Back to Journals » Breast Cancer: Targets and Therapy » Volume 13
FOXQ1 is Differentially Expressed Across Breast Cancer Subtypes with Low Expression Associated with Poor Overall Survival
Authors Elian FA, Are U, Ghosh S, Nuin P, Footz T, McMullen TPW, Brindley DN, Walter MA ![]()
Received 31 October 2020
Accepted for publication 16 December 2020
Published 1 March 2021 Volume 2021:13 Pages 171—188
DOI https://doi.org/10.2147/BCTT.S282860
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Pranela Rameshwar
Fahed A Elian,1 Ubah Are,1 Sunita Ghosh,2,3 Paulo Nuin,1 Tim Footz,1 Todd PW McMullen,4 David N Brindley,5,6 Michael A Walter1
1Department of Medical Genetics, Faculty of Medicine and Dentistry, University of Alberta, Edmonton, AB, Canada; 2Department of Medical Oncology, Faculty of Medicine and Dentistry, University of Alberta, Edmonton, AB, Canada; 3Department of Mathematical and Statistical Sciences, Faculty of Science, University of Alberta, Edmonton, AB, Canada; 4Department of Surgery, Faculty of Medicine and Dentistry, University of Alberta, Edmonton, AB, Canada; 5Department of Biochemistry, Faculty of Medicine and Dentistry, University of Alberta, Edmonton, AB, Canada; 6Cancer Research Institute of Northern Alberta, University of Alberta, Edmonton, AB, Canada
Correspondence: Michael A Walter
Department of Medical Genetics, Faculty of Medicine & Dentistry, University of Alberta, 8-32 Medical Sciences Building, Edmonton, AB, T6G 2H7, Canada
Tel +1 (780) 492-3028
Email [email protected]
Purpose: Forkhead box Q1 (FOXQ1) has been shown to contribute to the development and progression of cancers, including ovarian and breast cancer (BC). However, research exploring FOXQ1 expression, copy number variation (CNV), and prognostic value across different BC subtypes is limited. Our purpose was to evaluate FOXQ1 mRNA expression, CNV, and prognostic value across BC subtypes.
Materials and Methods: We determined FOXQ1 expression and CNV in BC patient tumors using RT-qPCR and qPCR, respectively. We also analyzed FOXQ1 expression and CNV in BC cell lines in the CCLE database using K-means clustering. The prognostic value of FOXQ1 expression in the TCGA-BRCA database was assessed using univariate and multivariate Cox’s regression analysis as well as using the online tools OncoLnc, GEPIA, and UALCAN.
Results: Our analyses reveal that FOXQ1 mRNA is differentially expressed between different subtypes of BC and is significantly decreased in luminal BC and HER2 patients when compared to normal breast tissue samples. Furthermore, analysis of BC cell lines showed that FOXQ1 mRNA expression was independent of CNV. Moreover, patients with low FOXQ1 mRNA expression had significantly poorer overall survival compared to those with high FOXQ1 mRNA expression. Finally, low FOXQ1 expression had a critical impact on the prognostic values of BC patients and was an independent predictor of overall survival when it was adjusted for BC subtypes and to two other FOX genes, FOXF2 and FOXM1.
Conclusion: Our study reveals for the first time that FOXQ1 is differentially expressed across BC subtypes and that low expression of FOXQ1 is indicative of poor prognosis in patients with BC.
Keywords: transcription factors, mRNA expression, copy number variation, predictive marker
Introduction
Breast cancer (BC) is well known as a highly heterogeneous disease comprising distinct molecular subtypes.1,2 Each BC subgroup differs from each other in terms of biological characteristics, risk factors, treatment responses, and patient survival outcomes.1,2 This variability in BC behaviours and characteristics presents major clinical challenges and implications with regard to prognosis and BC management.3,4 Therefore, much effort has been made to stratify heterogeneous BC subtypes, in order to increase our knowledge of the pathobiology of BC as well as to discover new treatments.5–8 Typically, BC tumors have been divided into subtypes according to immunohistochemical (IHC) markers of estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2), with the combination of differential gene expression.9 Hence, there are three major subtypes of BC, and these subtypes are luminal (ER positive), triple-negative breast cancer (TNBC), and HER2 BC.9 Clinicians can determine what therapeutic options are most effective by using well-defined standard IHC markers to identify BC tumor subtype. However, care is now increasingly shifting towards looking at genetic differences to develop more personalized medicine approaches.10 The Prediction Analysis of Microarray 50 (PAM50) predictor is a multigene signature that stratified traditional pathological classification of BC (TNBC, luminal, HER2) into intrinsic molecular subtypes—luminal A, luminal B, HER2 enriched, normal-like and basal-like (which commonly is TNBC) BC—based on multigene differential expression.11,12 With this move to precision medicine for cancer treatment, there has been an increasing need to identify new potential biomarkers across the different subtypes of BC.
Forkhead box (FOX) family members are highly conserved transcription factors that have a major role in a plethora of biological functions. These family members have a common DNA-binding domain (DBD) composed of 100 amino acids that is known as the forkhead box or “winged helix” domain.13 Many FOX transcription factors have been linked to tumorigenesis, where oncogenic and/or tumor-suppressive roles of FOX genes have been suggested in several cancers.14,15 Members of the FOXA, FOXC, FOXM, FOXO, and FOXP subclasses of FOX proteins, in particular, have direct effects on the initiation, maintenance, progression, and drug resistance of cancers.16 For example, overexpression of FOXM1 accelerates the proliferation and progression of prostate cancers in mouse models.17 The widely studied FOXO proteins are key regulators of tumour suppression, as the simultaneous deletion of FOXO1, FOXO3, and FOXO4 alleles in somatic cells invokes thymic lymphomas and systemic haemangiomas in mouse models.18 Moreover, FOXF2, FOXM1, FOXA1, and FOXC1 genes were suggested to play critical roles in BC initiation, proliferation, migration, and invasion.15,19–21 FOXQ1 is another member of the FOX protein superfamily of transcription factors that regulate the expression of genes necessary for embryonic development, cell proliferation, differentiation, and apoptosis.22 This single-exon gene is located on chromosomal region 6p25.3 and its encoded protein is characterized by a distinctive evolutionarily conserved DBD.23 Functionally, FOXQ1 plays a role in a diverse range of important biological processes such as angiogenesis, epithelial differentiation, smooth muscle differentiation, mucin secretion, and natural killer cell effector function activation.13,22,24,25 Moreover, FOXQ1 has been associated with the aggressive phenotype of cancers such as breast, colorectal, ovarian, and pancreatic cancers, since altered FOXQ1 expression has been previously detected in human tumor specimens.26–31 Increasing numbers of studies have examined the role of FOXQ1 in tumor progression and it has been suggested that suppressing FOXQ1 expression may decrease invasion and migration in TNBC cell lines.32,33 FOXQ1 has also been suggested as potentially being a driving force in the heterogeneity of BC.6 Thus, additional knowledge of the role of FOXQ1 in BC could have the potential to improve the diagnosis and treatment of BC tumors. In particular, the role and expression of FOXQ1 in luminal and HER2 BC needs to be determined.
In this study, we investigated FOXQ1 mRNA expression and copy number variation (CNV) across BC patient subtypes and cell lines. Furthermore, we assessed the prognostic significance of FOXQ1 for BC patients. Our results revealed for the first time that FOXQ1 mRNA is differentially expressed in TNBC, luminal, and HER2 BC patients and cell lines. We found that FOXQ1 expression is significantly lower in luminal and HER2-positive tumors and that low expression of FOXQ1 predicts poor overall survival in BC patients. We also found that FOXQ1 has more copies in tumors from TNBC patients compared to normal breast tissue samples. Interestingly, FOXQ1 mRNA expression is independent of its CNV in BC cell lines. Our findings highlight that low expression of FOXQ1 mRNA is associated with significantly poorer survival for different classes of BC.
Materials and Methods
Tissue Samples
BC patients’ samples were obtained with approval of the University of Alberta Health Research Ethics Board (Pro00018758) with written informed consent, in accordance with the Declaration of Helsinki. The tumor samples from BC patients were collected at surgery and frozen in liquid nitrogen within 20 min of devitalization for further experiments. Normal human breast tissue was obtained from breast reduction surgery. BC subtypes have been defined using immunohistochemistry (IHC) markers; ER, Estrogen receptor; PR, Progesterone; and HER2, Human Epidermal Receptor (Table 1).
