Back to Journals » International Journal of Nanomedicine » Volume 15
Formulation and Characterization of Cinnarizine Targeted Aural Transfersomal Gel for Vertigo Treatment: A Pharmacokinetic Study on Rabbits
Authors Abdelmonem R ![]() , Hamed RR
, Hamed RR ![]() , Abdelhalim SA, ElMiligi MF
, Abdelhalim SA, ElMiligi MF ![]() , El-Nabarawi MA
, El-Nabarawi MA ![]()
Received 18 April 2020
Accepted for publication 8 July 2020
Published 19 August 2020 Volume 2020:15 Pages 6211—6223
DOI https://doi.org/10.2147/IJN.S258764
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Anderson Oliveira Lobo
Rehab Abdelmonem,1 Raghda Rabe Hamed,1 Sally A Abdelhalim,2 Mohamed F ElMiligi,1,2 Mohamed A El-Nabarawi2
1Department of Industrial Pharmacy, College of Pharmaceutical Science and Drug Manufacturing, Misr University for Science and Technology, Cairo, Egypt; 2Department of Pharmaceutics, Faculty of Pharmacy, Cairo University, Cairo, Egypt
Correspondence: Rehab Abdelmonem
Department of Industrial Pharmacy, College of Pharmaceutical Science and Drug Manufacturing, Misr University for Science and Technology, Al-Motameyez District, 6th October City, Giza, Egypt
Tel +20 1222127127
Email [email protected]
Introduction and Aim: Cinnarizine is indicated orally for treating vertigo associated with Ménière’s syndrome and has a local anesthetic effect as well. The present study aims to develop an aural Cinnarizine mucoadhesive transfersomal gel to overcome the first-pass metabolism.
Methods: Eighteen Cinnarizine transfersomes were prepared by the thin-film hydration technique using different types of phosphatidylcholine and edge activators in different ratios. Formulae were tested for their appearance, entrapment efficiency, and in-vitro drug release after eight hours. F1, F4, F7, F9, F10, and F12 were selected to be examined for particle size, polydispersity index, and zeta potential. According to the previous parameters, F1 and F10 were incorporated into gels using different polymers according to factorial design 23. The eight gels were tested for appearance, pH, mucoadhesion, spreadability, drug content, in-vitro drug release after eight hours, and rheology. The transfersomal gel F1A was subjected to FTIR analysis and in-vivo pharmacokinetic study.
Results: The transfersomal dispersion colors were ranging between the white and yellow. Their EE % ranged from 64.36± 1.985% to 94.09± 1.74%, and their in-vitro release percentages were between 61.82± 1.92% and 95.92± 1.18%. Also, the vesicles PS ranged from 212.3± 30.05nm to 2150± 35.35nm, DI from 0.238± 0.134 to 1± 0.00 and zeta potential from − 57.5± 2.54 to +4.73± 1.57 mV. The transfersomal gels showed pseudoplastic behavior, pH range of 5.5 to 8, a mucoadhesive force of 169.188± 1.26 to 321.212± 6.94 (dyne/cm2× 102), spreadability of 40 ± 7.03mm to 138 ± 3.77mm, and in-vitro drug release of 81.63± 1.128% to 97.78± 0.102%. The IR spectra of the (drug-excipients) physical mixture revealed that there were no shifts of incompatibility. The in-vivo pharmacokinetic study illustrated that [AUC]0– 24 of F1A was significantly higher than that of tablets at (P< 0.05), equivalent to 703.563± 26.470 and 494.256± 9.621ɲg.hr/mL respectively.
Conclusion: The study revealed that Cinnarizine aural mucoadhesive targeted delivery provides an improved systemic bioavailability over the conventional oral route.
Keywords: targeted transfersomes, aural gel, Cinnarizine
Introduction
Targeted drug delivery is the process of delivering the therapeutic substance to the targeted tissues to enhance the therapeutic efficacy and decrease the potential side effects.1 Middle ear targeted drug delivery is challenging because of the presence of the tympanic membrane, which is considered the first barrier that resists drug entry to the middle and inner ear.2 Therefore, to achieve an efficient middle ear drug delivery, drugs should have a sufficient ability to diffuse through the intact tympanic membrane.3 As both the tympanic membrane and the middle ear are considered highly perfused tissues, the aural drug administration can provide the right choice for local and systemic drug delivery.4 Short residence time due to rapid spillage from the ear is the major drawback of the eardrops, while more precise dosing is the privilege of the aural gels because they have high viscosity.5 The formulation of a mucoadhesive gel depends on the presence of the suitable thermosensitive gelling polymers, including poloxamers, pluronics, and carbopols that undergo a phase transition and bio-adhesion.6 Transferosome is a targeted vesicular drug delivery system that has a highly elastic wall membrane. Therefore, those vesicles can squeeze themselves through a pore that could be smaller than their size.7
This system is designed to deliver drugs non-invasively through different membrane barriers without measurable loss. Transferosomes are recently used as carriers for the controlled and targeted delivery of drugs as proteins, peptides, hormones.8 Cinnarizine is indicated for the treatment of the symptoms of Ménière’s syndrome as vertigo.9 Moreover, it improves the cerebral vascularization and relieves the migraine symptoms. It is a sedative calcium antagonist that has a local anesthetic effect.10 Cinnarizine is extensively and quickly metabolized in the liver mainly by N-dealkylation.11 The present study’s focal intent is to develop a middle ear targeted transtympanic delivery system of Cinnarizine in the form of transfersomal gel to enhance its absorption and bioavailability.
