")
Back to Journals » Clinical, Cosmetic and Investigational Dermatology » Volume 12
From Clinical Phenotype to Genotypic Modelling: Incidence and Prevalence of Recessive Dystrophic Epidermolysis Bullosa (RDEB)
Authors Eichstadt S, Tang JY, Solis DC, Siprashvili Z, Marinkovich MP, Whitehead N , Schu M , Fang F, Erickson SW, Ritchey ME , Colao M, Spratt K , Shaygan A, Ahn MJ , Sarin KY
Received 26 September 2019
Accepted for publication 10 December 2019
Published 24 December 2019 Volume 2019:12 Pages 933—942
DOI https://doi.org/10.2147/CCID.S232547
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Jeffrey Weinberg
Shaundra Eichstadt, 1 Jean Y Tang, 1 Daniel C Solis, 1 Zurab Siprashvili, 1 M Peter Marinkovich, 1, 2 Nedra Whitehead, 3 Matthew Schu, 3 Fang Fang, 3 Stephen W Erickson, 3 Mary E Ritchey, 3 Max Colao, 4 Kaye Spratt, 4 Amir Shaygan, 5 Mark J Ahn, 5 Kavita Y Sarin 1
1Stanford University School of Medicine, Department of Dermatology, Redwood City, CA 94063, USA; 2Veterans Affairs Medical Center, Palo Alto, CA, USA; 3RTI International, Research Triangle Park, NC, USA; 4Abeona Therapeutics, New York, NY, USA; 5Department of Engineering and Technology Management, Portland State University, Portland, OR, USA
Correspondence: Mark J Ahn
Department of Engineering and Technology Management, Portland State University, 1900 SW 4th Avenue, Suite LL50-01, Portland, OR 97201, USA
Tel +1503961-4466
Email [email protected]
Background: Recessive dystrophic epidermolysis bullosa (RDEB) is an inherited genetic disorder characterized by recurrent and chronic open wounds with significant morbidity, impaired quality of life, and early mortality. RDEB patients demonstrate reduction or structural alteration type VII collagen (C7) owing to mutations in the gene COL7A1, the main component of anchoring fibrils (AF) necessary to maintain epidermal-dermal cohesion. While over 700 alterations in COL7A1 have been reported to cause dystrophic epidermolysis bullosa (DEB), which may be inherited in an autosomal dominant (DDEB) or autosomal recessive pattern (RDEB), the incidence and prevalence of RDEB is not well defined. To date, the widely estimated incidence (0.2– 6.65 per million births) and prevalence (3.5– 20.4 per million people) of RDEB has been primarily characterized by limited analyses of clinical databases or registries.
Methods: Using a genetic modelling approach, we use whole exome and genome sequencing data to estimate the allele frequency of pathogenic variants. Through the ClinVar and NCBI database of human genome variants and phenotypes, DEB Register, and analyzing premature COL7A1 termination variants we built a model to predict the pathogenicity of previously unclassified variants. We applied the model to publicly available sequences from the Exome Aggregation Consortium (ExAC) and Genome Aggregation Database (gnomAD) and identified variants which were classified as pathogenic for RDEB from which we estimate disease incidence and prevalence.
Results: Genetic modelling applied to the whole exome and genome sequencing data resulted in the identification of predicted RDEB pathogenic alleles, from which our estimate of the incidence of RDEB is 95 per million live births, 30 times the 3.05 per million live birth incidence estimated by the National Epidermolysis Bullosa Registry (NEBR). Using a simulation approach, we estimate a mean of approximately 3,850 patients in the US who may benefit from COL7A1-mediated treatments in the US.
Conclusion: We conclude that genetic allele frequency estimation may enhance the underdiagnosis of rare genetic diseases generally, and RDEB specifically, which may improve incidence and prevalence estimates of patients who may benefit from treatment.
Keywords: Dystrophic Epidermolysis Bullosa, genotype, phenotype, incidence, prevalence
Corrigendum for this paper has been published
Background
Recessive dystrophic epidermolysis bullosa (RDEB) is an inherited genetic disorder characterized by recurrent and chronic open wounds with significant morbidity, impaired quality of life, and early mortality. RDEB patients lack functional type VII collagen (C7) owing to mutations in the gene COL7A1, the main component of anchoring fibrils (AF) necessary to maintain epidermal-dermal cohesion. While over 700 alterations in COL7A1 have been reported to cause Dystrophic epidermolysis bullosa (DEB), which may be inherited in an autosomal dominant or autosomal recessive pattern,1 the incidence and prevalence of RDEB are not well defined. To date, the widely estimated incidence (0.2–6.65 per million births) and prevalence (3.5–20.4 per million people) of RDEB has been primarily characterized by limited analyses of clinical databases or registries (Table 1).