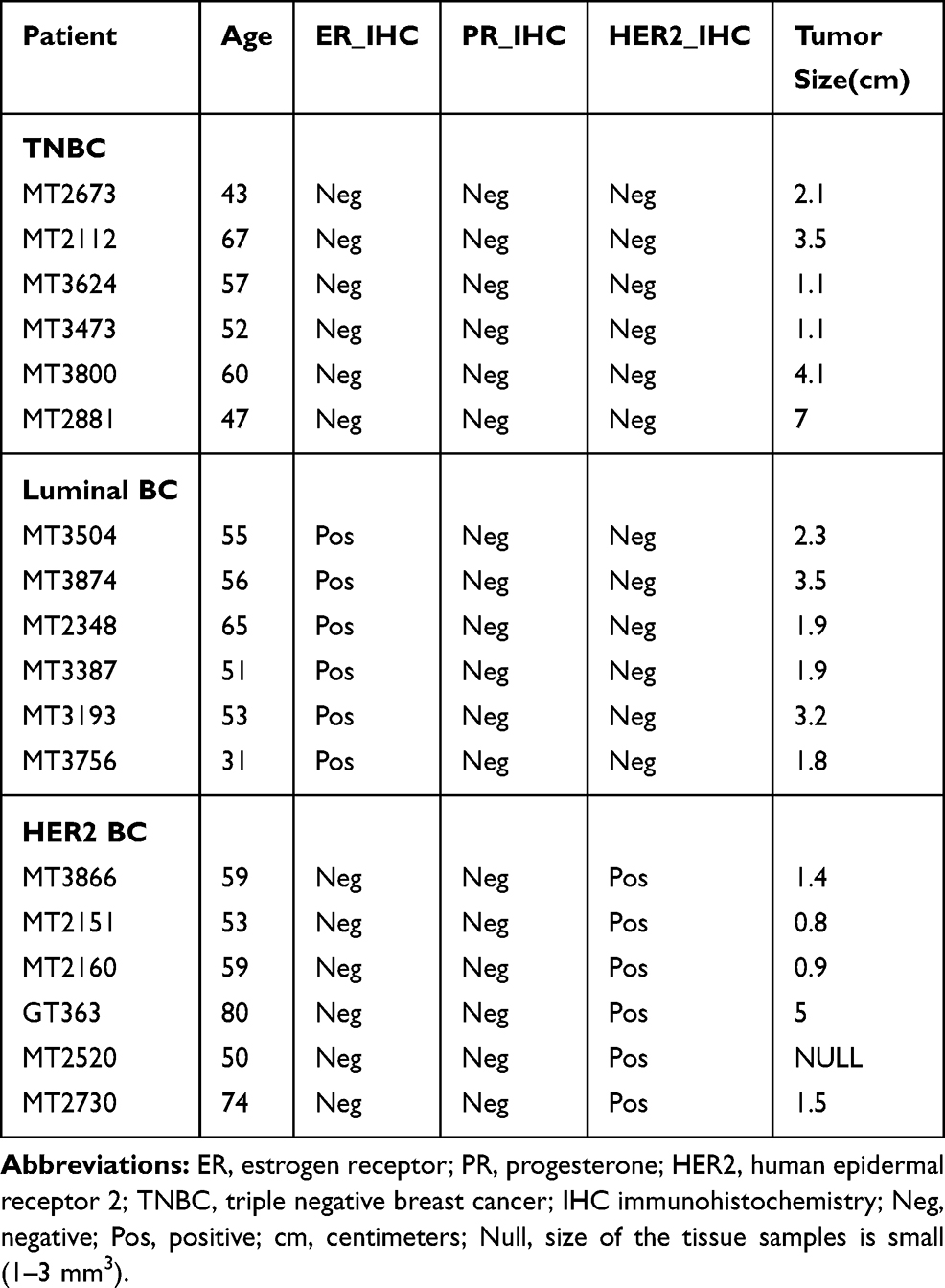 |
Table 1 BC Patient Demographics |
Overall Survival Analysis
Kaplan-Meier curves were generated using the online analysis tools OncoLnc (http://www.oncolnc.org)34 and Gene Expression Profiling Interactive Analysis (GEPIA, http://gepia.cancer-pku.cn/).35 OncoLnc and GEPIA use patient survival data and level 3 RNA sequencing expression data of BC tumor samples from the Cancer Genome Atlas (TCGA) invasive breast carcinoma project (BRCA) database (https://portal.gdc.cancer.gov/projects/TCGA-BRCA). Patient samples (n=1006) and (n=1055) from TCGA-BRCA were used to generate the Kaplan-Meier (KM) curves in OncoLnc and GEPIA online tools, respectively. Patient samples in OncoLnc and GEPIA were divided into high and low groups based on the median and the quartile of FOXQ1 expression in BRCA database. The median cut-off divided patients into two groups, high FOXQ1 expression group are above the 50th percentile, and low FOXQ1 expression group are below the 50th percentile. For the quartile cut-off analysis, patients were categorized into two groups, high FOXQ1 expression group are above the third quartile (higher than 75th percentile), and low FOXQ1 expression group are below the first quartile (lower than 25th percentile). More details on the mRNA expression data and their normalization can be found in (http://www.oncolnc.org).34 The long-rank test was used for the hypothesis evaluation. In regard to the censorship method, we used right censoring method in which the patients who did not experience the event (death) during the study duration and had no available further follow-up data were censored. The mean age at diagnosis of the BC patients in TCGA-BRCA database is 58.4± 13.2 years and the median follow-up time is 27.7 months where the median follow-up time for the overall survival time to event is 41.8 months and the median follow-up time for the overall survival time to censor is 25.0 months.36
Kaplan-Meier survival curves were generated of FOXQ1 expression in BC subtypes using UALCAN online tool (http://ualcan.path.uab.edu/index.html).37 TCGA-BRCA patient samples were divided into two groups, high and low FOXQ1 expression groups. Patients with FOXQ1 transcript per million (TPM) above the third quartile (higher than 75th percentile) were categorized as high FOXQ1 expression group, while patients with FOXQ1 TPM below the third quartile (lower than 75th percentile) were categorized as low/medium FOXQ1 expression group. Subsequently, patients within each group were further stratified into TNBC, luminal, and HER2 BC subtypes. Log rank test was used to generate the p value to test statistical significance of survival correlation between BC subtypes and FOXQ1 expression. UCSC Xena website: https://xenabrowser.net/38 was used to generate Kaplan-Meier curves for the TCGA-BRCA data with a cut-off time after 10 years. Log rank test was used to generate the p value to test statistical significance of survival correlation.
SPSS version 25 (IBM Corp. Released 2017. IBM SPSS Statistics for Windows, Version 25.0. Armonk, NY: IBM Corp.) was used to run the Cox’s regression hazard model. p-value <0.05 was used for statistical significance. We used the TCGA-BRCA data (https://portal.gdc.cancer.gov/projects/TCGA-BRCA) to conduct the univariate overall survival of Cox’s regression analysis for FOXQ1 expression. One thousand six TCGA-BRCA patients data with high and low FOXQ1 expression with respect to the median and quartile cut-off were downloaded from OncoLnc (http://www.oncolnc.org).34 OncoLnc includes only patients who have all the necessary clinical data for each cancer such as age, sex, and grade as well as gene expression. More details on TCGA_BRCA patient data used for FOXQ1 Cox regression analysis can be found in Supplementary Table 1. Unadjusted and adjusted Cox’s regression hazard model was used to determine the association of FOXQ1, FOXF2, and FOXM1 with overall survival, more details can be found in Supplementary Table 2.
Finally, we also used TCGA-BRCA data (https://portal.gdc.cancer.gov/projects/TCGA-BRCA) to conduct Cox’s regression analysis of FOXQ1 expression adjusted to BC subtypes. One thousand six BC patient data with high and low FOXQ1 expression with respect to the median were downloaded. Then, we identified each BC patient subtype as TNBC, luminal, and HER2 as previously described in Ref. 39. More details on the latter analysis can be found in Supplementary Table 3.
RT-qPCR
RT-qPCR was conducted according to the detailed protocol as previously described.40 Total RNA was extracted from BC patients’ tumor samples and human breast normal tissue using the RNAqueous kit (Ambion, Streetsville, ON, Canada) according to the manufacturer’s instruction. cDNA was synthesised from 1 µg RNA using Superscript II reverse transcriptase.41 RT-qPCR assays were performed with a QuantiTect SYBR Green PCR kit (Applied Biosystems Foster City, CA, United States) and analyzed on a LightCycler 96 Real-Time PCR System (Roche Life Science, Penzberg, Germany), at least three times with each reaction in triplicate. mRNA levels were normalized to TFRC (selected as an internal control gene) through the ΔΔCt method and changes in mRNA levels were described in fold change compared to the control samples. Primers for FOXQ1 and TFRC were designed using the Primer3 software (http://primer3.ut.ee/); FOXQ1: F: 5′-CGGAGATCAACGAGTACCTCA-3′, R: 5′-CAGTCGTTGAGCGAAAGGTT-3′; TFRC F: 5′-AACAACAGATTTCGGGAATGC-3′, R: 5′-CGTAGGGAGAGAGGAAGTGATA-3′. For statistical analyses, we first compared FOXQ1 expression in all four groups; normal breast tissue, TNBC, luminal, and HER2, using one-way ANOVA. Then, one-tailed unpaired Student’s t-tests and Bonferroni correction were used for multiple comparisons to assess statistical differences.
Copy Number Variation
FOXQ1 gene copy number in BC patients was quantified through Real-time qPCR, as previously described42 to determine if the altered dosage of FOXQ1 gene affected expression levels in BC tumors. Genomic DNA was extracted from human breast normal tissue and from TNBC, luminal, and HER2 BC patient tumor samples using the EZ‐10 Spin Column Genomic DNA Minipreps Kit (Bio Basic Inc., Markham, ON, Canada) according to the manufacturer’s instructions. Real-time qPCR was then conducted on the genomic DNA of all samples to measure FOXQ1 dosage. PCR reactions for normal breast tissue, TNBC, luminal, and HER2 BC patient tumor samples (Table 1) were analyzed in triplicate. Normal breast tissue samples were used as a control to normalize FOXQ1 dosage. We used the ΔΔCT method as our quantification strategy, with GJA5 selected as an internal control gene. Average CT values of triplicates were calculated for each sample. ΔCT for each sample was then calculated by subtracting the average CT number of FOXQ1 from that of GJA5. FOXQ1: F: 5′-CGGAGATCAACGAGTACCTCA-3′, R: 5′-CAGTCGTTGAGCGAAAGGTT-3′; GJA5 F: 5′-AGTTCCCAGCCAATAGACAGC-3′, R: 5′-AAGGCTGAGTAGAGGGAGGAG-3′. We used a two-tailed unpaired Student’s t-tests for comparisons of each BC subtype with normal breast tissues to assess statistical differences.
K-Means Clustering
K means clustering analysis was performed to investigate and compare FOXQ1 CNV and its mRNA expression. In order to conduct K-means clustering, we first identified the ideal number of clusters. Our initial approach was to compare the biological ideal number of clusters, which is 3 clusters, based on cell line annotation and classification; TNBC, luminal, and HER2 (supervised K-means clustering). For the unsupervised ideal number of clusters, we used the elbow method43 to determine the ideal statistical number of clusters for K-means with the scikit-learn’s method of the MinMaxScaler module used with a sum of squares distance and fit between 1 and 15 possible clusters.43 We then devised Python scripts to identify the cluster sets by applying standard supervised K-means clustering using both number of clusters (supervised and unsupervised) and FOXQ1 mRNA expression and copy number variation (CNV) as seeds as described.44 The mRNA and CNV data of FOXQ1 were obtained from Cancer Cell Line Encyclopedia online (CCLE) database.45 K-means clustering performed 1000 iterations using the Euclidean distance and using seeds between 1 and 3 clusters and 1 and 4 clusters. Thirty-six BC cell lines were used for the analysis. Cell lines were grouped as TNBC, luminal, and HER2 BC cell lines as described.46,47 CCLE CNV data of BC cell lines were obtained using genome-wide human Affymetrix SNP Array 6.0.45 The mRNA data on the CCLE website were produced by Affymetrix Human Genome U133 Plus 2.0 Array followed by Robust Multiarray Averaging (RMA) method of microarray normalization and were uploaded as log(2) gene expression signal.45 The log(2) FOXQ1 expression data for each of the 36 BC cell lines from CCLE were obtained and then scaled to values between 0 and 1 using the equation;
Zi = {xi-min(x)}/{max(x)-min(x)}
where x = (x1, … …,xn) producing zi as the scaled value for FOXQ1 mRNA expression in each of the 36 BC cell lines. CNV raw data created by Affymetrix (CEL file) were normalized to copy number estimates using a GenePattern pipeline.45 0.5 was used as a cut-off, where cell lines with values above the cut-off were considered as relative high expression of FOXQ1 cell lines, while cell lines with FOXQ1 expression below the cut-off were considered as relative low expression of FOXQ1 cell lines.
Breast Cancer Patient Publicly Available Dataset
RNAseq data for FOXQ1 expression in breast cancer patients from the TCGA-BRCA (https://portal.gdc.cancer.gov/projects/TCGA-BRCA) were obtained from UCSC Xena website: https://xenabrowser.net/38 to generate Boxplots analysis across breast cancer subtypes. One-way ANOVA was applied to generate the p value to test statistical significance. Microarray data of FOXQ1 expression in breast cancer patients from the Van’t Veer48 study were accessed using UCSC Xena website: https://xenabrowser.net/.38 Welch’s t-test was applied to generate p values to test statistical significance.