Materials and Methods
Materials
Cinnarizine was kindly supplied as a gift by The Arab Drug Co., Cairo, Egypt. Phosphatidylcholine of egg yolk, Phosphatidylcholine of soybean and Dipalmitoylphosphatidylcholine, Pluronic F-127, Sodium cholate, and sodium deoxycholate were purchased from Sigma Chemical Co., USA. Sodium dihydrogen phosphate, Citric acid, and Chloroform were purchased from El- Nasr Pharmaceutical Co., Cairo, Egypt. Methanol of HPLC grade was purchased from El-Shark al Awsat Company, Cairo, Egypt. Triethanolamine was purchased from E.Merk, Germany. Carbopol 934 was supplied by B.F. Goodrich Chemical Company, Ohio, USA. Hydroxypropyl methylcellulose (HPMC) was provided by Tama, Tokyo, Japan.
Methods
Cinnarizine Transfersomal Dispersion
Eighteen Cinnarizine transfersomes were prepared using the thin-film hydration technique (TFH). Cinnarizine, Phosphatidylcholine (PC), and the edge activators (EA) are the primarily used components, as shown in Table 1. In a round flask, the PC and the EA were dissolved in 10 mL of a chloroform-methanol mixture (1:1 v/v). The organic solvent was then slowly evaporated using a rotary evaporator at 50ᵒC and 90 rpm under vacuum (Heidolph VV 2000, Burladingen, Germany) until the formation of a thin, dry film of the components. The dried thin film was hydrated via adding 5mL of McIlvaine buffer (pH= 5.5) containing the required amount of drug. The flask was rotated in the water bath of the rotary evaporator at 50ᵒC under normal pressure. The obtained dispersions were then collected and sonicated for two minutes in a bath sonicator (Crest Ultrasonics Corp, Trenton, USA). Then the transfersomal dispersions were left overnight at 4ᵒC to mature.12
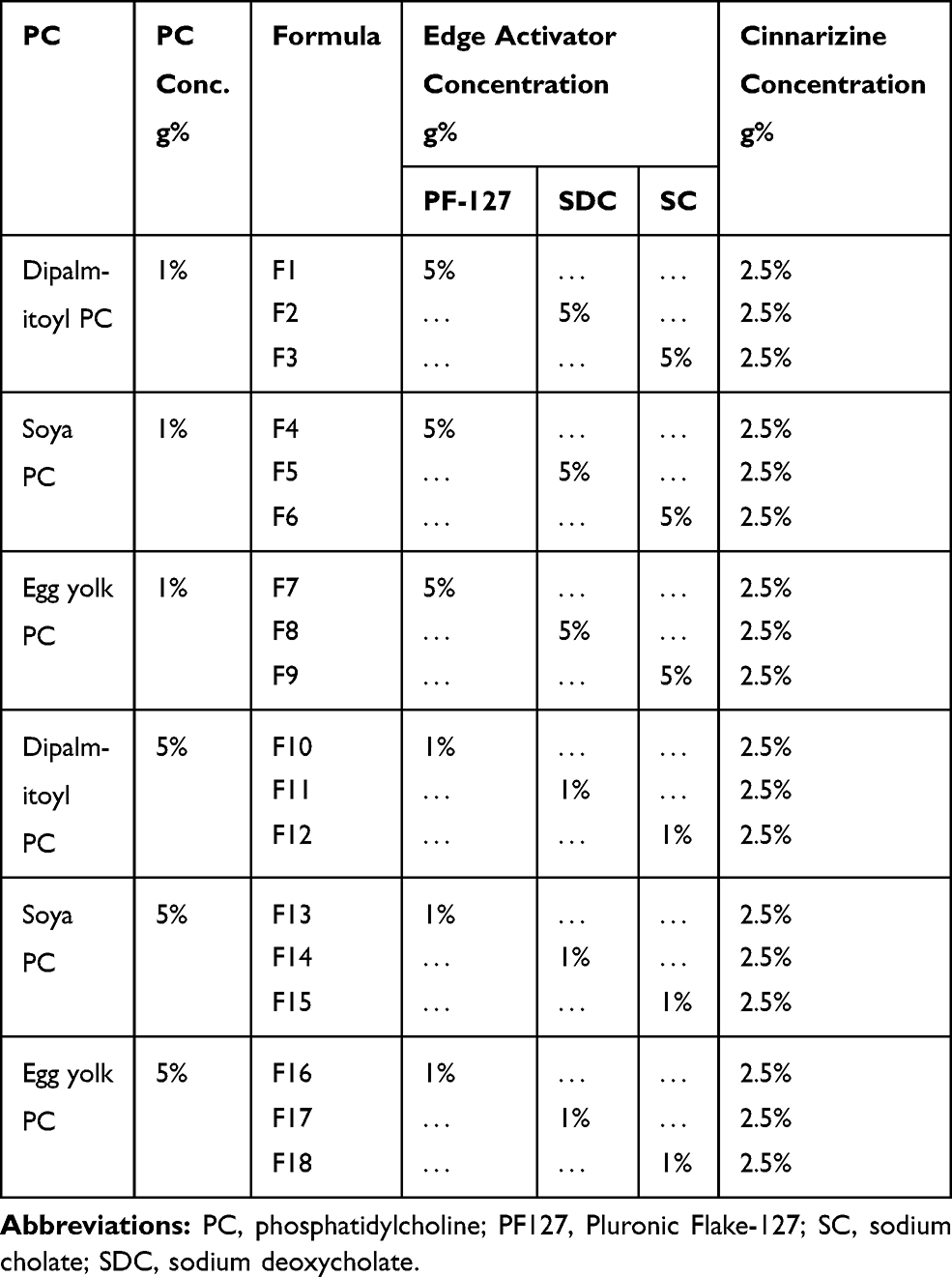 |
Table 1 The Composition of Cinnarizine Transfersomes |
The features and other properties, including color, turbidity, odor, and dispersibility of the freshly prepared transfersomal dispersion, were investigated. All formulae were examined visually above black and white backgrounds and then were rated as a slightly turbid (+), turbid (++), and a cloudy preparation (+++). Upon shaking for one-minute, all the preparations were evaluated as dispersible or non-dispersible.
