Back to Journals » Drug Design, Development and Therapy » Volume 13
Formulation, Characterization And Evaluation Of Curcumin- Loaded PLGA- TPGS Nanoparticles For Liver Cancer Treatment
Authors Chen XP, Li Y, Zhang Y, Li GW
Received 10 April 2019
Accepted for publication 27 September 2019
Published 16 October 2019 Volume 2019:13 Pages 3569—3578
DOI https://doi.org/10.2147/DDDT.S211748
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sukesh Voruganti
Xiao-ping Chen, Yi Li, Yu Zhang, Gao-wei Li
Department of Oncology, Beibei District Hospital of Traditional Chinese Medicine, Chongqing, People’s Republic of China
Correspondence: Yi Li
Beibei District Hospital of Traditional Chinese Medicine, No. 380, General Road, Beibei District, Chongqing 400711, People’s Republic of China
Tel/fax +86 23 68863618
Email [email protected]
Background: Liver cancer is a major health problem facing mankind. Currently, the focus of research is to improve the treatment of liver cancer using a variety of treatment options such as providing chemotherapy drugs through nanocarriers.
Purpose: The aim of this study was to prepare a curcumin-loaded (PLGA/TPGS) NPs delivery system by the emulsification-solvent evaporation method in order to achieve synergistic antitumor activity against liver cancer.
Methods: Curcumin-loaded (PLGA/TPGS) NPs were prepared by the emulsification and solvent evaporation method. The physical and chemical characteristics of NPs such as size, morphology, and release profiles were discussed. In vitro and in vivo studies were carried out to evaluate its anti-tumor activity in target cells.
Results: Curcumin-loaded (PLGA/TPGS) NPs could be successfully internalized by HepG2 cells and play a synergistic role in inhibiting the growth of hepatocellular carcinoma cells. They exhibited high target organ accumulation, superior antitumor efficiency, and lower toxicity in vivo.
Conclusion: The present study indicates that the curcumin-loaded (PLGA/TPGS) NPs provide a promising platform for the treatment of liver cancer.
Keywords: curcumin, PLGA, TPGS, nanoparticles, liver cancer
Introduction
Liver is the largest solid organ of the human body and bears some important functions. The occurrence of malignant liver tumors will lead to life-threatening consequences. Liver cancer is the third most fatal cancer in the world, which is usually treated by surgery.1 Since most patients are diagnosed at advanced stages, effective non-surgical treatment, such as systemic chemotherapy, is urgently needed.2 At present, the most active single drugs for liver cancer include adriamycin, sorafenib, 5-fluorouracil and cisplatin. However, with a response rate of only 10%, they have no significant effect on the overall survival rate. This may be due to the toxicity and poor response resulted from chemoresistance.3 Some plant extracts have shown significant inhibitory effects on cancer cells without serious adverse reactions. Among them, curcumin (CUR) has exhibited a variety of anti-cancer mechanisms in many basic studies, including inhibiting the initiation, progression, invasion and metastasis of cancer cells. It has been proved in the literature that CUR acts on protein kinases MAPK, Akt and Bcl-2; transcription factors NF-κB, AP-1 and STAT-3 and enzymes such as COX-2, matrix metalloproteinases (MMPs) and LOX.4–14 Although CUR has many pharmacological activities, its clinical application is still limited by many factors such as poor water solubility (≈0.125 mg/L), low bioavailability, fast metabolism and systemic clearance. The best pharmacological action requires an oral dose of more than 8.0g/day.15 Improving CUR bioavailability is a major challenge.
In recent years, in order to overcome poor absorption and other limitations of CUR, researchers have adopted new pharmaceutical methods such as liposomes, solid dispersions, complexes, emulsions, micelles, nanogels and microspheres.16–22 But these strategies also have some shortcomings, such as limited benefits and tedious preparation process. In this study, we aim to build a liver target system containing CUR in the form of nanoparticles (NPs), which could improve the stability of drugs by dispersing them into carrier material and reducing their contact with the outer environment.23 At the same time, by adjusting the particle size, surface charge and encapsulation materials, CUR-loaded NPs can perform liver targeting function.24 Therefore, targeted drug delivery based on NPs is used to accumulate drugs in tumors so as to improve the permeability, retention rate and pharmacokinetic characteristics of drugs and to reduce side effects.
Nanocarrier-based chemotherapeutic agent system has been considered as a new and promising strategy for cancer treatment. As we all know, PLGA (polylactic-co-glycolic acid) has been approved by the FDA (Food and Drug Administration) as a drug material for drug delivery systems and has been widely used.25 It is a macromolecule material with good biocompatibility and no immunogenicity and can be biodegraded into non-toxic natural products. D-a-Tocopheryl polyethylene glycol 1000 succinate (TPGS), an excellent emulsifier for preparing NPs, can improve drug permeability and reduce P-glycoprotein (P-gp)-mediated multiple drug resistance (MDR) in cancer cells.26,27
Song et al prepared a solid dispersion formulation of CUR with TPGS and mannitol to improve the solubility, dissolution rate, oral bioavailability and cell permeability of CUR.28 Since the system was administered orally, it has not been confirmed whether it has liver targeting functions. Additionally, because it has been applied only in intestinal cancer models, its effectiveness in liver cancer models is not clear yet.
This study aims to prepare a CUR-loaded (PLGA/TPGS) NPs delivery system by the emulsification-solvent evaporation method. NPs were characterized for physical properties, such as morphology and particle size, and in vitro drug release. In addition, cytotoxicity, uptake and in vivo status of NPs were also evaluated in HepG2 and HCA-F cells.
