinase inhibitors lithium (NCT00703677)107 and tideglusib (NCT01350362, NCT01049399);108,109 riluzole, a sodium channel blocker;110 coenzyme Q10, which improves mitochondrial function;111 rasagiline, a monoamine oxidase inhibitor (NCT01187888);112 and davunetide, a microtubule stabilizer (NCT01110720, NCT01056965).113,114 Beyond that, preclinical studies are under way that aim at the identification of agents with the potential to normalize GRN levels either by increased production or reduced clearance in FTLD-TDP cases that are caused by loss of function mutations in the GRN gene.106 Given all these efforts, hopefully a causal treatment for FTLD will be available sooner or later.
Disclosure
The authors report no conflicts of interest in this work.
References
Pick A. Über die Beziehungen der senilen Hirnatrophie zur Aphasie. [About the relations of senile brain atrophie and aphasia]. Prager Med Wochenschr. 1892;17(16):165–167. German. | |
Davatzikos C, Resnick SM, Wu X, Parmpi P, Clark CM. Individual patient diagnosis of AD and FTD via high-dimensional pattern classification of MRI. Neuroimage. 2008;41(4):1220–1227. | |
Hornberger M, Savage S, Hsieh S, Mioshi E, Piguet O, Hodges JR. Orbitofrontal dysfunction discriminates behavioral variant frontotemporal dementia from Alzheimer’s disease. Dement Geriatr Cogn Disord. 2010;30(6):547–552. | |
Seeley WW, Crawford R, Rascovsky K, et al. Frontal paralimbic network atrophy in very mild behavioral variant frontotemporal dementia. Arch Neurol. 2008;65(2):249–255. | |
McKhann GM, Albert MS, Grossman M, Miller B, Dickson D, Trojanowski JQ; Work Group on Frontotemporal Dementia and Pick’s Disease. Clinical and pathological diagnosis of frontotemporal dementia: report of the Work Group on Frontotemporal Dementia and Pick’s Disease. Arch Neurol. 2001;58(11):1803–1809. | |
Neary D, Snowden JS, Gustafson L, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology. 1998;51(6):1546–1554. | |
Rascovsky K, Hodges JR, Knopman D, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain. 2011;134(Pt 9):2456–2477. | |
[No authors listed]. Clinical and neuropathological criteria for frontotemporal dementia. The Lund and Manchester Groups. J Neurol Neurosurg Psychiatry. 1994;57(4):416–418. | |
Rascovsky K, Grossman M. Clinical diagnostic criteria and classification controversies in frontotemporal lobar degeneration. Int Rev Psychiatry. 2013;25(2):145–158. | |
Shamay-Tsoory SG. The neural bases for empathy. Neuroscientist. 2011;17(1):18–24. | |
Adenzato M, Cavallo M, Enrici I. Theory of mind ability in the behavioural variant of frontotemporal dementia: an analysis of the neural, cognitive, and social levels. Neuropsychologia. 2010;48(1):2–12. | |
Huey ED, Armstrong N, Momeni P, Grafman J. Challenges and new opportunities in the investigation of new drug therapies to treat frontotemporal dementia. Expert Opin Ther Targets. 2008;12(11):1367–1376. | |
Ikeda M, Brown J, Holland AJ, Fukuhara R, Hodges JR. Changes in appetite, food preference, and eating habits in frontotemporal dementia and Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 2002;73(4):371–376. | |
Piguet O, Petersén A, Yin Ka Lam B, et al. Eating and hypothalamus changes in behavioral-variant frontotemporal dementia. Ann Neurol. 2011;69(2):312–319. | |
Davies RR, Scahill VL, Graham A, Williams GB, Graham KS, Hodges JR. Development of an MRI rating scale for multiple brain regions: comparison with volumetrics and with voxel-based morphometry. Neuroradiology. 2009;51(8):491–503. | |
