Back to Journals » Journal of Multidisciplinary Healthcare » Volume 14
Friedreich Ataxia: Multidisciplinary Clinical Care
Authors Lynch DR, Schadt K, Kichula E, McCormack S, Lin KY
Received 14 April 2021
Accepted for publication 4 June 2021
Published 28 June 2021 Volume 2021:14 Pages 1645—1658
DOI https://doi.org/10.2147/JMDH.S292945
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Scott Fraser
David R Lynch,1 Kim Schadt,1 Elizabeth Kichula,1 Shana McCormack,2 Kimberly Y Lin3
1Division of Neurology, Departments of Pediatrics and Neurology, Children’s Hospital of Philadelphia and the Perelman School of Medicine, Philadelphia, PA, 19104, USA; 2Division of Endocrinology, Department of Pediatrics, Children’s Hospital of Philadelphia and the Perelman School of Medicine, Philadelphia, PA, 19104, USA; 3Division of Cardiology, Department of Pediatrics, Children’s Hospital of Philadelphia and the Perelman School of Medicine, Philadelphia, PA, 19104, USA
Correspondence: David R Lynch
Division of Neurology, Children’s Hospital of Philadelphia, 502 Abramson, Philadelphia, PA, 19104, USA
Tel +1 215-590-2242
Fax +1 215-590-3779
Email [email protected]
Abstract: Friedreich ataxia (FRDA) is a multisystem disorder affecting 1 in 50,000– 100,000 person in the United States. Traditionally viewed as a neurodegenerative disease, FRDA patients also develop cardiomyopathy, scoliosis, diabetes and other manifestation. Although it usually presents in childhood, it continues throughout life, thus requiring expertise from both pediatric and adult subspecialist in order to provide optimal management. The phenotype of FRDA is unique, giving rise to specific loss of neuronal pathways, a unique form of cardiomyopathy with early hypertrophy and later fibrosis, and diabetes incorporating components of both type I and type II disease. Vision loss, hearing loss, urinary dysfunction and depression also occur in FRDA. Many agents are reaching Phase III trials; if successful, these will provide a variety of new treatments for FRDA that will require many specialists who are not familiar with FRDA to provide clinical therapy. This review provides a summary of the diverse manifestation of FRDA, existing symptomatic therapies, and approaches for integrative care for future therapy in FRDA.
Keywords: triplet repeat, scoliosis, diabetes, cardiomyopathy
Friedreich ataxia (FRDA) is a slowly progressive degenerative disease of children and young adults whose features involve both neural and extraneural components. Individuals develop neurological features (ataxia, loss of coordination, sensory loss, dysarthria, visual dysfunction and hearing loss) as well progressive cardiomyopathy, scoliosis and an elevated risk of diabetes.1–5 An autosomal recessive disease resulting from mutations in the FXN gene, FRDA typically manifests in patients between ages 5y and 20y, but some individuals present outside this range. This constellation of features demands management by multiple different specialists for optimal clinical care.
Although sometimes classed either as a neuromuscular disorder or a neurodegenerative disease, the clinical features of FRDA require a management approach distinct from other neurological disorders. FRDA clinically is commonly grouped with the peripheral nerve disorders of childhood. However, there are substantial differences between FRDA and other disorders of this type (Spinal muscular atrophy, SMA; Duchenne muscular dystrophy, DMD; Charcot-Marie-tooth, CMT) (Table 1).6,7 These disorders, like FRDA, frequently require collaborative care among multiple specialists. Guidelines for both SMA and Duchenne have focused on multidisciplinary care as a cornerstone of management.8,9 Increasingly, such care exists mainly within centers of excellence for these disorders, where multidisciplinary care is a critical component and a necessity for getting the designation. However, there are distinct differences in the care of FRDA from that of those disorders. SMA and DMD present early with more aggressive loss of motor function whereas FRDA will more commonly presented in adolescence and with slower progression. Pulmonary compromise becomes a major issue in SMA and DMD, while pulmonary dysfunction is rarely an issue in FRDA until late in the course of disease.1–4 While FRDA has most in common with CMT as a disease, as both have a neuropathic component, in general CMT has a slower progression, and fewer systemic effects. CMT may present earlier than FRDA, but its progression is slower and depends on the length of axons, leading to a different pattern of progression than FRDA. Occasionally (10% or less of the time), FRDA will clearly differ from CMT by having retained reflexes. FRDA also differs from the typical adult degenerative disorders including other ataxias. Though they share neurological symptoms, FRDA presents more uniformly than spinocerebellar ataxias (SCA) of adulthood, with few extrapyramidal or cognitive features early in the course of FRDA.10 In addition, SCAs lack direct extra neurological features, making FRDA fundamentally different in its clinical management.1–5
 |
Table 1 Comparison of FRDA and Some Other Pediatric Onset Neurological Disorders |
Perhaps the most unique aspect of FRDA is the age of patients. Individuals with FRDA typically present after 5 years of age, although a few have presented earlier, and many families retrospectively identify symptoms earlier in life.1–5 In addition, although 80–90% of patients present by age 20y, some individuals have presented as late as their late 60’s.11,12 With a mean age of death at 37y (about 25 years or more after presentation even in the youngest presenting individuals), care of FRDA demands specialists from the pediatric and adult domains.13 In addition, this diverse age range covers many of the transitions of life: from childhood to adolescence, and adolescence to adult. Patients go from lower school to upper school to college to employment and finally retirement, requiring unique medical care through all those phases. The medical features of FRDA reflect this diversity: we increasingly appreciate that the developmental aspects of FRDA impact the early aspects of life, and the similarities of FRDA to accelerated aging affect the degenerative components.14–16
The diverse age range of FRDA patients renders interpretation of clinical data difficult. At times physical examination or laboratory markers (particularly related to the heart) become abnormal early but then pseudo-normalize later. Consequently, competent reading of the FRDA literature requires that one pay close attention to the age range of subjects included. Findings from early in the disease may not generalize to older individuals and vice versa.
After the identification of the causative mutations in 1996, the pace of research in FRDA has accelerated dramatically.17–19 As FRDA enters a new era of therapeutic development, clinical care must also advance in a manner covering all of these issues. In this manuscript we describe the individual components of care in FRDA, followed by a discussion of multidisciplinary interactions and how they may be managed.
Genetics and Diagnosis of FRDA
FRDA is an autosomal recessive disease resulting only from mutations in the FXN gene, all of which lead to decreased levels of functional frataxin protein. In 96% of patients, both mutations are expanded GAA repeats in the first intron of the gene; such mutations decrease transcription of the mRNA for frataxin to about 10% of normal levels. In 4% of patients, the second mutation is a point mutation or deletion, all of which lead to lack of functional frataxin by abnormal protein folding, premature truncation, loss of biological activity, or other methods.17–22 Frataxin maintains the iron sulfur clusters of enzymes involved in oxidative phosphorylation, the Krebs cycle, and other cellular processes.23–26 Consequently, FRDA has many features of mitochondrial diseases at a molecular level.
Many drugs are in development for FRDA, some of which are in late-stage trials and are likely to slow progression.1–5 Consequently, rapid identification of patients through genetic testing is a crucial part of care. As a recessive disease, most patients have no family history of FRDA and until therapies are developed, there is no role for widespread screening. Instead, diagnosis is typically made after symptoms develop by sub-specialists, usually pediatric neurologists, cardiologists or orthopedic surgeons rather than primary care physicians, genetic counselors or genetic specialists. The diagnosis of FRDA is confirmed by a positive genetic test for biallelic mutations in an appropriate clinical context (ataxia, cardiomyopathy, scoliosis, and/or rarely diabetes). Targeted testing directly assesses the length of the GAA repeat in FXN. This test alone will be diagnostic in 96% of patients with FRDA; 4% of patients will require FXN sequencing, identifying one expanded allele and one normal allele by GAA repeat sizing. Those with one abnormality can have the second mutation identified by follow up DNA sequencing. Only a single person has been found with 2 point mutations and no expanded alleles; thus, GAA expansion testing is the first step in genetic evaluation.27 In planning genetic testing, it is crucial to recognize that whole exome (WES) or next generation sequencing do not identify expansions; thus, most WES testing is normal in patients with FRDA. Consequently, GAA repeat testing is always the correct starting point in any patient in whom FRDA is a realistic possibility.
Specific Care Aspects
Neurological Features and Treatment
All patients with FRDA have some degree of neurological involvement, but the exact systems affected may vary between patients. Still, the phenotypic variability in neurological features of FRDA is more uniform than most other ataxias. Three major neurological systems degenerate in FRDA.28–34
1: Large fiber sensory systems (mainly proprioception), leading to loss of sensation and deep tendon reflexes, but more importantly creating a vision-dependent component to the ataxia in early disease.
2. Cerebellar coordination systems (mainly reflecting loss of cerebellar outflow neurons), leading to ataxia, loss of arm coordination, dysarthria.