Materials And Methods
Materials
CUR was a gift from Jiaye Biotechnology Co., Ltd. (Chengdu, China). PLGA-TPGS (MW 21–22 kD) was prepared in our own laboratory according to the references.29,30 HepG2 and HCa-F cell was purchased from Xiangshi Biotech Co., Ltd. (Shanghai, China). The other reagents were obtained from Chinese pharmaceutical chemical reagents (Shanghai, China). Polyvinyl alcohol, methanol and acetonitrile (chromatographic grade) were obtained from Sigma (Aldrich, USA). The deionized water used in the study was produced using Milli-Q gradient A10 system.
Preparation
CUR-loaded (PLGA/TPGS) NPs (CPTN) were formulated using the emulsification-solvent evaporation method.31 Simply, 30 mg of PLGA-TPGS was dissolved in chloroform. Free CUR was added to the PLGA-TPGS/chloroform solution and sonicated at 55 W for 1 min in a Branson Sonifier model W-350 to produce the primary emulsion. This emulsion was then added to a solution of 2% PVA and ethanol (1:1) and again sonicated at 55 W for 2 mins to form the final emulsion. Then, the emulsions were stirred for 24 hrs to volatilize the organic phase and centrifuged for 15 mins with a high-speed refrigerated centrifuge (20,000 r/min) to ensure that there were no free drug and carriers residues. Deionized distilled water was used to wash the obtained NPs three times. They were then freeze-dried and lyophilized for 24 hrs on an ATR FD 3.0 system. The NPs were stored at 4°C until further use. Drug-free empty PLGA/TPGS NPs (EPTN) were prepared by the same method. In addition, CUR-loaded PLGA NPs (CPN) were also prepared as described above, using PLGA instead of PLGA-TPGS. CUR/C6-loaded PLGA-TPGS NPs were prepared using the same method, and C6 was added to ethyl acetate quantitatively as a fluorescent label that also contained CUR and PLGA-TPGS.
Characterizations
The laser diffraction method was used to determine the particle size of the newly prepared NPs. The particle size and zeta potential were measured using the Zeta analyzer. A specific amount of CPTN lyophilized powder was dissolved in 10 mL of methanol using a probe sonicator and then analyzed by the HPLC method.
Drug loading and encapsulation efficiency were determined by Equations 1 and 2, respectively.
(1) (2)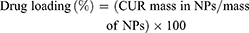
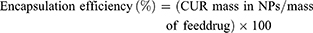
The surface morphology of the prepared CPTN was observed by TEM. Morphological examination was performed using TEM (Philips CM120, Netherlands).
Stability Study
Provisions of Chinese Pharmacopoeia were the main references for stability study. The NPs were placed in a stable chamber at room temperature and saturated with sodium chloride solution (relative humidity=75%±5%). At the end of 0, 1, 2, 3 and 6 months, the properties of the samples were evaluated to determine whether the particle size, zeta potential, encapsulation efficiency% and drug loading% had changed.
In Vitro Release
In this study, in vitro release of CUR NPs was assessed by the dialysis bag diffusion method. In brief, appropriate amounts of CPTN and CPN (≈10 mg CUR) were put dialysis bags (MWCO: 8000–10,000 Da) separately. The dialysis bags were then immersed in 50 mL of PBS (0.01 M; pH 7.4) containing Tween-80 (1%, w/v) and ethanol (20%, v/v) at 37°C in a QYC-200 shaker incubator at 100 rpm. One milliliter of the released medium was withdrawn after 0.125, 0.25, 0.5, 1, 2, 6, 8, 10, 12, 14, 16, 20 and 25 days and replaced with an equivalent volume of fresh release medium to maintain a constant volume. Released samples (CUR content) were determined by the HPLC method and carried out in triplicate.
In Vitro Cytotoxicity
The cytotoxicity of blank NPs, free CUR, CPN and CPTN in HepG2 cells was determined by MTT assay. In short, cells (1×105 cells/mL) were inoculated into 96-well plates in DMEM medium supplemented with 10% FBS and then treated with 100 μL of 0–35 μM of blank NPs, free CUR, CPN and CPTN for 48 hrs. Then, 25 μL of MTT solution (5 mg/mL) was added to each well and the plate was cultured at 37°C for 3 hrs. After incubation, DMSO was added and the absorbance was measured at 570 nm using a microplate reader. The inhibitory concentration (IC)50 values were calculated by the software GraphPad Prism 5.
Intracellular Uptake
The intracellular uptake of free CUR, CPN and CPTN was visualized by confocal microscopy using coumarin-6 as a fluorescent probe. HepG2 cancer cells were grown in DMEM medium supplemented with 10% (v/v) FBS and 5% antibiotics. HepG2 cells were inoculated in a cell culture dish at 1×107 cells per dish for initial density. Cells were then incubated with coumarin-6-adsorbed free CUR, CPN and CPTN (equivalent to 0.1 µg/mL of coumarin-6) for 2 hrs at 37°C±0.5°C.
Subsequently, PBS was used to wash the cells several times and fixed with 4% paraformaldehyde for 10 mins. Finally, the cells were observed under the confocal microscopy. For the quantitative estimation of CUR uptake, the density of cells inoculated on 24-well plates was 3×104. When they reached a confluence of 70–80%, cells were cultured with coumarin-6-adsorbed free CUR, CPN and CPTN (equivalent to 0.1 µg/mL coumarin-6). After 2 hrs of culture, cells were washed several times with cold PBS. Subsequently, Triton X-100 (0.1%) was added to dissolve the cells. Fluorescence intensities were measured at 440 nm excitation wavelength and 520 nm emission wavelength with a multimode microplate reader.