Gorno-Tempini ML, Hillis AE, Weintraub S, et al. Classification of primary progressive aphasia and its variants. Neurology. 2011;76(11):1006–1014. | |
Brambati SM, Rankin KP, Narvid J, et al. Atrophy progression in semantic dementia with asymmetric temporal involvement: a tensor-based morphometry study. Neurobiol Aging. 2009;30(1):103–111. | |
Rohrer JD, Warren JD, Modat M, et al. Patterns of cortical thinning in the language variants of frontotemporal lobar degeneration. Neurology. 2009;72(18):1562–1569. | |
Bozeat S, Gregory CA, Ralph MA, Hodges JR. Which neuropsychiatric and behavioural features distinguish frontal and temporal variants of frontotemporal dementia from Alzheimer’s disease? J Neurol Neurosurg Psychiatry. 2000;69(2):178–186. | |
Seeley WW, Bauer AM, Miller BL, etal. The natural history of temporal variant frontotemporal dementia. Neurology. 2005;64(8):1384–1390. | |
Josephs KA, Whitwell JL, Knopman DS, et al. Two distinct subtypes of right temporal variant frontotemporal dementia. Neurology. 2009;73(18):1443–1450. | |
Grossman M, Ash S. Primary progressive aphasia: a review. Neurocase. 2004;10(1):3–18. | |
Harris JM, Gall C, Thompson JC, et al. Classification and pathology of primary progressive aphasia. Neurology. 2013;81(21):1832–1839. | |
Sixel-Döring F, Schweitzer M, Mollenhauer B, Trenkwalder C. Polysomnographic findings, video-based sleep analysis and sleep perception in progressive supranuclear palsy. Sleep Med. 2009;10(4):407–415. | |
Bak TH, Hodges JR. Corticobasal degeneration: clinical aspects. Handb Clin Neurol. 2008;89:509–521. | |
Bruns MB, Josephs KA. Neuropsychiatry of corticobasal degeneration and progressive supranuclear palsy. Int Rev Psychiatry. 2013;25(2):197–209. | |
Kertesz A, McMonagle P, Blair M, Davidson W, Munoz DG. The evolution and pathology of frontotemporal dementia. Brain. 2005;128(Pt 9):1996–2005. | |
Lomen-Hoerth C, Murphy J, Langmore S, Kramer JH, Olney RK, Miller B. Are amyotrophic lateral sclerosis patients cognitively normal? Neurology. 2003;60(7):1094–1097. | |
Murphy JM, Henry RG, Langmore S, Kramer JH, Miller BL, Lomen-Hoerth C. Continuum of frontal lobe impairment in amyotrophic lateral sclerosis. Arch Neurol. 2007;64(4):530–534. | |
Rosso SM, Donker Kaat L, Baks T, et al. Frontotemporal dementia in The Netherlands: patient characteristics and prevalence estimates from a population-based study. Brain. 2003;126(Pt 9):2016–2022. | |
Ratnavalli E, Brayne C, Dawson K, Hodges JR. The prevalence of frontotemporal dementia. Neurology. 2002;58(11):1615–1621. | |
Johnson JK, Diehl J, Mendez MF, et al. Frontotemporal lobar degeneration: demographic characteristics of 353 patients. Arch Neurol. 2005;62(6):925–930. | |
Mackenzie IRA, Foti D, Woulfe J, Hurwitz TA. Atypical frontotemporal lobar degeneration with ubiquitin-positive, TDP-43-negative neuronal inclusions. Brain. 2008;131(Pt 5):1282–1293. | |
Hodges JR, Davies R, Xuereb J, Kril J, Halliday G. Survival in frontotemporal dementia. Neurology. 2003;61(3):349–354. | |
Nunnemann S, Last D, Schuster T, Förstl H, Kurz A, Diehl-Schmid J. Survival in a German population with frontotemporal lobar degeneration. Neuroepidemiology. 2011;37(3–4):160–165. | |
Diehl-Schmid J, Pohl C, Perneczky R, Hartmann J, Förstl H, Kurz A. Frühsymptome, Überlebenszeit und Todesursachen - Beobachtungen an 115 Patienten mit Demenz auf der Grundlage frontotemporaler lobärer Degenerationen. [Initial symptoms, survival and causes of death in 115 patients with frontotemporal lobar degeneration]. Fortschr Neurol Psychiatr. 2007;75(12):708–713. German. | |