FOXQ1 mRNA expression correlation with each of PDGFRα and β, Twist1, Zeb2, and DACH1 mRNA expression was analyzed in the TCGA-BRCA database using the co-expression analysis tool on cBioPortal.96,97 Spearman and Pearson were used for correlation analysis. FOXQ1, PDGFRα and β, Twist1, Zeb2, and DACH1 log2 mRNA values of normalized RSEM RNAseq V249 in 994 patient and normal samples were obtained from the PanCanAtlas 2018 of the TCGA-BRCA https://gdc.cancer.gov/about-data/publications/pancanatlas.50
Results
FOXQ1 mRNA Expression Varies Between BC Patient Subtypes
To evaluate FOXQ1 expression across the different subtypes of BC, RT-qPCR assays were conducted on TNBC, luminal, and HER2 BC patient samples and on normal breast tissue samples (Table 1). One-way ANOVA analysis showed that FOXQ1 mRNA expression varied between BC patients’ subtypes, F(30,52) = 3.8, p=0.026. Furthermore, we found that FOXQ1 expression was significantly lower in luminal BC when it was compared to normal breast tissue (Figure 1A). The mRNA level of FOXQ1 was lower in HER2 BC compared to normal breast tissue (p=0.02) (Figure 1A). Interestingly, FOXQ1 mRNA levels were lower in luminal and HER2 BC compared to TNBC (p=0.017 and p=0.029, respectively) (Figure 1A). We further confirmed our results in an expanded panel of BC patients (n = 522) using a publicly available dataset from the Cancer Genome Atlas (Figure 1A). Consistent with our RT-qPCR, we found that the expression of FOXQ1 mRNA varied across BC patient subtypes (Figure 1A, F = 24.64, p<0.0001). To substantiate our findings of significant low expression of FOXQ1 in luminal BC, we further investigated FOXQ1 expression in a public dataset of BC patients after they were stratified by ER status into ER positive and ER negative. Consistent with our RT-qPCR, we found that the expression of FOXQ1 was significantly lower in ER-positive BC patients compared to ER-negative BC patients (Figure 1B). These results indicate that FOXQ1 mRNA levels are differentially expressed among BC patients and that FOXQ1 could potentially have different roles based on the BC subtype.
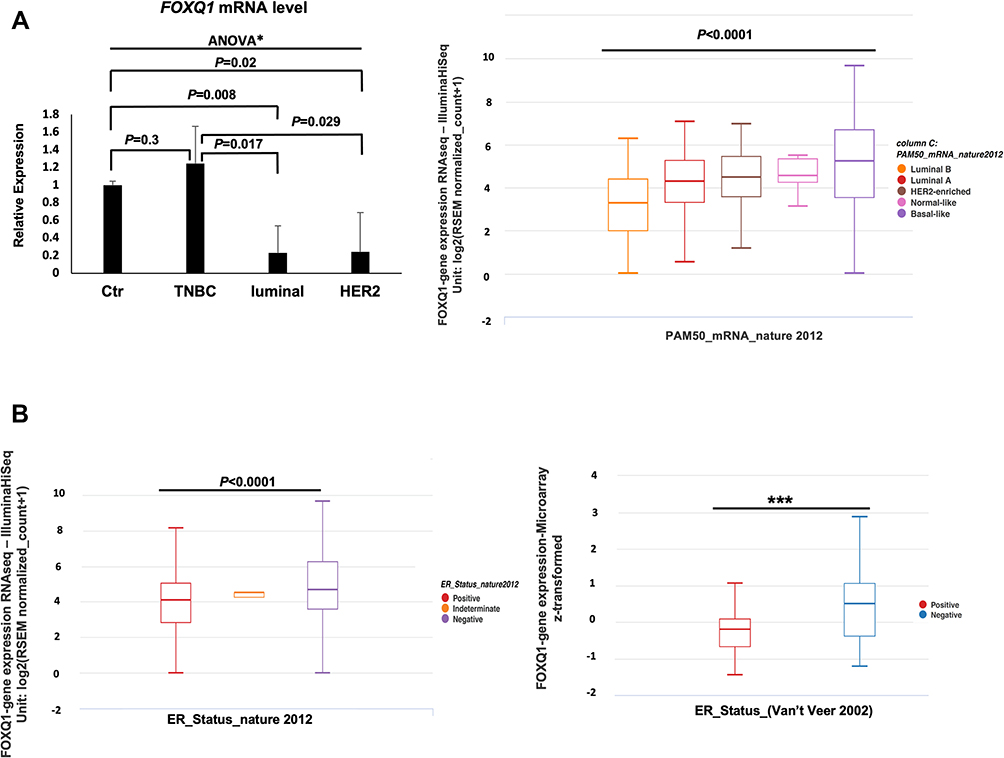 |
Figure 1 FOXQ1 is differentially expressed across breast cancer subtypes and is under-expressed in luminal and HER2 breast cancer patient samples. (A) Left, qPCR experiments were conducted to measure FOXQ1 mRNA levels in normal breast tissue (Ctr, n=6) acquired from reduction mammoplasties, TNBC (n=6), luminal (n=6), and HER2 (n=6) BC patient samples. One-way ANOVA analysis was used to asses statistical difference in all groups *F(30,52) = 3.8, p=0.026 followed by unpaired t-tests and Bonferroni corrections were used for multiple comparisons. α= 0.05, adjusted α= 0.0125. The ΔΔCT method was used for analysis and TFRC was used as a reference gene. Error bars represent standard error of the mean (SEM). (A) Right, box plots showing FOXQ1 expression across BC intrinsic subtypes (PAM50). FOXQ1 RNAseq data in BC subtypes from the TCGA-BRCA data95 were used. Luminal B (orange), n = 127, Minimum = 0.00, Q1 = 2.0, Median = 3.29, Q3 = 4.40, Maximum = 6.26; Luminal A (red), n = 231, Minimum = 0.527, Q1 = 3.31, Median = 4.32, Q3 = 5.26, Maximum = 7.08; HER2-enriched (brown), n = 58, Minimum = 1.17, Q1 = 3.58, Median = 4.50, Q3 = 5.44, Maximum = 6.94; Normal-like (pink), n = 8, Minimum = 3.13, Q1 = 4.25, Median = 4.58, Q3 = 5.35, Maximum = 5.49; Basal-like (purple), n = 98, Minimum = 0.00, Q1 = 3.54, Median = 5.25, Q3 = 6.68, Maximum = 9.65. One-way ANOVA was used for statistical significance (F = 21.25, p<0.0001). FOXQ1 expression is expressed in RSEM (RNA-seq by expectation-Maximization) format. (B) Left, box plot of FOXQ1 RNAseq data in BC patients stratified by ER status. ER-positive (red), n = 601, Minimum = 0.00, Q1 = 2.82, Median = 4.12, Q3 = 5.05, Maximum = 8.13; ER-intermediate (orange), n = 2, Minimum = 4.24, Q1 = 4.24, Median = 4.50, Q3 = 4.50, Maximum = 4.50; ER-negative (blue), n = 179, Minimum = 0.00, Q1 = 3.58, Median = 4.68, Q3 = 6.26, Maximum = 9.65. One-way ANOVA was used for statistical significance (F = 24.64, p<0.0001). (B) Right, box plot of microarray data of FOXQ1 expression in BC patients stratified by ER status from the Van’t Veer et al48 study. ER-positive (red), n = 71, Minimum = −1.46, Q1 = −0.67, Median = −0.20, Q3 = 0.08, Maximum = 1.06; ER-negative (blue), n = 46, Minimum = −1.22, Q1 = −0.38, Median = 0.5, Q3 = 1.06, Maximum = 2.87. Welch’s t-test was used for statistical significance (***p<0.0001). Whiskers represent minimum and maximum values. The TCGA-BRCA95 and the Van’t Veer et al48 data were accessed using UCSC XENA cancer genome browser: https://xenabrowser.net/. |
Low Expression of FOXQ1 is Associated with Poorer Overall Survival in Breast Cancer Patients
Kaplan-Meier curves were performed on the TCGA-BRCA dataset using the OncoLnc,34 GEPIA,35 and UALCAN37 online tools to investigate whether FOXQ1 mRNA expression is prognostic for life expectancy. BC patients were divided into two risk groups with high and low FOXQ1 expression levels with respect to the quartile and the median used as the cut-off values. BC patients with low FOXQ1 mRNA expression had significantly shorter overall survival time compared to those with high FOXQ1 mRNA expression (Figure 2, p=0.00473 and p=0.0479) and (Supplementary Figure 1, p=0.0087 and p=0.042), and after 10 years follow-up (Supplementary Figure 2).
 |
Figure 2 Low FOXQ1 expression predicts poor overall survival in BC patient. Kaplan-Meier (KM) analysis identified low and high-risk BC patient groups based upon significant differences of FOXQ1 mRNA in BRCA-TCGA database. BC patients were divided with high and low FOXQ1 expression levels with respect to the quartile and the median used as the cut-off value. This graph was generated using the bioinformatics online tool OncoLnc (http://www.oncolnc.org)34 and then modified to include the number of patients at risk at 0, 5 and 10 years. |
In addition, a univariate Cox’s regression analysis was conducted for overall survival on the TCGA-BRCA patient data with respect to the expression of FOXQ1. The results of the univariate Cox’s regression analysis in Table 2 indicate that BC patients who have high FOXQ1 expression (≥50th percentile) were significantly associated with 29% lower risk of death as compared to BC patients who have low FOXQ1 expression (p=0.048). Further analysis was conducted, comparing the highest quartile and the lowest quartile of FOXQ1 expression. The results indicate that FOXQ1 high expression (≥75th percentile) was significantly associated with about 51% lower risk of BC patient’s death as compared to the low FOXQ1 expression (below 25th percentile) with a p-value of 0.006 (Table 2).