Tissue Distribution Studies
Kunming strain mice were divided into three groups and administered with the same dose of free CUR, CPN and CPTN, respectively, via the tail vein (10 mg/kg). At predetermined time points (0.5, 1, 4, 8, 12, 24 and 36 hrs post i.v. dose administration), three animals from each group were anaesthetized and their blood was collected from the postorbital vein before the serum was separated by centrifugation. The hearts, livers, spleens, lungs and kidneys were rinsed with saline and dried with tissue paper. Serum and tissue samples were frozen at −20°C until analysis. After treatment, the mice were sacrificed, their main organs were weighed and the tissue sections were collected. The specimens were fixed with 4% paraformaldehyde and then paraffin embedded for hematoxylin and eosin (H-E) staining and histological examination. Animal experiments were approved by the ethics committee of the Beibei Traditional Chinese Medicinal Hospital and carried out in accordance with the guidelines provided by the National Institutes of Health.
In Vivo Anti-Tumor Study
Six-week-old male mice weighing 20±1.8 g were inoculated intraperitoneally with HCa-F cells, and ascites were formed about 7 days later. The extracted ascites were washed twice with PBS and adjusted to the cell suspension at a cellular concentration of 2×107 cells/mL. HCA-F cell suspension (0.01 mL/g) was subcutaneously implanted into the right axilla of mice. Treatments were started when the tumor in the nude mice reached a volume of ~15 mm3. This was designated as day 0.
On day 0, mice were randomly divided into four groups: saline, free CUR, CPN and CPTN (n=6). Samples at a dose of 10 mg/kg were injected intravenously via tail vein once per day for 7 days. The tumor size was measured once per for two days during the study. On day 20, all the mice were sacrificed followed by separation and weighing of the tumor block.
Tumor volume (V) was calculated according to the formula: (W2×L)/2, in which W was the widest point of tumor measurement, and L the longest point of tumor dimension. Relative tumor volume (R) was calculated by the formula Vi/V0, where V0 was the tumor volume on day 0, and Vi the tumor volume at the point of measuring. The anti-tumor activity was estimated by relative tumor inhibitory rate (IR, %) which was calculated by the formula: [1-[R(treatment group)/R(negative control group)]] × 100%.
Statistical Analysis
All values were presented as mean and standard deviation (mean ± SD), and the significance of differences was investigated using ANOVA or unpaired Student’s t-test at p<0.05.
Result And Discussion
Characterizations
In this study, an improved emulsion evaporation method was used to prepare CPTN. Emulsifier was added to separate oil and water phases, thus preventing the aggregation of NPs. As an emulsifier, PVA has been widely used in the preparation of NPs. During the formation of NPs, hydrophobic fragments of PVA permeated into the organic phase and remained in the polymer matrix of NPs (PLGA/TGPS). Then, by steric hindrance, the hydrophilic fragments of PVA surrounding the system stabilized the NPs. As shown in Table 1 and Figure 1, the prepared NPs were spherical with a uniform size, as well as high drug loading and encapsulation efficiencies. The average particle size was found to be 110.6±2.3 nm. The encapsulation efficiency% and drug loading% of the preparation were 83.2±2.7% and 10.1±1.5%, respectively. Usually, higher absolute zeta potential results in stronger repulsion between NPs in the dispersion process, which leads to higher stability. In this study, the zeta potential of CPTN was −23.6 mV, which might be related to the formation of molecular polarization and charge adsorption in water. The NPs in this study may have higher zeta potential due to their similar ionic double-layer structure. The absolute zeta potential of CPTN was higher than that of CPN, indicating that CPTN was more stable than CPN in dispersion.
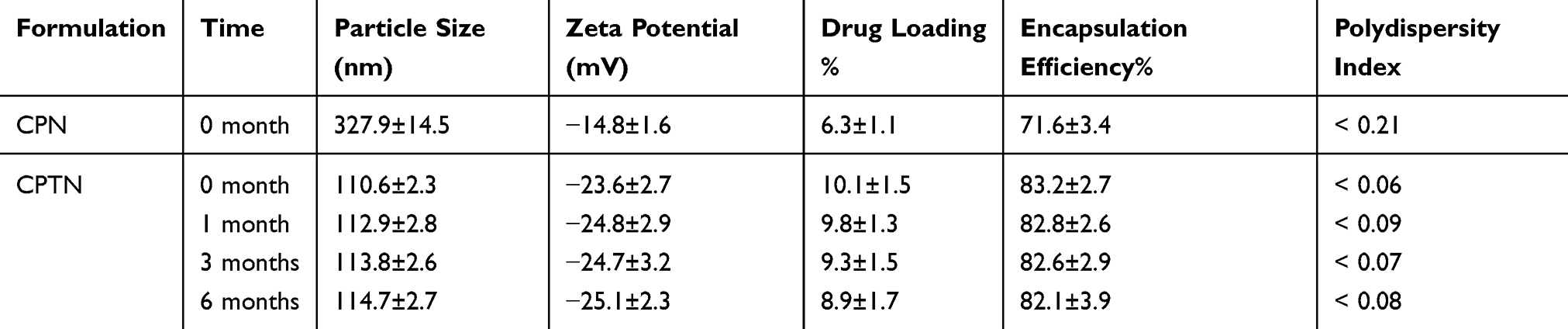 |
Table 1 Physical And Chemical Properties And Stability Data Of CPN And CPTN. Data Represents The Mean ± SD (n = 3) |
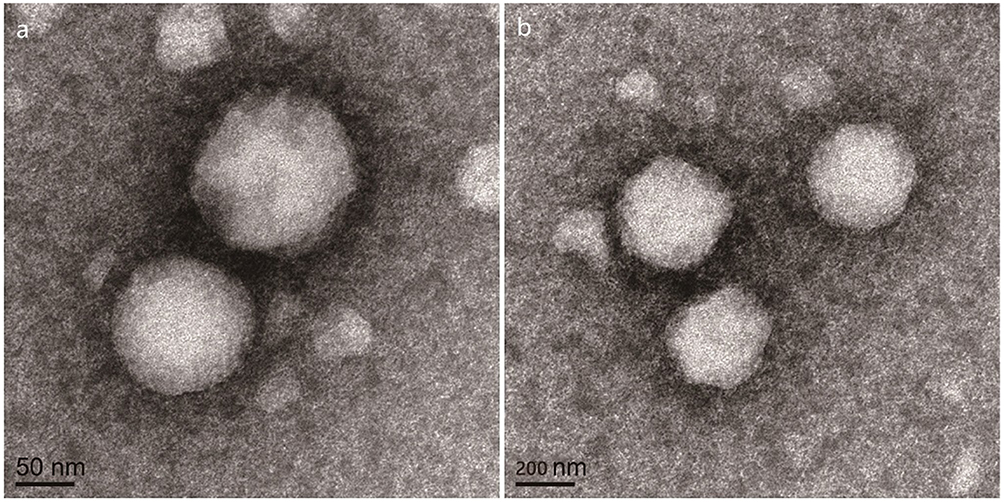 |
Figure 1 The transmission electron microscope of CPTN (A) and CPN (B) (75,000×). |
In Vitro Release
The release kinetics of CUR from the NPs were studied for 25 days and the results are shown in Figure 2. Free CUR exhibited a quicker drug release in PBS at a pH value of 7.4, and 99% of CUR was released in the first 12 hrs. In contrast, CUR release from NPs usually occurred in a biphasic manner, with an initial burst phase followed by a diffusion-controlled slower drug release phase. In this study, an initial burst phase corresponding to about 21% (CPTN) and 28% (CPN) was observed within 0.5 day due to the drug desorption and release from the NPs surface. A sustained CUR release to a total of about 56% (CPTN) and 73% (CPN) was found for different NPs over the entire period of study.