One mL of each prepared dispersion was first dissolved in methanol then measured at the predetermined λmax of Cinnarizine in methanol (253nm) to calculate the total drug content (Free + entrapped), using a UV spectrophotometer (Shimadzu UV 1650, Japan). One mL of each formula was centrifuged at 14,000 rpm for 50 min at 4°C using the cooling ultracentrifugation (Beckman, Fullerton, Canada) to separate the transfersomal pellets. The separated pellets were washed with a McIlvaine buffer and isolated carefully using the cooling ultracentrifuge then disrupted by sonication with methanol to estimate the amount of Cinnarizine entrapped spectrophotometrically at the same λmax. The entrapment efficiency was calculated according to the following equation, and each result was the mean of three determinations ±SD.13

The transfersomal pellets of one mL of each preparation were isolated, washed, and diluted in McIlvaine buffer of pH 5.5, forming one mL suspension (equivalent to 25mg of Cinnarizine). The drug released from that suspension was tested using the modified USP dissolution apparatus II (Diss 6000, Switzerland).14 The receiver vessel contained 100 mL McIlvaine buffer (pH 5.5) at 37±0.5°C with a paddles speed of 100 rpm.15 An aliquot of (1 mL) was withdrawn at predetermined time intervals over 8 hours; at 0.5, 1, 1.5, 2, 3, 4, 6, and 8 hours and immediately replaced with a fresh release medium. The withdrawn aliquots were spectrophotometrically analyzed at the predetermined λmax of Cinnarizine in McIlvaine buffer (253 nm) to determine the released drug amount. The results are the mean values of three runs ±SD. A cumulative correction factor was applied to compensate for diluting the samples in all processes.16,17
The average diameter in (nm), polydispersity index, and zeta potential of the selected formulae were determined by Malvern Zetasizer at 25оC, backscatter detection of 173ºC, and refractive index of 1.330 (Malvern Instruments, Ltd., UK). The formulations were appropriately diluted with distilled water, which provided suitable vesicles scattering intensity to measure the pellet size. The PDI was measured through dynamic light scattering (DLS), which detects vesicle distribution. The ZP was carried out in distilled water by observing the electrophoretic mobility of charged vesicles in an electrical field, and that indicates the vesicle permeation behavior by studying its colloidal property and stability.18–20 The results are the mean values of three runs ±SD. F1 and F10 were selected to be incorporated into gel formulae.