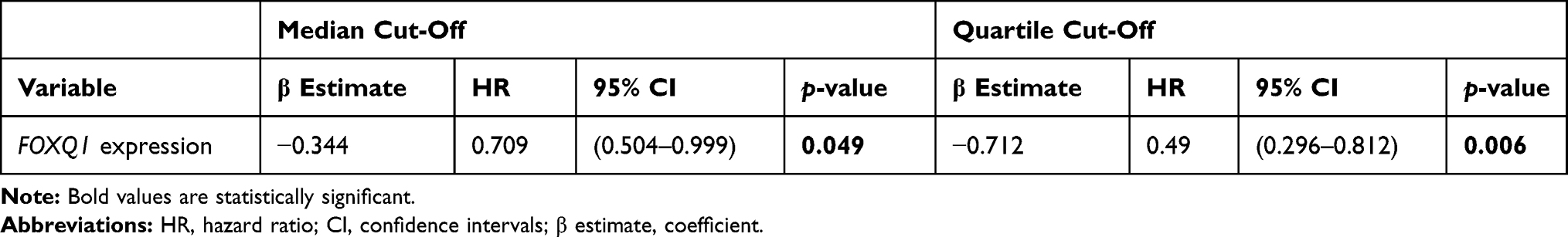 |
Table 2 Hazard Ratios from the Univariate Cox’s Regression Analysis for the TCGA-BRCA Database |
Furthermore, using UALCAN online tool, we examined the correlation of FOXQ1 expression and BC subtypes by dividing patients into two groups; high and low/medium FOXQ1 expression cohorts (Figure 3). Kaplan-Meier curves indicate that patients with low/medium FOXQ1 expression had significantly shorter overall survival time than those with high expression of FOXQ1 (Figure 3, p=0.011). Subsequently, using the TCGA-BRCA data, we conducted Cox’s regression analysis for FOXQ1 expression stratified by BC subtype as well as for FOXQ1 expression adjusted for BC subtype (Table 3). For HER2 subtype, high FOXQ1 expression was significantly associated with about 73% lower risk of death as compared to low FOXQ1 expression (Table 3, p=0.024). The same trend was observed for luminal and TNBC subtypes indicating that high FOXQ1 expression showed a reduced risk of death by 19% and 32%, respectively. However, these associations were not statistically significant (Table 3).
 |
Figure 3 Low FOXQ1 expression predicts poor overall survival in BC patient subtypes. Kaplan-Meier (KM) analysis of the effect of high and low/medium FOXQ1 expression on overall survival of HER2, luminal, and TNBC patients shows a cumulative significance of p=0.011. This graph was generated and modified using the bioinformatics online tool UALCAN online tool (http://ualcan.path.uab.edu/index.html)37. |
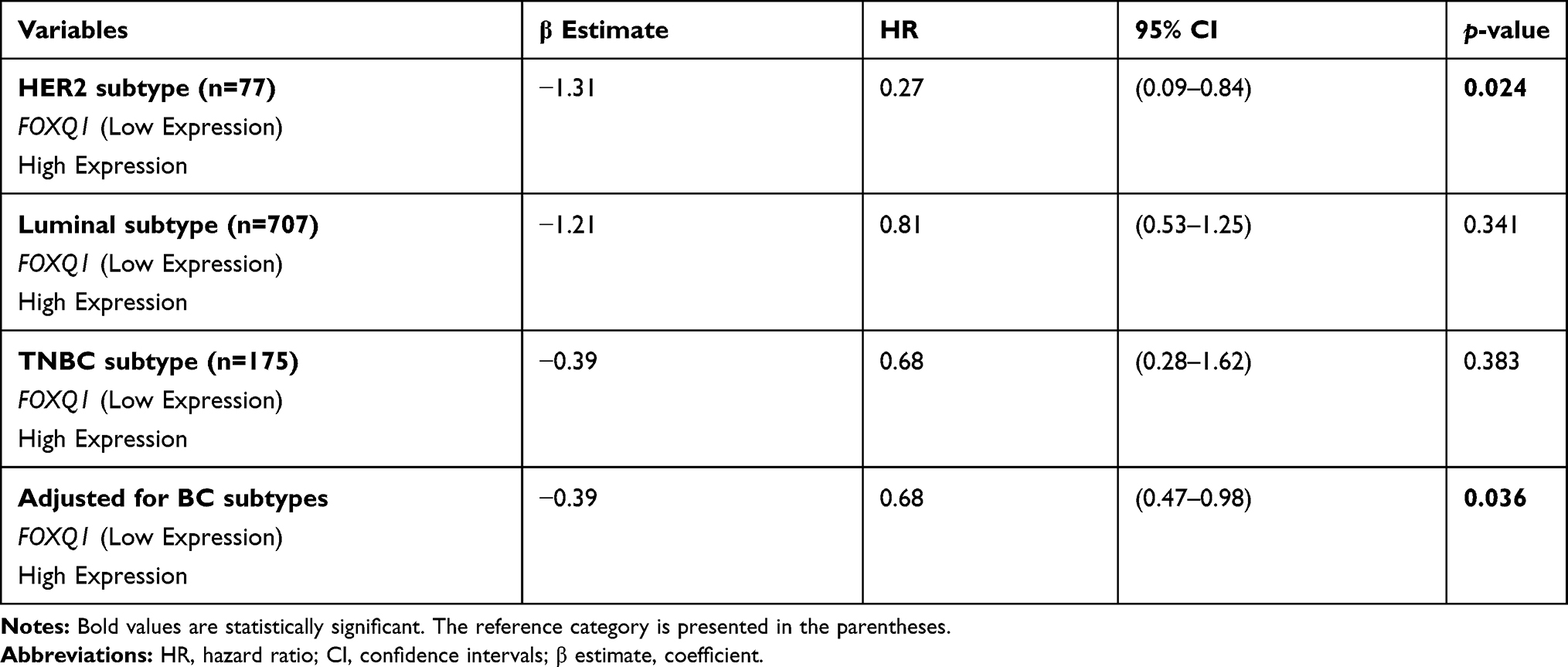 |
Table 3 Cox’s Regression Analysis for FOXQ1 Expression Based on Median Cut-off Stratified by BC Subtypes and Adjusted for BC Subtypes |
When adjusting for BC subtype, high FOXQ1 expression showed a 32% lower risk of death (Table 3, p=0.036). The unadjusted analysis from Table 2 and adjusted analysis from Table 3 indicate that FOXQ1 is an independent predictor of overall survival in BC patients. Together, these results demonstrate that low expression of FOXQ1 predicts poor overall survival in patients with BC.
Relation of FOXQ1 Copy Number in BC Subtypes to mRNA Levels
It has recently been suggested that CNV correlates with gene expression in BC cell lines and patient tissue.51 To determine FOXQ1 CNV in the subtypes of BC patients, qPCR assays were conducted on TNBC, luminal, and HER2 BC patient DNA samples (Table 1) and on normal breast tissue DNA samples. The number of copies of FOXQ1 was significantly higher in TNBC patients compared to normal breast tissue p=0.03 (Figure 4). Although FOXQ1 appeared to have a trend for more copies in HER2 BC patients, this did not reach statistical significance (Figure 4, p=0.08).
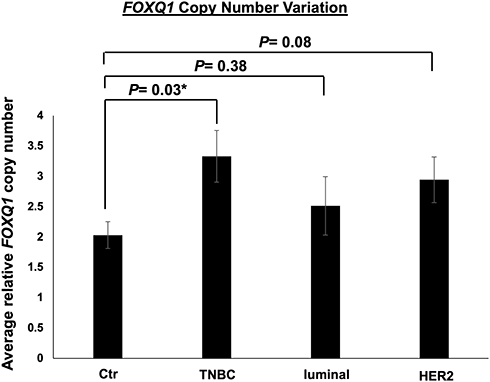 |
Figure 4 FOXQ1 has more copies in TNBC compared to control samples. qPCR experiments were conducted on genomic DNA to measure FOXQ1 dosage in TNBC, luminal, and HER2 BC patient tumor tissues. The ΔCT method was used for analysis and GJA5 was used as a reference gene. FOXQ1 dosage was calculated using 2 [ΔCT sample – ΔCT control]. Control (Ctr) samples are normal breast tissue samples acquired from reduction mammoplasties. Ctr (n=6), TNBC (n=6), luminal (n=6), and HER2 (n=6). Two-tailed unpaired Student’s t-tests were used for comparisons of each BC subtype with normal breast tissue to assess statistical differences. Error bars represent standard error of the mean (SEM). *p<0.05. |
Unsupervised and supervised K-means clustering analyses (Table 4 and Figure 5) were performed to identify clusters of BC cell lines as well as to investigate any associations between FOXQ1 mRNA expression and gene copy number. Unsupervised K-means clustering identified 4 clusters among BC cell lines for FOXQ1 mRNA compared to their copy number (Table 4 and Figure 5A).