3. Corticospinal motor systems, leading to lower extremity weakness, spasticity (only occasionally with increased reflexes)
Most textbooks cite the loss of proprioception as the most important neuroanatomical abnormality of FRDA. However, present views suggest that loss of proprioceptive pathways and deep tendon reflexes occurs early, probably before clinical presentation.14–16 Its severity also decreases as age of onset increases, such that proprioceptive loss may not be present in older onset patients, but such patients have similarly progressive ataxia. Thus, the importance of proprioceptive loss to progression in FRDA is limited although it clearly exacerbates the cerebellar ataxia early in the disease. Instead, the major sites of active progression in clinical FRDA likely are the dentate nucleus of the cerebellum (the output nucleus) and the corticospinal tract. Their loss gives rise to the progressive ataxia and later spasticity.
A few other locations are affected in FRDA over time: skeletal muscle (metabolic rather than degenerative), small sensory fibers (giving rise to neuropathic pain), auditory nerve and cochlear nucleus (hearing loss, without loss of primary tone hearing), autonomic fibers to the bladder (leading to urinary urgency), and retinal ganglion cells (causing optic neuropathy and visual loss).35–39 In contrast, cognitive involvement is an incompletely characterized aspect of FRDA at the clinical level, whose neuroanatomical substrate involves cerebellar features and forebrain aspects.40–42
There is no disease modifying therapy for FRDA at present, though many are being tested. Similarly, there are no symptomatic treatments for weakness and ataxia. Several of the clinical symptoms such as spasticity (baclofen, tizanidine, Botox), and neuropathic pain (gabapentin, pregabalin) have selective pharmacological treatments. Urinary urgency reflecting neurological issues affecting the bladder can be controlled with anticholinergic agents (oxybutynin). Formal urological evaluation is usually not necessary unless a person gives a history of true incontinence (which is uncommon in FRDA).43 Visual and hearing loss deserve evaluation for non-FRDA causes that potentiate FRDA-related issues. This usually involves referral to ophthalmology or audiology.
The major approaches for treatment also include rehabilitative services: physical therapy/occupational therapy/speech therapy. Their roles are discussed further below.
Cardiac Features
Sixty percent of individuals with FRDA die of cardiac disease, the main reason for premature mortality.13 Almost all individuals with FRDA have some degree of cardiac abnormality based on electrocardiogram or echocardiography.44–47 However, meaningful cardiac disease is almost always limited to those with onset before age 18y, who have more severe genetic abnormalities.44–47 In general, as neurological dysfunction and cardiac disease are both linked to the length of the GAA repeat, early onset individuals have both significant cardiac disease and severe neurological features. However, individuals with relatively more cardiac dysfunction or relative cardiac sparing do exist. In later onset individuals, ECG or echocardiographic abnormalities appear to change minimally, or possibly their evolution gets lost in the neurological dysfunction or other age dependent events (such as increasing amounts of non-FRDA cardiac dysfunction after age 50y).
Cardiac disease in FRDA is reasonably well defined pathologically, but the clinical manifestations are sometimes harder to decipher. The earliest features of cardiac disease reflect cardiac hypertrophy (usually concentric) with later evolution to fibrosis and myocyte dropout.48,49 From a structural point of view, cardiac imaging (echocardiography or cardiac magnetic resonance imaging) reveals early left ventricular hypertrophy along with decreases ventricular volumes and preserved systolic function; later in disease, wall thicknesses decrease, ventricular volumes increase, and systolic function falls.47–50 Thus, the overall pathophysiological course is often described as an early hypertrophic cardiomyopathy phenotype with presumed diastolic dysfunction that variably progresses to a later dilated cardiomyopathy phase with systolic dysfunction and classical heart failure.
This simplistic approach belies the complexity of the situation. While patients with early onset FRDA almost always have some degree of hypertrophy, it is usually not severe and is not commonly accompanied by demonstrable outflow tract obstruction. The neurologic disability limits the exercise abilities of FRDA patients, making it harder to demonstrate any direct effect of structural hypertrophy on daily activities; instead, the diastolic dysfunction becomes apparent only during times of volume overload (eg, from rapid IV fluid replacement) or volume depletion (eg, from gastrointestinal illness). In addition, the relative levels of hypertrophy and eventual change to fibrosis (if it is destined to occur) do not clearly correlate at a quantitative level. The evolution from hypertrophy to fibrosis occurs over the course of several decades in many individuals. Consequently, this process is poorly documented and bridges the gap from pediatric to adult cardiologist oversight. Such features all complicate the understanding of the course of cardiomyopathy in FRDA and its clinical features.
The management of FA-related cardiomyopathy is based on the generic management guidelines for patients with cardiomyopathy.51–55 There are currently no FRDA-specific medications to prevent or treat cardiac disease progression. Likewise, there are no specific medications indicated for hypertrophic cardiomyopathy in the absence of symptomatic outflow tract obstruction. However, when systolic dysfunction is present, medications such as angiotensin converting enzyme inhibitors, beta-blockers, and aldosterone antagonists may be indicated. Newer agents, such as SGLT2 inhibitors and combination angiotensin receptor blocker-neprilysin inhibitor therapy may also be considered. When fluid overload is present, diuretics may be warranted. For arrhythmia management, careful consideration for appropriate antithrombotic measures as well as medications and interventions to manage rate and/or rhythm should be made, most often with an electrophysiologist.
Heart failure symptoms may be subtle and difficult to distinguish from nonspecific symptoms of non-cardiac etiology in FRDA, especially when there are neuromuscular limitations to activity and physical exertion. At any stage of disease, however, it is important to recognize the potential for cardiac decompensation with stressors such as surgery and illness. Pre-operative cardiac evaluation and careful monitoring for the emergence of systolic or diastolic dysfunction as well as arrhythmias may be particularly helpful for multidisciplinary teams managing an FRDA patient before, during, and after sedated procedures. For those FRDA patients who progress to overt heart failure, their candidacy for advanced heart failure therapies should be carefully considered in the context of their individual well-being and non-cardiac quality of life.54,56 There are multiple published reports of individuals with FRDA who have undergone advanced heart failure therapies such as ventricular assist device implantation and heart transplantation with positive outcomes.56–62
Apart from overt heart failure, FRDA patients can have several other cardiac symptoms. Many develop chest pain sounding like classical angina, though not always linked to exertion. It is usually not associated with changes in ECG or troponin levels, though troponin levels are confounded by elevated troponin values at baseline.46 Anecdotally, we have seen individuals in whom this chest pain has responded to quinone containing neutriceutical (CoQ, idebenone), beta-blockers, calcium channel blockers, or nitrates. While early-onset coronary artery disease does not appear to be a common feature of FRDA, medications such as beta-blockers, calcium channel blockers, or nitrates may alleviate symptoms, presumably by decreasing myocardial oxygen demand or increasing microvascular blood supply to the hypertrophied heart.
Another prominent though understudied feature of cardiomyopathy in FRDA is the tendency to arrhythmias.63 Atrial arrhythmias are more common in FRDA, sometimes without correlation to structurally disease.63 Ventricular arrhythmias can occur, particularly late as fibrosis and end stage disease develop. In properly selected candidates, electrophysiological ablation and internal cardioverter-defibrillator implantation may be considered.54,64–66 As noted above, both medical and procedural management of atrial and ventricular arrhythmias in FRDA should include consultation with an electrophysiology specialist, as well as a multidisciplinary care team attuned to careful fluid and hemodynamic management when sedated procedures are pursued.
Orthopedic Conditions- Scoliosis and Pes Cavus
Scoliosis is found in a majority of FRDA patients while pes cavus is frequent but not always found. Scoliosis appears early (usually by time of presentation), progresses with growth during adolescent years, and tends not to progress after that time, thus mirroring idiopathic scoliosis.68–73 However, it has a more troublesome course than idiopathic scoliosis, progressing to surgery in a large number of the earliest onset patients. While its presentation is variable, it is more likely to have unusual aspects (such as left curve) than idiopathic scoliosis. Severe scoliosis is most common in early onset disease in subjects with long GAA repeat length, but some individuals present with scoliosis in teenage years while showing no ataxia until much later; this contrasts to most other neuromuscular conditions where scoliosis typically occurs once individuals have significant motor impairment, and often with the most rapid progression after they are non-ambulatory. In large natural history studies, scoliosis is a more common presenting symptom than overt cardiac disease, reflecting these individuals presenting with apparently isolated scoliosis. The high prevalence of scoliosis in FRDA may result from the high incidence of idiopathic scoliosis in the general population, which when combined with FRDA, may be more progressive.
The management of scoliosis in FRDA is similar to that in the general population: physical therapy to build core strength, bracing to stabilize the spine during growth, and should those failure to ameliorate progression, surgical intervention when Cobb angles reach high numbers (roughly greater than 45 degrees), though timing of surgery is individualized. Without such intervention, secondary neurological complications (pain, loss of stability) may occur along with eventual cardiopulmonary compromise and restrictive lung disease.