Pijnenburg YAL, Gillissen F, Jonker C, Scheltens P. Initial complaints in frontotemporal lobar degeneration. Dement Geriatr Cogn Disord. 2004;17(4):302–306. | |
Kertesz A, Davidson W, Fox H. Frontal behavioral inventory: diagnostic criteria for frontal lobe dementia. Can J Neurol Sci. 1997;24(1):29–36. | |
Grace J, Malloy P. Frontal Systems Behavior Scale: Professional Manual. Lutz, FL: PAR Inc; 2001 [cited September 7, 2013]. Available from: http://www4.parinc.com/Products/Product.aspx?ProductID=FRSBE. | |
Schroeter ML, Raczka K, Neumann J, Yves von Cramon D. Towards a nosology for frontotemporal lobar degenerations-a meta-analysis involving 267 subjects. Neuroimage. 2007;36(3):497–510. | |
Schroeter ML, Raczka K, Neumann J, von Cramon DY. Neural networks in frontotemporal dementia – a meta-analysis. Neurobiol Aging. 2008;29(3):418–426. | |
Kipps CM, Davies RR, Mitchell J, Kril JJ, Halliday GM, Hodges JR. Clinical significance of lobar atrophy in frontotemporal dementia: application of an MRI visual rating scale. Dement Geriatr Cogn Disord. 2007;23(5):334–342. | |
Barnes J, Whitwell JL, Frost C, Josephs KA, Rossor M, Fox NC. Measurements of the amygdala and hippocampus in pathologically confirmed Alzheimer disease and frontotemporal lobar degeneration. Arch Neurol. 2006;63(10):1434–1439. | |
Josephs KA, Dickson DW. Hippocampal sclerosis in tau-negative frontotemporal lobar degeneration. Neurobiol Aging. 2007;28(11):1718–1722. | |
Lindberg O, Walterfang M, Looi JC, et al. Hippocampal shape analysis in Alzheimer’s disease and frontotemporal lobar degeneration subtypes. J Alzheimers Dis. 2012;30(2):355–365. | |
Gorno-Tempini ML, Dronkers NF, Rankin KP, et al. Cognition and anatomy in three variants of primary progressive aphasia. Ann Neurol. 2004;55(3):335–346. | |
Josephs KA, Duffy JR, Strand EA, et al. Clinicopathological and imaging correlates of progressive aphasia and apraxia of speech. Brain. 2006;129(Pt 6):1385–1398. | |
Rogalski E, Cobia D, Harrison TM, Wieneke C, Weintraub S, Mesulam M-M. Progression of language decline and cortical atrophy in subtypes of primary progressive aphasia. Neurology. 2011;76(21):1804–1810. | |
Drzezga A, Grimmer T, Henriksen G, et al. Imaging of amyloid plaques and cerebral glucose metabolism in semantic dementia and Alzheimer’s disease. Neuroimage. 2008;39(2):619–633. | |
Rabinovici GD, Furst AJ, O’Neil JP, et al. 11C-PIB PET imaging in Alzheimer disease and frontotemporal lobar degeneration. Neurology. 2007;68(15):1205–1212. | |
Bibl M, Mollenhauer B, Lewczuk P, et al. Cerebrospinal fluid tau, p-tau 181 and amyloid-β38/40/42 in frontotemporal dementias and primary progressive aphasias. Dement Geriatr Cogn Disord. 2011;31(1):37–44. | |
Hutton M, Lendon CL, Rizzu P, et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393(6686):702–705. | |
Rademakers R, Cruts M, van Broeckhoven C. The role of tau (MAPT) in frontotemporal dementia and related tauopathies. Hum Mutat. 2004;24(4):277–295. | |
Brandt R, Hundelt M, Shahani N. Tau alteration and neuronal degeneration in tauopathies: mechanisms and models. Biochim Biophys Acta. 2005;1739(2–3):331–354. | |
Le Ber I. Genetics of frontotemporal lobar degeneration: An up-date and diagnosis algorithm. Rev Neurol (Paris). 2013;169(10):811–819. | |
Baker M, Mackenzie IR, Pickering-Brown SM, et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442(7105):916–919. | |
Sieben A, Van Langenhove T, Engelborghs S, et al. The genetics and neuropathology of frontotemporal lobar degeneration. Acta Neuropathol. 2012;124(3):353–372. | |