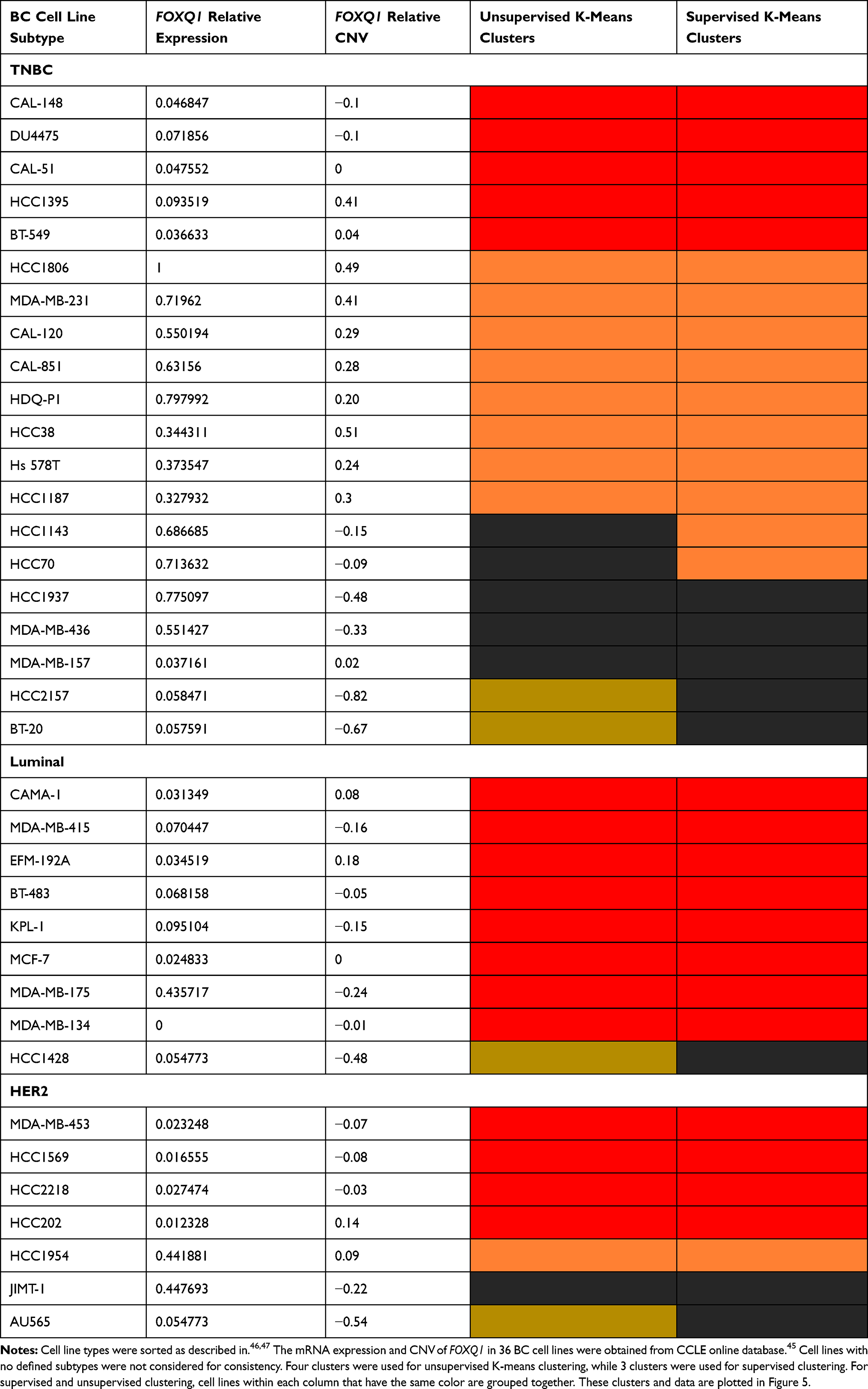 |
Table 4 BC Cell Line Names and Types That Were Used for Standard K-Means Clustering |
 |
Figure 5 FOXQ1 expression is independent of its CNV in BC cell lines. (A) unsupervised and (B) supervised K-means clustering analyses show different clusters of BC cell lines that have similar ranges of CNV but different FOXQ1 expression (the orange cluster vs the red cluster). Unsupervised clustering identified an extra cluster in BC cell lines compared to supervised clustering (the olive cluster, lower left). |
We found some cell lines clusters that have similar copy number (orange cluster vs red cluster, Figure 5A) but different mRNA expression (Figure 5A). We also found similar results after we conducted supervised K-means clustering, where some cell line clusters had high and/or low FOXQ1 mRNA expression but similar ranges of variation in relative copy number (Figure 5B). For the supervised K-means clustering, we chose 3 as the number of clusters to correspond to the 3 different subtypes of BC; TNBC, luminal, and HER2 (see K-means clustering in method’s section). Interestingly, unsupervised and supervised K-means clustering analyses suggest that the steady state level of FOXQ1 mRNA expression appears to be independent of gene copy number in BC cell lines. Thus, FOXQ1 mRNA expression levels are not correlated with FOXQ1 CNV in BC subtypes. However, consistent with our results where FOXQ1 expression was lower in luminal and HER2 BC patient tumors (Figure 1), we found that FOXQ1 expression was low in all luminal and HER2 cell lines (Figure 5A and B). Similar to our observation of increased expression of FOXQ1 in TNBC patient tumors compared to luminal and HER2 (Figure 1), we found that FOXQ1 has elevated expression in some TNBC cell lines as compared to luminal and HER2 BC cell lines (Figure 5A and B). Interestingly, we found numerous TNBC cell lines cluster with other luminal and/or HER2 cell lines and have low FOXQ1 expression. This difference of FOXQ1 mRNA expression within the TNBC classification might be attributed to the distinct subgroups (basal A and basal B) that exists within TNBC cell lines.47
FOXQ1 Expression is an Independent Predictor of Overall Survival in BC Patients When Adjusted to FOXF2 and FOXM1 Expression
We also investigated the overall survival of FOXQ1 adjusted for FOXF2 and FOXM1 expression. We evaluated FOXF2 and FOXM1 genes due to their critical role in BC initiation, proliferation, migration, and invasion.19,20,52
TCGA-BRCA patients were divided into low and high expressing FOXF2, FOXM1, and FOXQ1 groups using the medians as cut-off values, followed by a univariate and multivariate Cox’s regression overall survival analysis. FOXM1 high expression was associated with 1.5 times higher risk of BC patient death as compared to FOXM1 low expression values (p=0.013; Table 5). Adjusted Cox’s regression model with FOXQ1, FOXM1, and FOXF2 expression levels indicated that FOXQ1 and FOXM1 were significantly associated with overall survival (Table 5; p=0.05 and p=0.011, respectively). The results indicate that FOXQ1 and FOXM1 were independent predictors of overall survival in BC patients. Having high expression of FOXQ1 was associated with 32% lower risk for overall survival when adjusted for FOXF2 and FOXM1 (p=0.05; Table 5). Higher expression of FOXM1 was associated with 1.6 times higher risk of death when adjusted for FOXQ1 and FOXF2 expressions, this association was statistically significant (p=0.011; Table 5). Our results show that FOXQ1 expression correlates inversely with overall survival in BC patients and that FOXQ1 expression is an independent predictor of BC overall survival when adjusted to the expression of FOXF2 and FOXM1 in BC patients.
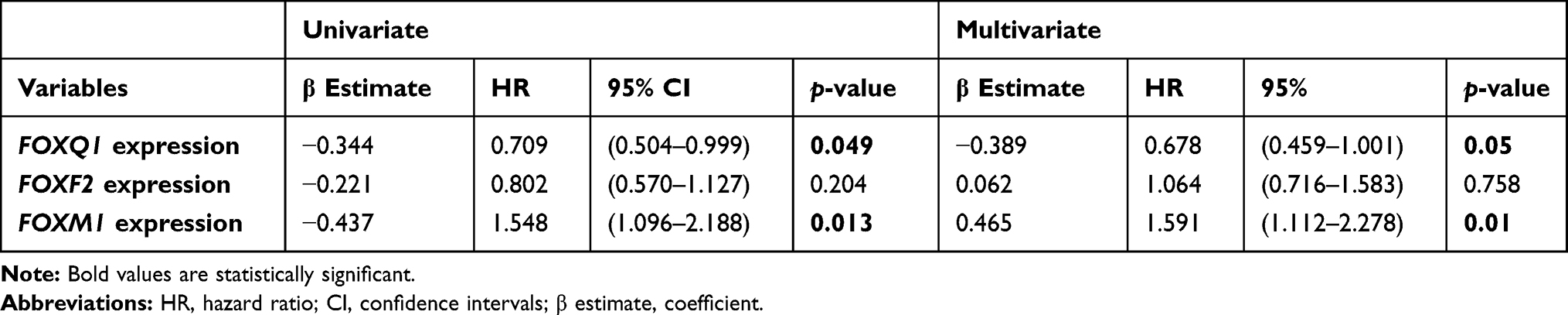 |
Table 5 Cox’s Regression Analysis for FOXQ1, FOXF2, and FOXM1 Expression Levels from the TCGA-BRCA dataset Based on Median Cut-off |
Discussion
FOXQ1 plays an important role in BC tumor re-initiation,53 stemness and chemoresistance,54 epithelial–mesenchymal transition (EMT), invasion, and metastasis.28 Increasing evidence supports the hypothesis that FOXQ1 is a biomarker for cancer prognosis and diagnosis. Indeed, high levels of FOXQ1 expression can predict poor overall survival in hepatocellular carcinoma, gastric, colorectal, pancreatic, and non-small cell lung cancers.29,55–57 However, the prognostic value of FOXQ1 in BC is not well known. In particular, the expression of FOXQ1 in other subtypes of BC, specifically luminal and HER2 BC, is not well studied. Thus, a molecular understanding of the mechanism of altered expression of FOXQ1 warrants further investigation. In this study, we report for the first time that FOXQ1 mRNA is differentially expressed across different types of BC patients and cell lines. In contrast to the earlier studies of a limited number of BC cell lines,28,30,32,58 our studies of an expanded panel of thirty-six BC cell lines show that FOXQ1 has low expression in many TNBC cell lines and in all luminal, HER2 BC patients and cell lines. We also show that this low expression of FOXQ1 is associated with poor prognosis in BC subtypes and that FOXQ1 expression is an independent predictor of overall survival in BC patients.
In order to understand FOXQ1 expression across BC subtypes, we investigated FOXQ1 expression in BC tumor tissues (Table 1). FOXQ1 expression across BC subtypes was analyzed in TNBC, luminal, and HER2 tumor tissues (Figure 1A). Two previous papers6,30 studied FOXQ1 expression data of BC tumors from the TCGA-BRCA database and found an overexpression of FOXQ1 in TNBC compared with luminal BC30 and with HER2 BC.6 Similarly, it was suggested that FOXQ1 is overexpressed in BC tumor tissue compared with normal adjacent tissue;32 however, it was not clear what BC subtypes were used for this analysis. Consistent with these studies, we found that FOXQ1 mRNA levels were higher in TNBC tumor tissues when compared with luminal BC tumor tissues (p=0.017) (Figure 1A). As well, FOXQ1mRNA levels were higher in TNBC compared with HER2 BC (p=0.029) (Figure 1A). Importantly, we reveal for the first time that FOXQ1 is expressed at lower levels in luminal BC (p=0.008) and in HER2 BC (p=0.029) when each subtype was compared with unmatched normal breast tissue (Figure 1A). A limitation of our RT-qPCR analysis was the lack of available samples of matched normal breast tissue. However, our RT-qPCR analysis results were substantiated by our analyses of publicly available data from the Cancer Genome Atlas (Figure 1A and B). Interestingly, we found lower expression of FOXQ1 in luminal B than in luminal A (Figure 1A). Luminal B has been suggested to have higher levels of proliferation and cell cycle markers genes, but lower luminal markers compared to luminal A.59–61 In addition, luminal A has a good prognosis compared to luminal B, where the latter has a higher recurrence rate, lower overall survival rate, and more invasive and proliferative cancers.62,63 Whether FOXQ1 has a subtype-specific tumor-suppressive role in luminal BC, in particular in luminal B similar to its neighbour gene, FOXC1,64 has yet to be determined. Nevertheless, our results reveal that significant differences in FOXQ1 expression occur in BC subtypes, with high FOXQ1 expression in TNBC and low FOXQ1 expression in luminal and HER2.
We further investigated the impact of FOXQ1 expression on BC patients’ overall survival. Using the OncoLnc,34 and GEPIA35 online tools, overall survival curves identified two risk groups with high and low FOXQ1 mRNA expression levels (Figure 2 and Supplementary Figures 1 and 2). We found that BC patients with low FOXQ1 expression had significantly shorter overall survival than BC patients with high FOXQ1 expression (Figure 2, and Supplementary Figures 1 and 2). Univariate Cox regression analysis for FOXQ1 expression, where BC patients who have high FOXQ1 expression were found to be significantly associated with lower risk of death as compared to BC patients who have low FOXQ1 expression (Table 2). Kaplan-Meier curves also showed that TNBC, luminal, and HER2 BC patients with low FOXQ1 expression have significantly poorer overall survival compared to TNBC, luminal, and HER2 BC patients with high FOXQ1 expression (Figure 3). When adjusting for BC subtype, high FOXQ1 expression showed significantly lower risk of death (Table 3) indicating that FOXQ1 is an independent predictor of overall survival in BC patients. This suggests that low expression of FOXQ1 in BC patients is significantly correlated with poor prognosis.