While surgical techniques are beyond the scope of this review, there are several aspects of scoliosis surgery that require multidisciplinary care. Timing of surgery to growth is crucial; one goal is to perform surgery after an individual has reached skeletal maturation, as assessed by bone age x-ray. Monitoring of surgery is also complicated as somatosensory evoked potential- (SSEP) based monitoring is not useful; patients with FRDA have lost such potentials before presentation.74 Finally, the prolonged anesthesia time without oral intake makes monitoring of fluid status and understanding of baseline cardiomyopathy important. These considerations all establish the need for an integrated anesthesia team.
Diabetes and Growth-Related Issues
Diabetes in FRDA is a highly variable and age dependent feature in FRDA. Estimates of the incidence of diabetes in FRDA range from 10% to 65% (if one also includes abnormal glucose tolerance).28,75–77 However, its driving pathogenic mechanism and prevalence vary with age. Individuals with early onset FRDA and diabetes can present similar to individuals with autoimmune “Type 1” diabetes, with diabetic ketoacidosis and low insulin and c-peptide levels, and usually require insulin as soon as DM is diagnosed.78 The main pathogenic factor of diabetes in early-onset FRDA-related diabetes thus appears to be pancreatic β cell insulin deficiency.79. In contrast, later in life, peripheral insulin resistance also contributes to development of FRDA-related diabetes.75,80,81 Diabetes development is more insidious and can be related to advancing age and increasing adiposity in individuals with later onset FRDA. However, pancreatic β cell insulin deficiency is still a prominent feature, with diabetes occurring at younger ages and at lower levels of adiposity than in common “Type 2” diabetes.78,80,82 Also, a disproportionate share of individuals with later onset FRDA and diabetes will require insulin.77
Although there is relatively little evidence to guide screening strategies for FRDA-related diabetes, at minimum individuals with FRDA should be educated about symptoms of diabetes and be screened at least once annually with hemoglobin A1c (HbA1c) and fasting blood glucose. Of note, screening with HbA1c alone may miss individuals with acute-onset diabetes. Usual consensus diagnostic criteria for diabetes should be applied for FRDA-related diabetes.82 Oral glucose tolerance testing and/or serial blood glucose measurements can be considered in those whose initial screening results are equivocal.
For those diagnosed with FRDA-related diabetes, we recommend self-monitoring blood glucose using a glucometer and/or continuous glucose monitoring (CGM). CGM is particularly useful for individuals who are at risk for hypoglycemia related to insulin or secretagogues and/or for those for whom manual dexterity limits the ability to independently measure glucose by fingerstick. Given the risk of ketoacidosis, individuals should also have the ability to test for ketones. We recommend using blood instead of urine ketone measurements, both for ease of use and for improved accuracy in the setting of potential secondary mitochondrial deficits in ketone oxidation, where urinary acetoacetate may underestimate the degree of ketosis relative to blood beta-hydroxybutyrate.83
With respect to therapies, insulin remains the cornerstone of therapy for individuals with clinical evidence of significant insulin deficiency (eg, ketoacidosis, hyperglycemia with weight loss), especially in children.84 There may be a role for other glucose-lowering therapies with careful consideration of individualized risk-benefit assessment in light of comorbidities. In contrast to other forms of diabetes related to mitochondrial impairment, most individuals with FRDA do not have a personal history of lactic acidosis, so metformin is not strictly contraindicated and may be helpful, though its use should be monitored closely, and as with all individuals, discontinued with illness.85 Novel glucose lowering medications may have a role not only in the management of FRDA-related diabetes, but also for other problems, such as glucagon-like peptide 1 receptor (GLP1R) agonists for neurologic and/or cardiac benefits, eg, exenatide and sodium-glucose co-transporter 2 (SGLT2) inhibitors for heart failure with or without diabetes.67,86 These exciting potential benefits warrant additional study in FRDA, with attention to risk for associated adverse effects. Particularly relevant for individuals with FRDA is the increased incidence of euglycemic diabetic ketoacidosis with SGLT2 inhibitor use.87 Finally, in FRDA related diabetes, glycemic targets should be individualized, and screening for complications as in other forms of diabetes, although with attention to the overlap with complications of FRDA itself.
Linear growth and weight gain should be monitored closely in children with FRDA, as in any child with a chronic, multi-system disorder. Specifically, in FRDA, scoliosis is expected to impact height measurements, in particular during and after the pubertal growth spurt. Also, more severe cardiac disease could also adversely impact growth, as in other pediatric heart disorders.88 Many children with FRDA have low body mass index (BMI) when commonly used thresholds for assessing nutritional status are applied.89 However, it is not clear the extent to which BMI truly reflects body composition in FRDA, and whether more nutrition would produce clinical benefit as low BMI values most likely do not directly lead to neurological or cardiac dysfunction. Indeed, in adults with FRDA, BMI may underestimate the degree of excess visceral adiposity that predisposes to diabetes.81 In both children and adults, height and weight are frequently not recorded at clinical visits, likely due to difficulties obtaining measurements in individuals with limited mobility.28 We recommend that increasing attention be paid to making these measurements, and using alternate assessments (eg, recumbent length, arm span, or tibial length) be considered where needed.
Children and adults with FRDA also have multiple risk factors for impaired bone health. In children, both chronic disease and decreased mobility may impact acquisition of peak bone mass, and thus predispose to development of adult osteoporosis.90 In one small study of adults with FRDA, nearly 20% had low areal bone mineral density as assessed by dual energy x-ray absorptiometry (DXA) scanning.91 Mobility impairment in combination with low bone density may be expected to increase the risk for falls and associated fractures, though this phenomenon has not been studied in detail in FRDA. In the absence of FRDA-specific evidence, we recommend optimization of calcium and vitamin D status in all individuals with FRDA, and individualized consideration of screening using DXA and treatment with anti-osteoporosis medications according to general age-specific consensus guidelines.92–96 Also, we recommend counseling individuals regarding risk for occult fracture and that physical therapy prescriptions take into account the potential for bone fragility.
Mental Health in FRDA
While not well characterized, depression is a component of FRDA. Many neurological case series have documented depression, but few psychiatric studies have provided a better-characterized discussion.97–105 Thus, it is unclear if this is a disease-specific pattern or if this is part of the reaction to the diagnosis and its challenges. Having a long-term condition in itself a cause of psychological and adaptive issues in children and young adults. Ideally, multidisciplinary care team possesses a trained mental health professional who is adept at counseling patients on adapting to the evolving features of their disorder. This becomes particularly difficult in FRDA considering that the patients span a large age range; the psychological issues of children do not match those of adults who are diagnosed after age 25.
In some situations, psychiatric intervention may be necessary. From a pharmacological perspective, both classical medications and electroconvulsive therapy have been used to treat depression in FRDA.
Genetic Counseling
While specialized genetic counseling questions can arise in FRDA, most genetic counseling questions are straightforward. There are few (if any) new mutations, so parents are obligate carriers. Siblings have a 1 in 4 chance of being affected, and, if unaffected, a 2 in 3 chance of being a carrier. While GAA repeat length correlates with age of onset, the correlation is not sufficiently precise that one can use it to predictively test age of onset in patients. Other relatives seeking to investigate carrier status can be easily be assessed through commercial testing. Prenatal testing has been done but predicting exact phenotype based on GAA repeat length is not readily possible.106
Rare Associations and Treatments with FRDA
The presence of large natural history studies has facilitated the search for uncommon associations that might be parts of FRDA. Ferritin levels in FRDA tend to be low, suggestive of systemic iron mishandling, while heterozygosity for hemochromatosis may worsen the severity of FRDA.107–109 The effect of iron supplementation in FRDA remains somewhat controversial. While FRDA involves iron overload in mitochondria, cells behave as if they are cytosolically depleted of iron. Consequently, iron supplementation may be with beneficial or harmful depending on the context. In some situations, individuals with FRDA have worsened with iron administration, and iron chelation itself with deferiprone decreases cardiac hypertrophy but is associated with adverse events at higher doses.110–112 Such a complex relationship with systemic iron levels suggests that any iron supplementation in FRDA should judicious, based on measures like hemoglobin levels rather than ferritin or blood iron levels.
Within the FACOMS cohort in the United States, growth hormone deficiency and inflammatory bowel disease appear more common than expected among FRDA patients.113 In addition, case reports suggest that nephrotic syndrome and Graves’ disease may be associated at other times.113–116 Cancer does not seem to be an association of FRDA.117,118 Whether these represent true biological associations will be revealed over time.
