Back to Journals » Drug Design, Development and Therapy » Volume 13
Formulation and clinical investigation of optimized vinpocetine lyoplant-tabs: new strategy in development of buccal solid dosage form
Authors Ahmed TA
Received 28 September 2018
Accepted for publication 4 December 2018
Published 28 December 2018 Volume 2019:13 Pages 205—220
DOI https://doi.org/10.2147/DDDT.S189105
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Jianbo Sun
Tarek A Ahmed1,2
1Department of Pharmaceutics, Faculty of Pharmacy, King Abdulaziz University, Jeddah, Kingdom of Saudi Arabia; 2Department of Pharmaceutics and Industrial Pharmacy, Faculty of Pharmacy, Al-Azhar University, Cairo, Egypt
Background: This work aimed to develop a new solid dosage formulation of vinpocetine (VPN) in the form of buccal freeze-dried pullulan-based tablets (lyoplant-tabs) loaded with physically modified drug binary system.
Methods: Different polyvinyl pyrrolidone (PVP) grades were studied to prepare an efficient VPN binary system characterized by enhanced equilibrium saturation solubility, solubilization efficiency, thermodynamic stability, and permeation through oral mucosal cell lines. The concentrations of pullulan and swelling-aid polymer that affect the quality attributes of lyoplant-tabs were optimized. Clinical pharmacokinetics study on human volunteers for the optimized lyoplant-tabs compared to marketed product was accomplished.
Results: A promising drug binary system with polyvinyl pyrrolidone vinyl acetate (PVP-VA64) utilizing the lyophilization technique was developed. Solid-state characterization confirmed transformation of VPN completely into the amorphous form. The concentrations of pullulan and swelling-aid polymer were significantly affecting the characteristics of the tablets. Compared to the commercial VPN tablets, pullulan-based buccal tablets demonstrated enhancement in the studied pharmacokinetic parameters with positive impact on the drug bioavailability.
Conclusion: These VPN lyoplant-tabs containing lyophilized PVP-VA64-VPN binary system can be considered as an alternative to currently available marketed tablets; however, further preclinical investigations using large number of volunteers are required.
Keywords: vinpocetin, freeze-drying, binary system, buccal tablets, permeation, clinical pharmacokinetics
Introduction
Different pharmaceutical formulation strategies have been used to enhance the bioavailability of drugs that are suffering from poor aqueous solubility. These include size reduction by nanosizing and micronization,1,2 use of cosolvent and surfactants,3,4 solid dispersion,5 and inclusion complexation.6,7 Moreover, formation of colloidal drug delivery systems such as solid lipid nanoparticles (NP),8 polymeric NP,9 and lipid-based NP,10 microemulsion formation, and self-microemulsifying drug delivery systems have attracted increasing attention.5,11–13 Solid dispersion technique, which was first introduced in the early 1970s, is the distribution of a hydrophobic drug(s) in and around a hydrophilic polymer(s) at solid state.14 Melting (fusion), solvent evaporation, melt extrusion, and supercritical antisolvent process are commonly used to prepare solid dispersion.6,13,15 Polyvinyl pyrrolidone (PVP), namely poly-[1-(2-oxo-1-pyrrolidinyl)-ethylene], is a quite water-soluble polymer that has been utilized in solid dispersion preparation to improve solubility and dissolution of many water-insoluble drugs.16,17
Vinpocetine (VPN) is a derivative of the alkaloid vincamine, which has been reported to have vasodilator, anti-ischemic, anticonvulsant, anti-inflammatory, and neuroprotective activity.18–22 It is a weak basic drug (pKa =7.31) with a limited water solubility of 2.4 μg/mL. The main mechanism of drug action is attributed to inhibition of the enzyme phosphodiesterase type 1 that results in increase in the level of 3,4-dihydroxyphenylacetic acid, a metabolic breakdown product of dopamine, which selectively enhances the brain circulation and oxygen utilization without significant modification in systemic circulation parameters. It facilitates blood flow redistribution toward ischemic areas and enhances cerebral circulation and oxygen utilization.23 The major concerns about VPN are the poor aqueous solubility, short elimination half-life (1–2 hours), and the extensive first-pass metabolism (75% metabolism in liver) that limit the drug bioavailability.24,25 VPN is currently administered as an oral tablet containing 5 mg drug with a poor oral bioavailability. Accordingly, it would be of widely applicable implication to develop a formulation that enhances the aqueous solubility and bioavailability of the drug for effective treatment of cerebral degenerative diseases. Several drug delivery systems have been employed to overcome VPN problems such as mixed polymeric micelles, solid lipid NP, and self-microemulsifying drug delivery systems.24,26,27
Pullulan, a linear homopolysaccharide of glucose composed of maltotriose units, is a naturally occurring polymer that is obtained from starch by the fungus Aureobasidium pullulans.28 This polymer does not require any chemical modification and provides highly clear and homogeneous formulations, such as oral films, that are characterized by low oxygen permeability and low water content. Pullulan-based preparations readily dissolve in water, and so immediately melt in the mouth, which makes pullulan the polymer of choice for production of oral films.28,29 Recently, pullulan-based capsule shells, Plantcaps™ capsules, have been introduced to the market as a new natural alternative. These capsules characterized by unique features make these dosage forms of great benefits for manufacturers, health care professionals, and customers. Plantcaps™ capsules offer all the advantages of gelatin capsules, yet they are starch-free, gluten-free, preservative-free, of natural origin, and non-genetically modified.30 Moreover, they meet the requirements for certain group of consumers who are looking for products that are generally regarded as safe and Halal and kosher certified. For companies, Plantcaps™ capsules are made from the only plant-derived polymer suitable for “made with organic ingredients” label language in the United States. Their elegant crystal-clear transparent and high luster appearance adds extra benefits.
The buccal route of administration offers improved bioavailability and fast onset of action when compared to orally administered drugs. These advantages are attributed to avoidance of the first-pass metabolism because the drug does not go through the gastrointestinal system.31 In this route, the drug is held in the cheek, diffuses through the oral mouth mucosal tissues, and enters the blood stream. Different pharmacologically active compounds that belong to antipsychotic, opioid, cardiovascular, antiemetic, nonsteroidal anti-inflammatory, and anticonvulsant drugs have been successfully formulated in buccal formulations.31–34 As far as we know, no pullulan-based tablets are available in the market, under clinical trials, or have been mentioned in the literature. Moreover, development of solid dosage form using lyophilization method is beneficial especially for active pharmaceutical ingredients (APIs) characterized by poor solubility and limited dissolution rate. The technique increases the product stability, drug wetting, and solubility.35 It is also useful for APIs that are sensitive to high force of impaction currently used in the direct compression.
In this study, the goal was to enhance VPN bioavailability by formulation of pullulan-based buccal tablets (lyoplant-tabs) using the freeze-drying technique. To accomplish this goal first, the aqueous solubility of the crystalline drug, VPN, has been improved by development of solid binary systems using different PVP grades, of several viscosity and copolymer blends, utilizing the solvent evaporation and freeze-drying techniques. Second, optimization of the formulation factors that affect the quality attributed of the developed lyoplant-tabs. Permeation of the binary system through oral mucosal cell lines has been conducted. Clinical pharmacokinetic study of the developed tablets was also studied to provide an alternative VPN delivery system for the currently available marketed product.