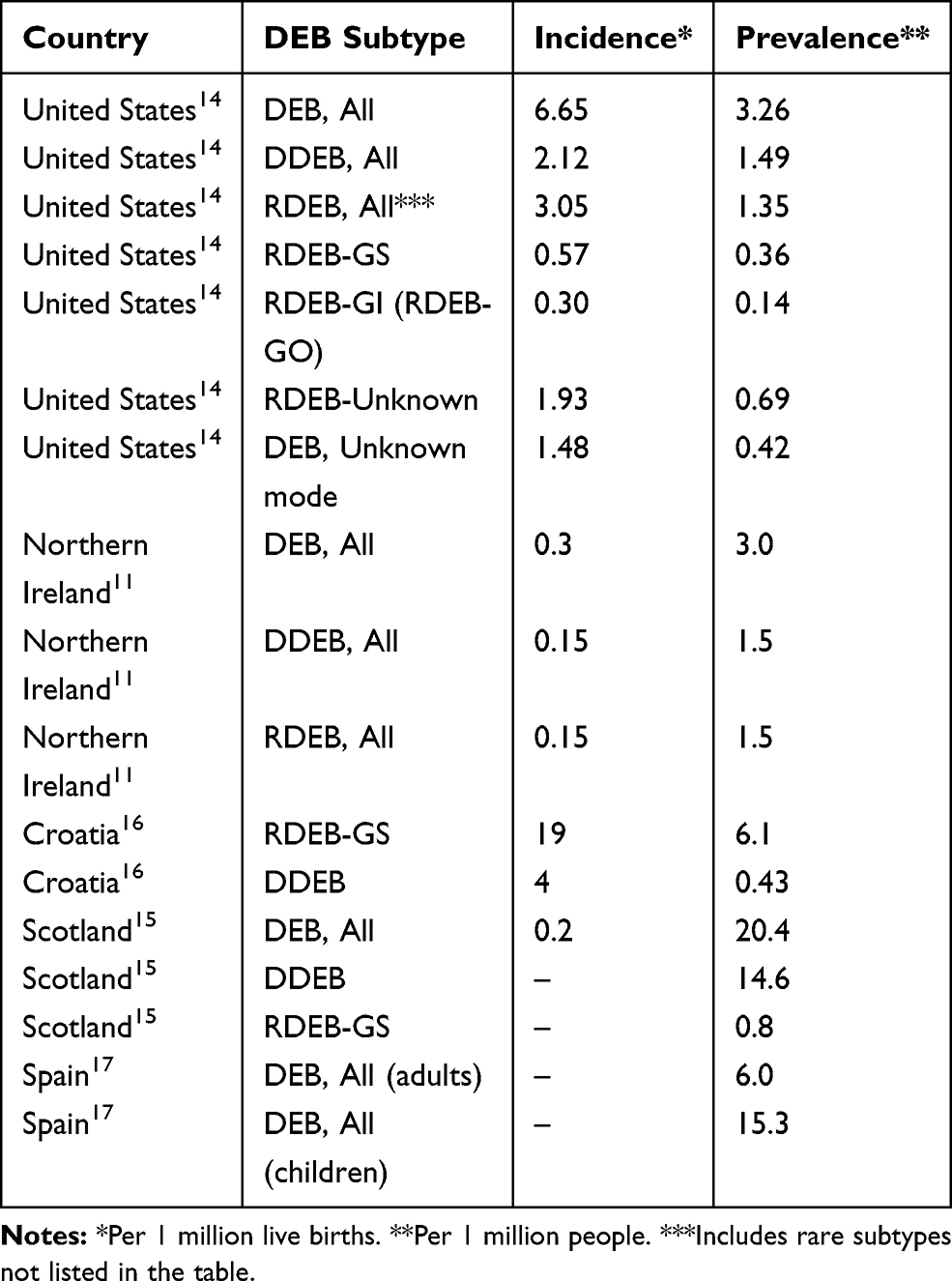 |
Table 1 Reported Incidence and Prevalence of Dystrophic Epidermolysis Bullosa |
Of note, patients face significant cost and delays in receiving an accurate and timely diagnosis due to the cost and availability of genetic testing, as well as a limited number of rare disease specialists. In the US, rare disease patients visit an average of 7.3 physicians over 7.6 years before receiving a diagnosis.2,3 Also, a growing number of population genome sequencing efforts such as the 100,000 Genome Project in the UK are highlighting cases of missed diagnoses and developing more accurate genotype-phenotype correlations,4 including mutations associated with various degrees of pathogenicity.5 In this context, we explore whether genetic allele frequency estimation may enhance the under-diagnosis of rare genetic diseases generally, and RDEB specifically, towards improving the incidence and prevalence estimates of patients who may benefit from treatment.
Using a genetic modelling approach, we use whole exome and genome sequencing data to estimate the allele frequency of recessive pathogenic variants in the COL7A1 gene. Through ClinVar, the NCBI database of human genome variants and phenotypes, DEB Register, and analyzing premature COL7A1 termination variants, we identified 270 variants with documented clinical significance. From these, we built a model to predict the pathogenicity of previously unclassified variants. Applying the model to variant data in the Exome Aggregation Consortium (ExAC) database and the Genome Aggregation Database (gnomAD) in Appendix A, we identified the aggregate frequency of variants predicted to be pathogenic for RDEB and estimated the incidence of the disease.
Dystrophic Epidermolysis Bullosa (DEB)
Dystrophic epidermolysis bullosa (DEB) is one of four major types of epidermolysis bullosa (EB), a group of genetic disorders of the skin and mucous membranes which arise from the defects of basal keratinocyte attachment to the underlying dermis. The skin layer in which the separation occurs defines the EB type. In DEB, separation occurs in the sublamina densa, which attaches the epidermis to the papillary dermis. The separation results from reduction or alteration of C7, a major component of the anchoring fibrils that mediate dermal-epidermal cohesion.1,6
Gene and Protein
Structure
The gene that encodes C7 is COL7A1. Each C7 molecule is composed of 3 procollagen α1 chains; each procollagen chain contains 3 domains: an amino-terminal noncollagenous domain (NC1), a central collagenous, triple-helical domain, and a carboxyl-terminal noncollagenous domain (NC2).