Cinnarizine Aural Transfersomal Gels
Eight aural gel formulae were prepared by 23 factorial design at two levels taking the type of the selected transfersomal formulae, type of gel-forming polymer, and the polymer concentration into consideration.21 They were prepared by the gradual addition of 4 mL of the selected transfersomal pellets suspension of F1 and F10 to 1/3 of the required amount of McIlvaine buffer of pH 5.5 at 80°C. After that, the calculated amount of the selected polymer was added while stirring (LMS-1003, Kyonggi-Do, Korea). Then, the final volume was adjusted to100ml by McIlvaine buffer. Finally, the prepared gels were stored overnight in the refrigerator. Any air bubbles entrapped in the gels were removed by centrifugation.22 The composition of the prepared gels is listed in Table 3.
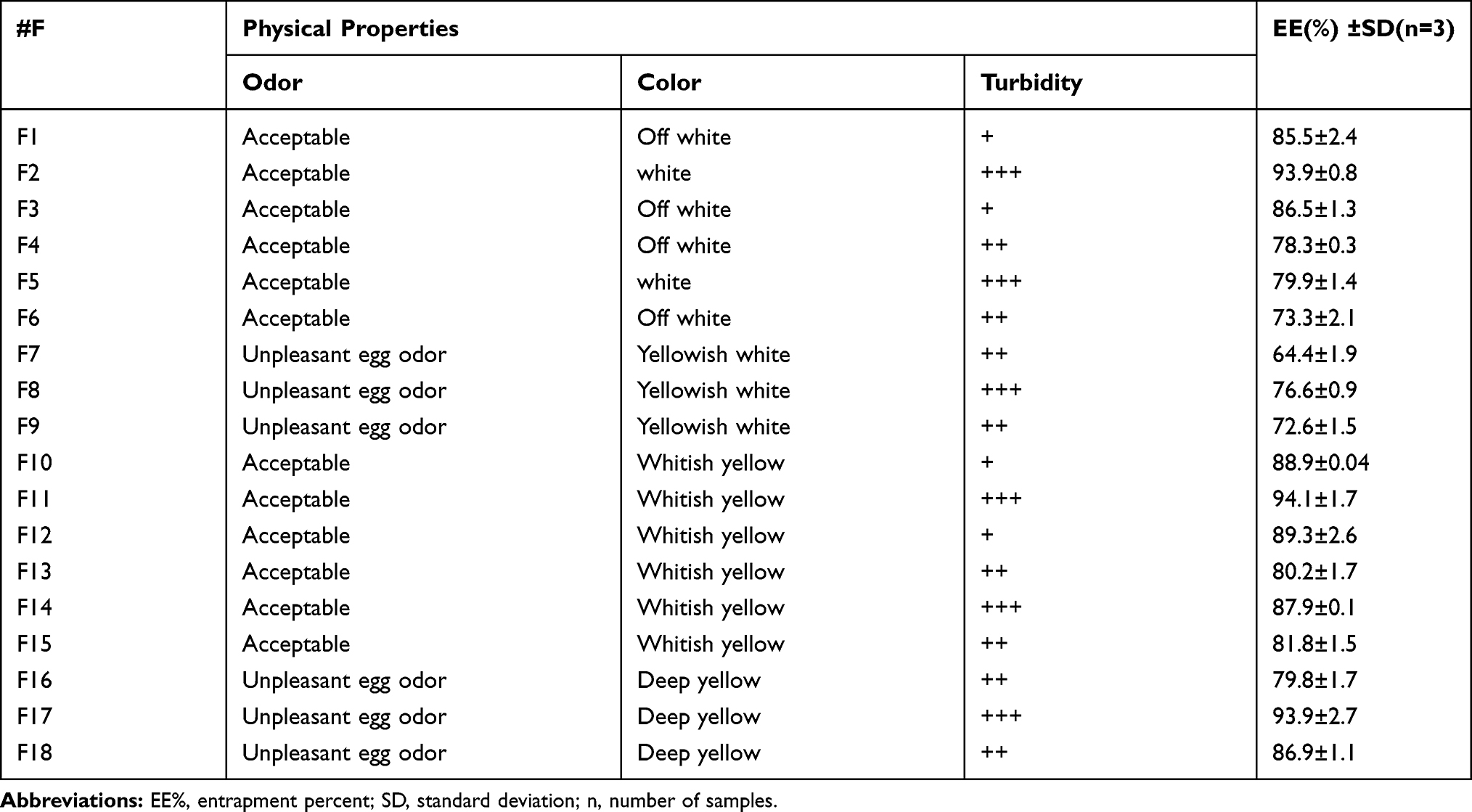 |
Table 2 The Physical Properties, EE% of Cinnarizine Freshly Prepared Transfersomal Dispersion |
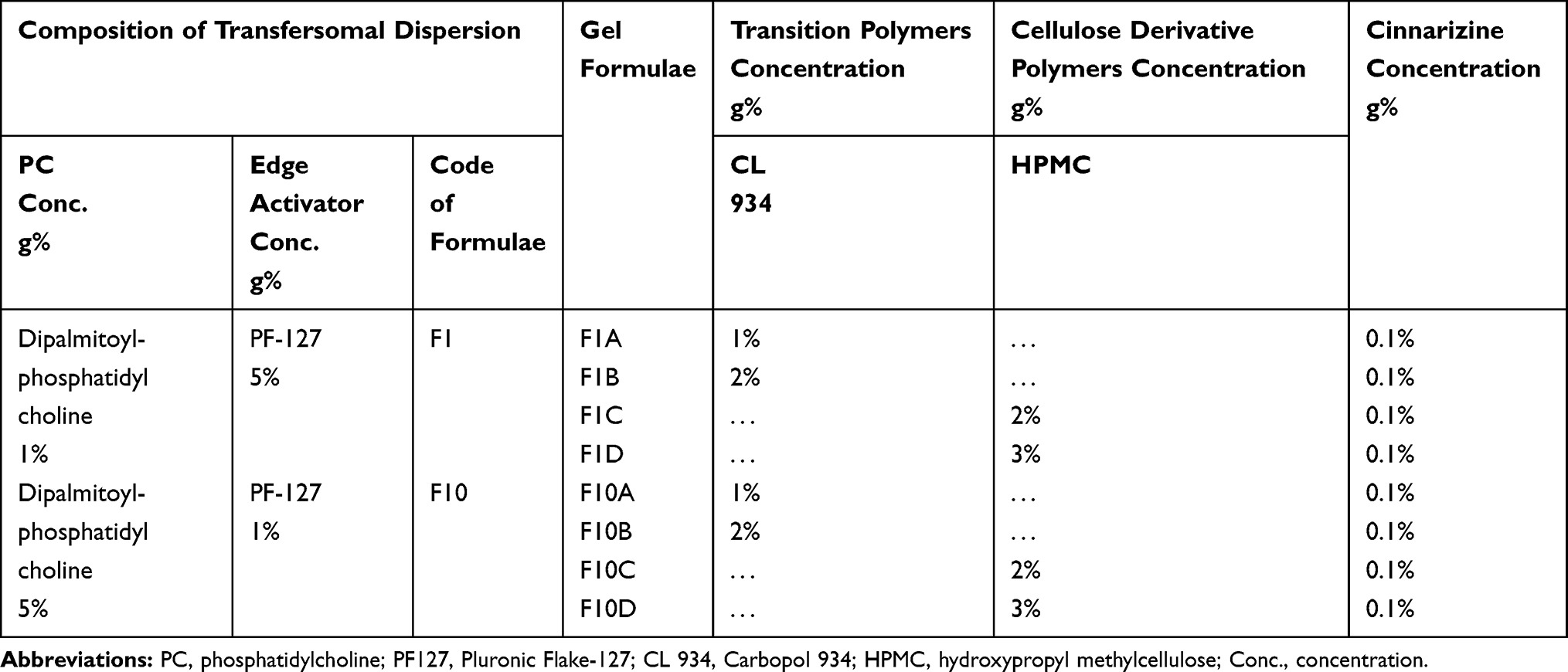 |
Table 3 The Composition of the Cinnarizine Transfersomal Gels Using 23 Factorial Designs |
The physical properties as clarity, turbidity, precipitation, and syneresis of freshly prepared gels were inspected visually over black and white backgrounds then were graded as turbid, clear, and very clear.23
The pH of the freshly formed gels was investigated by mixing one gm of each gel with distilled water to get a ten mL solution. The pH was determined using the pH meter. The mean of the three results was taken.24
In a 100mL volumetric flask, 1gm of each gel was withdrawn and diluted then sonicated with McIlvaine buffer of pH 5.5. Further dilution of one mL of that solution up to ten was done when necessary. The absorbance of the solution was estimated at 253nm using a UV visible spectrophotometer (Shimadzu UV 1650, Japan).25
Mucoadhesion was estimated using a laboratory adjusted balance by placing 250mL beaker on the right side of the balance, and on the left side, a 100 gm weight was hung using a thin steel wire. The balance was equalized, making the hanging weight resting over the base of a 250mL inverted beaker. Two slices of a fresh rabbit small intestine were cleaned with normal saline. One was attached to the undersurface of the 100gm weight, while the other was fixed on the base of an inverted beaker of 250mL capacity using cyanoacrylate adhesive. 0.5 gm of the gel was applied between the two sections and left for two minutes to ensure good contact. An infusion device was opened to drop water into the beaker of the right side at a fixed flow rate of 13–15 drops per minute. The weight of the beaker continued increasing until the two slices at the opposite side were just separated. The weight of water was recorded to calculate the mucoadhesive force; (ie, the detachment stress) (dyne/cm2) using the following equation.21 The results are the mean values of three runs ±SD.