Materials
VPN was purchased from Wuhan Trustchem Fine Chemical Co., Ltd. (Wuhan, China). Pullulan was a kind gift from Hayashibara Co., Ltd. (Okayama, Japan). PVP with an average molecular weight of 24,000 (PVP-K25), 44,000 (PVP-K30), and 360,000 (PVP-K90) were obtained from Spectrum Chemicals & Laboratory Products (New Brunswick, NJ, USA). PVP-VA64, PVP-XL10, and PVP-PXL-USP32 were procured from Shanghai Yuking Water Soluble Material Tech Co., Ltd. (Shanghai, China). Aspartame was procured from Sigma-Aldrich Co. (St Louis, MO, USA). Hydroxypropyl methyl cellulose (HPMC) 4,000 cp was obtained from Spectrum Chemical Manufacturing Corporation (Gardena, CA, USA). Xylitol was procured from Acros organics (Fair Lawn, NJ, USA). 1-Ethenylpyrrolidin-2-one (Plasdone XL) was provided by ISP (Baar, Switzerland). All chemicals were of analytical grade.
Methods
Preparation of binary system
VPN polymeric solid dispersions were prepared by either solvent evaporation or lyophilization methods using different PVP grades, namely PVP-K25, PVP-K30, PVP-K90, PVP-VA64, PVP-XL10, and PVP-PXLUSP32 in drug-to-polymer ratios of 1:1, 1:2, and 1:4. Also, mixture of PVP-K25, PVP-K30, and PVP-VA64 in 1:1:1 ratio was prepared and mixed with the drug in 1:4 drug-to-polymer mixture ratio. The specified quantities of the drug and the studied polymer(s) were weighed, dissolved in 10% aqueous acetic acid solution, and shaken well until complete mixing and formation of homogeneously colorless solution that was transferred into a hot air oven at 40°C until complete evaporation of the solvent and formation of transparent glassy mass. The dried masses obtained were pulverized, passed through sieve no 100, and stored in a desiccator until further evaluation.
For the lyophilization method, drug polymeric solutions were prepared, as previously described, transferred to a freezer at −80°C for 24 hours, and then subjected to freeze-drying for 48 hours using Christ Alpha 1-2 LD Plus lyophilizer (Martin Christ Gefriertrocknungsanlagen GmbH, Osterode am Harz, Germany). Table 1 illustrates the polymers and the drug-to-polymer ratios used to develop solid dispersion by the solvent evaporation and lyophilization methods.
 | Table 1 Aqueous solubility and solubility enhancement of vinpocetine in the prepared solid dispersions |
Equilibrium saturation solubility
To study the effects of the prepared solid dispersions on the solubility of VPN, the equilibrium solubility was determined as previously reported.6 An excess amount of either pure drug or the prepared solid dispersions was added to screw-capped vials containing 10 mL of distilled water. The vials were placed in a thermostatically controlled shaking water bath (Model 1031; GFL Corporation, Burgwedel, Germany) at 25°C±0.5°C for 48 hours. Samples from the vials were taken and analyzed for VPN concentrations every day until equilibrium was reached (the drug solubility values on two consecutive days did not vary by more than 5%). Aliquots withdrawn were filtered through a 0.22 μm pore size Millipore filter and assayed for drug content using high-performance liquid chromatography (HPLC) method mentioned earlier by Ding et al,36 except for slight modifications. An isocratic HPLC method was performed using Agilent 1200 series equipped with UV diode array detector. Reversed phase C18 analytical column (4×250 mm, 5 μm; Thermo Fisher Scientific, Waltham, MA, USA) was used. The mobile phase composed of a mixture of methanol and 0.05 ammonium acetate buffer of pH 5.5, 80:20 (v/v). The flow rate of the mobile phase was 1 mL/min, the injection volume was 20 μL, and the detection wavelength was 273 nm. Each experiment was performed in triplicate.
Solubilization efficiency
The solubility of VPN, in the prepared solid dispersions, was compared to the intrinsic drug solubility and the resulting ratios of solubilization efficiency (solubility enhancement) were calculated.15 These ratios were used to compare the relative solubilization efficiency of VPN in the solid dispersion using both techniques.
Phase solubility study and thermodynamic parameter of solubility
An excess amount of pure VPN was added separately into screw-capped vials containing 10 mL of aqueous polyvinyl pyrrolidone vinyl acetate (PVP-VA64) solution (0%–10% w/v). The vials were sealed and kept shaken on a shaking water bath at 37°C for 72 hours. Aliquots (n=3) from each vial were filtered through a 0.22 μm pore size Millipore filter, and the filtrates were assayed by HPLC method previously described. The change in the solubility of VPN resulting from the addition of different PVP-VA64 concentrations was used to construct phase-solubility plot and to evaluate the stoichiometry and the stability constant of complexation (Kst), which exemplifies the strength of the interaction and stability of the complex formed.
Calculation of the apparent stability constant, Kst, was achieved from the phase solubility diagram according to the equation described by Higuchi and Connors,37 based on the assumption that 1:1 complex was initially formed.38
|

where “So” represents the drug solubility in absence of the studied polymer (the intercept of the phase solubility diagram constructed).
To evaluate the solubilizing efficiency of the studied polymer for the drug, the complexation efficiency (CE) was determined. It was calculated according to the following equation:
|
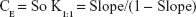
Gibbs free energy (ΔGotr) for the transfer of VPN from pure water to aqueous solutions of PVP-VA64 was calculated using the following equation:
|

where Sc/So is the ratio of the molar drug solubility in aqueous solution of PVP-VA64 to that of pure water. R is the gas rate constant (8.314 J/°C). T is the temperature in Kelvin at which the study was conducted.
Gibbs free energy could also be referred to as the thermodynamic potential that is minimized when a system reaches chemical equilibrium from an initial state to a final state at constant pressure and temperature.
Dissolution of the prepared solid dispersion
In vitro dissolution study of the pure drug and vinpocetin-PVP-VA64 inclusion complex (equivalent to 5 mg VPN) was performed in triplicate, using the United States Pharmacopeia (USP) dissolution test apparatus II paddle type, DT 700 LH device, Erweka GmbH DT 700 (Heusenstamm, Germany) at 37°C for 2 hours, with a stirring rate of 50 rpm, in 900 mL of enzyme-free simulated intestinal fluid (pH 6.8). Aliquots were collected at 5, 10, 15, 20, 25, 30, 45, 60, 90, and 120 minutes, with immediate replacement of an equal volume, and the collected samples were filtered through a 0.22 μm pore-size Millipore filter and assayed for drug content using HPLC. From the dissolution profile curve constructed, DP5 min, DP30 min, and DP120 min, which are the percent of drug dissolved within 5, 30, and 120 minutes, respectively, were estimated. Also, the dissolution efficiency within 10 and 120 minutes, (DE10 min) and (DE120 min), respectively, and the mean dissolution time were calculated. For each formulation, the dissolution efficiency was determined as the percent ratio of area under the dissolution profile curve up to the time, t, to that of the area of the rectangle described by 100% dissolution at the same time.