Importantly, we found FOXQ1 has lower expression in HER2 BC patient tissues (Figure 1A) and that HER2 BC patients with low FOXQ1 expression had significantly shorter overall survival compared to HER2 BC patients with high FOXQ1 expression (Table 3, Figure 3). This is an interesting result giving that HER2 BC, in comparison to other BC subtypes, is normally known to respond well to numerous targeted therapies that favourably impact patients overall survival.65 On the other hand, TNBC lacks targeted therapy and is considered to have a clinically aggressive phenotype compared to other BC subtypes.66 Although TNBC patient tissue had higher FOXQ1 mRNA expression compared to luminal and HER2 (Figure 1A), those TNBC patients with low expression of FOXQ1 appeared to have poorer overall survival compared to those with high FOXQ1 expression (Table 3 and Figure 3). This suggests that, clinically and case-by-case, BC patients in the TNBC cohort who have low FOXQ1 expression could have poorer clinical outcomes. Further studies that focus on stratifying the TNBC subtype further using gene signatures and cancer stages could reveal better understating of FOXQ1 expression in TNBC.
Intriguingly, while it has been reported that high FOXQ1 expression predicts poor overall survival in hepatocellular carcinoma, gastric, colorectal, pancreatic, and non-small cell lung cancers,29,55–57 we found that high expression of FOXQ1 favourably impacts overall survival in BC (Tables 2 and 3), suggesting a dual roles of FOXQ1 across human cancers. Dual role of FOXQ1 and other FOX genes in cancers has been reported before. For instance, it was suggested that the ectopic expression of FOXQ1 promotes tumour growth in colorectal cancer by inhibiting apoptosis.29 At the same time, FOXQ1 over-expression did not impact BC cell survival and/or proliferation in the highly metastatic 4T1 mouse cell line and it induced cell death in epithelial BC cell lines, MCF7 (labeled as luminal BC cell line) and BT-20 (labeled as TNBC cell line)28 supporting a tumor-suppressive role of FOXQ1. High expression of FOXF2 enhanced EMT, migration and invasion of lung cancer cells67 and in TNBC,68 which directly contrasts other reports where low expression of FOXF2-induced EMT and was associated with poor overall survival in BC.19 Furthermore, high expression of FOXA1 was associated with poor overall survival in prostate cancer,69 while high expression of the same protein favourably impacted BC prognosis.70,71 In addition, FOXA1 activated luminal genes and repressed basal genes in ER-positive and ER-negative BC cells, respectively,72 indicating different roles of FOXA1 in different BC subtypes. This further highlights the complex and differing roles of FOX proteins in cancer. Our data also contrast the findings of Qiao et al who suggested that overall survival was significantly poorer in BC patients with high FOXQ1 expression.58 Qiao et al58 used the van de Vijver cohort to generate Kaplan-Meier plots of their overall survival analysis but the number of patients within that cohort who had high and low levels of FOXQ1 was not indicated.58 Moreover, Van de Vijver et al73 included microarray data of 295 BC tumors, some of which were lymph-node/ER-positive and some of which were lymph-node/ER-negative. OncoLnc,34 GEPIA,35 and UALCAN,37 on the other hand, use up-to-date RNA sequencing data of the TCGA-BRCA database. In our study, we applied OncoLnc and GEPIA (respectively) to obtain Kaplan-Meier curves on BC tumors (n=1006 and n=1055), with high (n=503 and n=524), and low (n=503 and n=531) levels of FOXQ1 expression (Figure 2 and Supplementary Figure 1). The TCGA-BRCA data were also used to perform the adjusted and unadjusted Cox regression (see methods and Supplementary Tables 1, 2, and 3). The differences in numbers of BC tumor tissue samples, BC subtypes, and assays used could explain why we obtained different clinical outcomes of FOXQ1 expression compared to Qiao et al58.
We also measured FOXQ1 CNV in BC samples (Table 1) to determine if altered copy number could underlie the differences in mRNA expression levels in TNBC, luminal, and HER2 tumors (Figure 4). Although there are numerous studies that have identified an association between copy number alteration and altered gene expression, how CNVs alter gene expression in BC is not well understood.51,74,75 We found that FOXQ1 copy number is significantly amplified in TNBC compared with normal breast tissue (Figure 4). In contrast, no significant changes in FOXQ1 copy number were found in luminal and HER2 BC compared to control samples (Figure 4). To explore the possible ramifications of these findings further, we performed K-means clustering to investigate and compare FOXQ1 CNV and its mRNA expression. Our K-means clustering analyses took two approaches; unsupervised (Figure 5A) and supervised (Figure 5B). For the supervised K-means clustering, we determined the biological ideal number of clusters based on BC cell lines classification, TNBC, luminal, and HER2. For the unsupervised K-means, we used the elbow method43 to identify the ideal statistical number of clusters. For both methods, the numbers of clusters and the FOXQ1 expression and CNV were used as seeds. We found that some clusters have high copy number with low mRNA expression, while other clusters have high mRNA expression with no CNV (Figure 5A and B). Intriguingly, this suggests that FOXQ1 expression is independent from FOXQ1 copy number in BC cell lines.
Several interesting patterns, however, emerge from our K-means clustering analysis. Our supervised analysis investigated if FOXQ1 expression and CNV correlate within a specific BC subtype more than others. Intriguingly, while it was expected that TNBC cell lines would cluster together based on FOXQ1 expression and CNV (Figure 5B), we found some TNBC cell lines also cluster with HER2 and with HER2 and luminal BC cell lines (red and black clusters, Figure 5B). Furthermore, our unsupervised analysis identified 4 clusters (Table 4 and Figure 5A), an extra cluster compared to the supervised method (Table 4 and Figure 5B). This suggests that FOXQ1 expression and CNV could potentially identify a sub-population of BC within TNBC, HER2, and luminal BC cell lines. This supports the findings of Yang et al, which suggest that FOXQ1 expression could drive the heterogeneity of BC subtypes.6 Consistent with our findings of low levels of FOXQ1 expression in luminal and HER2 BC patient tumors (Figure 1), we found FOXQ1 mRNA to be low in all luminal and HER2 BC cell lines (Figure 5A and B). Interestingly, while other studies reported FOXQ1 overexpression in TNBC cell lines,28,32 our results show that FOXQ1 has low expression in many TNBC cell lines (Figure 5A and B), but FOXQ1 is overexpressed in some TNBC cell lines (Figure 5A and B). The difference in FOXQ1 overexpression across TNBC cell lines is striking. It has been previously suggested that, on the basis of molecular features and morphology and invasion potential, TNBC might be further subdivided into basal A and basal B types.47 Basal A has a less differentiated and more epithelial-like morphology, whereas basal B has a more mesenchymal-like morphology which is more invasive.47 Basal B has additionally been characterized as exhibiting more stem-cell-like characteristics (“stemness”), as cells of this subgroup have been found to possess the CD44+/CD24−/low phenotype47 normally associated with mammary cancer stem cells (CSCs).76,77 Moreover, a role of FOXQ1 in stemness,54,78 and EMT28 has been reported. This could provide an explanation of why FOXQ1 is overexpressed in certain TNBC cell lines rather than other FOXQ1 TNBC cell lines (Figure 5A and B). Further classification of TNBC cell lines into basal A and B could provide a clearer picture of the different consequences of FOXQ1 expression between these two TNBC subtypes.
Very few direct targets of FOXQ1 are known. These genes may be positively regulated by FOXQ1 such as PDGFRα and β, Twist1 and Zeb254 or negatively regulated by FOXQ1 such as DACH1 gene.30 Consistent with these studies, we found in preliminary studies of BC gene expression databases a significant positive correlation between FOXQ1 and PDGFRα or β, Twist1 and Zeb2 and a significant negative correlation between FOXQ1 and DACH1 (Supplementary Figure 3). While it has been suggested that FOXQ1 regulates these genes that play roles in BC stemness and EMT, there is no specific transcriptional signature that has been proposed for FOXQ1 in either development or cancer. The underlying mechanisms altering FOXQ1 expression in BC subtypes, as well remain elusive. This is not surprising given the lack of knowledge on the regulatory machinery controlling the expression of transcription factors such as FOXQ1. The actual number of genes regulated by FOXQ1 is unknown but could number in the thousands based upon similar analyses of the direct targets of the related FOXC1 transcription factor.79 Thus, while a “transcriptional addiction in cancer” has been proposed,80 it remains a challenge to precisely identify in cancer how and when transcription factors are themselves regulated and activated, and then to understand the target genes regulated by the activity of these transcription factors in normal and disease states.81 Epigenetics, gene function redundancy, tissue specificity, predicting enhancer-promoter regions, and the role of coactivators are a few among many other challenges to studying transcription factors in cancer.81 Future studies that reveal the regulation of FOXQ1 expression, together with the identification of direct transcriptional targets of FOXQ1, might reveal how FOXQ1 expression is controlled and the consequences of its dysregulation of this regulation in BC.
Our research, and that from other groups,29,55–58 clearly demonstrate that FOXQ1 is another member of the FOX class of transcription factors with a key emerging role in cancer.14,15 Previous studies have investigated the role of FOXQ1 expression, regulation, and function in relation to the expression of FOXF2 and FOXM1 genes, function and roles in BC and other diseases. Our findings that FOXQ1 is an independent predictor of overall survival in BC when adjusted to FOXM1 and FOXF2 expressions highlight the prognostic significance of FOXQ1 expression in BC patients (Table 5). FOXQ1 and FOXM1 expression levels have previously been shown to have prognostic value in colorectal cancer patients.56 Moreover, a recent study suggested that FOXQ1 expression is negatively regulated by FOXF2 in BC cells.82 FOXQ1, FOXF2, and FOXC1 are all located within a 300 kb region of chromosome 6, physically linking in close proximity three FOX transcription factors with substantial roles in BC,15,19,83,84 development,13,24,25,85–87 and in several diseases.88–94 Our results suggest that FOXQ1 expression in BC patients could have significant prognostic value for survival from BC.
Conclusion
In our study, we show that FOXQ1 is differentially expressed in BC subtypes. Low expression of FOXQ1 predicts poor overall survival in BC and is an independent prognostic predictor. Moreover, FOXQ1 CNV alterations are not associated with its expression in BC. Understanding the consequences of FOXQ1 expression in BC subtypes could have clinical value.
Acknowledgments
We would like to thank Dr. Ing Swie Goping for her critical feedback, suggestions, and comments during the process of writing the manuscript. We would also like to thank Dr. Xiaoyun Tang for his help with RNA and DNA extraction from breast cancer patient tissues and normal breast tissues, and Dr. David Eisenstat for providing access to LightCycler 96 Real-Time PCR System.
Funding
This work was supported by the Canadian Institute for Health Research (M.A. Walter, [grant number G118160216]) and from the University of Alberta.
Disclosure
Todd PW McMullen is a consultant with Eli-Lilly and Ipsen. The authors report no other potential conflicts of interest for this work.