Rademakers R, Baker M, Gass J, et al. Phenotypic variability associated with progranulin haploinsufficiency in patients with the common 1477C – >T (Arg493X) mutation: an international initiative. Lancet Neurol. 2007;6(10):857–868. | |
Renton AE, Majounie E, Waite A, et al; ITALSGEN Consortium. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72(2):257–268. | |
van der Zee J, Gijselinck I, Dillen L, et al; European Early-Onset Dementia Consortium. A pan-European study of the C9orf72 repeat associated with FTLD: geographic prevalence, genomic instability, and intermediate repeats. Hum Mutat. 2013;34(2):363–373. | |
Loy CT, Schofield PR, Turner AM, Kwok JB. Genetics of dementia. Lancet. Epub August 5, 2013. | |
Snowden JS, Rollinson S, Thompson JC, et al. Distinct clinical and pathological characteristics of frontotemporal dementia associated with C9ORF72 mutations. Brain. 2012;135(Pt 3):693–708. | |
Watts GD, Wymer J, Kovach MJ, et al. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat Genet. 2004;36(4):377–381. | |
Skibinski G, Parkinson NJ, Brown JM, et al. Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat Genet. 2005;37(8):806–808. | |
Goldman JS, Hahn SE, Catania JW, et al; American College of Medical Genetics and the National Society of Genetic Counselors. Genetic counseling and testing for Alzheimer disease: joint practice guidelines of the American College of Medical Genetics and the National Society of Genetic Counselors. Genet Med. 2011;13(6):597–605. | |
Neumann M, Tolnay M, Mackenzie IRA. The molecular basis of frontotemporal dementia. Expert Rev Mol Med. 2009;11:e23. | |
Mackenzie IR, Neumann M, Bigio EH, et al. Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: an update. Acta Neuropathol. 2010;119(1):1–4. | |
Neumann M, Sampathu DM, Kwong LK, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314(5796):130–133. | |
Mackenzie IR, Neumann M, Baborie A, et al. A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol. 2011;122(1):111–113. | |
Rademakers R, Neumann M, Mackenzie IR. Advances in understanding the molecular basis of frontotemporal dementia. Nat Rev Neurol. 2012;8(8):423–434. | |
Ash PEA, Bieniek KF, Gendron TF, et al. Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron. 2013;77(4):639–646. | |
Mori K, Weng SM, Arzberger T, et al. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science. 2013;339(6125):1335–1338. | |
Neumann M, Bentmann E, Dormann D, et al. FET proteins TAF15 and EWS are selective markers that distinguish FTLD with FUS pathology from amyotrophic lateral sclerosis with FUS mutations. Brain. 2011;134(Pt 9):2595–2609. | |
Procter AW, Qurne M, Francis PT. Neurochemical features of frontotemporal dementia. Dement Geriatr Cogn Disord. 1999;10 Suppl 1:80–84. | |
Huey ED, Putnam KT, Grafman J. A systematic review of neurotransmitter deficits and treatments in frontotemporal dementia. Neurology. 2006;66(1):17–22. | |
Sjögren M, Minthon L, Passant U, Blennow K, Wallin A. Decreased monoamine metabolites in frontotemporal dementia and Alzheimer’s disease. Neurobiol Aging. 1998;19(5):379–384. | |
Rinne JO, Laine M, Kaasinen V, Norvasuo-Heilä MK, Någren K, Helenius H. Striatal dopamine transporter and extrapyramidal symptoms in frontotemporal dementia. Neurology. 2002;58(10):1489–1493. | |
Diehl-Schmid J, Last D, Schuster T, Förstl H, Schneider-Schelte H, Kurz A. Health care utilization in frontotemporal lobar degeneration. Alzheimer Dis Assoc Disord. 2012;26(2):166–170. | |