Genetics
Over 700 alterations in COL7A1 have been reported to cause DEB, which may be inherited in an autosomal dominant or autosomal recessive pattern.7 Less common genetic mechanisms have also been observed. A patient carrying only one COL7A1 mutation not present in the parents’ peripheral blood leukocytes suggested de novo mutation or parental germline mosaicism. Such cases recurred in at least one family, strongly suggesting parental mosaicism.8 In two cases with apparently recessive inheritance, only one heterozygous mutation was detected after sequencing all 118 exons and flanking exon–intron borders, while the parent carrying the identified mutation was unaffected.9 Finally, one case of RDEB-GS was homozygous for a frameshift mutation, 345insG, carried by the mother due to maternal isodisomy of chromosome 3.8 Most mutations are family-specific, but a few recurrent mutations have been noted.6,8
RDEB Subtypes
Individuals with RDEB-GS produce little to no C7. They frequently have two COL7AI alleles with premature stop codons, resulting in mRNA decay, absence of mRNA expression, or formation of truncated C7 polypeptides that are structurally defective or rapidly degraded in cells. They often have generalized blistering from birth that results in extensive scarring, and alopecia. Patients with RDEB-GS also suffer from extracutaneous manifestations, resulting in an increased risk of corneal abrasions, mucous membrane blistering, oesophagal strictures, kidney problems and cardiomyopathy. Scarring can lead to difficulty eating, vision impairment, painful stools, and constipation. RDEB patients have a high risk of developing aggressive squamous cell carcinomas, which is associated with early mortality.6
Patients with RDEB-other (RDEB-O) have a similar, though less severe, phenotype compared to those with RDEB-GS. They produce some functional, albeit abnormal, C7. They also have a better prognosis, with some affected women capable of giving birth. RDEB-O patients are frequently compound heterozygotes: one allele is a missense, in-frame, or splice site mutation, while the second often contains a premature stop codon.8,10
Genotype-Phenotype Correlation
RDEB generalized severe cases often result from null mutations. Most reported null mutations are nonsense or frameshift mutations that cause premature stop codons, but others destroy the methionine initiation code or completely disrupt splicing.11,12 Approximately 12% of RDEB-GS cases are compound heterozygotes for one premature termination mutation and one missense mutations, or two missense mutations.12
Premature stop codons (PTCs) also play a significant role in RDEB-O. In one study, 34% of cases had PTC mutations in both alleles.12 Over half of the remaining cases were compound heterozygotes for a PTC mutation and a missense mutation, primarily glycine substitutions. Glycine or arginine substitutions in the collagenous domain appear to cause RDEB inversa (RDEB-I). Some of these substitutions are specific to RDEB-I, but others have also been reported in non-inversa subtypes.13 Cases with one or two missense mutations usually have a milder phenotype than those with a PTC mutation. The presentation may be similar to DDEB in some cases.11 It is not always clear why a case with two PTC alleles may present with the milder RDEB-O phenotype. Exon skipping, resulting in a shortened but functional protein, explains this phenomenon in some, but not all, cases.12
Epidemiology
Several authors have reported on the epidemiology of DEB (Table 1).11,14–17 The incidence of DEB mutations reported in these studies ranged from 0.2 to 6.65 per million live births; the prevalence ranged from 3.0 to 20.4 per million people. All studies used multiple methods of ascertainment, including records from hospitals, dermatology clinics, paediatricians and general practitioners, newspaper advertisements, announcements in patient newsletters, and family networks. The two studies that examined ascertainment by source found that the majority of cases were not followed at referral centres or recognized as having EB by their primary care physician, highlighting the difficulty of complete ascertainment.18,19
Methods
In this study, we estimate the incidence of recessive dystrophic epidermolysis bullosa cases by leveraging publicly available whole-exome sequencing (WES) and whole-genome sequencing (WGS) data to estimate the allele frequency of recessive pathogenic variants in the COL7A1 gene among healthy adult carriers.
Pathogenic Genetic Variants Observed in Cases
We developed an inventory of reported pathogenic COL7A1 variants from ClinVar20 and the DEB Register,12,21 an international registry for DEB patients and their COL7A1 mutations. The primary goal was to build a model to predict whether an allele could result in an RDEB phenotype when harboured in a homozygous state or compound heterozygous state with another pathogenic allele. To build our model, we constructed a training data set composed of established nonpathogenic variants in COL7A1 and variants that were clearly associated with some form of dystrophic EB. We classified alleles as pathogenic for RDEB if the majority of sources report the allele as causing or associated with RDEB; DDEB if 50% or more of the sources report the allele causing or in association with DDEB; and DEB, not otherwise specified (NOS) if no sources report inheritance pattern.
We considered variants as causal if they were consistently classified as “Pathogenic” or “Likely Pathogenic” in ClinVar or the majority of DEB Register participants that carried the allele had a dystrophic EB phenotype and there was no conflicting evidence in ClinVar. We identified a total of 86 variants that cause DDEB, 155 variants causing RDEB, and 15 DEB-NOS variants. All variants that were removed from the training set due to conflicting evidence annotations were retained in the test set and subsequently evaluated for their potential pathogenic influence with respect to RDEB.
Population Genetics Database
In order to estimate the allele frequency of pathogenic variants, we leveraged the Exome Aggregation Consortium (ExAC) database and the Genome Aggregation Database (gnomAD).22 ExAC contains over 60,000 genotypes from 14 datasets and is ethnically diverse (Appendix A). The only individuals excluded from the final database are those with severe pediatric diseases, such as DEB and other genetic diseases, and those without adequate informed consent. The inclusion criteria allow for the detection of heterozygous carriers of COL7A1 variants, but should exclude affected individuals with homozygous alleles for pathogenic variants. Therefore, COL7A1 variants that are homozygous within ExAC samples are unlikely to be causal for RDEB.
The ExAC database is a subset of gnomAD. GnomAD contains 123,136 exomes and 15,496 genomes from healthy human donors. As the first degree relatives of individuals with severe congenital disorders are excluded in gnomAD, using it risks a downward bias in the estimated allele frequency of pathogenic COL7A1 variants. Nonetheless, as RDEB is a rare disorder, the likelihood that any carrier has a first degree relative with the disease is still quite low. Therefore, while we preferentially report estimates derived from ExAC when a pathogenic variant is listed in both databases, pathogenic and predicted pathogenic variants found only in gnomAD are still reported.
Predicted Pathogenic Alleles
We predicted the pathogenicity of previously unclassified missense variants observed in whole-exome sequences using two automated classifiers, one for missense mutations and one for frameshift mutations. The missense classifier predicts the clinical impact of amino acid substitutions caused by missense mutations by synthesizing the predictions from six variant functional prediction tools: FATHMM,23 MutationAssessor,24 PolyPhen-2,25 PROVEAN,18 SIFT,26 and CONDEL.27 Variants for training the model were selected from variants with known clinical relationships to dystrophic EB as described in section 2.1.
We first compiled a training set of 256 known pathogenic and 14 known benign missense mutations based on well-established pathogenic status from ClinVar28 or the DEB Register.29 We applied six prediction tools to these variants, and from their scores fitted a multivariable logistic regression model to predict the disease state of a hypothetical homozygous carrier for a given variant (Figure 1).