References
1. Feng Y, Spezia M, Huang S, et al. Breast cancer development and progression: risk factors, cancer stem cells, signaling pathways, genomics, and molecular pathogenesis. Genes Dis. 2018;5(2):77–106. doi:10.1016/J.GENDIS.2018.05.001
2. Badve S, Dabbs DJ, Schnitt SJ, et al. Basal-like and triple-negative breast cancers: a critical review with an emphasis on the implications for pathologists and oncologists. Nat Rev. 2011;157–167. doi:10.1038/modpathol.2010.200.
3. Polyak K. Heterogeneity in breast cancer. J Clin Invest. 2011;121(10):3786–3788. doi:10.1172/JCI60534
4. Blows FM, Driver KE, Schmidt MK, et al. Subtyping of breast cancer by immunohistochemistry to investigate a relationship between subtype and short and long term survival: a collaborative analysis of data for 10,159 cases from 12 studies. PLoS Med. 2010;7(5):e1000279. doi:10.1371/journal.pmed.1000279
5. Prat A, Adamo B, Cheang MCU, Anders CK, Carey LA, Perou CM. Molecular characterization of basal-like and non-basal-like triple-negative breast cancer. Oncologist. 2013;18(2):123–133. doi:10.1634/theoncologist.2012-0397
6. Li Y, Tang X-Q, Bai Z, Dai X. Exploring the intrinsic differences among breast tumor subtypes defined using immunohistochemistry markers based on the decision tree. Sci Rep. 2016;6(1):35773. doi:10.1038/srep35773
7. Buffa FM, Camps C, Winchester L, et al. microRNA-associated progression pathways and potential therapeutic targets identified by integrated mRNA and microRNA expression profiling in breast cancer. Cancer Res. 2011;71(17):5635–5645. doi:10.1158/0008-5472.CAN-11-0489
8. Blenkiron C, Goldstein LD, Thorne NP, et al. MicroRNA expression profiling of human breast cancer identifies new markers of tumor subtype. Genome Biol. 2007;8(10):R214. doi:10.1186/gb-2007-8-10-r214
9. Walker RA. Immunohistochemical markers as predictive tools for breast cancer. J Clin Pathol. 2008;61(6):689–696. doi:10.1136/JCP.2006.041830
10. Perez EA. Breast cancer management: opportunities and barriers to an individualized approach. Oncologist. 2011;16(S1):20–22. doi:10.1634/theoncologist.2011-s1-20
11. Perou CM, Sørile T, Eisen MB, et al. Molecular portraits of human breast tumours. Nature. 2000;406(6797):747–752. doi:10.1038/35021093
12. Sørlie T, Perou CM, Tibshirani R, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001;98(19):10869–10874. doi:10.1073/pnas.191367098
13. Hannenhalli S, Kaestner KH. The evolution of Fox genes and their role in development and disease. Nat Rev. 2009;10(4):233–240. doi:10.1038/nrg2523
14. Lam EW, Brosens JJ, Gomes AR, Koo CY. Forkhead box proteins: tuning forks for transcriptional harmony. Nat Rev Cancer. 2013;13(7):482–495. doi:10.1038/nrc3539
15. Elian FA, Yan E, Walter MA, et al. FOXC1, the new player in the cancer sandbox. Oncotarget. 2018;9(8):8165–8178. doi:10.18632/oncotarget.22742
16. Bach D-H, Long N, Luu TTT, et al. The dominant role of forkhead box proteins in cancer. Int J Mol Sci. 2018;19(10):3279. doi:10.3390/ijms19103279
17. Kalin TV, Wang IC, Ackerson TJ, et al. Increased levels of the FoxM1 transcription factor accelerate development and progression of prostate carcinomas in both TRAMP and LADY transgenic mice. Cancer Res. 2006;66(3):1712–1720. doi:10.1158/0008-5472.CAN-05-3138
18. Paik JH, Kollipara R, Chu G, et al. FoxOs are lineage-restricted redundant tumor suppressors and regulate endothelial cell homeostasis. Cell. 2007;128(2):309–323. doi:10.1016/j.cell.2006.12.029
19. Wang Q-S, Kong P-Z, Li X-Q, Yang F, Feng Y-M. FOXF2 deficiency promotes epithelial-mesenchymal transition and metastasis of basal-like breast cancer. Breast Cancer Res. 2015;17(1):30. doi:10.1186/s13058-015-0531-1
20. Song X, Fiati Kenston SS, Zhao J, Yang D, Gu Y. Roles of FoxM1 in cell regulation and breast cancer targeting therapy. Med Oncol. 2017;34(3). doi:10.1007/s12032-017-0888-3
21. Hurtado A, Holmes KA, Ross-Innes CS, Schmidt D, Carroll JS. FOXA1 is a key determinant of estrogen receptor function and endocrine response. Nat Genet. 2011;43(1):27–33. doi:10.1038/ng.730
22. Li Y, Zhang Y, Yao Z, Li S, Yin Z, Xu M. Forkhead box Q1: a key player in the pathogenesis of tumors (Review). Int J Oncol. 2016;49(1):51–58. doi:10.3892/ijo.2016.3517
23. Katoh M, Katoh M. Human FOX gene family (Review). Int J Oncol. 2004;25(5):1495–1500.
24. Hoggatt AM, Kriegel AM, Smith AF, Herring BP. Hepatocyte nuclear factor-3 homologue 1 (HFH-1) represses transcription of smooth muscle-specific genes. J Biol Chem. 2000;275(40):31162–31170. doi:10.1074/jbc.M005595200
25. Jonsson H, Peng SL. Forkhead transcription factors in immunology. Cell Mol Life Sci. 2005;62(4):397–409. doi:10.1007/s00018-004-4365-8
26. Abba M, Patil N, Rasheed K, et al. Unraveling the role of FOXQ1 in colorectal cancer metastasis. Mol Cancer Res. 2013;11(9):1017–1028. doi:10.1158/1541-7786.MCR-13-0024
27. Gao M, Shih IM, Wang TL. The role of forkhead box Q1 transcription factor in ovarian epithelial carcinomas. Int J Mol Sci. 2012;13:13881–13893. doi:10.3390/ijms131113881
28. Zhang H, Meng F, Liu G, et al. Forkhead transcription factor Foxq1 promotes epithelial–mesenchymal transition and breast cancer metastasis. Cancer Res. 2011;71(4):1292–1301. doi:10.1158/0008-5472.CAN-10-2825
29. Kaneda H, Arao T, Tanaka K, et al. FOXQ1 is overexpressed in colorectal cancer and enhances tumorigenicity and tumor growth. Cancer Res. 2010;70(5):2053–2063. doi:10.1158/0008-5472.CAN-09-2161
30. Kim S-H, Kaschula CH, Priedigkeit N, Lee AV, Singh SV. Forkhead box Q1 is a novel target of breast cancer stem cell inhibition by diallyl trisulfide. J Biol Chem. 2016;291(26):13495–13508. doi:10.1074/jbc.M116.715219
31. Zhan H, Xu J, Wang L, Wu D, Zhang G, Hu S. FoxQ1 is a novel molecular target for pancreatic cancer and is associated with poor prognosis. Curr Mol Med. 2015;15(5):469–477. doi:10.2174/1566524015666150630125247
32. Han X, Guo X, Zhang W, Cong Q. MicroRNA-937 inhibits the malignant phenotypes of breast cancer by directly targeting and downregulating forkhead box Q1. Onco Targets Ther. 2019;12:4813–4824. doi:10.2147/OTT.S207593
33. Wu X, Gardashova G, Lan L, et al. Targeting the interaction between RNA-binding protein HuR and FOXQ1 suppresses breast cancer invasion and metastasis. Commun Biol. 2020;3(1):193. doi:10.1038/s42003-020-0933-1
34. Anaya J. OncoLnc: linking TCGA survival data to mRNAs, miRNAs, and lncRNAs. PeerJ Comput Sci. 2016;2(6):e67. doi:10.7717/peerj-cs.67
35. Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45:W98–W102. doi:10.1093/nar/gkx247
36. Liu J, Lichtenberg T, Hoadley KA, et al. An integrated TCGA pan-cancer clinical data resource to drive high-quality survival outcome analytics. Cell. 2018;173(2):400–416.e11. doi:10.1016/j.cell.2018.02.052
37. Chandrashekar DS, Bashel B, Balasubramanya SAH, et al. UALCAN: a portal for facilitating tumor subgroup gene expression and survival analyses. Neoplasia (United States). 2017;19(8):649–658. doi:10.1016/j.neo.2017.05.002
38. Zhu J, Sanborn JZ, Benz S, et al. The UCSC cancer genomics browser. Nat Methods. 2009;6(4):239–240. doi:10.1038/nmeth0409-239
39. Berger AC, Korkut A, Kanchi RS, et al. A comprehensive pan-cancer molecular study of gynecologic and breast cancers. Cancer Cell. 2018;33(4):690–705.e9. doi:10.1016/j.ccell.2018.03.014
40. Rasnitsyn A, Doucette L, Seifi M, Footz T, Raymond V, Walter MA. FOXC1 modulates MYOC secretion through regulation of the exocytic proteins RAB3GAP1, RAB3GAP2 and SNAP25. PLoS One. 2017;12(6):e0178518. doi:10.1371/journal.pone.0178518
41. Gaetano CG, Samadi N, Tomsig JL, Macdonald TL, Lynch KR, Brindley DN. Inhibition of autotaxin production or activity blocks lysophosphatidylcholine-induced migration of human breast cancer and melanoma cells. Mol Carcinog. 2009;48(9):801–809. doi:10.1002/mc.20524
42. Seifi M, Footz T, Taylor SAM, Elhady GM, Abdalla EM, Walter MA. Novel PITX2 gene mutations in patients with Axenfeld-Rieger syndrome. Acta Ophthalmol. 2016;94(7):e571–e579. doi:10.1111/aos.13030
43. Pedregosa F, Varoquaux G, Gramfort A, et al. Scikit-learn: machine learning in python. J Mach Learn Res. 2011;12:2825–2830.