In addition, such large natural history studies also identify individuals with FRDA who have tolerated medical treatment for other disorders. The multisystem nature of FRDA might lead some to exclude patients from high-risk therapies for coincidental disorders. However, this need not necessarily be in the best interest of the patient. Individuals with FRDA have undergone liver transplantation and bone marrow transplantation without ill effects, and in contrast to CMT, no form of chemotherapy has yet been shown to worsen FRDA.119–121 Similarly, at least one individual with FRDA has tolerated doxorubicin, a potentially cardiotoxic chemotherapeutic agent, in treatment of osteogenic sarcoma without intermediate-term cardiac decompensation.117 Furthermore, pregnancy in FRDA does not have an increased number of risks compared to the general population.122 Thus, the presence of FRDA does not rule out a bold approach to treatment of other disorders.
Rehabilitation in FRDA
One area of medicine involving all of the aspects of FRDA is rehabilitation. While studies suggest that systematic rehabilitation is beneficial in FRDA, the details are not yet well developed.123–132 Physical therapy and occupation therapy are beneficial for restoration during acute events as well as possibly to ameliorate long-term changes. Speech therapy is also useful in the context of eventual speech dysfunction and swallowing difficulty. In addition, exercise is anecdotally extremely beneficial in FRDA, perhaps reflecting underlying stimulation of mitophagy. Still there have been few systematic studies directed to the type of rehabilitative therapies that are most appropriate. In addition, integrative rehabilitation is best performed local to a person’s place of residence, and not commonly performed at distant tertiary care sites. As a result, it is not currently well integrated into the multidisciplinary care of many FRDA patients.
Coordination of Multidisciplinary Care
So, who is in charge of the multidisciplinary approach? Ideally a single physician such as a primary care practitioner would act as the organizer and triage physician for chronic and acute medical care in FRDA. Unfortunately, the advancement in understanding of FRDA and its care make management of FRDA the domain of tertiary care specialists, even at the triage level. Indeed, even among tertiary care centers, centers specialized for FRDA are found in only 10–20 locations in the United States. Such locations follow >15 people with FRDA, which is still a modest number. Consequently, the global knowledge needed to manage the complete disease is localized. In such centers, the neurologist usually takes a dominant role because neurological progression invariably occurs each year in every patient, distinct from the cardiomyopathy and endocrinopathy that are relegated to subgroups of individuals. In addition, they are most commonly the ones to make the diagnosis initially. Clinical trials at present are almost always are based on neurological function, with loss of neurological abilities being the primary outcome measure. Cardiac measures remain either secondary efficacy measures or more commonly safety measures. Thus, multidisciplinary care tends to be organized around neurological programs for FRDA with close communication to the other subspecialists.
Still, excellent communication between specialists provides the essential element of high quality multidisciplinary care. Traditional mechanisms of communication work best in typical paradigms in which all the specialists are located in one institution. In FRDA, such specialists frequently employ both pediatric and adult subspecialists, often across multiple health care systems. In addition, the great distance patients travel for FRDA-centered care center can further complicate communication.133 Three major advancements in medical care help overcome such limitations. First, the creation of electronic medical records with sharing capability allows local specialists to communicate readily with those at FRDA centers. Secondly, the use of telemedicine and video visits may at some point allow better integration with local specialists. While COVID has decreased the comfort of patients in traveling to FRDA centers, and the need for social distancing in clinical settings has led to a decrease in the number of patients who can be seen, the pandemic has also amplified the development and need for mechanisms which enable remote care delivery. The final change in medical care is the increased emphasis on transition from the pediatric to the adult medical world. Models exist in many diseases for this process, and health care professionals are now trained to address the evolving needs of this period.
Clinical Trials
With no disease-modifying clinical therapies presently available for FRDA, may discussions in FRDA coalesce around clinical trials of new agents (Table 2). Such agents uniformly attempt to treat the pathophysiology of FRDA by improving mitochondrial function or by increasing expression of frataxin. Ideally, these therapies would be effective against all aspects FRDA pathology. However, as most are being directly tested only for slowing the neurological progression, there is no assurance of this. Understanding whether new therapies act on all components of the disease or simply the neurological aspect will require multidisciplinary studies, most likely as post marketing protocols. Thus the need for integrated care in FRDA will continue to expand.
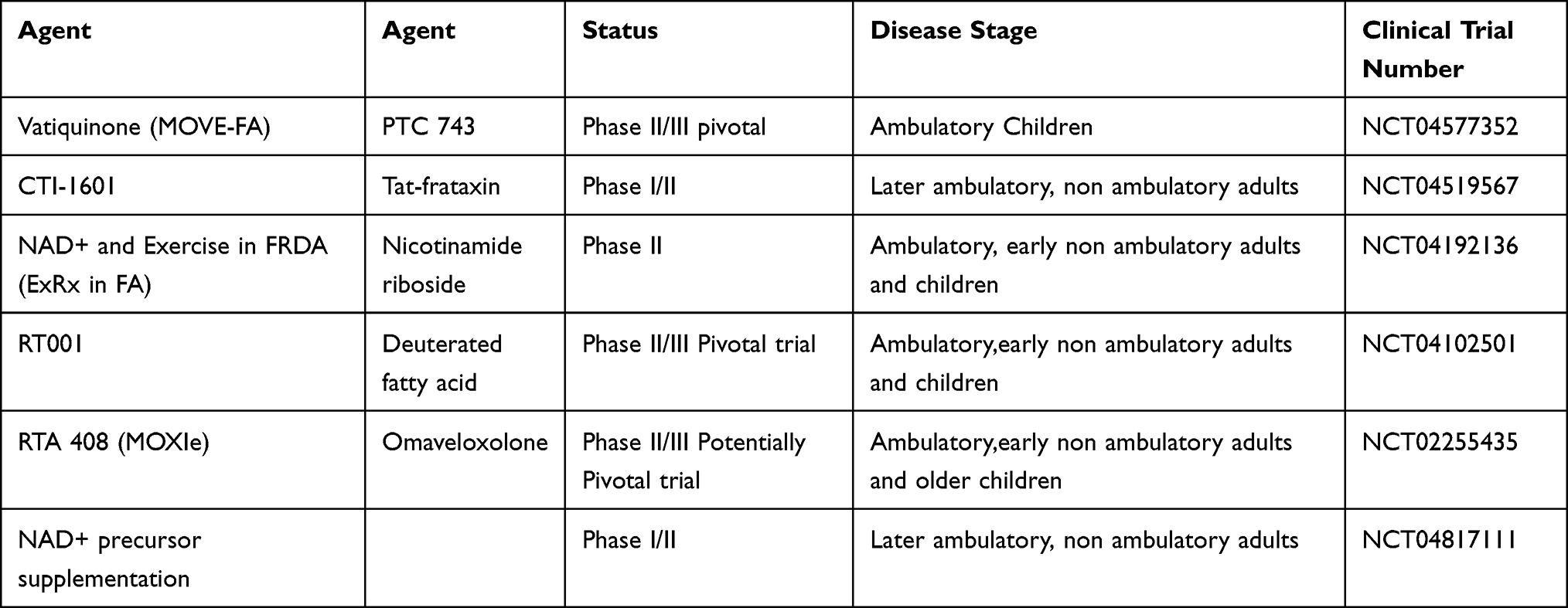 |
Table 2 Active Clinical Trials in FRDA |
Abbreviations
FRDA, Friedreich Ataxia; GAA, Guanine adenine, adenine; SMA, spinal muscular atrophy; DMD, Duchenne’s muscular dystrophy; SCA, spinocerebellar atrophy; WES, whole exome sequencing; CMT, Charcot Marie Tooth; DNA, deoxyribonucleic acid; CGM, continuous glucose monitoring; DXA, Dual-energy X-ray absorptiometry; SGLT2, Sodium glucose co-transporter 2; FXN, frataxin gene; GLP1R, Glucagon Like Peptide 1 Receptor; BMI, body mass index; HbA1C- hemoglobin A1C; BG, blood glucose; OGTT, oral glucose tolerance test.
Acknowledgments
We would like to acknowledgement the contribution of Dr. Jacqueline Tamaroff in preparation of Figure 1.