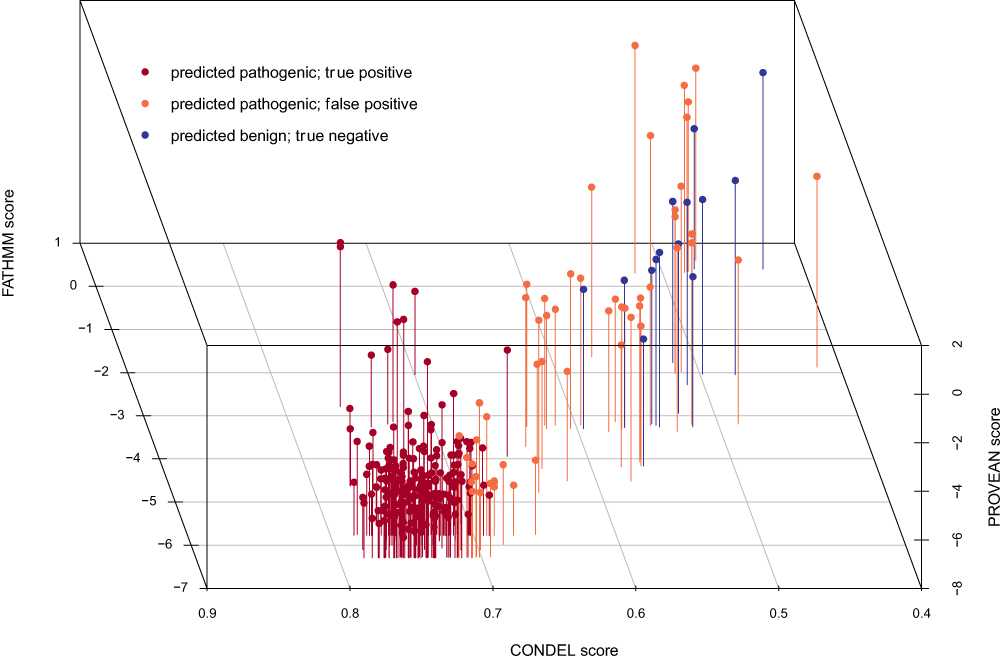 |
Figure 1 Model Classification of Training Data. The 3D scatter plot shown here demonstrates the ability of the classifier and three components to stratify the variants into similar functional clusters. |
Based on the clustering of pathogenic variants and distribution of prediction scores (Figure 2) in the training database, we classified variants as pathogenic if the prediction score was 0.95 or higher. With this cut-point, the model correctly classified 14 of 14 benign variants and 225 of 256 pathogenic variants, resulting in a positive predictive value of 100%, sensitivity of 88% and specificity of 100%. To guard against the final estimate of net pathogenic allele frequency being biased upward by likely false positives we filtered our list of previously reported and newly predicted pathogenic variants to remove variants for which homozygous carriers are observed in either ExAC or gnomAD (both of which should only contain unaffected individuals), variants with low confidence loss of function flags in either sequencing database, and any variant with an allele frequency greater than 0.0001 as it would suggest a disease prevalence that is unrealistically high for this rare disorder.
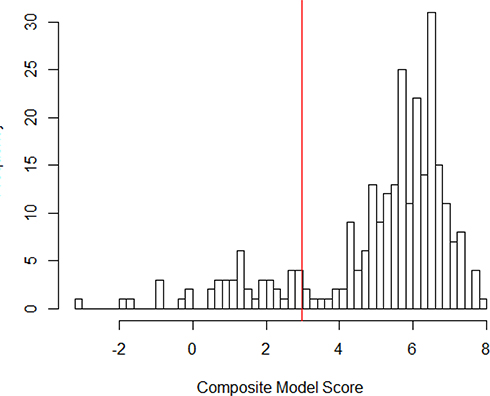 |
Figure 2 Distribution of Pathogenicity Scores in Training Data. |
The splice site classifier predicts the impact of non-coding variants on intron-exon splicing. The classifier is based on a logistic regression model, which integrates the functional annotation scores from the CADD19 and EIGEN28 algorithms into a composite score. As with the missense classifier, the predictions from the splice site model are refined using a maximum cutoff 0.0001 for allele frequency. We applied the model to a training dataset of 16 known pathogenic variants and 17 known benign variants. We set the threshold of pathogenicity conservatively at a composite score of 0.8, resulting in a sensitivity of 0.44 and a specificity of 1.
We estimated the net carrier allele frequency as the sum of the individual pathogenic allele frequencies from the ExAC database, or when a variant was not found in ExAC, from the larger gnomAD database. We calculated the frequency estimates for pathogenic alleles using all available genotypes in each respective database, as we found no differences in the net carrier allele frequency by ancestry.
The assumption was made that individuals with two null alleles would have RDEB-GS and those with any other two pathogenic alleles would have RDEB-O. We estimated genotype frequency from observed allele frequencies assuming homozygote/heterozygote proportions expected under Hardy–Weinberg equilibrium.
The incidence of RDEB was estimated as the number of births in the US per year times the total expected frequency of pathogenic genotypes. Next, the number of cases per group were summed to get the total number of cases for a birth cohort.29 We estimated prevalence using the estimated number of cases born in a birth cohort and the cumulative probability of death for RDEB subtype (Table 2).30 The calculations were done using cases born in 1960 or later.