Bei Hu, Ross L, Neuhaus J, et al. Off-label medication use in frontotemporal dementia. Am J Alzheimers Dis Other Demen. 2010;25(2):128–133. | |
Manoochehri M, Huey ED. Diagnosis and management of behavioral issues in frontotemporal dementia. Curr Neurol Neurosci Rep. 2012;12(5):528–536. | |
O’Brien JT, Burns A; BAP Dementia Consensus Group. Clinical practice with anti-dementia drugs: a revised (second) consensus statement from the British Association for Psychopharmacology. J Psychopharmacol. 2011;25(8):997–1019. | |
Franco KN, Messinger-Rapport B. Pharmacological treatment of neuropsychiatric symptoms of dementia: a review of the evidence. J Am Med Dir Assoc. 2006;7(3):201–202. | |
Curtis RC, Resch DS. Case of pick’s central lobar atrophy with apparent stabilization of cognitive decline after treatment with risperidone. J Clin Psychopharmacol. 2000;20(3):384–385. | |
Fellgiebel A, Müller MJ, Hiemke C, Bartenstein P, Schreckenberger M. Clinical improvement in a case of frontotemporal dementia under aripiprazole treatment corresponds to partial recovery of disturbed frontal glucose metabolism. World J Biol Psychiatry. 2007;8(2):123–126. | |
Moretti R, Torre P, Antonello RM, Cazzato G, Griggio S, Bava A. Olanzapine as a treatment of neuropsychiatric disorders of Alzheimer’s disease and other dementias: a 24-month follow-up of 68 patients. Am J Alzheimers Dis Other Demen. 2003;18(4):205–214. | |
Pijnenburg YAL, Sampson EL, Harvey RJ, Fox NC, Rossor MN. Vulnerability to neuroleptic side effects in frontotemporal lobar degeneration. Int J Geriatr Psychiatry. 2003;18(1):67–72. | |
Gill SS, Bronskill SE, Normand SL, et al. Antipsychotic drug use and mortality in older adults with dementia. Ann Intern Med. 2007;146(11):775–786. | |
Kortte KB, Rogalski EJ. Behavioural interventions for enhancing life participation in behavioural variant frontotemporal dementia and primary progressive aphasia. Int Rev Psychiatry. 2013;25(2):237–245. | |
Hall GR, Shapira J, Gallagher M, Denny SS. Managing differences: care of the person with frontotemporal degeneration. J Gerontol Nurs. 2013;39(3):10–14. | |
Merrilees J. A model for management of behavioral symptoms in frontotemporal lobar degeneration. Alzheimer Dis Assoc Disord. 2007;21(4):S64–S69. | |
Merrilees J, Ketelle R. Advanced practice nursing: meeting the caregiving challenges for families of persons with frontotemporal dementia. Clin Nurse Spec. 2010;24(5):245–251. | |
Romero B, Wenz M. Konzept und Wirksamkeit eines Behandlungsprogrammes für Demenzkranke und deren Angehörige Ergebnisse aus dem Alzheimer Therapiezentrum Bad Aibling. [Concept and effectiveness of a treatment program for patients with dementia and their relatives. Results from the Bad Aibling Alzheimer Disease Therapy Center]. Z Gerontol Geriatr. 2002;35(2):118–128. German. | |
Croot K, Nickels L, Laurence F, Manning M. Impairment- and activity/participation-directed interventions in progressive language impairment: Clinical and theoretical issues. Aphasiology. 2009;23(2):125–160. | |
Jokel R, Cupit J, Rochon E, Leonard C. Relearning lost vocabulary in nonfluent progressive aphasia with MossTalk Words®. Aphasiology. 2009;23(2):175–191. | |
Newhart M, Davis C, Kannan V, Heidler-Gary J, Cloutman L, Hillis AE. Therapy for naming deficits in two variants of primary progressive aphasia. Aphasiology. 2009;23(7–8):823–834. | |
Khayum B, Wieneke C, Rogalski E, Robinson J, O’Hara M. Thinking Outside the Stroke: Treating Primary Progressive Aphasia (PPA). Perspect Gerontol. 2012;17(2):37–49. | |