44. Witten DM, Tibshirani RA. Framework for feature selection in clustering. J Am Stat Assoc. 2010;105(490):713–726. doi:10.1198/jasa.2010.tm09415
45. Barretina J, Caponigro G, Stransky N, et al. The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483(7391):603–607. doi:10.1038/nature11003
46. Dai X, Cheng H, Bai Z, Li J. Breast cancer cell line classification and its relevance with breast tumor subtyping. J Cancer. 2017;8(16):3131–3141. doi:10.7150/jca.18457
47. Neve RM, Chin K, Fridlyand J, et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell. 2006;10(6):515–527. doi:10.1016/j.ccr.2006.10.008
48. Van’t Veer LJ, Dai H, Van de Vijver MJ, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415(6871):530–536. doi:10.1038/415530a
49. Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011;12(1):323. doi:10.1186/1471-2105-12-323
50. Hoadley KA, Yau C, Hinoue T, et al. Cell-of-origin patterns dominate the molecular classification of 10,000 tumors from 33 types of cancer. Cell. 2018;173(2):291–304.e6. doi:10.1016/j.cell.2018.03.022
51. Shao X, Lv N, Liao J, et al. Copy number variation is highly correlated with differential gene expression: a pan-cancer study. BMC Med Genet. 2019;20(1):175. doi:10.1186/s12881-019-0909-5
52. Wang S, Li GX, Tan CC, et al. FOXF2 reprograms breast cancer cells into bone metastasis seeds. Nat Commun. 2019;10(1). doi:10.1038/S41467-019-10379-7
53. Ross JB, Huh D, Noble LB, Tavazoie SF. Identification of molecular determinants of primary and metastatic tumour re-initiation in breast cancer. Nat Cell Biol. 2015;17(5):651–664. doi:10.1038/ncb3148
54. Meng F, Speyer CL, Zhang B, et al. PDGFRα and β play critical roles in mediating Foxq1-driven breast cancer stemness and chemoresistance. Cancer Res. 2015;75(3):584–593. doi:10.1158/0008-5472.CAN-13-3029
55. Cui X, Zhang J, Lv J, et al. Prognostic value of FOXQ1 in patients with malignant solid tumors: a meta-analysis. Onco Targets Ther. 2017;10:1777–1781. doi:10.2147/OTT.S130905
56. Weng W, Okugawa Y, Toden S, Toiyama Y, Kusunoki M, Goel A. FOXM1 and FOXQ1 are promising prognostic biomarkers and novel targets of tumor-suppressive miR-342 in human colorectal cancer. Clin Cancer Res. 2016;22(19):4947–4957. doi:10.1158/1078-0432.CCR-16-0360
57. Liang SH, Yan XZ, Wang BL, et al. Increased expression of FOXQ1 is a prognostic marker for patients with gastric cancer. Tumor Biol. 2013;34(5):2605–2609. doi:10.1007/s13277-013-0808-x
58. Qiao Y, Jiang X, Lee ST, Karuturi RKM, Hooi SC, Yu Q. FOXQ1 regulates epithelial-mesenchymal transition in human cancers. Cancer Res. 2011;71(8):3076–3086. doi:10.1158/0008-5472.CAN-10-2787
59. Martín M, González-Rivera M, Morales S, et al. Abstract P6-08-10: prospective study of the impact of the ProsignaTM assay on adjuvant clinical decision-making in women with estrogen receptor-positive, HER2-negative, node-negative breast cancer: a GEICAM study. Cancer Res. 2015. doi:10.1158/1538-7445.sabcs14-p6-08-10
60. Prat A, Cheang MCU, Martín M, et al. Prognostic significance of progesterone receptor-positive tumor cells within immunohistochemically defined luminal a breast cancer. J Clin Oncol. 2013;31(2):203–209. doi:10.1200/JCO.2012.43.4134
61. Chia SK, Bramwell VH, Tu D, et al. A 50-gene intrinsic subtype classifier for prognosis and prediction of benefit from adjuvant tamoxifen. Clin Cancer Res. 2012;18(16):4465–4472. doi:10.1158/1078-0432.CCR-12-0286
62. Creighton CJ. The molecular profile of luminal B breast cancer. Biol Targets Ther. 2012;6:289–297. doi:10.2147/BTT.S29923
63. Ellis MJ, Tao Y, Luo J, et al. Outcome prediction for estrogen receptor-positive breast cancer based on postneoadjuvant endocrine therapy tumor characteristics. J Natl Cancer Inst. 2008;100(19):1380–1388. doi:10.1093/jnci/djn309
64. Hirukawa A, Smith HW, Zuo D, et al. Targeting EZH2 reactivates a breast cancer subtype-specific anti-metastatic transcriptional program. Nat Commun. 2018;9(1):1–15. doi:10.1038/s41467-018-04864-8
65. Mendes D, Alves C, Afonso N, et al. The benefit of HER2-targeted therapies on overall survival of patients with metastatic HER2-positive breast cancer - a systematic review. Breast Cancer Res. 2015;17(1):1–14. doi:10.1186/s13058-015-0648-2
66. Dai X, Li T, Bai Z, et al. Breast cancer intrinsic subtype classification, clinical use and future trends. Am J Cancer Res. 2015;5(10):2929–2943.
67. Kundu ST, Byers LA, Peng DH, et al. The miR-200 family and the miR-183~96~182 cluster target Foxf2 to inhibit invasion and metastasis in lung cancers. Oncogene. 2016;35(2):173–186. doi:10.1038/onc.2015.71
68. Lo P-K, Lee JS, Liang X, Sukumar S. The dual role of FOXF2 in regulation of DNA replication and the epithelial-mesenchymal transition in breast cancer progression. Cell Signal. 2016;28(10):1502–1519. doi:10.1016/j.cellsig.2016.06.021
69. Sahu B, Laakso M, Ovaska K, et al. Dual role of FoxA1 in androgen receptor binding to chromatin, androgen signalling and prostate cancer. EMBO J. 2011;30(19):3962–3976. doi:10.1038/emboj.2011.328
70. Badve S, Turbin D, Thorat MA, et al. FOXA1 expression in breast cancer - Correlation with luminal subtype A and survival. Clin Cancer Res. 2007;13(15):4415–4421. doi:10.1158/1078-0432.CCR-07-0122
71. Albergaria A, Paredes J, Sousa B, et al. Expression of FOXA1 and GATA-3 in breast cancer: the prognostic significance in hormone receptor-negative tumours. Breast Cancer Res. 2009;11(3). doi:10.1186/bcr2327
72. Bernardo GM, Bebek G, Ginther CL, et al. FOXA1 represses the molecular phenotype of basal breast cancer cells. Oncogene. 2013;32(5):554–563. doi:10.1038/onc.2012.62
73. van de Vijver MJ, He YD, van ’T Veer LJ, et al. A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med. 2002;347(25):1999–2009. doi:10.1056/NEJMoa021967
74. Park HS, Jang MH, Kim EJ, et al. High EGFR gene copy number predicts poor outcome in triple-negative breast cancer. Mod Pathol. 2014;27(9):1212–1222. doi:10.1038/modpathol.2013.251
75. Hyman E, Kauraniemi P, Hautaniemi S, et al. Impact of DNA amplification on gene expression patterns in breast cancer. Cancer Res. 2002;62(21):6240–6245.
76. Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100(7):3983–3988. doi:10.1073/pnas.0530291100
77. Dontu G, Al-Hajj M, Abdallah WM, Clarke MF, Wicha MS. Stem cells in normal breast development and breast cancer. Cell Prolif. 2003;36(1):59–72. doi:10.1046/j.1365-2184.36.s.1.6.x
78. Hardy K, Wu F, Tu W, et al. Identification of chromatin accessibility domains in human breast cancer stem cells. Nucleus. 2016;7(1):50–67. doi:10.1080/19491034.2016.1150392
79. Berry FB, Skarie JM, Mirzayans F, et al. FOXC1 is required for cell viability and resistance to oxidative stress in the eye through the transcriptional regulation of FOXO1A. Hum Mol Genet. 2008;17(4):490–505. doi:10.1093/hmg/ddm326
80. Bradner JE, Hnisz D, Young RA. Transcriptional Addiction in Cancer. Cell. 2017;168(4):629–643. doi:10.1016/j.cell.2016.12.013
81. Lambert SA, Jolma A, Campitelli LF, et al. The human transcription factors. Cell. 2018;172(4):650–665. doi:10.1016/j.cell.2018.01.029
82. Kang L-J, Yu Z-H, Cai J, et al. Reciprocal transrepression between FOXF2 and FOXQ1 controls basal-like breast cancer aggressiveness. FASEB J. 2019;33:6564–6573. doi:10.1096/fj.201801916R
83. Ray PS, Wang J, Qu Y, et al. FOXC1 is a potential prognostic biomarker with functional significance in basal-like breast cancer. Cancer Res. 2010;70(10):3870–3877. doi:10.1158/0008-5472.CAN-09-4120
84. Kong P-Z, Yang F, Li L, Li X-Q, Feng Y-M. Decreased FOXF2 mRNA expression indicates early-onset metastasis and poor prognosis for breast cancer patients with histological grade II tumor. PLoS One. 2013;8(4):e61591. doi:10.1371/journal.pone.0061591
85. Doucette LP, Rasnitsyn A, Seifi M, Walter MA. The interactions of genes, age, and environment in glaucoma pathogenesis. Surv Ophthalmol. 2015;60(4):310–326. doi:10.1016/j.survophthal.2015.01.004
86. Ormestad M, Astorga J, Landgren H, et al. Foxf1 and Foxf2 control murine gut development by limiting mesenchymal Wnt signaling and promoting extracellular matrix production. Development. 2006;133(5):833–843. doi:10.1242/dev.02252
87. Bademci G, Abad C, Incesulu A, et al. FOXF2 is required for cochlear development in humans and mice. Hum Mol Genet. 2018. doi:10.1093/hmg/ddy431
88. Chanda B, Asai-Coakwell M, Ye M, et al. A novel mechanistic spectrum underlies glaucoma-associated chromosome 6p25 copy number variation. Hum Mol Genet. 2008;17(22):3446–3458. doi:10.1093/hmg/ddn238
89. Mears AJ, Jordan T, Mirzayans F, et al. Mutations of the forkhead/winged-helix gene, FKHL7, in patients with axenfeld-rieger anomaly. Am J Hum Genet. 1998;63(5):1316–1328. doi:10.1086/302109
90. Mirzayans F, Mears AJ, Guo SW, Pearce WG, Walter MA. Identification of the human chromosomal region containing the iridogoniodysgenesis anomaly locus by genomic-mismatch scanning. Am J Hum Genet. 1997;61(1):111–119. doi:10.1086/513894
91. Mears AJ, Mirzayans F, Gould DB, Pearce WG, Walter MA. Autosomal dominant iridogoniodysgenesis anomaly maps to 6p25. Am J Hum Genet. 1996;59(6):1321–1327.
92. Walter MA, Mirzayans F, Mears AJ, Hickey K, Pearce WG. Autosomal-dominant iridogoniodysgenesis and Axenfeld-Rieger syndrome are genetically distinct. Ophthalmology. 1996;103(11):1907–1915. doi:10.1016/S0161-6420(96)30408-9
93. Reis LM, Tyler RC, Volkmann Kloss BA, et al. PITX2 and FOXC1 spectrum of mutations in ocular syndromes. Eur J Hum Genet. 2012;20(12):1224–1233. doi:10.1038/ejhg.2012.80
94. Anderlid BM, Schoumans J, Hallqvist Å, et al. Cryptic subtelomeric 6p deletion in a girl with congenital malformations and severe language impairment. Eur J Hum Genet. 2003;11(1):89–92. doi:10.1038/sj.ejhg.5200907
95. Koboldt DC, Fulton RS, McLellan MD, et al. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. doi:10.1038/nature11412
96. Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–404. doi:10.1158/2159-8290.CD-12-0095
97. Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):1. doi:10.1126/scisignal.2004088
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.