Permeation study
Before formulation of a buccal drug delivery system, buccal permeation study must be carried out to determine the feasibility and the impact of administration the drug formulation via this route. In this study, a human oral epithelial cell culture (OEC; Applied Biological Materials Inc., Richmond, BC, Canada) was used. The cells were cultured, seeded on T25 flasks, and the culture medium was replaced every day until the cells were tested to be suitable for the permeation experiment. This was confirmed by examination of the electrical resistance and the apparent permeability coefficient. Cells were divided into two groups. Group I was exposed to 0.1 mg/mL VPN in the form of inclusion complex in dimethyl sulfoxide (DMSO), while group II was subjected to the same concentration of pure VPN solution in DMSO. Blank cells containing only the culture medium without VPN were used as a reference. Cells (n=3) were incubated, collected after 1, 2, 4, 6, and 24 hours, and washed twice with ice-cold PBS. The cell pellets collected were exposed to two repeated cycles of freezing and thawing, and finally to ultrasonication for 10 minutes for complete rupture of the cells. Cell lysates were subjected to centrifugation using 3K30 sigma laboratory centrifuge (Osterode am Harz, Germany) at 15,000× g for 1 hour at 4°C. The concentration of VPN in the supernatant was determined using HPLC method described earlier. VPN standards containing known amounts of the drug in the studied cells were prepared, treated as described above, and analyzed before estimation of the unknown drug concentrations in the samples.
Physicochemical characterization of VPN in the solid dispersion mixture
Differential scanning calorimetry (DSC)
The thermal behavior of VPN, PVP-VA64, and the lyophilized solid dispersion was studied using Shimadzu DSC TA-50 ESI DSC apparatus (Tokyo, Japan) calibrated with indium. Analysis of all the studied samples was performed from 10°C to 300°C with a 10°C/min heating rate.
Fourier transform infrared (FT-IR) spectroscopy
Spectra of samples used in the DSC study were collected using Nicolet iS10 (Thermo Fisher Scientific) at a resolution of 4 cm−1, and the number of scans was 36. Frequency was in the range of 4,000–500 cm−1.
X-ray powder diffraction (XRPD)
Change in the crystalline state of the prepared VPN-PVP-VA64 solid dispersion compared to that of pure VPN powder was studied using powder X-ray diffractometer (D/max 2500, Rigaku, Tokyo, Japan). The diffraction patterns of the pure drug and solid dispersion were recorded at a scan speed of 0.5000°/min.
Formulation of lyoplant-tabs
Experimental design
A three-level factorial design was used to study the effect of the concentration of the aqueous pullulan solution (X1) and the concentration of the wetting-aid polymer, plasdone XL (X2), on the cumulative drug released (Y1) and the wetting index (Y2) of the lyoplant-tabs utilizing StatGraphics Centurion XV version 15.2.05 software (StatPoint Technologies, Inc., Warrenton, VA, USA). Concentrations of 5%–15% and 4%–6% were used for X1 and X2, respectively. The study was conducted to maximize Y1 and Y2. Table 2 illustrates the composition of the formulations suggested by the experimental design.
 | Table 2 Composition of the lyoplant-tab formulations along with the observed, fitted values and statistical ANOVA of the studied responses Y1 and Y2 |
Preparation of the lyoplant-tabs
Buccal freeze-dried pullulan-based tablets (lyoplant-tabs) were prepared according to the method previously described in our laboratory for development of Vitamin K and finasteride lyophilized tablets.11,12 Briefly, three different pullulan solutions (5%, 10%, and 15%) were prepared by dissolving the specified quantity of pullulan in distilled water over a magnetic stirrer. A quantity of the lyophilized binary system equivalent to 100 mg VPN, HPMC (2% w/v), aspartame (0.125% w/v), xylitol (1.6% w/v), and plasdone XL (4%–6% w/v) was homogeneously dispersed in 10 mL of the specified pullulan solution. A weight equivalent to 5 mg VPN of the prepared mixture was carefully poured into an empty pocket of tablet blister packs that were frozen at −80°C for 24 hours and finally subjected to freeze-drying for 48 hours using Christ Alpha 1-2 LD Plus lyophilizer (Martin Christ Gefrier-trocknungsanlagen GmbH, Osterode am Harz, Germany). The obtained freeze-dried tablets were kept at 25°C in a desiccator containing calcium chloride until further study.
Evaluation of the prepared tablets
All the prepared tablets were characterized for weight variation, thickness, drug content, friability, wetting time, wetting index, and in vitro drug release based on the specifications mentioned in USP 28/NF23.39 Ten tablets from each run were weighed individually using Mettler Toledo AJ100, electric balance (Greifensee, Switzerland) and the average weight was calculated. Tablet thickness was estimated using Mitutoyo dial thickness gage (Kawasaki, Japan) for the same number of tablets. For determination of the drug content, six tablets from each run were randomly selected and placed separately in screw cap glass bottles containing 100 mL of 10% acetic acid solution. The mixtures were thoroughly mixed in a thermostatically controlled shaking water bath, Model 1031; GFL Corporation (Burgwedel, Germany), at 25°C for 24 hours, then homogenized using UltraTurax, IKA® T18 basic Homogenizer (Campinas, Brazil). The content of the bottles was subsequently filtered using Acrodisc® syringe filter of 0.45 mm. The drug concentration in the filtrate was determined using HPLC method previously stated.
Friability of the prepared tablets was assessed using Erweka Friabilator type PTF1, Pharma-test (Hainburg, Germany). Ten tablets from each run were de-dusted, weighed, and allowed to rotate for 4 minutes at 25 rpm, after which the tablets were de-dusted and weighed again. Friability was expressed as the loss of weight and calculated as a fraction of the original weight of the tablets using the following equation:
|

Wetting time and wetting index of the tablets were evaluated by weighing three individual tablets from each run, placing each tablet in the center of a petri dish containing a tissue paper that was folded twice and kept wetted with a small volume (8 mL) of rhodamine dye solution. The time taken until first appearance of the dye on the tablet surface was taken as the wetting time. The wetting index was estimated by reweighing the wetted tablet and then applying the following equation:
|

In vitro drug release study
The release of VPN from the lyoplant-tab formulations was carried out using USP dissolution test apparatus type II (Paddle type), DT 700 LH device, Erweka GmbH DT 700 (Heusenstamm, Germany). The study was conducted in 900 mL phosphate buffer of pH 6.8 at 37°C. Paddles were rotated at 50 rpm and aliquots of 3 mL were withdrawn, with immediate replacement, for 120 minutes and analyzed for VPN content using HPLC method described earlier. The results obtained were the average of three measurements. The condition of the experiment was selected to achieve the sink conditions.
Experimental design statistical analysis
Data obtained for Y1 and Y2 were statistically analyzed (P-value <0.05) to determine the significant factors that affect each studied response. This was achieved by introducing the obtained results into the response column of the StatGraphics software after which the model was run.
Preparation and characterization of the optimized tablets
The proposed optimized tablet formulation was prepared and characterized for the same quality attributes that have been previously described, and the data obtained for the cumulative drug release and swelling index were compared with the predicted values.
Evaluation of the clinical pharmacokinetic behavior
The pharmacokinetic parameters and the plasma concentration–time curve for VPN in the optimized lyoplant-tabs formulation, pure drug lyophilized buccal tablets, and commercial oral drug tablets were studied following administration to healthy volunteers. Pure VPN buccal tablets were prepared as previously described except that pure VPN was used instead of the drug binary system.