Boutoleau-Bretonnière C, Vercelletto M, Volteau C, Renou P, Lamy E. Zarit burden inventory and activities of daily living in the behavioral variant of frontotemporal dementia. Dement Geriatr Cogn Disord. 2008;25(3):272–277. | |
Mioshi E, Bristow M, Cook R, Hodges JR. Factors underlying caregiver stress in frontotemporal dementia and Alzheimer’s disease. Dement Geriatr Cogn Disord. 2009;27(1):76–81. | |
Riedijk SR, De Vugt ME, Duivenvoorden HJ, et al. Caregiver burden, health-related quality of life and coping in dementia caregivers: a comparison of frontotemporal dementia and Alzheimer’s disease. Dement Geriatr Cogn Disord. 2006;22(5–6):405–412. | |
Nunnemann S, Kurz A, Leucht S, Diehl-Schmid J. Caregivers of patients with frontotemporal lobar degeneration: a review of burden, problems, needs, and interventions. Int Psychogeriatr. 2012;24(9):1368–1386. | |
Bakker C, de Vugt ME, van Vliet D, et al. Unmet needs and health-related quality of life in young-onset dementia. Am J Geriatr Psychiatry. Epub July 17, 2013. | |
Diehl-Schmid J, Schmidt E-M, Nunnemann S, et al. Caregiver burden and needs in frontotemporal dementia. J Geriatr Psychiatry Neurol. 2013;26(4):221–229. | |
Shnall A, Agate A, Grinberg A, Huijbregts M, Nguyen MQ, Chow TW. Development of supportive services for frontotemporal dementias through community engagement. Int Rev Psychiatry. 2013;25(2):246–252. | |
TauRx Therapeutics Ltd. Safety and Efficacy Study Evaluating TRx0237 in Subjects With Behavioral Variant Frontotemporal Dementia (bvFTD). Available from: http://clinicaltrials.gov/show/NCT01626378. NLM identifier: NCT01626378. Accessed December 9, 2013. | |
Vaccaro A, Patten SA, Ciura S, et al. Methylene blue protects against TDP-43 and FUS neuronal toxicity in C. elegans and D. rerio. PloS One. 2012;7(7):e42117. | |
Boxer AL, Gold M, Huey E, et al. Frontotemporal degeneration, the next therapeutic frontier: molecules and animal models for frontotemporal degeneration drug development. Alzheimers Dement. 2013;9(2):176–188. | |
Westat. A Pilot Trial of Lithium in Subjects With Progressive Supranuclear Palsy or Corticobasal Degeneration, Available from: http://clinicaltrials.gov/show/NCT00703677. NLM identifier: NCT00703677. Accessed December 9, 2013. | |
Noscira SA. Efficacy, Safety and Tolerability of Tideglusib to Treat Mild-to-Moderate Alzheimer’s Disease Patients (ARGO). Available from: http://clinicaltrials.gov/show/NCT01350362. NLM identifier: NCT01350362. Accessed December 9, 2013. | |
Noscira SA. Safety, Tolerability, and Efficacy of Two Different Oral Doses of NP031112 Versus Placebo in the Treatment of Patients With Mild-to-Moderate Progressive Supranuclear Palsy (Tauros). Available from: http://clinicaltrials.gov/show/NCT01049399. NLM identifier: NCT01049399. Accessed December 9, 2013. | |
Bensimon G, Ludolph A, Agid Y, Vidailhet M, Payan C, Leigh PN; NNIPPS Study Group. Riluzole treatment, survival and diagnostic criteria in Parkinson plus disorders: the NNIPPS study. Brain. 2009;132(Pt 1):156–171. | |
Stamelou M, Reuss A, Pilatus U, et al. Short-term effects of coenzyme Q10 in progressive supranuclear palsy: a randomized, placebo-controlled trial. Mov Disord. 2008;23(7):942–949. | |
Lorenzl S. Efficacy, Tolerability and Safety of Azilect in Subjects With Progressive Supranuclear Palsy (PROSPERA). Available from: http://clinicaltrials.gov/show/NCT01187888. NLM identifier: NCT01187888. Accessed December 9, 2013. | |