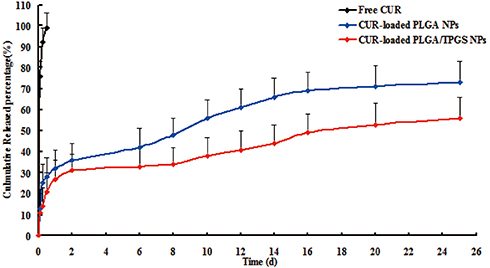 |
Figure 2 The release profile of free CUR, CPN and CPTN (n=6). |
The drug release data obtained were fitted into release kinetic models, including zero order, first order, Higuchi, and Ritger–Peppas equations. The release of CUR from CPTN and CPN calculated in the Ritger–Peppas equations was better than that calculated in others (r=0.9912 and 0.9821 for Ritger–Peppas, respectively). In regard to the in vitro drug release, the rapid initial release was attributed more to the drug adsorbed or weakly bound to the surface area of the NPs, than to the drug incorporated inside NPs. According to the release kinetic models, the release mechanism of CUR from NPs involved diffusion and matrix erosion.32
In Vitro Cytotoxicity
The cytotoxicity results of blank NPs, free CUR, CPN and CPTN in HepG2 cells are shown in Figure 3. Untreated cells served as controls. We observed 90% cell viability in HepG2 with the treatment of blank NPs at 20 μM. These results suggested that blank NPs were nontoxic. By contrast, under the same incubation condition (concentration), the cellular cytotoxicity of CPN increased significantly, which might result from the increase of intracellular drug concentration through the transportation of NPs. In addition, at the same concentration, CPTN exhibited a stronger trend of cellular cytotoxicity. The IC50 of CPTN was found to be 16.3 μM while that of free CUR was 32.2 μM and CPN 25.4 μM in HepG2 cancer cell lines (Table 2). This accounted for almost 35% reduction in the IC50 value of CPTN. The assay demonstrated that CPTN were more effective in arresting cell growth as compared to free CUR and CPN.
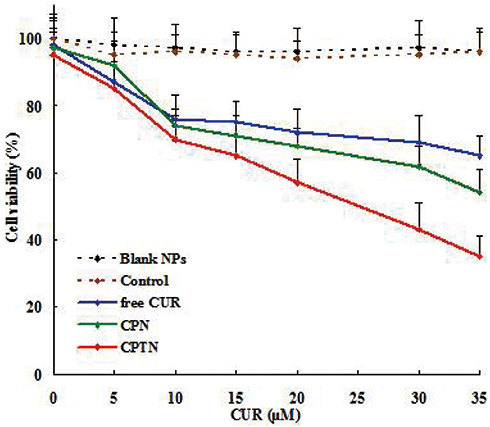 |
Figure 3 In vitro cytotoxicity analysis of free CUR, CPN and CPTN on HepG2 cancer cell lines. Cell viability assay was performed by MTT assay. |
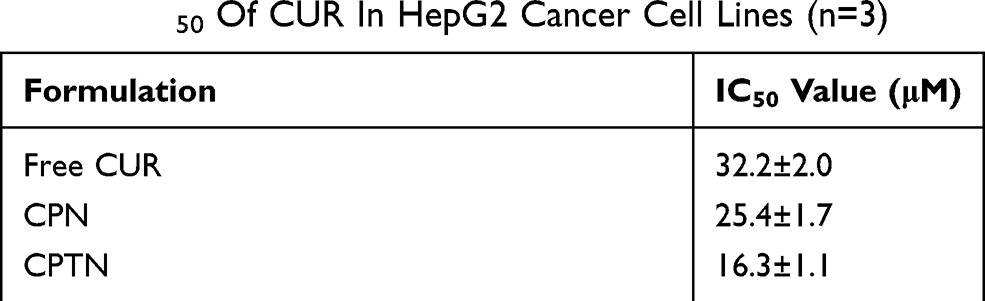 |
Table 2 IC50 Of CUR In HepG2 Cancer Cell Lines (n=3) |
Intracellular Uptake
The intracellular uptakes of free CUR, CPN and CPTN in HepG2 cells were observed by confocal microscopy. As shown in Figure 4, a strong green fluorescence was observed in the cytoplasmic region after incubation of CPTN for 2 hrs. Cell internalization in HepG2 cells was higher for CPTN than for others. The results showed that the surface modification enabled cells to internalize and more drugs to enter cells