The health status of nine male Egyptian volunteers, aged 21–30 years with a body mass index of 20–30 kg/m2, was confirmed after considering their medical history and carrying out physical examinations and laboratory investigations. Subjects participating in the study were asked to read, understand, and sign a written consent about the nature of the research. Before administration of the formulations, volunteers were kept in-house and were not allowed to get any medication for 2 days. Subsequently, blood samples were withdrawn at predetermined periods for 24 hours. The study was performed in accordance with European Medicines Agency, International Conference on Harmonization, Good Clinical Practice, Food and Drug Administration (FDA) guidelines, and the Declaration of Helsinki. The protocol was approved by the Ethics Committee of the Egyptian Research and Development Company (ERDC) on July 2nd, 2018.
A single-dose one-period parallel design was conducted. Volunteers were classified into three groups (n=3); group I (test group) was given the optimized lyoplant-tabs formulation, group II (positive control group) was administered the buccal tablets containing pure drug, while group III (reference group) was given the commercial VPN tablets, namely Vinporal® 5 mg (Amriya Pharmaceutical Industries Company, Alexandria, Egypt). Volunteers of group I and II were asked to hold the tablets in the cheek. Venus blood samples of 5 mL were collected from every volunteer for 24 hours and the concentration of VPN in the collected samples was determined as described below.
Chromatographic condition
A high-performance liquid chromatographic method coupled with MS/MS detection was developed, optimized, and validated at the ERDC laboratories for the determination of VPN in human plasma. An Agilent HPLC series 1200, Agilent Technologies, Deutschland GmbH (Waldbronn, Germany) equipped with G1311A quaternary pump, G1329A autosampler, G1322A vacuum degasser and mass hunter software was used. The mobile phase consisted of 75% acetonitrile: 25% (10 mM ammonium acetate and 50 μL formic acid for each 100 mL water). It was pumped at a flow rate of 0.30 mL/min and a reverse-phase column Intersil ODS-3 (4.6 × 50 cm, dp 5 μm Sigma-Aldrich Co.) temporized at 25°C. Retention times of 1.74 and 3.47 minutes were detected for galantamine (internal standard) and VPN, respectively. The method was fully validated according to the “FDA Bio-analytical Method Validation Guidelines 2003”. Linearity of the assay method was verified within the concentration range of 0.125–25 ng/mL with a regression coefficient (R2) =0.9983. All the results were within the acceptance criteria as stated in the recommended guidelines. The mean recovery of VPN was 103.2 at 0.125 ng/mL (low limit of quantification) and 94.4% at 25 ng/mL (upper limit of quantification).
Plasma calibration curve and sample collection
VPN and IS stock methanolic solutions were prepared by dissolving an accurately weighed amount of each drug in a separate volumetric flask. Calibration standards in blank human heparinized plasma were freshly prepared in duplicate at final VPN plasma concentrations of 0.125, 0.250, 0.5, 0.750, 1.5, 2.5, 5, 7.5, 10, 15, 20, and 25 ng/mL for every analytical run. A fixed volume of 50 μL IS (4 μg/mL) was added for each VPN sample. Quality control (QC) samples were prepared independently from the calibration standards, using separately prepared master stock solutions. These solutions were diluted with control human plasma to produce the following QC samples: LLOQ 0.125 ng, QC low 0.375 ng, QC mid 12.5 ng, and QC high 22.5 ng.
Pharmacokinetic data analysis
A noncompartmental pharmacokinetic model using Kinetica™ pharmacokinetic software (version 4; Thermo Fisher Scientific) was utilized to determine the following pharmacokinetic parameters: maximum plasma concentration over the time span (Cmax), time point of maximum plasma concentration (Tmax), the apparent first-order absorption rate constant (Kabs), the absorption half-life (t½ abs), the apparent first-order elimination rate constant (Kelm), the elimination half-life (t½ elm), apparent volume of distribution (Vd), total clearance rate, area under the plasma concentration–time curve from time zero to the last measurable drug concentration (AUC0-last), area under the moment curve (AUMC0-last), and mean residence time. The data obtained were expressed as mean ± SD and statistically analyzed using GraphPad Prism 6 (GraphPad Software, Inc., La Jolla, CA, USA). To indicate the significant difference between the optimized lyoplant-tabs, the drug commercial oral tablet, and pure drug-loaded buccal lyophilized tablets, two-way ANOVA followed by Tukey’s multiple comparisons test was used. P-value <0.05 was considered to be statistically significant.
Results and discussion
Equilibrium saturation solubility and solubilization efficiency
The solubility of VPN in water at 25°C was found to be 2.31 μg/mL. Accordingly, VPN is considered a practically water-insoluble drug. Different PVP grades were used as hydrophilic carriers to enhance the drug aqueous solubility. Solvent evaporation and lyophilization were the two techniques employed to prepare drug binary systems. Results of the equilibrium solubility and solubility enhancement ratio revealed that the lyophilization was superior to the solvent evaporation technique and drug binary system with PVP-VA64 in a 1:4 ratio was the best among all the studied carriers as illustrated in Table 1.
PVPs have been extensively used as hydrophilic carriers in the preparation of solid dispersion systems with a wide variety of drug classes.40 PVP K25, K30, and K90 are the most common grades that have been utilized for this purpose.16,41,42 The solubility and dissolution of various drugs such as mefenamic acid, glimepiride, sulindac, tenoxicam, tacrolimus, fenofibrate, and flurinazine have been increased following preparation of solid dispersions with PVPs.6,7,17 Enhancement of the drug solubility following complexation with PVPs could be attributed to conversion of the drug from the crystalline into the amorphous state, dispersion of the drug particles into the hydrophilic PVPs’ large surface area with subsequent enhancement in the drug wettability, and decrease in the drug particles’ tendency for aggregation and agglomeration.43
Three drug-to-polymer ratios were studied to evaluate the effect of increasing the polymer concentration on the drug solubility. Results indicated that the drug solubility was augmented as the concentration of the polymer was increased, the effect that could be attributed to uniform distribution of VPN on the polymer surface. Further discussion of this behavior will be illustrated during discussion of the type of complexation in the phase solubility study section.