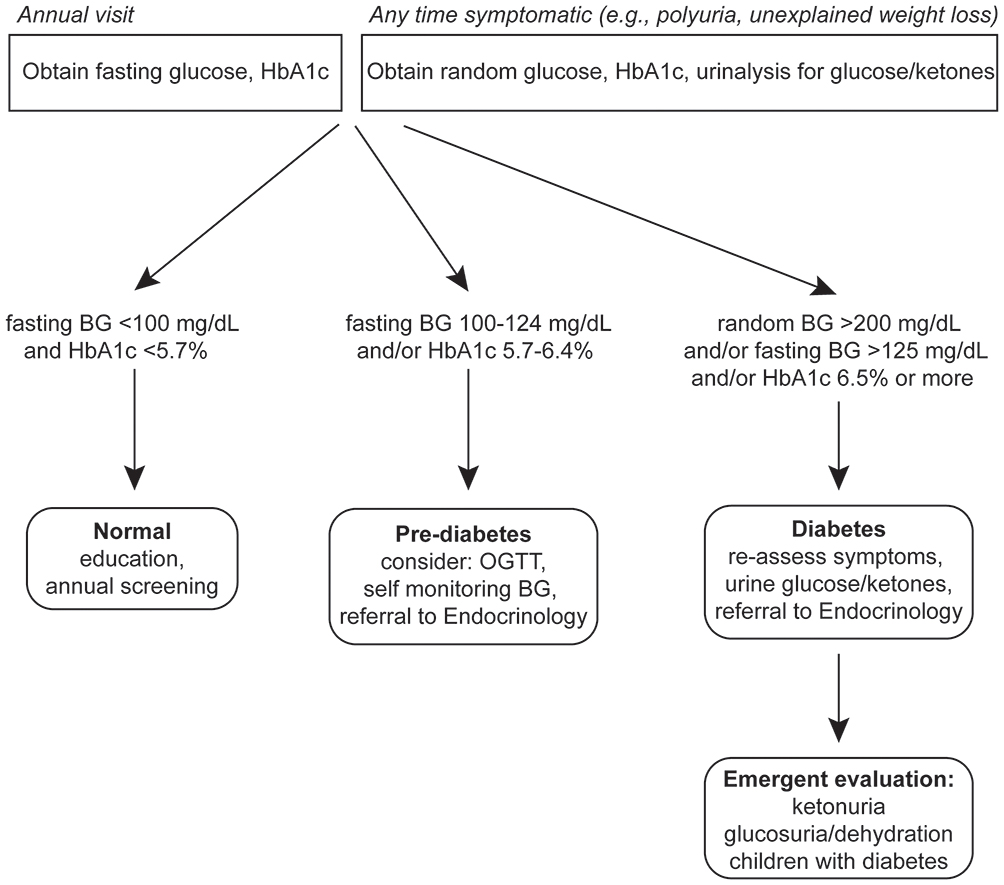 |
Figure 1 Scheme for diagnosis of diabetes in FRDA. Abbreviations: HbA1C, hemoglobin A1C; BG, blood glucose; OGTT, oral glucose tolerance test. |
Disclosure
The authors have no conflicts of interest with the present work. Dr Lynch receives grant support from the NIH, MDA, FDA, Friedreich Ataxia Research Alliance, Reata Pharmaceuticals, PTC Therapeutics and Retrotope Pharmaceuticals. Dr Lin receives grant money from Friedreich Ataxia Research Alliance. Dr McCormack receives grant support from the NIH, Friedreich Ataxia Research Alliance, Chromatex and Metro-Biotech. Dr Kichula serves on advisory boards for Sarepta, PTC Therapeutics, Novartis, Biogen and Roche and a speaker’s bureau for Roche.
References
1. Clay A, Hearle P, Schadt K, Lynch DR. New developments in pharmacotherapy for Friedreich ataxia. Expert Opin Pharmacother. 2019;20(15):1855–1867. doi:10.1080/14656566.2019.1639671
2. Strawser C, Schadt K, Hauser L, et al. Pharmacological therapeutics in Friedreich ataxia: the present state. Expert Rev Neurother. 2017;17(9):895–907. doi:10.1080/14737175.2017.1356721
3. Strawser CJ, Schadt KA, Lynch DR. Therapeutic approaches for the treatment of Friedreich’s ataxia. Expert Rev Neurother. 2014;14(8):949–957. doi:10.1586/14737175.2014.939173
4. Pandolfo M. Friedreich ataxia: the clinical picture. J Neurol. 2009;256(Suppl 1):3–8. doi:10.1007/s00415-009-1002-3
5. Pandolfo M. Friedreich ataxia. Handb Clin Neurol. 2012;103:275–294.
6. Chua K, Tan CY, Chen Z, et al. Long-term follow-up of pulmonary function and scoliosis in patients with duchenne’s muscular dystrophy and spinal muscular atrophy. J Pediatr Orthop. 2016;36(1):63–69. doi:10.1097/BPO.0000000000000396
7. Bureau MA, Ngassam P, Lemieux B, Trias A. Pulmonary function studies in Friedreich’s ataxia. Can J Neurol Sci. 1976;3(4):343–347. doi:10.1017/S0317167100025579
8. Mercuri E, Finkel RS, Munotni F, et al. Diagnosis and management of Spinal muscular atrophy: part 1: recommendations for diagnosis, rehabilitation, orthopaedic and nutritional care. Neuromuscular Disord. 2018;28:103–115. doi:10.1016/j.nmd.2017.11.005
9. Birnkrant DJ, Bushby K, Bann CM, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol. 2018;17(3):251–267. doi:10.1016/S1474-4422(18)30024-3
10. Kuo SH. Ataxia. Continuum (Minneap Minn). 2019;25(4):1036–1054. doi:10.1212/CON.0000000000000753
11. Stolle CA, Frackelton EC, McCallum J, et al. Novel, complex interruptions of the GAA repeat in small, expanded alleles of two affected siblings with late-onset Friedreich ataxia. Mov Disord. 2008;23(9):1303–1306. doi:10.1002/mds.22012
12. McDaniel DO, Keats B, Vedanarayanan VV, Subramony SH. Sequence variation in GAA repeat expansions may cause differential phenotype display in Friedreich’s ataxia. Mov Disord. 2001;16(6):1153–1158. doi:10.1002/mds.1210
13. Tsou AY, Paulsen EK, Lagedrost SJ, et al. Mortality in Friedreich ataxia. J Neurol Sci. 2011;307(1–2):46–49. doi:10.1016/j.jns.2011.05.023
14. Marty B, Naeije G, Bourguignon M, et al. Evidence for genetically determined degeneration of proprioceptive tracts in Friedreich ataxia. Neurology. 2019;93(2):e116–e124. doi:10.1212/WNL.0000000000007750
15. Lynch DR, Deutsch EC, Wilson RB, Tennekoon G. Unanswered questions in Friedreich ataxia. J Child Neurol. 2012;27(9):1223–1229. doi:10.1177/0883073812453498
16. Naeije G, Bourguignon M, Wens V, et al. Electrophysiological evidence for limited progression of the proprioceptive impairment in Friedreich ataxia. Clin Neurophysiol. 2020;131(2):574–576. doi:10.1016/j.clinph.2019.10.021
17. Babady NE, Carelle N, Wells RD, et al. Advancements in the pathophysiology of Friedreich’s ataxia and new prospects for treatments. Mol Genet Metab. 2007;92(1–2):23–35. doi:10.1016/j.ymgme.2007.05.009
18. Campuzano V, Montermini L, Moltò MD, et al. Friedreich’s ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science. 1996;271(5254):1423–1427. doi:10.1126/science.271.5254.1423
19. Dürr A, Cossee M, Agid Y, et al. Clinical and genetic abnormalities in patients with Friedreich’s ataxia. N Engl J Med. 1996;335(16):1169–1175. doi:10.1056/NEJM199610173351601
20. Galea CA, Huq A, Lockhart PJ, et al. Compound heterozygous FXN mutations and clinical outcome in Friedreich ataxia. Ann Neurol. 2016;79(3):485–495. doi:10.1002/ana.24595
21. Cossée M, Dürr A, Schmitt M, et al. Friedreich’s ataxia: point mutations and clinical presentation of compound heterozygotes. Ann Neurol. 1999;45(2):200–206. doi:10.1002/1531-8249(199902)45:2<200::AID-ANA10>3.0.CO;2-U
22. Anheim M, Mariani LL, Calvas P, et al. Exonic deletions of FXN and early-onset Friedreich ataxia. Arch Neurol. 2012;69(7):912–916. doi:10.1001/archneurol.2011.834
23. Puccio H, Koenig M. Recent advances in the molecular pathogenesis of Friedreich ataxia. Hum Mol Genet. 2000;9(6):887–892. doi:10.1093/hmg/9.6.887
24. Rodden LN, Chutake YK, Gilliam K, et al. Methylated and unmethylated epialleles support variegated epigenetic silencing in Friedreich ataxia. Hum Mol Genet. 2021;29(23):3818–3829. doi:10.1093/hmg/ddaa267
25. Lazaropoulos M, Dong Y, Clark E, et al. Frataxin levels in peripheral tissue in Friedreich ataxia. Ann Clin Transl Neurol. 2015;2(8):831–842. doi:10.1002/acn3.225
26. Campuzano V, Montermini L, Lutz Y, et al. Frataxin is reduced in Friedreich ataxia patients and is associated with mitochondrial membranes. Hum Mol Genet. 1997;6(11):1771–1780. doi:10.1093/hmg/6.11.1771