successfully. In quantitative cell uptake studies, coumarin-6 on the three formulations was quantified by recovering the drug NPs from cells and measuring their fluorescence (normalization to the total cell protein content per milligram). Mean fluorescence intensities for CPTN were approximately 2 folds higher than those for CPN (data not shown). The high cell uptake of CPTN might be due to the absorption-promoting effect of TPGS. TPGS-based nanocarriers showed enhanced circulation behavior and helped drugs target mitochondria. In addition, TPGS was shown to be the most effective inhibitor among P-gp inhibitory surfactants. Moreover, it could induce cell apoptosis and develop a synergistic effect with chemotherapy drugs.27,33,34
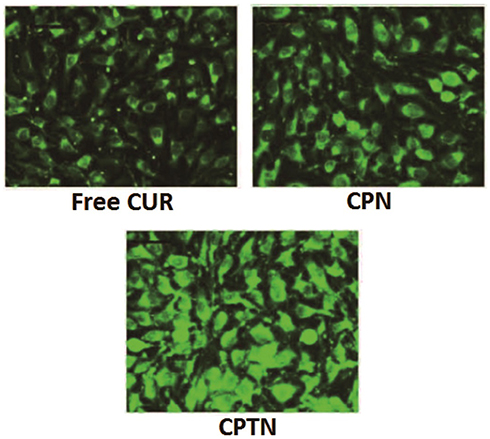 |
Figure 4 Confocal images of cellular uptake of free CUR, CPN and CPTN on HepG2 cancer cell lines. Incubation time was 2 hrs. |
Tissue Distribution Studies
The serum and tissue CUR concentrations versus time after intravenous administration of free CUR, CPN and CPTN were shown in Figure 5. After intravenous injection of free CUR, CPN and CPTN preparations, the distribution of CUR with time was estimated in the heart, liver, spleen, lung, kidney (μg/g) and blood (μg/mL). For CPTN, it did not dissolve rapidly, that was the drug NPs could circulate in the blood circulation as submicron particles for a certain time period. CUR concentration in mice livers (47 μg/g, 4 hrs) of the CPTN group was appreciably higher than in other tissues and plasma. CUR showed the highest value of AUC (805.75 μgh/mL) and target index (TI, 3.27 and 14.27) in liver, and the difference was statistically significant (p<0.05). The TI of different formulations is shown in Table 3. CUR concentration in livers was considered a vital factor in achieving an effective clinical treatment. The tissue tolerability was a major concern in NPs-based targeted system due to the accumulation of the drug and excipients in the targeted organs. The histopathology of the mice liver (Figure 6) did not show any degenerative changes in the CPTN formulation. This proved the safety and biocompatibility of the CPTN as a parenteral formulation for liver targeting. The results showed that CPTN could not only prolong the detention time in vivo, but also increase drug concentration in the liver. Therefore, in theory, it should be ideal for the treatment of liver tumors.