Where (m) is the weight of water, (g) is the acceleration due to gravity taken as 981 cm/sec2, and (A) is the area of the rabbit’s small intestine (area of contact). The experiment was performed at room temperature in triplicates.
0.5 gm of each gel was located between two glassy slides; the upper slide was moveable while the lower one was fixed. A fixed pressure was applied above the upper slide. The gel spreading diameter of three replicates was recorded.26
The drug release profile of each gel was studied with the same procedure of studying that of Transfersomes using one mL of each gel (equivalent to 1mg of Cinnarizine).
The aural gels viscosity was evaluated using a rotational Brookfield viscometer of cone and plate structure, spindle 52 at 25±1oC (Cone and Plate viscometer; model III, Brookfield, USA). The measurements were taken over the acceleration of speed from 10 to 70 rpm, with 10 seconds between every two successive speeds, and then over the deceleration. The individual rheological data [η min, η max, Farrow’s constant (N), and hysteresis loop area (HA)] for each tested gel were calculated. The rheological data were analyzed using Farrow’s equation and the Power-law equation to predict each formula’s rheological behavior.

Farrow’s equation
Where D is the Shear rate (sec −1), S is Shear Stress (dyne/cm2), N is Farrow’s constant, and η is the viscosity (cp).
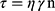
Power law equation
Where γ is Shear rate (sec −1), τ is Shear Stress (dyne/cm2), n is Power constant, and η is the consistency index (apparent viscosity).27 F1A was subjected to spectral analysis FTIR and in-vivo pharmacokinetic study.
Spectral analysis FTIR of the pure drug and the physical mixture of drug with F1A excipients were reported using an FTIR Spectrophotometer, (FTIR-8300, Shimadzu, Japan). The samples were prepared and scanned in the range of 4000 cm−1 to 400 cm−1. The IR spectrums were compared to explore whether there were any interactions between Cinnarizine and the used excipients.28
Eighteen healthy albino rabbits, weighing between 2000 and 2500 gm, were randomly allocated to one of three groups of equal sizes (ie, six rabbits in each group) according to parallel experimental design. The experiment protocol is coded as (PI-1562). The study was approved by the ethical committee of the Faculty of Pharmacy of Cairo University (Cairo, Egypt). According to the Egyptian National Laboratory Animal Center guidelines, the animals were allowed to acclimate to the approved environmental conditions under the supervision of the Laboratory Animal Center of Cairo University. The first group of rabbits was regarded as the control group, as subjects did not receive any treatment. The second group’s rabbits received one mL volume of a pre-dissolved Cinnarizine 25mg tablet® (equivalent to 1mg of Cinnarizine) via needleless syringe orally. The third group’s rabbits received one mL of the selected transfersomal gel FIA via a specific applicator ototopically directly over its tympanic membrane after being carefully cleaned (equivalent to 1mg of Cinnarizine). Blood samples were collected into heparinized-coated micro-centrifuge tubes at zero time, then after 0.5, 1, 2, 3, 4, 6, 8, 12, and 24 h following the drug administration. The plasma was collected by centrifuging the blood samples at 15,000 rpm for 15 min then was stored at –20°C.
The drug level in the prepared plasma samples was assessed by high-performance liquid chromatography Agilent 1260 Infinity II HPLC; Quaternary pump; UV detector; (Agilent 1260 Infinity II G4782A, Hewlett-Packard, Waldron, Germany) using manual injector of 20 µL loop and Hypersil BDS RP C18 column (5 µm, 200 × 4.6 mm) (Thermo Fisher, Bellefonte, PA, USA). The Chromatographic separation run consumed about 26 minutes at ambient temperature. Gradient elution was performed using two mobile phases. The first phase was Acetonitrile: Phosphate buffer pH 6 (60:40 v/v), while the second was Acetonitrile: Phosphate buffer pH 6 (80:20 v/v). Both were pumped through the column at a rate of 2 mL/min. The system was equipped using a UV-visible detector at 254 nm.
The plasma samples were prepared as follows: First, a five-hundred microliter of each plasma sample was added to a clean Eppendorf tube. Next, a double equivalent volume of acetonitrile was added and mixed well, then centrifuged at 5000 rpm for 10 min. Later, the supernatant was injected with the 20 µL loop injector to the HPLC.