27. Candayan A, Yunisova G, Çakar A, et al. The first biallelic missense mutation in the FXN gene in a consanguineous Turkish family with Charcot-Marie-Tooth-like phenotype. Neurogenetics. 2020;21(1):73–78. doi:10.1007/s10048-019-00594-1
28. Patel M, Isaacs CJ, Seyer L, et al. Progression of Friedreich ataxia: quantitative characterization over 5 years. Ann Clin Transl Neurol. 2016;3(9):684–694. doi:10.1002/acn3.332
29. Harding IH, Lynch DR, Koeppen AH, Pandolfo M. Central nervous system therapeutic targets in Friedreich ataxia. Hum Gene Ther. 2020;31(23–24):1226–1236. doi:10.1089/hum.2020.264
30. Koeppen AH, Becker AB, Qian J, Gelman BB, Mazurkiewicz JE. Friedreich ataxia: developmental failure of the dorsal root entry zone. J Neuropathol Exp Neurol. 2017;76(11):969–977. doi:10.1093/jnen/nlx087
31. Koeppen AH, Becker AB, Qian J, Feustel PJ. Friedreich ataxia: hypoplasia of spinal cord and dorsal root ganglia. J Neuropathol Exp Neurol. 2017;76(2):101–108. doi:10.1093/jnen/nlw111
32. Koeppen AH, Mazurkiewicz JE. Friedreich ataxia: neuropathology revised. J Neuropathol Exp Neurol. 2013;72(2):78–90. doi:10.1097/NEN.0b013e31827e5762
33. Koeppen AH, Davis AN, Morral JA. The cerebellar component of Friedreich’s ataxia. Acta Neuropathol. 2011;122(3):323–330. doi:10.1007/s00401-011-0844-9
34. Rummey C, Farmer JM, Lynch DR. Predictors of loss of ambulation in Friedreich’s ataxia. EClinicalMedicine. 2020;18:100213. doi:10.1016/j.eclinm.2019.11.006
35. DeBrosse C, Nanga RPR, Wilson N, et al. Muscle oxidative phosphorylation quantitation using creatine chemical exchange saturation transfer (CrCEST) MRI in mitochondrial disorders. JCI Insight. 2016;1(18):e88207. doi:10.1172/jci.insight.88207
36. Rance G, Fava R, Baldock H, et al. Speech perception ability in individuals with Friedreich ataxia. Brain. 2008;131(8):2002–2012. doi:10.1093/brain/awn104
37. Seyer LA, Galetta K, Wilson J, et al. Analysis of the visual system in Friedreich ataxia. J Neurol. 2013;260(9):2362–2369. doi:10.1007/s00415-013-6978-z
38. Afsharian P, Nolan-Kenney R, Lynch AE, Balcer LJ, Lynch DR. Correlation of visual quality of life with clinical and visual status in Friedreich ataxia. J Neuroophthalmol. 2020;40(2):213–217. doi:10.1097/WNO.0000000000000878
39. Hamedani AG, Hauser LA, Perlman S, et al. Longitudinal analysis of contrast acuity in Friedreich ataxia. Neurol Genet. 2018;4(4):e250. doi:10.1212/NXG.0000000000000250
40. Corben LA, Klopper F, Stagnitti M, et al. Measuring Inhibition and Cognitive Flexibility in Friedreich Ataxia. Cerebellum. 2017;16(4):757–763. doi:10.1007/s12311-017-0848-7
41. Hernández-Torres A, Montón F, Hess Medler S, de Nóbrega É, Nieto A. Longitudinal study of cognitive functioning in Friedreich’s ataxia. J Int Neuropsychol Soc. 2021;27(4):343–350. doi:10.1017/S1355617720000958
42. Naeije G, Rai M, Allaerts N, Sjogard M, De Tiège X, Pandolfo M. Cerebellar cognitive disorder parallels cerebellar motor symptoms in Friedreich ataxia. Ann Clin Transl Neurol. 2020;7(6):1050–1054. doi:10.1002/acn3.51079
43. Musegante AF, Almeida PN, Monteiro RT, Barroso U. Urinary symptoms and urodynamic findings in patients with Friedreich’s ataxia. Int Braz J Urol. 2013;39(6):867–874. doi:10.1590/S1677-5538.IBJU.2013.06.14
44. Lynch DR, Regner SR, Schadt KA, Friedman LS, Lin KY, St John Sutton MG. Management and therapy for cardiomyopathy in Friedreich’s ataxia. Expert Rev Cardiovasc Ther. 2012;10(6):767–777. doi:10.1586/erc.12.57
45. Schadt KA, Friedman LS, Regner SR, Mark GE, Lynch DR, Lin KY. Cross-sectional analysis of electrocardiograms in a large heterogeneous cohort of Friedreich ataxia subjects. J Child Neurol. 2012;27(9):1187–1192. doi:10.1177/0883073812448461
46. Friedman LS, Schadt KA, Regner SR, et al. Elevation of serum cardiac troponin I in a cross-sectional cohort of asymptomatic subjects with Friedreich ataxia. Int J Cardiol. 2013;167(4):1622–1624. doi:10.1016/j.ijcard.2012.04.159
47. Dedobbeleer C, Rai M, Donal E, Pandolfo M, Unger P. Normal left ventricular ejection fraction and mass but subclinical myocardial dysfunction in patients with Friedreich’s ataxia. Eur Heart J Cardiovasc Imaging. 2012;13(4):346–352. doi:10.1093/ejechocard/jer267
48. Isnard R, Kalotka H, Dürr A, et al. Correlation between left ventricular hypertrophy and GAA trinucleotide repeat length in Friedreich’s ataxia. Circulation. 1997;95(9):2247–2249. doi:10.1161/01.CIR.95.9.2247
49. Koeppen AH, Ramirez RL, Becker AB, et al. The pathogenesis of cardiomyopathy in Friedreich ataxia. PLoS One. 2015;10(3):e0116396. doi:10.1371/journal.pone.0116396
50. Pousset F, Legrand L, Monin ML, et al. A 22-year follow-up study of long-term cardiac outcome and predictors of survival in Friedreich ataxia. JAMA Neurol. 2015;72(11):1334–1341. doi:10.1001/jamaneurol.2015.1855
51. Writing Committee M; Yancy CW, Jessup M, Jessup M, et al. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation. 2013;128(16):e240–327. doi:10.1161/CIR.0b013e31829e8776.
52. Writing Committee M; Yancy CW, Jessup M, Jessup M, et al. 2016 ACC/AHA/HFSA focused update on new pharmacological therapy for heart failure: an update of the 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Failure Society of America. Circulation. 2016;134(13):e282–293. doi:10.1161/CIR.0000000000000435.