Different techniques have been reported for preparation of drug PVP binary system, namely kneading,5,16,17 solvent evaporation,44 lyophilization,45 and supercritical antisolvent process.46 Previous studies illustrated the successfulness of the freeze-drying (lyophilization) technique in development of binary system formulations. The process was efficiently used to prepare atorvastatin solid dispersion using skimmed milk and was found to improve the drug oral bioavailability.45 In another study, Kharshoum et al developed vinpocetin-poly ethylene glycol 4000 solid dispersion using freeze-drying technique and reported enhancing the bioavailability of the studied drug.47 Also, aripiprazole binary system, using naturally and chemically modified forms of cyclodextrins, has been developed by kneading, microwave irradiation, and freeze-drying techniques, and authors demonstrated the effectiveness of the freeze-drying process in improving aripiprazole dissolution.48
In this study, PVP-VA64 was superior to all the studied carriers in enhancing the solubility and solubilization efficiency of VPN following development of binary system by either the solvent evaporation or the lyophilization method (Table 1). This behavior could be attributed to the presence of the vinyl acetate group that increases the polymer surface area and may facilitate more physical attraction between the carbonyl group of the vinyl acetate and the drug amino group. Our results are in good agreement with previously published work by Kim et al, who prepared atorvastatin solid dispersions based on HPMC, PVP K30, and PVP-VA64 and reported that drug solid dispersion prepared using PVP-VA64 exhibited the best dissolution behavior and attributed this behavior to the higher specific surface area.46 In another study, Goddeeris et al reported faster dissolution for the anti-HIV drug UC 781 from solid dispersion of d-alpha-tocopheryl polyethylene glycol succinate 1000-PVP-VA64 than from d-alpha-tocopheryl polyethylene glycol succinate 1000-HPMC.49
Phase solubility study and thermodynamic parameter of solubility
To study the interaction of VPN with PVP-VA64, phase solubility was studied, and the diagram obtained is represented in Figure 1A. It was clear that the drug solubility was increased upon increasing the polymer concentration. The following chart equation was obtained:
|

 | Figure 1 Phase solubility study diagram of VPN in aqueous solution of PVP-VA64 (A), and dissolution profile of VPN solid dispersion and pure drug (B). |
The values of the stability constant Kst and the complexation efficiency CE were found to be 7 mg/mL−1 and 0.0014, respectively. These results are in good agreement and accordance with the well-established formation of drug soluble complexes between water-soluble polymeric carriers such as PVP-VA64 and poorly water-soluble drugs such as VPN.50
Carrier drug complexes have been classified according to the effect of the carrier on the drug solubility.37 Type A is obtained when the drug solubility is increased upon increasing the polymer concentration. When this complex is first order with respect to the carrier and first or higher order with respect to the drug, then AL type is obtained. If the obtained complex is first order with respect to the drug, but second or higher order with respect to the carrier, then AP type is obtained. B-type complexes are obtained when there is a limited solubility of the drug in the aqueous complexation medium, that is, increase solubility of the drug with the increase of the polymer concentration up to a certain limit followed by plateau. In this study, upon construction of VPN phase solubility profile (Figure 1A), AP-type complexation was obtained.
The values for ΔGotr give information about the process of drug solubilization whether it is favorable or not. Negative values for ΔGotr indicate favorable conditions and the lower the negative values obtained the more promising the condition.51 Results for VPN ΔGotr for 1%, 2%, 3%, 4%, 5%, 6%, 7%, 8%, 9%, and 10% polymeric solution were −4,789.515, −6,330.538, −7,390.998, −8,413.708, −8,775.631, −9,159.485, −9,686.917, −10,097.66, −10,278.36, and −10,658.79 J/mol, respectively. All the obtained values were negative and decreased as the polymeric solution increased. This indicates increased solubility of VPN in the presence of PVP-VA64 and demonstrated the spontaneous solubilization tendency of the drug in the polymeric solution, which becomes more satisfactory when the concentration of the polymer increased.
Dissolution of the prepared solid dispersion
The in vitro dissolution of pure VPN and the prepared drug PVP-VA64 binary system is demonstrated in Figure 1B. Data obtained from the dissolution profile curve were used to estimate DP5 min, DP30 min, and DP120 min in which the obtained values were 5.62%±2.45%, 16.46%±3.58%, and 22.41%±6.04% for the pure drug, and 31.09%±3.92%, 62.52%±5.34%, and 74.61%±3.19% for the binary system, respectively. Also, DE10 min was calculated and found to be 2.17%±0.23% and 0.428%±0.159% for the complex and the pure drug, respectively. The DE120 for the complex and the pure drug were 65.35%±2.5% and 18.67%±3.73%, respectively.
From the results obtained, it is evident that the binary system considerably enhanced the in vitro dissolution when compared to pure drug. In general, this behavior could be attributed to improve drug solubility through conversion of VPN from crystalline to amorphous state, reduction of drug crystal size, and absence of aggregation of drug crystals. The dissolution rate of VPN from the binary system was found to be higher than that of the corresponding pure drug (Figure 1B), owing to better wettability and prevention of particle aggregation.
Permeation across the buccal mucosa
This part aims to evaluate the ability of VPN to permeate through the oral buccal mucosal cells. VPN in the form of binary system with PVP-VA64 or pure drug was used. Effective permeation of VPN from the human OEC is expected to have a positive impact in the drug bioavailability and the therapeutic response.
Results of the permeation study revealed that drug binary system enhanced the rate but not the extent of drug permeation (data not shown). This finding indicates that complexation does not alter but accelerate the process of drug transport across the OEC. Previous reports illustrated that VPN is well absorbed from the gastrointestinal tract particularly from the small intestine.52 In this study, the transport of VPN, in the form of both pure VPN solution and binary system, was also possible from the oral epithelial cells with preference for the binary system due to the fast rate of drug permeation and suitability of the binary system for buccal tablets formulation.
Physicochemical characterization
The thermal behavior of pure VPN (Figure 2) illustrated a sharp endothermic peak at 152.28°C, typically of a crystalline anhydrous drug structure as previously reported.36,53 An endothermic peak was observed in the polymer thermogram at the range 80°C–100°C, which is attributed to the water loss from the hygroscopic polymer upon heating. Another peak corresponding to polymer glass transition temperature was also detected at 186°C. The prepared solid dispersion showed no crystalline peak of VPN, which implies that the drug is present in completely amorphous form and is molecularly dispersed in the polymeric matrix. This finding is in good agreement with the work done by Kharshoum et al, for VPN solid dispersion with poly ethylene glycol 4000 and VPN inclusion complex with 2-hydroxypropyl β-cyclodextrins.47 The same thermal behavior was previously illustrated for solid dispersion of ketoconazole with PVP-K30,51 atorvastatin solid dispersion using skimmed milk,45 glimepiride solid dispersions of pregelatinized starch, gelucire, and lactose,54 and celecoxib solid dispersions of poloxamer-188 and PVP-K30.16
 | Figure 2 Differential scanning calorimetry thermograms of vinpocetine, polyvinyl pyrrolidone vinyl acetate, and lyophilized binary mixture. |
The DSC spectra of the prepared VPN solid dispersion also indicated no chemical interaction because no new peaks for any new chemical moiety were observed. Further investigation for any possible interaction among components is required but with another tool such as FT-IR.
The FT-IR spectrum of VPN (Figure 3) illustrated a characteristic sharp peak at 1,720 cm−1 equivalent to carbonyl stretching. Another distinct absorption band was detected in the region 2,840–3,000 cm−1, which is assigned to aromatic stretching. No new peaks or substantial shifting in the position of the drug or PVP functional groups were observed in the developed solid dispersion except for some overlapping between both, VPN and PVP, at specified positions owing to presence of common functional groups.