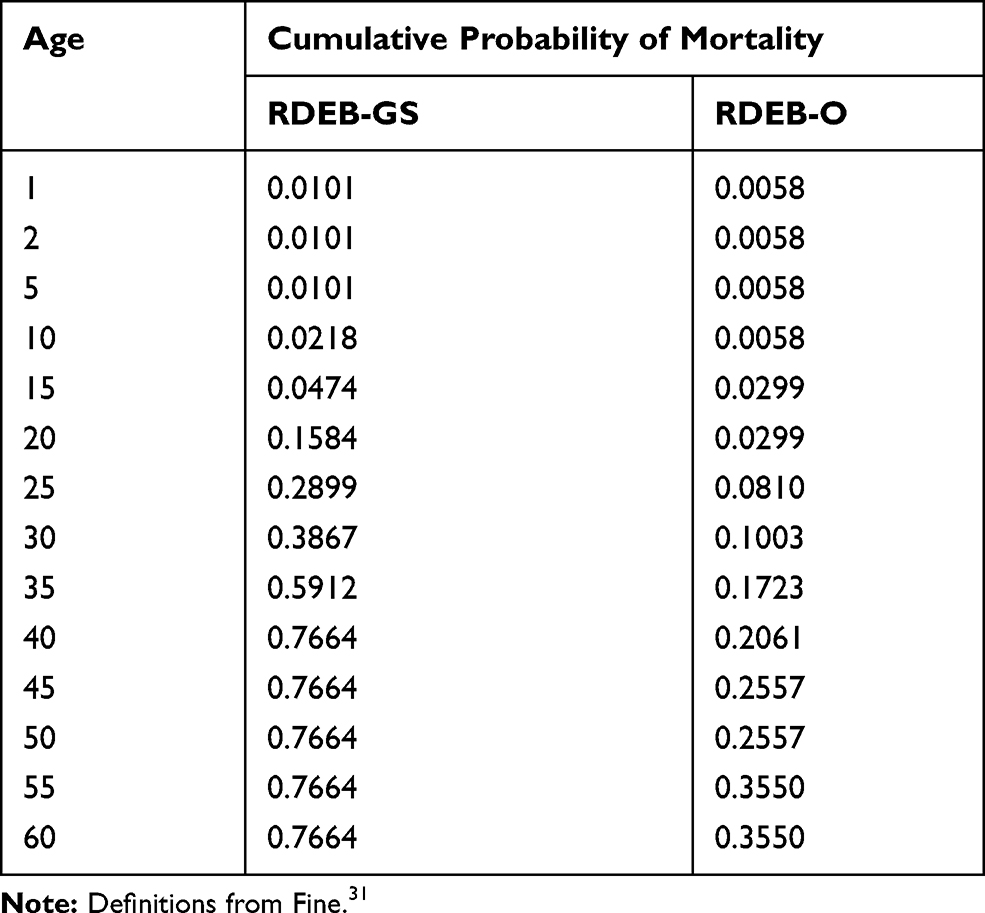 |
Table 2 Cumulative Probability of Mortality by Age and RDEB Subtype |
Results
Frequency of RDEB Variants
The ExAC and gnomAD databases contained 1620 COL7A1 exonic variants that cause amino acid substitutions absent in our training set. In total, 523 variants were classified as pathogenic for RDEB, encompassing 193 previously reported pathogenic variants, including 5 dominant mutations. The variants included 128 premature termination codons, 323 missense and 67 splice site mutations. The vast majority of both known and predicted pathogenic alleles had frequencies of less than the 0.0001 inclusion criteria (Figure 3). In all, we excluded 12 variants from our final inventory of pathogenic variants (2 previously reported and 10 newly predicted pathogenic alleles) whose observed allele frequencies exceeded this cutoff.
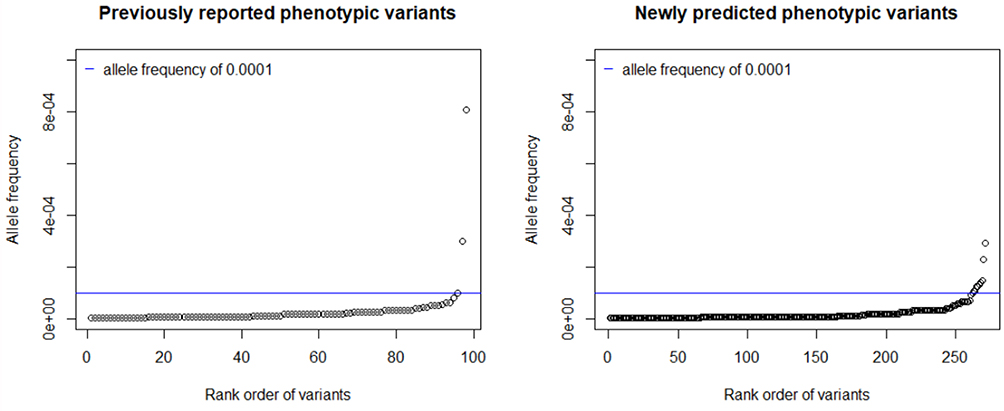 |
Figure 3 Distribution of Allele Frequencies. |
After adjusting the model to account for the overrepresentation of pathogenic variants in the training dataset, variant classification was more conservative and less sensitive to the threshold used to classify a variant as pathogenic (Table 3). To adjust for the pathogenic over-representation in the training set, we first estimated the true proportion of pathogenic variants in the population. We estimate this proportion as a function of the mean pathogenicity scores as follows:
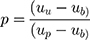
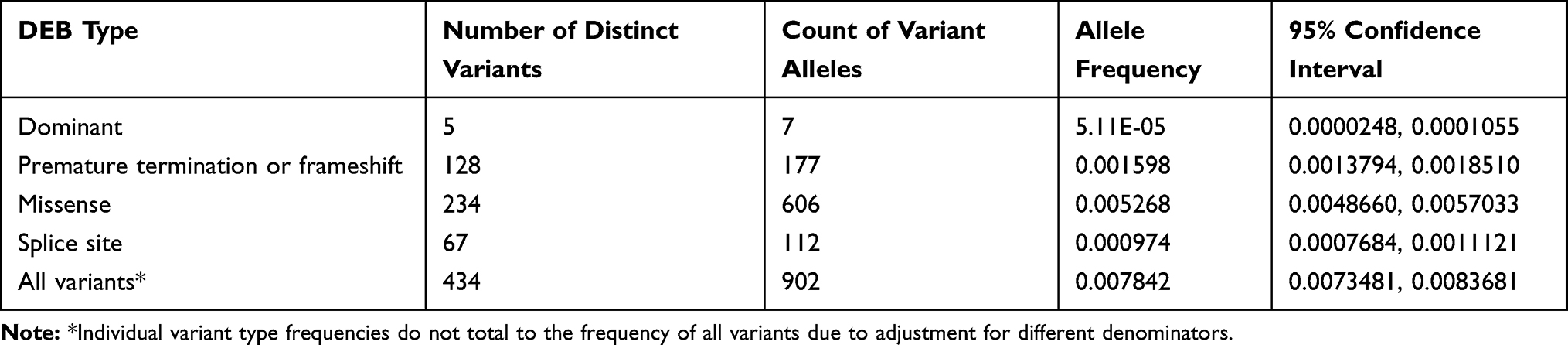 |
Table 3 Allele Frequency of DEB Pathogenic Variants in Col7A1 Gene |
Where
µb = mean composite score of the benign variants;
µp = mean composite score of the pathogenic variants;
µu = mean composite score of the unclassified variants; and
p = proportion of pathogenic variants in the unclassified set
We then performed weighted logistic regression similar to the methods previously described by Rose et al32 Since we are controlling for an overrepresentation of variants as opposed to patients, we modified Rose’s approach by treating pathogenic variants as cases, benign variants as controls, and the weight for each variant as the ratio of the proportion of the variant class in the unclassified set to the proportion of the variant class in the training set. After adjusting the model to account for the overrepresentation of pathogenic variants in the training dataset, variant classification was more conservative and less sensitive to the threshold used to classify a variant as pathogenic (Table 3). The adjustment also resulted in less dispersal in the distribution of the pathogenicity scores.