53. Yancy CW, Jessup M, Bozkurt B, et al. 2017 ACC/AHA/HFSA focused update of the 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Failure Society of America. Circulation. 2017;136(6):e137–e161. doi:10.1161/CIR.0000000000000509
54. Feingold B, Mahle WT, Auerbach S, et al. Management of cardiac involvement associated with neuromuscular diseases: a scientific statement from the American Heart Association. Circulation. 2017;136(13):e200–e231. doi:10.1161/CIR.0000000000000526
55. Ommen SR, Mital S, Burke MA, et al. 2020 AHA/ACC guideline for the diagnosis and treatment of patients with hypertrophic cardiomyopathy: executive summary: a report of the American College of Cardiology/American Heart Association Joint Committee on clinical practice guidelines. Circulation. 2020;142(25):e533–e557. doi:10.1161/CIR.0000000000000938
56. Ivak P, Zumrova A, Netuka I. Friedreich’s ataxia and advanced heart failure: an ethical conundrum in decision-making. J Heart Lung Transplant. 2016;35(9):1144–1145. doi:10.1016/j.healun.2016.06.021
57. Yoda M, El-Banayosy A, Arusoglu L, Tendrich G, Minami K, Korfer R. Permanent use of a ventricle assist device for dilated cardiomyopathy in Friedreich’s ataxia. J Heart Lung Transplant. 2006;25(2):251–252. doi:10.1016/j.healun.2005.04.020
58. McCormick A, Shinnick J, Schadt K, et al. Cardiac transplantation in Friedreich ataxia: extended follow-up. J Neurol Sci. 2017;375:471–473. doi:10.1016/j.jns.2017.01.027
59. Sedlak TL, Chandavimol M, Straatman L. Cardiac transplantation: a temporary solution for Friedreich’s ataxia-induced dilated cardiomyopathy. J Heart Lung Transplant. 2004;23(11):1304–1306. doi:10.1016/j.healun.2003.09.015
60. Leonard H, Forsyth R. Friedreich’s ataxia presenting after cardiac transplantation. Arch Dis Child. 2001;84(2):167–168. doi:10.1136/adc.84.2.167
61. Yoon G, Soman T, Wilson J, et al. Cardiac transplantation in Friedreich ataxia. J Child Neurol. 2012;27(9):1193–1196. doi:10.1177/0883073812448229
62. Segovia J, Alonso-Pulpon L, Burgos R, et al. Heart transplantation in Friedreich’s ataxia and other neuromuscular diseases. J Heart Lung Transplant. 2001;20(2):169. doi:10.1016/S1053-2498(00)00313-2
63. Mejia E, Lynch A, Hearle P, et al. Ectopic burden via holter monitors in Friedreich ataxia. Pediatr Neurol. 2021;117:29–33. doi:10.1016/j.pediatrneurol.2021.01.004
64. Coskun KO, Popov AF, Schmitto JD, et al. Feasibility of implantable cardioverter defibrillator treatment in five patients with familial Friedreich’s ataxia–a case series. Artif Organs. 2010;34(11):1061–1065. doi:10.1111/j.1525-1594.2010.01140.x
65. Zipse MM, Aleong RG. Acquired pulmonary vein isolation in a patient with Friedreich ataxia. Card Electrophysiol Clin. 2016;8(1):151–153. doi:10.1016/j.ccep.2015.10.016
66. Postigo A, Datino T, Espinosa MA, Fernandez-Aviles F, Arenal A. Epicardial ablation for ventricular tachycardia in Freidreich’s ataxia cardiomyopathy. Europace. 2019;21(7):1095. doi:10.1093/europace/euz030
67. Brown E, Wilding JP, Alam U, Barber TM, Karalliedde J, Cuthbertson DJ. The expanding role of SGLT2 inhibitors beyond glucose-lowering to cardiorenal protection. Ann Med. 2020;1–32. doi:10.1080/07853890.2020.1841281
68. Allard P, Dansereau J, Duhaime M, Geoffroy G. Scoliosis assessment in Friedreich’s ataxia by means of intrinsic parameters. Can J Neurol Sci. 1984;11(S4):582–587. doi:10.1017/S0317167100035095
69. Simon AL, Meyblum J, Roche B, et al. Scoliosis in patients with Friedreich ataxia: results of a consecutive prospective series. Spine Deform. 2019;7(5):812–821. doi:10.1016/j.jspd.2019.02.005
70. Agámez Medina GL, Pantin EJ, Lorthé J, Therrien PJ. Anaesthesia for correction of scoliosis in pediatric patient with Friedreich’s ataxia. Rev Esp Anestesiol Reanim. 2015;62(1):42–45. doi:10.1016/j.redar.2014.03.001
71. Corben LA, Lynch D, Pandolfo M, Schulz JB, Delatycki MB; Clinical Management Guidelines Writing Group. Consensus clinical management guidelines for Friedreich ataxia. Orphanet J Rare Dis. 2014;9:184. doi:10.1186/s13023-014-0184-7
72. Tsirikos AI, Smith G. Scoliosis in patients with Friedreich’s ataxia. J Bone Joint Surg Br. 2012;94(5):684–689. doi:10.1302/0301-620X.94B5.28391
73. Milbrandt TA, Kunes JR, Karol LA. Friedreich’s ataxia and scoliosis: the experience at two institutions. J Pediatr Orthop. 2008;28(2):234–238. doi:10.1097/BPO.0b013e318164fa79
74. Pelosi L, Lamb J, Grevitt M, Mehdian SM, Webb JK, Blumhardt LD. Combined monitoring of motor and somatosensory evoked potentials in orthopaedic spinal surgery. Clin Neurophysiol. 2002;113(7):1082–1091. doi:10.1016/S1388-2457(02)00027-5
75. Lynch DR, Willi SM, Wilson RB, et al. A0001 in Friedreich ataxia: biochemical characterization and effects in a clinical trial. Mov Disord. 2012;27(8):1026–1033. doi:10.1002/mds.25058
76. Cnop M, Mulder H, Igoillo-Esteve M. Diabetes in Friedreich ataxia. J Neurochem. 2013;126(Suppl 1):94–102. doi:10.1111/jnc.12216
77. McCormick A, Farmer J, Perlman S, et al. Impact of diabetes in the Friedreich ataxia clinical outcome measures study. Ann Clin Transl Neurol. 2017;4(9):622–631. doi:10.1002/acn3.439
78. Chakraborty PP, Ray S, Bhattacharjee R, et al. First presentation of diabetes as diabetic ketoacidosis in a case of Friedreich’s ataxia. Clin Diabetes. 2015;33(2):84–86. doi:10.2337/diaclin.33.2.84
79. Pappa A, Hausler MG, Veigel A, et al. Diabetes mellitus in Friedreich ataxia: a case series of 19 patients from the German-Austrian diabetes mellitus registry. Diabetes Res Clin Pract. 2018;141:229–236.
80. Igoillo-Esteve M, Gurgul-Convey E, Hu A, et al. Unveiling a common mechanism of apoptosis in beta-cells and neurons in Friedreich’s ataxia. Hum Mol Genet. 2015;24(8):2274–2286. doi:10.1093/hmg/ddu745
81. Cnop M, Igoillo-Esteve M, Rai M, et al. Central role and mechanisms of beta-cell dysfunction and death in friedreich ataxia-associated diabetes. Ann Neurol. 2012;72(6):971–982. doi:10.1002/ana.23698
82. American Diabetes Association. 2. Classification and diagnosis of diabetes: standards of medical care in diabetes-2021. Diabetes Care. 2021;44(Suppl 1):S15–S33. doi:10.2337/dc21-S002
83. Sharma R, Reinstadler B, Engelstad K, et al. Circulating markers of NADH-reductive stress correlate with mitochondrial disease severity. JCI. 2021;131(2). doi:10.1172/JCI136055.
84. American Diabetes Association. 9. Pharmacologic approaches to glycemic treatment: standards of medical care in diabetes-2021. Diabetes Care. 2021;44(Suppl 1):S111–S24. doi:10.2337/dc21-S009
85. Yeung RO, Al Jundi M, Gubbi S, et al. Management of mitochondrial diabetes in the era of novel therapies. J Diabetes Complications. 2020;107584.
86. Igoillo-Esteve M, Oliveira AF, Cosentino C, et al. Exenatide induces frataxin expression and improves mitochondrial function in Friedreich ataxia. JCI Insight. 2020;5(2):e134221. doi:10.1172/jci.insight.134221
87. Bamgboye AO, Oni IO, Collier A. Predisposing factors for the development of diabetic ketoacidosis with lower than anticipated glucose levels in type 2 diabetes patients on SGLT2-inhibitors: a review. Eur J Clin Pharmacol. 2020;77(5):651–657. doi:10.1007/s00228-020-03051-3
88. Wang T, Liu J, McDonald C, et al. GDF15 is a heart-derived hormone that regulates body growth. EMBO Mol Med. 2017;9(8):1150–1164. doi:10.15252/emmm.201707604
89. Greeley NR, Regner S, Willi S, Lynch DR. Cross-sectional analysis of glucose metabolism in Friedreich ataxia. J Neurol Sci. 2014;342(1–2):29–35. doi:10.1016/j.jns.2014.04.015
90. Weber DR. Bone health in childhood chronic disease. Endocrinol Metab Clin North Am. 2020;49(4):637–650. doi:10.1016/j.ecl.2020.07.002
91. Eigentler A, Nachbauer W, Donnemiller E, Poewe W, Gasser RW, Boesch S. Low bone mineral density in Friedreich ataxia. Cerebellum. 2014;13(5):549–557. doi:10.1007/s12311-014-0568-1
92. Watts NB, Adler RA, Bilezikian JP, et al. Osteoporosis in men: an Endocrine Society clinical practice guideline. J Clin Endo Met. 2012;97(6):1802–1822. doi:10.1210/jc.2011-3045
93. Force USPST; Curry SJ, Krist AH, Owens DK, et al. Screening for osteoporosis to prevent fractures: US preventive services task force recommendation statement. JAMA. 2018;319(24):2521–2531. doi:10.1001/jama.2018.7498.
94. Eastell R, Rosen CJ, Black DM, Cheung AM, Murad MH, Shoback D. Pharmacological management of osteoporosis in postmenopausal women: an endocrine society* clinical practice guideline. J Clin Endo Met. 2019;104(5):1595–1622. doi:10.1210/jc.2019-00221
95. Ward LM, Weber DR, Munns CF, Hogler W, Zemel BS, Contemporary A. View of the definition and diagnosis of osteoporosis in children and adolescents. The Journal of clinical endocrinology and metabolism. 2019. Management of primary and secondary osteoporosis in children. Ther Adv Musculoskelet Dis. 2020;12:1759720X20969262.