 | Figure 3 Fourier transform infrared spectra of vinpocetine, polyvinyl pyrrolidone vinyl acetate, and lyophilized binary mixture. |
X-ray diffraction pattern of pure VPN showed distinct characteristic peaks of the crystalline drug structure as illustrated in Figure 4, while XRPD of the solid dispersion was different from that of the pure drug, which is an indication of the crystalline state transformation into the amorphous form. This transformation may result in enhancement in the aqueous drug solubility as previously reported for finasteride and atorvastatin.55,56
 | Figure 4 X-ray powder diffraction patterns of pure vinpocetine and lyophilized binary mixture. |
Lyoplant-tabs formulation development
Lyoplant-tabs were prepared utilizing pullulan, HPMC, aspartame, xylitol, and plasdone XL. HPMC was used as a binder,11 and also it acts as a mucoadhesive polymer because buccal dosage forms are supposed to exhibit sufficient adhesion to the mucosa and resist the salivation and mechanical movements in the mouth for a sufficient period of time as previously described.57 Aspartame (0.125%) was added as a sweetener, while xylitol (1.6%) was incorporated to enhance the quality attributes of the prepared tablets because it has more negative heat of solution and lesser hygroscopicity when compared to mannitol and sorbitol that are commonly used in the pharmaceutical formulations designed to disintegrate in the oral cavity.58 Plasdone XL was used to facilitate swelling of the prepared tablets (swelling-aid polymer). Faster water diffusion and wetting have been reported to enhance the formation of adhesive bonds (rapid adhesion) and so mucoadhesion in buccal tablets.57 Pullulan, a naturally derived polysaccharide polymer, possesses unique characteristics that make it ideal for pharmaceutical dosage forms. It is very stable and matching the performance properties of gelatin.30 Capsules prepared using pullulan (Plantcaps™) have very low permeability to oxygen, have fast in vitro disintegration (less than 15 minutes), are suitable to encapsulate oxygen-sensitive ingredients with no need for antioxidants, are used to mask the taste and odor of their contents, and are compatible with a wide variety of ingredients. Also, Plantcaps™ are manufactured and filled using process identical to that used for hard gelatin capsules and possess the same moisture tolerance as gelatin capsules.30 Similarly, pullulan has been studied as a film-forming substance during development of fast-dissolving oral films.29,59–61
In this work, we have studied the effect of pullulan and the wetting-aid polymer concentrations to develop buccal pullulan-based tablets utilizing the lyophilization technique. To the best of our knowledge, no previous studies have investigated or developed similar formulation particularly using the same variables.
Characterization of lyoplant-tabs
The studied characteristics of the prepared lyoplant-tabs are represented in Tables 2 and 3. Weight and thickness were in the range of 108.05±1.94 to 167.03±6.07 mg and 3.93±0.20 to 4.91±0.42 mm, respectively. Friability ranged from 0.046% to 0.560%, which is <1%. Wetting time was in the range 4.33±0.22 to 14.51±1.57 minutes, while results of the drug content showed an average value of 98.04%±0.34% to 102.9%±0.21%. All the studied characteristics are in good agreement and met the USP specifications.39
 | Table 3 Quality attributes of the prepared pullulan-tabs formulations |
Effect of the studied variables on Y1 and Y2
Results for the cumulative drug release and wetting index were statistically analyzed using multiple regression analysis and two-way ANOVA utilizing the StatGraphics software. Table 2 illustrates the values for estimated effects, F-ratios, and P-values obtained. Positive values for estimated effect are an indication of a synergistic effect for the studied variable on this response and vice versa. To compare the actual and expected variations in the variable, average F-ratio is used, in which an F-ratio >1 is a sign of a location effect and hence P-value is used to indicate the significant effect if its value differs from 0 and is <0.05.
The equations obtained of the fit model were:
|
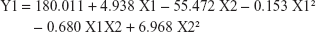
|

The observed values for Y1 and Y2 were in the range of 67.35%±4.49% to 98.54%±2.63% and 57.85%±7.34% to 144.03%±12.5%, respectively, as shown in Table 2. Aqueous pullulan solution concentration (X1), the concentration of the wetting-aid polymer, plasdone XL (X2), and the quadratic effect of X2 were significantly affecting Y1 at P-values of 0.0037, 0.0039, and 0.0217, respectively, while X1 and X2 were significantly affecting Y2 at P-values of 0.0114 and 0.0482, respectively. Pareto charts obtained, which are depicted in Figure 5, also confirmed the above finding. A vertical reference line at P-value of 0.05 is denoted in the chart in which an effect that exceeds this reference line is statistically significant. The X1 was negatively (antagonistically) affecting both Y1 and Y2, while X2 was positively (synergistically) affecting the same responses.
 | Figure 5 Standardized Pareto charts and estimated response surface plots for the effect of the studied factors on Y1 and Y2. |
As the pullulan concentration was increased, the cumulative drug release and the swelling index decreased owing to the formation of a water swollen gel-like structure that prevents the penetration of the dissolution medium or rhodamine dye solution into the matrix. The concentration of the wetting-aid polymer was found to be critical. Plasdone XL possesses hygroscopic or water-attracting characteristics with excellent swelling properties that promote water absorption, rapid water uptake, and so wetting of the tablet due to formation of porous structure. The effect of changing the concentrations of X1 and X2 on Y1 and Y2 is illustrated in the estimated response surfaces (Figure 5).
To develop an optimized lyoplant-tab characterized by a maximized cumulative drug release and swelling index over the indicated region, the optimum desirability that accomplishes this goal was identified. The suggested values for X1 and X2 were found to be 5% and 6%, respectively, and the predicted values for Y1 and Y2 were calculated to be 98.51% and 143.92%, respectively. The observed values were close to the predicted ones with standard errors not greater than 5%.
Clinical pharmacokinetic study
Plasma concentration–time curves of VPN following administration of a single dose of 5 mg VPN in the form of oral commercial drug tablets and lyophilized drug buccal tablets are shown in Figure 6. Maximum VPN plasma concentrations of 9.658±1.074, 6.672±1.234, and 5.050±0.239 ng/mL were observed after 1, 1.25, and 2.5 hours for the optimized lyoplant-tabs, buccal lyophilized tablets containing pure drug, and commercial VPN tablets, respectively. These findings confirmed the advantage of the lyoplant-tabs formulation in the rate and extent of VPN absorption. Other studied pharmacokinetic parameters are depicted in Table 4. Lyoplant-tabs demonstrated higher AUC and AUMC compared to the other formulations. Moreover, lyoplant-tab showed relative bioavailability of 123.37% and 159.64% with the buccal lyophilized tablets containing pure drug and commercial VPN tablets, respectively. Overall, enhanced pharmacokinetic parameters and drug bioavailability were observed with the group administered the optimized lyoplant-tabs formulation, which indicates that enhancement of the VPN aqueous solubility by complexation with PVP-VA64 using the lyophilization technique and administration of the drug binary system through the buccal route following development of pullulan-based solid dosage form utilizing the lyophilization technique could be considered as a promising formulation and an excellent alternative for the currently available VPN marketed product.
 | Figure 6 Plasma concentration–time curves for VPN after oral administration of commercial drug product and buccal formulations to human volunteers. |
 | Table 4 Pharmacokinetic parameters of the oral commercial and buccal vinpocetine tablets |
Conclusion
In this work, different hydrophilic carriers were studied for their effect on the preparation of VPN solid dispersion in order to enhance the drug aqueous solubility. Drug/PVP-VA64 binary system developed using lyophilization technique was superior when compared to the other studied carriers. Drug-to-polymer ratio was significantly affecting the solubility of the drug from the prepared solid dispersion systems. Physicochemical characterization confirmed presence of the drug in completely amorphous form and it was molecularly dispersed in the polymeric matrix. The concentrations of pullulan and the wetting-aid polymer (plasdone XL) were significantly affecting the development of buccal pullulan-based tablets. The lyophilization technique, which was utilized in this work to develop an optimized lyo-plant-tab, was successfully implemented to develop solid dosage form with acceptable quality attributes. The technique has been found to augment the bioavailability of VPN, a drug characterized by poor aqueous solubility and limited bioavailability, especially when a binary system of VPN has been used.