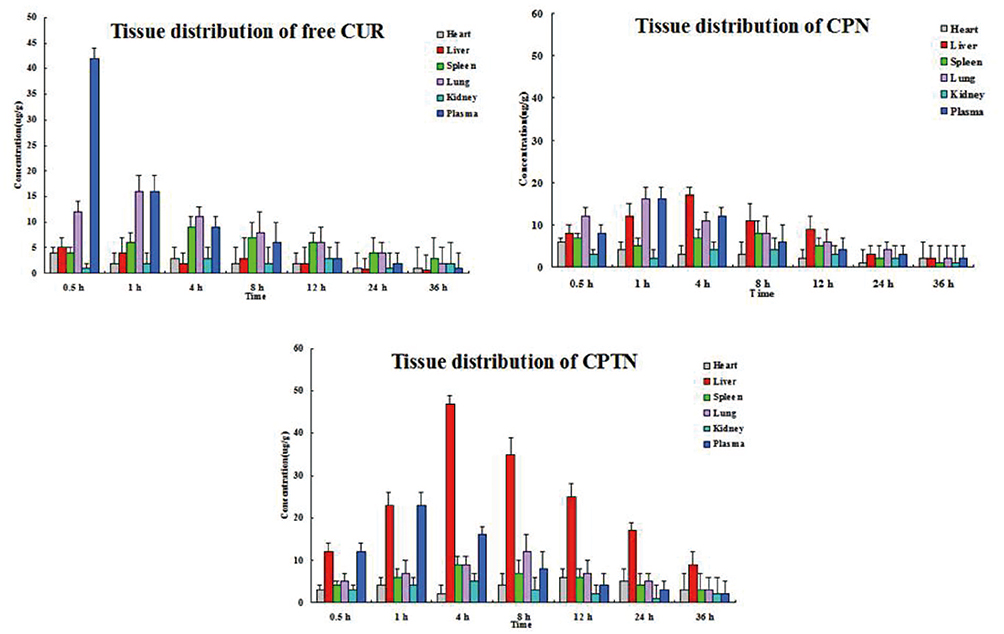 |
Figure 5 The serum and tissue CUR concentrations versus time after intravenous administration of free CUR, CPN and CPTN (n=6). |
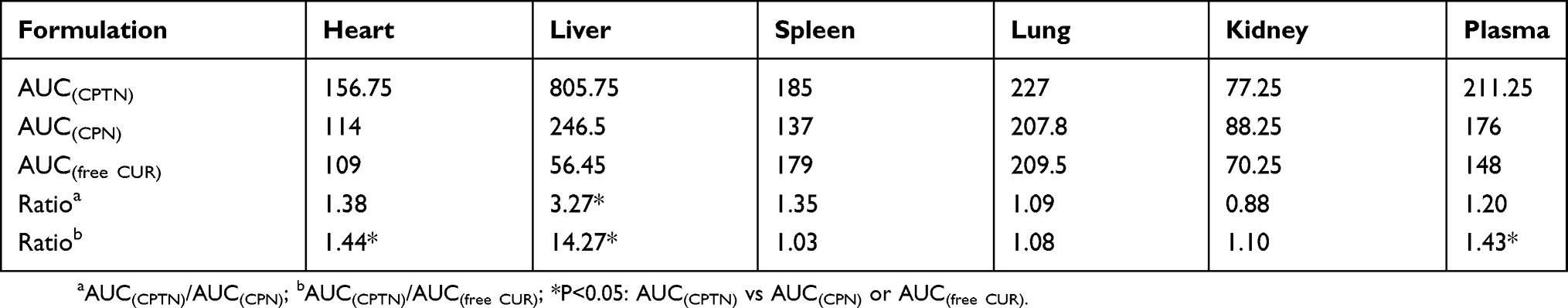 |
Table 3 AUC And TI Values Of CUR In The Plasma And Different Tissues Of Mice After Tail-Intravenous Administration (μgh/mL, N=6, Mean ± SD) |
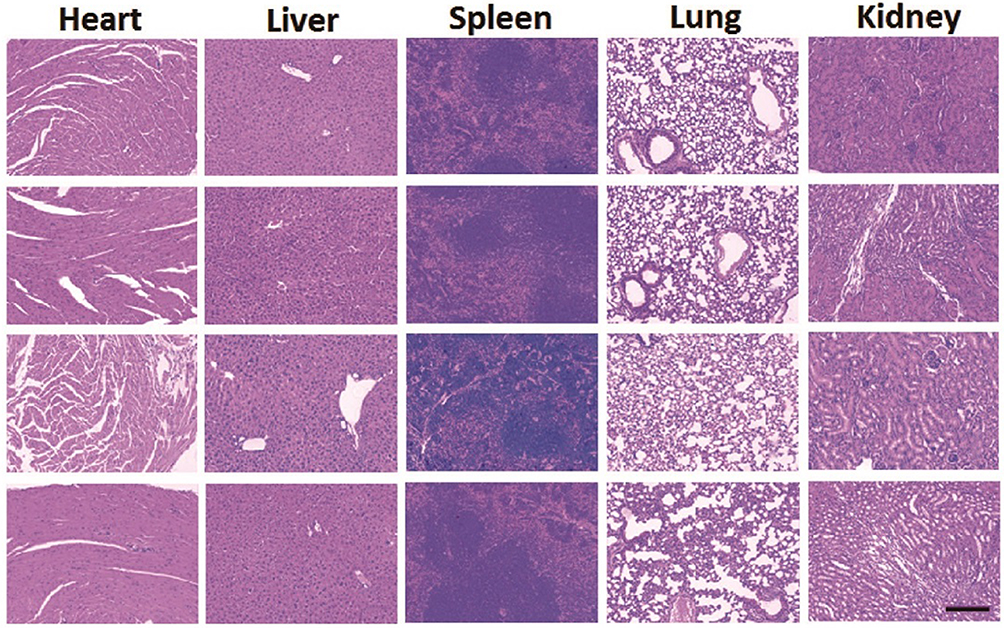 |
Figure 6 Histopathological studies of heart, liver, spleen, lung and kidney among four groups. (From top to bottom was control, free CUR, CPN and CPTN, respectively). |
In Vivo Anti-Tumor Study
To test the antitumor activity of CPTN, mice bearing HCa-F cells were administered with CUR in NPs ten times in 20 days. As shown in Figure 7A, compared with the control group, all NP groups resulted in growth inhibition of HCa-F cells transplanted solid tumor and obvious decrease in tumor size. However, compared with the saline group, the tumor volume of CPTN group decreased significantly, while that of CPN and free CUR group decreased not as much. Tumor parameters were consistent with the statistical analysis of tumor volume data, which showed that all treatments significantly inhibited tumor volume compared with the control group (P < 0.05), and the CPTN group demonstrated the greatest effect (Table 4). After treatment, the weights of mice in four groups showed no remarkable changes, whereas in the free CUR group, the mice weight reduced by 20%, likely due to CUR toxicity (Figure 7B). All the results indicated that CPTN had a better anti-tumor effect in vivo than CPN and free CUR. Pathological studies showed that there were no obvious pathological changes in the tissues of the treatment groups (data not shown). These data demonstrated that CPTN not only enhanced the antitumor effects but also reduced its toxicity in vivo, suggesting the potential of CPTN for clinical application. Taken together, the present in vivo results clearly demonstrated the potential of CPTN as a novel antitumor therapy. The advantages of CPTN over free CUR in terms of the EPR effect, intracellular uptake, improved tissue distribution behavior and tumor treatment had significantly strengthened its anticancer effect against liver tumors. Further investigations should focus on its in vivo antitumor mechanisms, and its reversing effect of MDR should be verified in tumor models.