96. Galindo-Zavala R, Bou-Torrent R, Magallares-Lopez B, et al. Expert panel consensus recommendations for diagnosis and treatment of secondary osteoporosis in children. Pediatr Rheumatol Online J. 2020;18(1):20. doi:10.1186/s12969-020-0411-9
97. Xiong E, Lynch AE, Corben LA, et al. Health related quality of life in Friedreich ataxia in a large heterogeneous cohort. J Neurol Sci. 2020;410:116642. doi:10.1016/j.jns.2019.116642
98. Nieto A, Hernández-Torres A, Pérez-Flores J, Montón F. Depressive symptoms in Friedreich ataxia. Int J Clin Health Psychol. 2018;18(1):18–26. doi:10.1016/j.ijchp.2017.11.004
99. Reetz K, Dogan I, Hohenfeld C, et al. Nonataxia symptoms in Friedreich ataxia: report from the registry of the European Friedreich’s Ataxia Consortium for Translational Studies (EFACTS). Neurology. 2018;91(10):e917–e930. doi:10.1212/WNL.0000000000006121
100. Costabile T, Capretti V, Abate F, et al. Emotion recognition and psychological comorbidity in Friedreich’s ataxia. Cerebellum. 2018;17(3):336–345. doi:10.1007/s12311-018-0918-5
101. Sayah S, Rotgé JY, Francisque H, et al. Personality and neuropsychological profiles in Friedreich ataxia. Cerebellum. 2018;17(2):204–212. doi:10.1007/s12311-017-0890-5
102. Silva CB, Yasuda CL, D’Abreu A, Cendes F, Lopes-Cendes I, França MC. Neuroanatomical correlates of depression in Friedreich’s ataxia: a voxel-based morphometry study. Cerebellum. 2013;12(3):429–436. doi:10.1007/s12311-012-0424-0
103. Singh G, Binstadt BA, Black DF, Corr AP, Rummans TA. Electroconvulsive therapy and Friedreich’s ataxia. J ECT. 2001;17(1):53–54. doi:10.1097/00124509-200103000-00011
104. Casey DA. Electroconvulsive therapy and Friedreich’s ataxia. Convuls Ther. 1991;7(1):45–47.
105. Flood MK, Perlman SL. The mental status of patients with Friedreich’s ataxia. J Neurosci Nurs. 1987;19(5):251–255. doi:10.1097/01376517-198710000-00006
106. Pandolfo M, Montermini L. Prenatal diagnosis of Friedreich ataxia. Prenat Diagn. 1998;18(8):831–833. doi:10.1002/(SICI)1097-0223(199808)18:8<831::AID-PD437>3.0.CO;2-N
107. Delatycki MB, Tai G, Corben L, et al. HFE p.C282Y heterozygosity is associated with earlier disease onset in Friedreich ataxia. Mov Disord. 2014;29(7):940–943. doi:10.1002/mds.25795
108. Wilson RB, Lynch DR, Farmer JM, Brooks DG, Fischbeck KH. Increased serum transferrin receptor concentrations in Friedreich ataxia. Ann Neurol. 2000;47(5):659–661. doi:10.1002/1531-8249(200005)47:5<659::AID-ANA17>3.0.CO;2-T
109. Wilson RB, Lynch DR, Fischbeck KH. Normal serum iron and ferritin concentrations in patients with Friedreich’s ataxia. Ann Neurol. 1998;44(1):132–134. doi:10.1002/ana.410440121
110. Pandolfo M, Arpa J, Delatycki MB, et al. Deferiprone in Friedreich ataxia: a 6-month randomized controlled trial. Ann Neurol. 2014;76(4):509–521. doi:10.1002/ana.24248
111. Boddaert N, Le Quan Sang KH, Rötig A, et al. Selective iron chelation in Friedreich ataxia: biologic and clinical implications. Blood. 2007;110(1):401–408. doi:10.1182/blood-2006-12-065433
112. Kakhlon O, Manning H, Breuer W, et al. Cell functions impaired by frataxin deficiency are restored by drug-mediated iron relocation. Blood. 2008;112(13):5219–5227. doi:10.1182/blood-2008-06-161919
113. Shinnick JE, Schadt K, Strawser C, et al. Comorbid medical conditions in Friedreich Ataxia: association with inflammatory bowel disease and growth hormone deficiency. J Child Neurol. 2016;31(9):1161–1165. doi:10.1177/0883073816643408
114. Shinnick JE, Isaacs CJ, Vivaldi S, Schadt K, Lynch DR. Friedreich Ataxia and nephrotic syndrome: a series of two patients. BMC Neurol. 2016;16:3. doi:10.1186/s12883-016-0526-2
115. Schweiger B, Klingensmith GJ, Wadwa RP. Graves’ disease in a patient with Friedreich’s ataxia and diabetes mellitus. Curr Opin Pediatr. 2010;22(4):536–538. doi:10.1097/MOP.0b013e32833a4511
116. Snyder M, Seyer L, Lynch DR, Resnick A, Zesiewicz TA. Cardiac dysfunction exacerbated by endocrinopathies in Friedreich ataxia: a case series. J Child Neurol. 2012;27(10):1316–1319. doi:10.1177/0883073811435242
117. Deutsch EC, Seyer LA, Perlman SL, Yu J, Lynch DR. Clinical monitoring in a patient with Friedreich ataxia and osteogenic sarcoma. J Child Neurol. 2012;27(9):1159–1163. doi:10.1177/0883073812448460
118. Martelli A, Friedman LS, Reutenauer L, et al. Clinical data and characterization of the liver conditional mouse model exclude neoplasia as a non-neurological manifestation associated with Friedreich’s ataxia. Dis Model Mech. 2012;5(6):860–869. doi:10.1242/dmm.009829
119. Cherian D, Schadt K, Park C, Veasey S, Goldberg D, Lynch D. Coexistence of tyrosinemia and Friedreich ataxia in a single patient: treatment with liver transplantation. Ann Case Rep. 2021. doi:10.1093/ehjcr/ytaa553
120. Aghajan Y, Yoon JM, Crawford JR. Severe vincristine-induced polyneuropathy in a teenager with anaplastic medulloblastoma and undiagnosed Charcot-Marie-Tooth disease. BMJ Case Rep. 2017;bcr2016218981. doi:10.1136/bcr-2016-218981
121. Ichikawa M, Suzuki D, Inamoto J, et al. Successful alternative treatment containing vindesine for acute lymphoblastic leukemia with Charcot-Marie-Tooth disease. J Pediatr Hematol Oncol. 2012;34(3):239–241. doi:10.1097/MPH.0b013e3182352cf5
122. Friedman LS, Paulsen EK, Schadt KA, et al. Pregnancy with Friedreich ataxia: a retrospective review of medical risks and psychosocial implications. Am J Obstet Gynecol. 2010;203(3):
123. Bonnechère B, Jansen B, Haack I, et al. Automated functional upper limb evaluation of patients with Friedreich ataxia using serious games rehabilitation exercises. J Neuroeng Rehabil. 2018;15(1):87. doi:10.1186/s12984-018-0430-7
124. Portaro S, Russo M, Bramanti A, et al. The role of robotic gait training and tDCS in Friedrich ataxia rehabilitation: a case report. Medicine (Baltimore). 2019;98(8):e14447. doi:10.1097/MD.0000000000014447
125. Doğan-Aslan M, Büyükvural-şen S, Nakipoğlu-Yüzer GF, Özgirgin N. Demographic and clinical features and rehabilitation outcomes of patients with Friedreich ataxia: a retrospective study. Turk J Phys Med Rehabil. 2018;64(3):230–238. doi:10.5606/tftrd.2018.2213
126. Milne SC, Corben LA, Roberts M, et al. Can rehabilitation improve the health and well-being in Friedreich’s ataxia: a randomized controlled trial? Clin Rehabil. 2018;32(5):630–643. doi:10.1177/0269215517736903
127. Maring J, Croarkin E, Morgan S, Plack M. Perceived effectiveness and barriers to physical therapy services for families and children with Friedreich ataxia. Pediatr Phys Ther. 2013;25(3):305–313. doi:10.1097/PEP.0b013e31828ed7cb
128. Daker-White G, Greenfield J, Ealing J. “Six sessions is a drop in the ocean”: an exploratory study of neurological physiotherapy in idiopathic and inherited ataxias. Physiotherapy. 2013;99(4):335–340. doi:10.1016/j.physio.2013.02.001
129. Milne SC, Campagna EJ, Corben LA, et al. Retrospective study of the effects of inpatient rehabilitation on improving and maintaining functional independence in people with Friedreich ataxia. Arch Phys Med Rehabil. 2012;93(10):1860–1863. doi:10.1016/j.apmr.2012.03.026
130. Goulipian C, Bensoussan L, Viton JM, Milhe-de Bovis V, Ramon J, Delarque A. Orthopedic 5 shoes improve gait in Friedreich’s ataxia: a clinical and quantified case study. Eur. J Phys Rehabil Med. 2008;44(1):93–98.
131. Maring JR, Croarkin E. Presentation and progression of Friedreich ataxia and implications for physical therapist examination. Phys Ther. 2007;87(12):1687–1696. doi:10.2522/ptj.20060232
132. Delatycki MB, Holian A, Corben L, et al. Surgery for equinovarus deformity in Friedreich’s ataxia improves mobility and independence. Clin Orthop Relat Res. 2005;430:138–141. doi:10.1097/01.blo.0000150339.74041.0e
133. Isaacs CJ, Farmer JM, Schadt KA, et al. Geographic and socio-demographic features of Friedreich ataxia: implications for clinical research. J Rare Dis. 2016;4(1):34–43.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.