We further explored whether the performance of the bias-corrected model varied due to the choice of benign and pathogenic variants in the training datasets using 3-fold cross-validation. We divided the training datasets of known benign and pathogenic variants into three distinct subsets. In each of the three cross-validations, two subsets formed the training dataset and one subset formed the testing dataset. Because the number of benign variants (n=14) and pathogenic variants (n=256) are not divisible by three, 2 of 3 cross-validation testing datasets will contain five benign variants and one testing dataset will contain only four benign variants, with an additional pathogenic variant. We conducted 100 iterations of the 3-fold cross-validation, maintaining the same weights and pathogenicity score cutoff (0.95) for each cross-validation. For each iteration of the 3-fold cross-validation, we calculated the sensitivity, specificity, positive predictive value and negative predictive value of the model. We also calculated the average sensitivity, specificity, positive predictive value and negative predictive value across all 100 iterations.
Approximately 4 million infants are born in the United States annually.33 Based on this number of births and the frequency of pathogenic allele variants, we estimate that 251 infants with RDEB are born each year in the US (Table 4). Of these, 10 are predicted to have the severe form of RDEB. During the last decade, the European Union averaged a total of 5.23 million births per year.33 Based on the above frequency of RDEB pathogenic alleles, an estimated 326 cases of RDEB are born in the European Union each year, of which 13 are predicted to be RDEB-GS (Table 4).
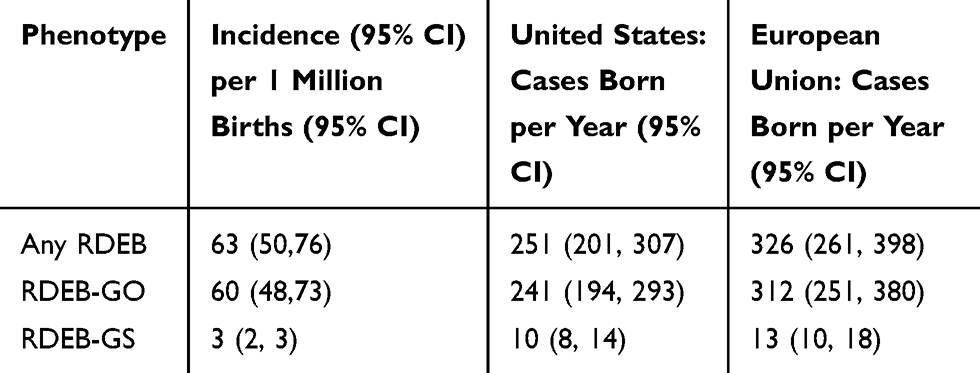 |
Table 4 Estimated Incidence of Recessive Dystrophic Epidermolysis Bullosa |
Applying life table analysis to the number of cases born per year, an estimated 12,562 individuals affected with RDEB who were born since 1960 are living in the US, and 16,290 are living in the European Union (Table 5). Fewer than 2% of those individuals are predicted to have the RDEB-GS subtype.
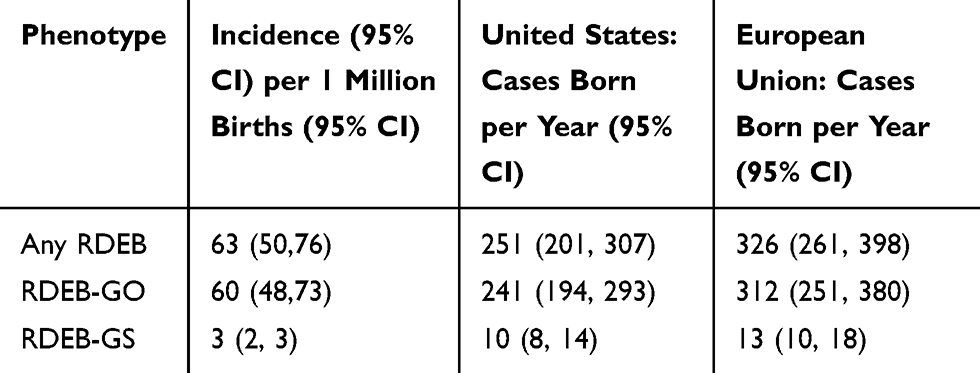 |
Table 5 Estimated Prevalence of Recessive Dystrophic Epidermolysis Bullosa |
Simulation
Given the challenges noted in clinical RDEB ascertainment,18,19 we further performed a Monte Carlo simulation to estimate the expected number of RDEB cases who may benefit from COL7A1-mediated treatments (e.g., gene therapy, gene editing, mRNA) using MATLAB® and Simulink®. The Monte Carlo method uses computational algorithms based on iterations of random sampling from a positively skewed probability distribution (i.e., the probability is inversely related to larger population-based inferences versus known validated patient registries) in order to acquire numerical results for the goals of sampling, optimization, and estimations.34–36 In other words, our population confidence decreased as we increased our reliance on sequencing point estimates, resulting in positive skewness for the probability distribution. Using a minimum amount defined as the mean of RDEB-GS of 343 and a maximum population estimate of 12,562 of any RDEB, we conducted 10,000 repetitions resulting in a mean estimate of approximately 3850 patients in the US who may benefit from COL7A1-mediated treatments in the US.