Allon Therapeutics. Study to Evaluate the Safety and Efficacy of Davunetide for the Treatment of Progressive Supranuclear Palsy. Available from: http://clinicaltrials.gov/show/NCT01110720. NLM identifier: NCT01110720. Accessed December 9, 2013. | |
University of California, San Francisco. Davunetide (AL-108) in Predicted Tauopathies – Pilot Study. Available from: http://clinicaltrials.gov/show/NCT01056965. NLM identifier: NCT01056965. Accessed December 9, 2013. | |
Moretti R, Torre P, Antonello RM, Cattaruzza T, Cazzato G, Bava A. Rivastigmine in frontotemporal dementia: an open-label study. Drugs Aging. 2004;21(14):931–937. | |
Mendez MF, Shapira JS, McMurtray A, Licht E. Preliminary findings: behavioral worsening on donepezil in patients with frontotemporal dementia. Am J Geriatr Psychiatry. 2007;15(1):84–87. | |
Lampl Y, Sadeh M, Lorberboym M. Efficacy of acetylcholinesterase inhibitors in frontotemporal dementia. Ann Pharmacother. 2004;38(11):1967–1968. | |
Kertesz A, Morlog D, Light M, et al. Galantamine in frontotemporal dementia and primary progressive aphasia. Dement Geriatr Cogn Disord. 2008;25(2):178–185. | |
Diehl-Schmid J, Förstl H, Perneczky R, Pohl C, Kurz A. A 6-month, open-label study of memantine in patients with frontotemporal dementia. Int J Geriatr Psychiatry. 2008;23(7):754–759. | |
Vercelletto M, Boutoleau-Bretonnière C, Volteau C, et al; French research network on Frontotemporal dementia. Memantine in behavioral variant frontotemporal dementia: negative results. J Alzheimers Dis. 2011;23(4):749–759. | |
Chow TW, Graff-Guerrero A, Verhoeff NP, et al. Open-label study of the short-term effects of memantine on FDG-PET in frontotemporal dementia. Neuropsychiatr Dis Treat. 2011;7:415–424. | |
Boxer AL, Knopman DS, Kaufer DI, et al. Memantine in patients with frontotemporal lobar degeneration: a multicentre, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2013;12(2):149–156. | |
Deakin JB, Rahman S, Nestor PJ, Hodges JR, Sahakian BJ. Paroxetine does not improve symptoms and impairs cognition in frontotemporal dementia: a double-blind randomized controlled trial. Psychopharmacology (Berl). 2004;172(4):400–408. | |
Mendez MF, Shapira JS, Miller BL. Stereotypical movements and frontotemporal dementia. Mov Disord. 2005;20(6):742–745. | |
Lebert F, Stekke W, Hasenbroekx C, Pasquier F. Frontotemporal dementia: a randomised, controlled trial with trazodone. Dement Geriatr Cogn Disord. 2004;17(4):355–359. | |
Ikeda M, Shigenobu K, Fukuhara R, et al. Efficacy of fluvoxamine as a treatment for behavioral symptoms in frontotemporal lobar degeneration patients. Dement Geriatr Cogn Disord. 2004;17(3):117–121. | |
Herrmann N, Black SE, Chow T, Cappell J, Tang-Wai DF, Lanctôt KL. Serotonergic function and treatment of behavioral and psychological symptoms of frontotemporal dementia. Am J Geriatr Psychiatry. 2012;20(9):789–797. | |
Adler G, Teufel M, Drach LM. Pharmacological treatment of frontotemporal dementia: treatment response to the MAO-A inhibitor moclobemide. Int J Geriatr Psychiatry. 2003;18(7):653–655. | |
Huey ED, Garcia C, Wassermann EM, Tierney MC, Grafman J. Stimulant treatment of frontotemporal dementia in 8 patients. J Clin Psychiatry. 2008;69(12):1981–1982. | |
Reed DA, Johnson NA, Thompson C, Weintraub S, Mesulam MM. A clinical trial of bromocriptine for treatment of primary progressive aphasia. Ann Neurol. 2004;56(5):750. | |
Rahman S, Robbins TW, Hodges JR, et al. Methylphenidate (‘Ritalin’) can ameliorate abnormal risk-taking behavior in the frontal variant of frontotemporal dementia. Neuropsychopharmacology. 2006;31(3):651–658. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.