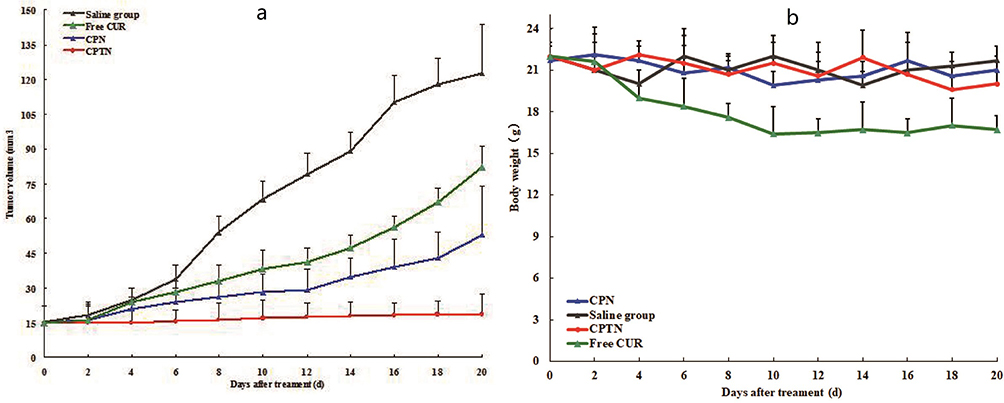 |
Figure 7 Changes of tumor volume (A) and body weight (B) in nude mice transplanted with HCa-F cell line during 20 days (n=6). |
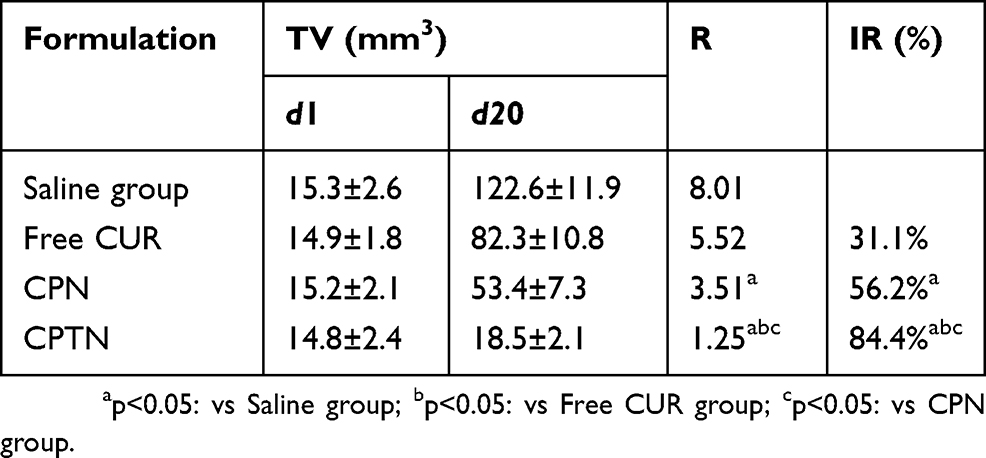 |
Table 4 Effect Of Free CUR, CPN And CPTN On The Mice Bearing HCa-F Cells |
Conclusion
This study aimed to prepare a CUR-loaded (PLGA/TPGS) NPs delivery system by the emulsification-solvent evaporation method. NPs were characterized for physical properties, such as morphology and particle size, and in vitro drug release. CUR-loaded (PLGA/TPGS) NPs could be successfully internalized by HepG2 cells and played a synergistic role in inhibiting the growth of hepatocellular carcinoma cells. They exhibited higher target organ accumulation, superior antitumor efficiency, and lower toxicity in vivo. The present study found that the CUR-loaded (PLGA/TPGS) NPs provided a promising platform for the treatment of liver cancer.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Altekruse SF, McGlynn KA, Reichman ME. Hepatocellular carcinoma incidence, mortality, and survival trends in the United States from 1975 to 2005. J Clin Oncol. 2009;27(9):1485–1491. doi:10.1200/JCO.2008.20.7753
2. Villanueva A, Llovet JM. Targeted therapies for hepatocellular carcinoma. Gastroenterology. 2011;140(5):1410–1426. doi:10.1053/j.gastro.2011.03.006
3. Asghar U, Meyer T. Are there opportunities for chemotherapy in the treatment of hepatocellular cancer? J Hepatol. 2012;56(3):686–695. doi:10.1016/j.jhep.2011.07.031
4. Ren J, Xu Y, Huang Q, et al. Chabamide induces cell cycle arrest and apoptosis by the Akt/MAPK pathway and inhibition of P-glycoprotein in K562/ADR cells. Anticancer Drugs. 2015;26(5):498–507. doi:10.1097/CAD.0000000000000209
5. Tham CL, Hazeera HH, Wai LK, et al. The synthetic curcuminoid BHMC restores endotoxin-stimulated HUVEC dysfunction: specific disruption on enzymatic activity of p38 MAPK. Eur J Pharmacol. 2015;749:1–11. doi:10.1016/j.ejphar.2014.12.015
6. Akkoc Y, Berrak Ö, Arisan ED, et al. Inhibition of P13k signaling triggered apoptotic potential of curcumin which is hindered by Bcl-2 through activation of autophagy in MCF-7 cells. Biomed Pharmacother. 2015;71:161–171. doi:10.1016/j.biopha.2015.02.029
7. Troselj KG, Kujundzic RN. Curcumin in combined cancer therapy. Curr Pharm Des. 2014;20(42):6682–6696. doi:10.2174/1381612820666140826154601
8. Vallianou NG, Evangelopoulos A, Schizas N, Kazazis C. Potential anticancer properties and mechanisms of action of curcumin. Anticancer Res. 2015;35(2):645–651.
9. Hu A, Huang JJ, Jin XJ, et al. Curcumin suppresses invasiveness and vasculogenic mimicry of squamous cell carcinoma of the larynx through the inhibition of JAK-2/STAT-3 signaling pathway. Am J Cancer Res. 2014;5(1):278–288.
10. Zhang C, Li B, Zhang X, et al. Curcumin selectively induces apoptosis in cutaneous T-cell lymphoma cell lines and patients’ PBMCs: potential role for STAT-3 and NF-κB signaling. J Invest Dermatol. 2010;130(8):2110–2119. doi:10.1038/jid.2010.86
11. Chiablaem K, Lirdprapamongkol K, Keeratichamroen S, Surarit R, Svasti J. Curcumin suppresses vasculogenic mimicry capacity of hepatocellular carcinoma cells through STAT3 and PI3K/AKT inhibition. Anticancer Res. 2014;34(4):1857–1864.