The Cinnarizine plasma calibration curve was constructed using the data of 11 different concentrations using the least-squares regression method as the Cinnarizine drug was spiked and vortexed for a few minutes 500μL of the blank plasma then subjected to the same preparation conditions of the plasma samples before being analyzed by the HPLC. The mean concentrations of Cinnarizine in (ɲg/mL) of the rabbit’s plasma samples were plotted versus time in hours after being estimated using the straight-line equation of the constructed calibration curve.
The pharmacokinetic parameters, (the maximum plasma concentration (Cmax) in ɲg/mL, the time to reach the peak concentration (Tmax) in hr, the elimination rate constant (Kel), the terminal elimination half-life (te1/2), the absorption rate constant (kabs), the absorption half-life (ta1/2), and the area under the concentration versus time curve over 24 hours [AUC0-24] in ɲg.h/mL), were estimated for both formulae. The [AUC0-24] was calculated using the trapezoidal rule. The elimination and absorption rate constants in hr−1 were estimated using the method of residuals, using the data of the last 3–4 points of the plasma concentration-time curve. The terminal elimination and absorption half-lives (t1/2) in hr were calculated as ln2/kel and ln2/kabs, respectively. All data are presented as mean values with their standard deviations (mean ±SD). The relative bioavailability (FRel.) can be assessed, using the following formula considering the aural gel as the test and the oral tablet as the standard:

Pairs of groups were compared by performing Student’s t-test, and multiple group comparison was conducted by the one-way analysis of variance (ANOVA) using the statistical MS Excel 2010® software. Differences were considered significant when P < 0.0529–31.
Results and Discussion
Evaluation of the Prepared Cinnarizine Transfersomal Dispersions
Visual Inspection
The prepared formulae were suspensions with color ranged from white to yellow according to their content. All formulae were dispersible after one minute shaking; Table 2.
Determination of Drug Content and Entrapment Efficiency (EE %)
The EE % was ranged between 64.36±1.985% and 94.09±1.74%, which could be related to the PC nature, PC: EA ratio, and differences among the used EA; Table 2. It was found that vesicles contained SDC EAs showed the highest EE %, as these surfactants have longer alkyl chain (ie, low HLB value and low molecular weight), which enhances the EE %. The HLB values were 22, 18, and 16, and the molecular weights were 12,600 Da, 430.6 Da, and 416.6 Da for pluronic F-127, sodium cholate, and sodium deoxycholate, respectively. EAs of higher molecular weights may disturb the entity of the vesicle’s bilayer membrane that raises the drug diffusion to the aqueous medium during the Transfersomes centrifugation for separation. Although sodium cholate and sodium deoxycholate are anionic, the presence of an additional hydroxyl group in sodium cholate structure is another cause of the observed differences in their entrapment efficiency. The stated results showed that the Dipalmetoyl PC vesicles have the highest EE % compared to the (Soybean PC) and (Egg yolk PC) vesicles as the synthetic PC provides higher EE % than the natural types. Moreover, it was reported that lower ratios of EA create more stable vesicles with higher EE % because the destabilization of the lipid bilayer and drug leakage outside the vesicles occurred when higher ratios were used. It was noticed that negatively charged Transfersomes exhibited the highest EE.29–32
Study of in-vitro Cinnarizine Release Profile from the Prepared Transfersomes