Acknowledgments
This project was funded by the Deanship of Scientific Research (DSR) at King Abdulaziz University, Jeddah, under grant no. G: 94-166-1439. The authors, therefore, acknowledge with thanks DSR for technical and financial support.
Disclosure
The authors report no conflicts of interest in this work.
References
Ahmed TA, Aljaeid BM. Preparation, characterization, and potential application of chitosan, chitosan derivatives, and chitosan metal nanoparticles in pharmaceutical drug delivery. Drug Des Devel Ther. 2016;10:483–507. | ||
Ahmed TA, El-Say KM. Transdermal film-loaded finasteride microplates to enhance drug skin permeation: two-step optimization study. Eur J Pharm Sci. 2016;88:246–256. | ||
Bauduin P, Renoncourt A, Kopf A, Touraud D, Kunz W. Unified concept of solubilization in water by hydrotropes and cosolvents. Langmuir. 2005;21(15):6769–6775. | ||
Ghebremeskel AN, Vemavarapu C, Lodaya M. Use of surfactants as plasticizers in preparing solid dispersions of poorly soluble API: selection of polymer-surfactant combinations using solubility parameters and testing the processability. Int J Pharm. 2007;328(2):119–129. | ||
Noolkar SB, Jadhav NR, Bhende SA, Killedar SG. Solid-state characterization and dissolution properties of meloxicam-moringa coagulant-PVP ternary solid dispersions. AAPS Pharm Sci Tech. 2013;14(2):569–577. | ||
Ahmed TA, Ibrahim HM, Ibrahim F, Samy AM, Fetoh E, Nutan MT. In vitro release, rheological, and stability studies of mefenamic acid coprecipitates in topical formulations. Pharm Dev Technol. 2011;16(5):497–510. | ||
Ahmed TA, Suhail MAA, Hosny KM, Abd-Allah FI. Clinical pharmacokinetic study for the effect of glimepiride matrix tablets developed by quality by design concept. Drug Dev Ind Pharm. 2017;44(1):66–81. | ||
Kalaycioglu GD, Aydogan N. Preparation and investigation of solid lipid nanoparticles for drug delivery. Colloids Surf A Physicochem Eng Asp. 2016;510:77–86. | ||
Ahmed TA, Aljaeid BM. A potential in situ gel formulation loaded with novel fabricated poly(lactide-co-glycolide) nanoparticles for enhancing and sustaining the ophthalmic delivery of ketoconazole. Int J Nanomedicine. 2017;12:1863–1875. | ||
Harbi I, Aljaeid B, El-Say KM, Zidan AS. Glycosylated sertraline-loaded liposomes for brain targeting: QbD study of formulation variabilities and brain transport. AAPS Pharm Sci Tech. 2016;17(6):1404–1420. | ||
Ahmed TA, El-Say KM, Hosny KM, Aljaeid BM. Development of optimized self-nanoemulsifying lyophilized tablets (SNELTs) to improve finasteride clinical pharmacokinetic behavior. Drug Dev Ind Pharm. 2018;44(4):652–661. | ||
El-Say KM, Ahmed TA, Ahmed OAA, Hosny KM, Abd-Allah FI. Self-nanoemulsifying lyophilized tablets for flash oral transmucosal delivery of vitamin K: development and clinical evaluation. J Pharm Sci. 2017;106(9):2447–2456. | ||
Krishnaiah YSR. Pharmaceutical technologies for enhancing oral bioavailability of poorly soluble drugs. J Bioequiv Availab. 2010;2(2):28–36. | ||
Chiou WL, Riegelman S. Pharmaceutical applications of solid dispersion systems. J Pharm Sci. 1971;60(9):1281–1302. | ||
Aggarwal AK, Jain S. Physicochemical characterization and dissolution study of solid dispersions of ketoconazole with nicotinamide. Chem Pharm Bull (Tokyo). 2011;59(5):629–638. | ||
Homayouni A, Sadeghi F, Nokhodchi A, Varshosaz J, Garekani HA. Preparation and characterization of celecoxib solid dispersions; comparison of poloxamer-188 and PVP-K30 as carriers. Iran J Basic Med Sci. 2014;17(5):322–331. | ||
Modi A, Tayade P. Enhancement of dissolution profile by solid dispersion (kneading) technique. AAPS Pharm Sci Tech. 2006;7(3):68. | ||
Dézsi L, Kis-Varga I, Nagy J, Komlódi Z, Kárpáti E. Neuroprotective effects of vinpocetine in vivo and in vitro. Apovincaminic acid derivatives as potential therapeutic tools in ischemic stroke. Acta Pharm Hung. 2002;72(2):84–91. Hungarian. | ||
Szilágyi G, Nagy Z, Balkay L, et al. Effects of vinpocetine on the redistribution of cerebral blood flow and glucose metabolism in chronic ischemic stroke patients: a PET study. J Neurol Sci. 2005;229–230:275–284. | ||
Medina AE. Vinpocetine as a potent antiinflammatory agent. Proc Natl Acad Sci USA. 2010;107(22):9921–9922. | ||
Jeon KI, Xu X, Aizawa T, et al. Vinpocetine inhibits NF-B-dependent inflammation via an IKK-dependent but PDE-independent mechanism. Proc Natl Acad Sci USA. 2010;107(21):9795–9800. | ||
Schmidt J. Comparative studies on the anticonvulsant effectiveness of nootropic drugs in kindled rats. Biomed Biochim Acta. 1990;49(5):413–419. | ||
Bönöczk P, Panczel G, Nagy Z. Vinpocetine increases cerebral blood flow and oxygenation in stroke patients: a near infrared spectroscopy and transcranial Doppler study. Eur J Ultrasound. 2002;15(1–2):85–91. | ||
Luo Y, Chen D, Ren L, Zhao X, Qin J. Solid lipid nanoparticles for enhancing vinpocetine’s oral bioavailability. J Control Release. 2006;114(1):53–59. | ||
Liu M, Zhang S, Cui S, et al. Preparation and evaluation of Vinpocetine self-emulsifying pH gradient release pellets. Drug Deliv. 2017;24(1):1598–1604. | ||
Cui SX, Nie SF, Li L, Wang CG, Pan WS, Sun JP. Preparation and evaluation of self-microemulsifying drug delivery system containing vinpocetine. Drug Dev Ind Pharm. 2009;35(5):603–611. | ||
El-Dahmy RM, Elsayed I, Elshafeey AH, Gawad NA, El-Gazayerly ON. Optimization of long circulating mixed polymeric micelles containing vinpocetine using simple lattice mixture design, in vitro and in vivo characterization. Int J Pharm. 2014;477(1–2):39–46. | ||
Leathers TD. Biotechnological production and applications of pullulan. Appl Microbiol Biotechnol. 2003;62(5–6):468–473. | ||
Dixit RP, Puthli SP. Oral strip technology: overview and future potential. J Control Release. 2009;139(2):94–107. | ||
Lowery M. New pullulan-based capsules offer unique benefits for supplement manufacturers and healthy; 2012. Available from: https://www.nutraceuticalsnow.com/articles/2012/08/27/new-pullulan-based-capsules-offer-unique-benefits-supplement-manufacturers-healthy/. Accessed May 9, 2018. | ||