12. Jung KT, Lim KJ. Curcumin, COX-2 and protein p300/CBP. Korean J Pain. 2014;27(4):365–366. doi:10.3344/kjp.2014.27.4.365
13. Shishodia S. Molecular mechanisms of curcumin action: gene expression. Biofactors. 2013;39(1):37–55. doi:10.1002/biof.1041
14. Zheng J, Zhou Y, Li Y, Xu D-P, Li S, Li H-B. Spices for prevention and treatment of cancers. Nutrients. 2016;8(8):495. doi:10.3390/nu8080495
15. Heath DD, Pruitt MA, Brenner DE, Rock CL. Curcumin in plasma and urine: quantitation by high-performance liquid chromatography. J Chromatogr B Analyt Technol Biomed Life Sci. 2003;783(1):287–295. doi:10.1016/s1570-0232(02)00714-6
16. Singh SP, Sharma M, Gupta PK. Cytotoxicity of curcumin silica nanoparticle complexes conjugated with hyaluronic acid on colon cancer cells. Int J Biol Macromol. 2015;74:162–170. doi:10.1016/j.ijbiomac.2014.11.037
17. Shinde RL, Devarajan PV. Docosahexaenoic acid-mediated, targeted and sustained brain delivery of curcumin microemulsion. Drug Deliv. 2017;24(1):152. doi:10.1080/10717544.2016.1267822
18. Wei X, Senanayake TH, Bohling A, Vinogradov SV. Targeted nanogel conjugate for improved stability and cellular permeability of curcumin: synthesis, pharmacokinetics, and tumor growth inhibition. Mol Pharm. 2014;11(9):3112–3122. doi:10.1021/mp500290f
19. Jyoti K, Bhatia RK, Martis EAF, et al. Soluble curcumin amalgamated chitosan microspheres augmented drug delivery and cytotoxicity in colon cancer cells: in vitro, and in vivo, study. Colloids Surf B Biointerfaces. 2016;148:674–683. doi:10.1016/j.colsurfb.2016.09.044
20. Kim S, Diab R, Joubert O, Canilho N, Pasc A. Core-shell microcapsules of solid lipid nanoparticles and mesoporous silica for enhanced oral delivery of curcumin. Colloids Surf B Biointerfaces. 2016;140:161–168. doi:10.1016/j.colsurfb.2015.12.040
21. Kotcherlakota R, Barui AK, Prashar S, et al. Curcumin loaded mesoporous silica: an effective drug delivery system for cancer treatment. Biomater Sci. 2016;4(3):448. doi:10.1039/C5BM00552C
22. Bollu VS, Barui AK, Mondal SK, et al. Curcumin-loaded silica-based mesoporous materials: synthesis, characterization and cytotoxic properties against cancer cells. Mater Sci Eng C Mater Biol Appl. 2016;63:393–410. doi:10.1016/j.msec.2016.03.011
23. Spencer DS, Puranik AS, Peppas NA. Intelligent nanoparticles for advanced drug delivery in cancer treatment. Curr Opin Chem Eng. 2015;7:84–92. doi:10.1016/j.coche.2014.12.003
24. Truong NP, Whittaker MR, Mak CW, Davis TP. The importance of nanoparticle shape in cancer drug delivery. Expert Opin Drug Deliv. 2015;12:129–142. doi:10.1517/17425247.2014.950564
25. Jain RA. The manufacturing techniques of various drug loaded biodegradable poly (lactide-co-glycolide) (PLGA) devices. Biomaterials. 2000;21:2475–2490. doi:10.1016/s0142-9612(00)00115-0
26. Collnot EM, Baldes C, Wempe MF, et al. Mechanism of inhibition of P-glycoprotein mediated efflux by vitamin E TPGS: influence on ATPase activity and membrane fluidity. Mol Pharm. 2007;4:465–474. doi:10.1021/mp060121r
27. Guo Y, Luo J, Tan S, Otieno BO, Zhang Z. The applications of Vitamin E TPGS in drug delivery. Eur J Pharm Sci. 2013;49(2):175–186. doi:10.1016/j.ejps.2013.02.006
28. Song IS, Cha JS, Choi MK. Characterization, in vivo and in vitro evaluation of solid dispersion of curcumin containing d-α-Tocopheryl polyethylene glycol 1000 succinate and mannitol. Molecules. 2016;21(10):1386. doi:10.3390/molecules21101386
29. Chen HB, Zheng Y, Tian G, et al. Oral delivery of DMABmodified docetaxel-loaded PLGA-TPGS nanoparticles for cancer chemotherapy. Nanoscale Res Lett. 2011;6(1):4.
30. Ma Y, Zheng Y, Liu K, et al. Nanoparticles of poly(lactide-coglycolide)-d-a-tocopheryl polyethylene glycol 1000 succinate random copolymer for cancer treatment. Nanoscale Res Lett. 2010;5:1161–1169. doi:10.1007/s11671-010-9620-3
31. Braden ARC, Vishwanatha JK Formulation of active agentloaded activated PLGA nanoparticles for targeted cancer nanotherapeutics. US Patent Application Publication 0253961 A1. 2008.
32. Ritger PL, Peppas NA. A simple equation for description of solute release II. Fickian and anomalous release from swellable devices. J Control Release. 1987;5(1):37–42. doi:10.1016/0168-3659(87)90035-6
33. Song J, Huang H, Xia Z, et al. TPGS/Phospholipids mixed micelles for delivery of Icariside II to multidrug-resistant breast cancer. Integr Cancer Ther. 2016;15(3):390–399. doi:10.1177/1534735415596571
34. Assanhou AG, Li W, Zhang L, et al. Reversal of multidrug resistance by co-delivery of paclitaxel and lonidamine using a TPGS and hyaluronic acid dual-functionalized liposome for cancer treatment. Biomaterials. 2015;73:284–295. doi:10.1016/j.biomaterials.2015.09.022
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.