Discussion
Genetic modelling applied to the whole exome and genome sequencing data resulted in the identification of predicted RDEB pathogenic alleles, from which our estimate of the incidence of RDEB is 95 per million live births, 30 times the 3.05 per million live birth incidence estimated by the National Epidermolysis Bullosa Registry (NEBR). The NEBR may have under-ascertained less severely affected cases, because at least two studies have reported that a majority of the cases in their studies were not evaluated and treated at EB referral centres.18,19 Alternatively, we may have overestimated the incidence of RDEB. Our estimates are based on allele frequencies, not clinical symptoms. Also, while a robust database of pathogenic mutations is available, there are a limited number of benign missense mutations available23 to utilize as a training set. The pathogenicity of splice site mutations is also difficult to predict, making them particularly susceptible to misclassification; even though a conservative threshold was used, some benign variants may be misclassified as pathogenic. Our methodology also overestimates the incidence of RDEB for those genotypes that are incompletely penetrant37 and if fetuses with RDEB are at an increased risk of fetal death. DEB is a complex disorder; clinical symptoms manifest along a continuum of mild to extremely severe. Genotype does not always predict either protein function or disease severity well.13 Our estimates of incidence and prevalence apply to a range of RDEB phenotypes and may be limited in the ability to estimate the prevalence of specific symptoms.
A further complication to estimating the incidence of RDEB is the reproductive potential of moderately affected patients. Shinkuma6 noted that some RDEB-GO patients are capable of giving birth, but we did not find any information on male fertility in RDEB patients. The methodology we used assumes alleles are in Hardy–Weinberg equilibrium. It remains valid if cases of either sex reproduce, as long as this assumption is met. Notwithstanding, consensus panels recognize that RDEB-GO is a diverse group of RDEB subtypes ranging from RDEB generalized intermediate, RDEB pruiginosa, RDEB bullous dermolysis of the newborn, RDEB pretibial, RDEB centripetalis, RDEB inversa and RDEB localized which may be explored further.38
Conclusion
In sum, this study evaluates the incidence and prevalence of RDEB using publicly available whole-exome sequencing and whole-genome sequencing databases. We established a range estimate, as well as a simulation model, of RDEB patients who may benefit from COL7A1-directed treatments. We conclude that genetic allele frequency estimation may enhance the underdiagnosis of rare genetic diseases generally, and RDEB specifically, which may improve incidence and prevalence estimates of patients who may benefit from treatment.
Abbreviations
AF, anchoring fibrils; C7, type VII collagen; ExAC,Exome Aggregation Consortium; gnomAD, Genome Aggregation Database; NC1,Amino-terminal noncollagenous domain; NC2, carboxyl-terminal noncollagenous domain; PTC, premature stop codons; WES, whole-exome sequencing; WGS, whole-genome sequencing.
Data Sharing Statement
All databases referenced in the manuscript are publicly available, including ExAC (Exome Aggregation Consortium) and gnomAD (Genome Aggregation Database).
Author Contributions
All authors contributed to data analysis, drafting and revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
Dr Shaundra Eichstadt reports grants from Epidermolysis Bullosa Research Partnershipand Epidermolysis Bullosa Medical Research Foundation during the conduct of the study. Dr Zurab Siprashvili reports a patent: Gene Therapy for Recessive Dystrophic Epidermolysis Bullosa using Genetically Corrected Autologous Keratinocytes, licensed to Abeona Therapeutics. Dr Mary Beth Ritchey reports contracted work for assessment of the incidence of RDEB for Abeona Therapeutics during the conduct of the study. Mr Max Colao reports he is employed by Abeona Therapeutics. The authors declare that they have no other competing interests.
References
1. Marinkovich MP. Inherited epidermolysis bullosa. In: Wolff K, Johnson R, Saavedra A, Roh E, editors. Fitzpatrick’s Color Atlas and Synopsis of Clinical Dermatology. New York, NY: McGraw-Hill; 2017:649–665.
2. Engel P, Bagal S, Broback M, Boice N. Physician and patient perceptions regarding physician training in rare diseases: the need for stronger educational initiatives for physicians. J Rare Dis. 2013;1(2):1–14.
3. NIH National Center for Advancing Translational Sciences. Genetic and rare disease information center. How to find a disease specialist [Internet]; 2017 [
4. Harvey K, Burke D, Heals S. The evolving role of enzymology and metabolomics in the diagnosis of lysosomal disorders in the post-genomic era. Mol Genet Metab. 2019;126(2):S69. doi:10.1016/j.ymgme.2018.12.164
5. Nakano A, Chao SC, Pulkkinen L, et al. Laminin 5 mutations in junctional epidermolysis bullosa: molecular basis of Herlitz vs non-Herlitz phenotypes. Hum Genet. 2002;110(1):41–51. doi:10.1007/s00439-001-0630-1
6. Shinkuma S. Dystrophic Epidermolysis Bullosa: A Review. Vol. 8, Clinical, Cosmetic and Investigational Dermatology. Dove Medical Press Ltd.; 2015:275–284.