Sattar M, Sayed OM, Lane ME. Oral transmucosal drug delivery – current status and future prospects. Int J Pharm. 2014;471(1–2):498–506. | ||
Brigo F, Nardone R, Tezzon F, Trinka E. Nonintravenous midazolam versus intravenous or rectal diazepam for the treatment of early status epilepticus: a systematic review with meta-analysis. Epilepsy Behav. 2015;49:325–336. | ||
Nazari K, Kontogiannidou E, Ahmad RH, et al. Development and characterisation of cellulose based electrospun mats for buccal delivery of non-steroidal anti-inflammatory drug (NSAID). Eur J Pharm Sci. 2017;102:147–155. | ||
Nazari K, Kontogiannidou E, Haj Ahmad R, et al. Fibrous polymeric buccal film formulation, engineering and bio-interface assessment. Eur Polym J. 2017;97:147–157. | ||
Teagarden DL, Baker DS. Practical aspects of lyophilization using non-aqueous co-solvent systems. Eur J Pharm Sci. 2002;15(2):115–133. | ||
Ding J, Li J, Mao S. Development and evaluation of vinpocetine inclusion complex for brain targeting. Asian J Pharm Sci. 2015;10(2):114–120. | ||
Higuchi T, Connoras K. Phase-solubility techniques. Adv Anal Chem Instrum. 1965;4:117–210. | ||
Nicolescu C, Aramă C, Nedelcu A, Monciu CM. Phase solubility studies of the inclusion complexes of repaglinide with β-cyclodextrin and β-cyclodextrin derivatives. Farmacia. 2010;58:5. | ||
The United States Pharmacopeia TNF. USP 28/NF 23. Rockville, MD: US Pharmacopoeial Convention Inc; 2005. | ||
Leuner C, Dressman J. Improving drug solubility for oral delivery using solid dispersions. Eur J Pharm Biopharm. 2000;50(1):47–60. | ||
Rajpurohit VS, Rakha P, Goyal S, Dureja H, Arorac G, Nagpal M. Formulation and characterization of solid dispersions of glimepiride through factorial design. Iran J Pharm Sci. 2011;7(1):7–16. | ||
Ahmed OAA, Ahmed TA, Abdel-Naim AB, Khedr A, Banjar ZM, Afouna MI. Enhancement of in vitro skin transport and in vivo hypoglycemic efficacy of glimepiride transdermal patches. Trop J Pharm Res. 2014;13:1207–1213. | ||
Dong Z, Chatterji A, Sandhu H, Choi DS, Chokshi H, Shah N. Evaluation of solid state properties of solid dispersions prepared by hot-melt extrusion and solvent co-precipitation. Int J Pharm. 2008;355(1–2):141–149. | ||
Shinde VR, Shelake MR, Shetty SS, Chavan-Patil AB, Pore YV, Late SG. Enhanced solubility and dissolution rate of lamotrigine by inclusion complexation and solid dispersion technique. J Pharm Pharmacol. 2008;60(9):1121–1129. | ||
Choudhary A, Rana AC, Aggarwal G, Kumar V, Zakir F. Development and characterization of an atorvastatin solid dispersion formulation using skimmed milk for improved oral bioavailability. Acta Pharmaceutica Sinica B. 2012;2(4):421–428. | ||
Kim MS, Kim JS, Cho W, Park HJ, Hwang SJ. Oral absorption of atorvastatin solid dispersion based on cellulose or pyrrolidone derivative polymers. Int J Biol Macromol. 2013;59:138–142. | ||
Kharshoum RM, Sanad RA, Abdelhaleem Ali AM. Comparative Pharmacokinetic Study of Two Lyophilized Orally Disintegrating Tablets Formulations of Vinpocetine in Human Volunteers. Vol 5; 2013. Available from: http://www.arjournals.org/index.php/ijdd/index. Accessed August 25, 2018. | ||
Badr-Eldin SM, Ahmed TA, Ismail HR. Aripiprazole-cyclodextrin binary systems for dissolution enhancement: effect of preparation technique, cyclodextrin type and molar ratio. Iran J Basic Med Sci. 2013;16(12):1223–1231. | ||
Goddeeris C, Willems T, Van den Mooter G. Formulation of fast disintegrating tablets of ternary solid dispersions consisting of TPGS 1000 and HPMC 2910 or PVPVA 64 to improve the dissolution of the anti-HIV drug UC 781. Eur J Pharm Sci. 2008;34(4–5):293–302. | ||
Singh OPB, Biswal S, Sahoo J, Murthy PN. Physicochemical properties of glimepiride in solid dispersions with polyethylene glycol 20000. Int J Pharm Sci Nanotechnol. 2009;2(2):537–543. | ||
Kumar P, Mohan C, Kanamsrinivasan Uma Shankar M, Gulati M. Physiochemical characterization and release rate studies of solid dispersions of ketoconazole with pluronic F127 and PVP K-30. Iran J Pharm Res. 2011;10(4):685–694. | ||
Pudleiner P, Vereczkey L. Study on the absorption of vinpocetine and apovincaminic acid. Eur J Drug Metab Pharmacokinet. 1993;18(4):317–321. | ||
Zhuang CY, Li N, Wang M, et al. Preparation and characterization of vinpocetine loaded nanostructured lipid carriers (NLC) for improved oral bioavailability. Int J Pharm. 2010;394(1–2):179–185. | ||
Makar RR, Latif R, Hosni EA, El Gazayerly ON. Optimization for glimepiride dissolution enhancement utilizing different carriers and techniques. J Pharm Invest. 2013;43(2):115–131. | ||
Ahmed TA. Preparation of finasteride capsules-loaded drug nanoparticles: formulation, optimization, in vitro, and pharmacokinetic evaluation. Int J Nanomedicine. 2016;11:515–527. | ||
Kurakula M, Ahmed TA. Co-delivery of atorvastatin nanocrystals in PLGA based in situ gel for anti-hyperlipidemic efficacy. Curr Drug Deliv. 2016;13(2):211–220. | ||
Esim O, Savaser A, Ozkan CK, Bayrak Z, Tas C, Ozkan Y. Effect of polymer type on characteristics of buccal tablets using factorial design. Saudi Pharm J. 2018;26(1):53–63. | ||
Dinge A, Nagarsenker M. Formulation and evaluation of fast dissolving films for delivery of triclosan to the oral cavity. AAPS PharmSciTech. 2008;9(2):349–356. | ||
Mital M, Panchal S, Patel H, Bagada A, Vadalia KR. Formulation and evaluation of mouth dissolving film of ropinirole hydrochloride by using pullulan polymers. Int J Pharm Res Allied Sci. 2012;1(3):60–72. | ||
Gherman S, Zavastin D, Ochiuz L, Biliuta G, Coseri S. Enalapril maleate loaded pullulan film for mucoadhesive buccal drug delivery applications. Cellul Chem Technol Cellul Chem Technol. 2016;50:5–6. | ||
Cilurzo F, Cupone IE, Minghetti P, Selmin F, Montanari L. Fast dissolving films made of maltodextrins. Eur J Pharm Biopharm. 2008;70(3):895–900. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.