7. Solis D, Nazaroff J, Dutt-Singkh Y, et al. Natural history of wounds in recessive dystrophic epidermolysis bullosa.
8. Saeidian AH, Youssefian L, Moreno Trevino MG, et al. Seven novel COL7A1 mutations identified in patients with recessive dystrophic epidermolysis bullosa from Mexico. Clin Exp Dermatol. 2018;43(5):579–584. doi:10.1111/ced.2018.43.issue-5
9. van den Akker PC, van Essen AJ, Kraak MMJ, et al. Long-term follow-up of patients with recessive dystrophic epidermolysis bullosa in the Netherlands: expansion of the mutation database and unusual phenotype-genotype correlations. J Dermatol Sci. 2009;56(1):9–18. doi:10.1016/j.jdermsci.2009.06.015
10. Varki R, Sadowski S, Uitto J, Pfendner E. Epidermolysis bullosa. II. Type VII collagen mutations and phenotype-genotype correlations in the dystrophic subtypes. J Med Genet. 2007;44(3):181–192. doi:10.1136/jmg.2006.045302
11. McKenna KE, Walsh MY, Bingham EA. Epidermolysis bullosa in Northern Ireland. Br J Dermatol. 1992;127(4):318–321. doi:10.1111/bjd.1992.127.issue-4
12. van den Akker PC, Jonkman MF, Rengaw T, et al. The international dystrophic epidermolysis bullosa patient registry: an online database of dystrophic epidermolysis bullosa patients and their COL7A1 mutations. Hum Mutat. 2011;32(10):1100–1107. doi:10.1002/humu.21551
13. Van Den Akker PC, Mellerio JE, Martinez AE, et al. The inversa type of recessive dystrophic epidermolysis bullosa is caused by specific arginine and glycine substitutions in type VII collagen. J Med Genet. 2011;48(3):160–167. doi:10.1136/jmg.2010.082230
14. Fine JD. Epidemiology of inherited epidermolysis bullosa based on incidence and prevalence estimates from the national epidermolysis Bullosa registry. JAMA Dermatol. 2016;152(11):1231–1238. doi:10.1001/jamadermatol.2016.2473
15. Horn HM, Priestley GC, Eady RAJ, Tidman MJ. The prevalence of epidermolysis bullosa in Scotland. Br J Dermatol. 1997;136(4):560–564. doi:10.1111/j.1365-2133.1997.tb02141.x
16. Pavičić Ž, Kmet‐Vižintin P, Kansky A, Dobrić I. Occurrence of hereditary bullous epidermolyses in croatia. Pediatr Dermatol. 1990;7(2):108–110. doi:10.1111/j.1525-1470.1990.tb00664.x
17. Hernandez-Martín A, Aranegui B, Escámez MJ, et al. Prevalence of dystrophic epidermolysis bullosa in Spain: a population-based study using the 3-source capture-recapture method. Evidence of a need for improvement in care. Actas Dermosifiliogr. 2013;104(10):890–896. doi:10.1016/j.ad.2013.03.006
18. Choi Y, Sims GE, Murphy S, Miller JR, Chan AP. Predicting the functional effect of amino acid substitutions and indels. de Brevern AG, editor. PLoS One. 2012;7(10):e46688. doi:10.1371/journal.pone.0046688
19. Kircher M, Witten DM, Jain P, O’roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46(3):310–315. doi:10.1038/ng.2892
20. Landrum MJ, Lee JM, Benson M, et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018;46(D1):D1062–7. doi:10.1093/nar/gkx1153
21. Wertheim-Tysarowska K, Sobczyńska-Tomaszewska A, Kowalewski C, et al. The COL7A1 mutation database. Hum Mutat. 2012;33(2):327–331. doi:10.1002/humu.21651
22. Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285–291. doi:10.1038/nature19057
23. Shihab HA, Gough J, Cooper DN, et al. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using Hidden Markov Models. Hum Mutat. 2013;34(1):57–65. doi:10.1002/humu.22225
24. Reva B, Antipin Y, Sander C. Determinants of protein function revealed by combinatorial entropy optimization. Genome Biol. 2007;8(11):R232. doi:10.1186/gb-2007-8-11-r232
25. Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi:10.1038/nmeth0410-248
26. Ng PC. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31(13):3812–3814. doi:10.1093/nar/gkg509
27. González-Pérez A, López-Bigas N. Improving the assessment of the outcome of nonsynonymous SNVs with a consensus deleteriousness score, Condel. Am J Hum Genet. 2011;88(4):440–449. doi:10.1016/j.ajhg.2011.03.004
28. Ionita-Laza I, Mccallum K, Xu B, Buxbaum JD. A spectral approach integrating functional genomic annotations for coding and noncoding variants. Nat Genet. 2016;48(2):214–220. doi:10.1038/ng.3477
29. Martin JA, Hamilton BE, Osterman MJK, Driscoll AK, Drake P. Births: final data for 2016. Natl Vital Stat Rep. 2018;67(1):1–55.
30. Fine J-D. Inherited epidermolysis bullosa. Orphanet J Rare Dis. 2010;5(12):1–17. doi:10.1186/1750-1172-5-12
31. Fine J-D. Epidermolysis Bullosa: Clinical, Epidemiologic, and Laboratory Advances, and the Findings of the National Epidermolysis Bullosa Registry. Johns Hopkins University Press; 1999.
32. Rose S, Van Der Laan MJ. Simple optimal weighting of cases and controls in case-control studies. Int J Biostat. 2008;4:1. doi:10.2202/1557-4679.1115
33. EurostatLive. Live births and crude birth rate [Internet]. [
34. Binder K, Heermann D, Roelofs L, Mallinckrodt AJ, Mckay S. Monte carlo simulation in statistical physics. Computers in Physics.1993;7(2):156–157.
35. Kroese DP, Rubinstein RY. Monte carlo methods. Computational Statistics. 2012;4(1):48–58.
36. Watnik M. Early computational statistics. J Comput Graph Stat. 2011;20(4):811–817. doi:10.1198/jcgs.2011.204b
37. Yang CS, Lu Y, Farhi A, et al. An incompletely penetrant novel mutation in COL7A1 causes epidermolysis bullosa pruriginosa and dominant dystrophic epidermolysis bullosa phenotypes in an extended kindred. Pediatr Dermatol. 2012;29(6):725–731. doi:10.1111/pde.2012.29.issue-6
38. Fine JD, Bruckner-Tuderman L, Eady RAJ, et al. Inherited epidermolysis bullosa: updated recommendations on diagnosis and classification. J Am Acad Dermatol. 2014;70:1103–1126. doi:10.1016/j.jaad.2014.01.903.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.