The in-vitro released percentages of Cinnarizine from its prepared Transfersomes ranged between 61.82±1.92% and 95.92±1.18%. Taking the EA type into consideration, an inversely proportional relationship between the EE % and the percentage of the drug released was observed, ie (PF127 > SC>SDC). The Q8 % revealed that using lower ratios of EA gives more stable vesicles with fewer drug release percentages; Figure 1. The six formulae that gave the highest in-vitro drug release percentages (F1, F10, F12, F4, F7, and F9) were selected for further investigations as particle size (PS), polydispersity index (PDI) and zeta potential (ZP) determination.30–33
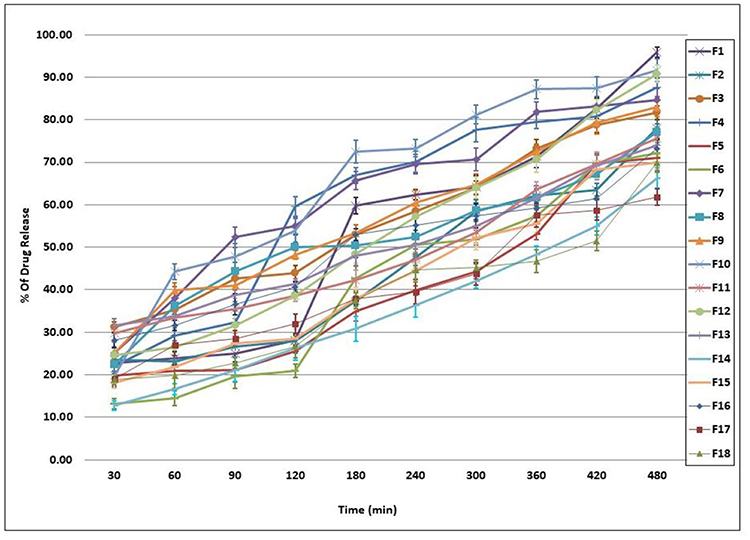 |
Figure 1 In-vitro release profile of Cinnarizine transfersomes ±SD (n=3). |
Determination of Particle Size (PS), Polydispersity Index (PDI), and Zeta Potential (ZP)
It was found that; the PS in (nm) was ranging from (212.3±30.05 to 2150±35.35), DI from (0.238±0.134 to 1±0.00), and the zeta potential in (mV) from (−57.5±2.54 to +4.73±1.57); Figure 2. The crucial factor that enables transfersomal vesicles to cross the biological membranes is their charge, size, and elasticity. It was apparent that PC: EA molar ratio affects the PS of the prepared formulae. For example, vesicles of smaller size were formed at high EA ratios as the high level of EA lowers the interfacial tension leading to the formation of smaller Nano-vesicles. Besides, the used type of EA affects the particle size, as size increases along with the decrease in the HLB value (ie, SC>PF127). EA with low HLB value interacts with the lipid membrane head groups that raise the packing density of the vesicle membrane and the surface free energy. As a result, vesicle’s size increased due to the fusion between the lipid bilayer. The net surface charge of Transfersomes is thought to be a combination of drug, lipid, and surfactant charges, so it was greatly affected by the used type of surfactant. EA of anionic nature as SC imparts a negative charge on the vesicles that lead to the repulsion between the vesicles’ lamellae (ie, increasing the vesicle’s size). On the other hand, using cationic or nonionic surfactants decrease the negativity of the vesicles. Baring negative charge is considered advantageous, because of the enhancement of the Transfersomes permeability due to the repulsive forces between the vesicles and the biological barrier’s surface, which help them diffuse deeper and faster through the stratum corneum than the positive vesicles at the initial time stage after application.34 It was reported that sodium cholate-based Transfersomes exhibited the highest negative zeta potential value as well as the largest size F9 and F12. Zeta potential plays a vital role in the vesicle’s stability by causing either repulsion or agglomeration among vesicles. The size of the vesicles detects their ability to pass through the small pores of the biological membranes. Therefore, factors as charge and size should be compromised to get the optimum vesicle. PDI is an indication of homogeneity of the vesicular dispersions (value of 0 specifies a monodispersed system and value of 1 specifies a highly polydispersed system). The reported PDI result indicates a fair size distribution and homogeneity of the formulations.35,36 The most successful Transfersomes were selected according to the following parameters: charge negativity (F1, F9, and F10& F12), then the highest EE % (F1, F10 & F12), and finally the smallest in size (F1&F10). So, F1 and F10 were selected to be incorporated into gel forms.
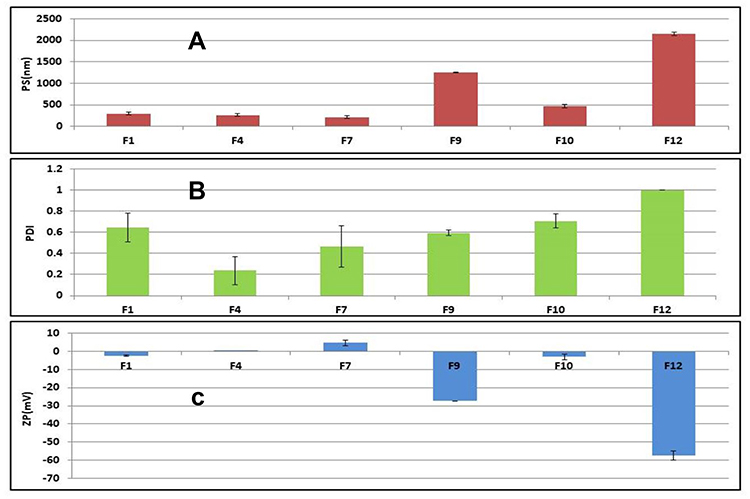 |
Figure 2 (A) Size distribution profile (nm) ±SD, (B) Polydispersity index ±SD and (C) Zeta potential (Mv) ±SD of the selected transfersomes, (n=3). |
Evaluation of Cinnarizine Prepared Transfersomal Gels
Visual Inspection
The colors of the prepared gels from the transfersomal formula F1 and F10 reflect the colors of the used mother transfersomal formula. The physical examination of the freshly prepared gels revealed that the formulae with higher polymer concentrations exhibited gels of higher viscosity. The degree of clarity is reported as (+) for highly translucent, (++) for slightly translucent, (+++) for clear and (++++) for crystal clear formulae; Table 4.
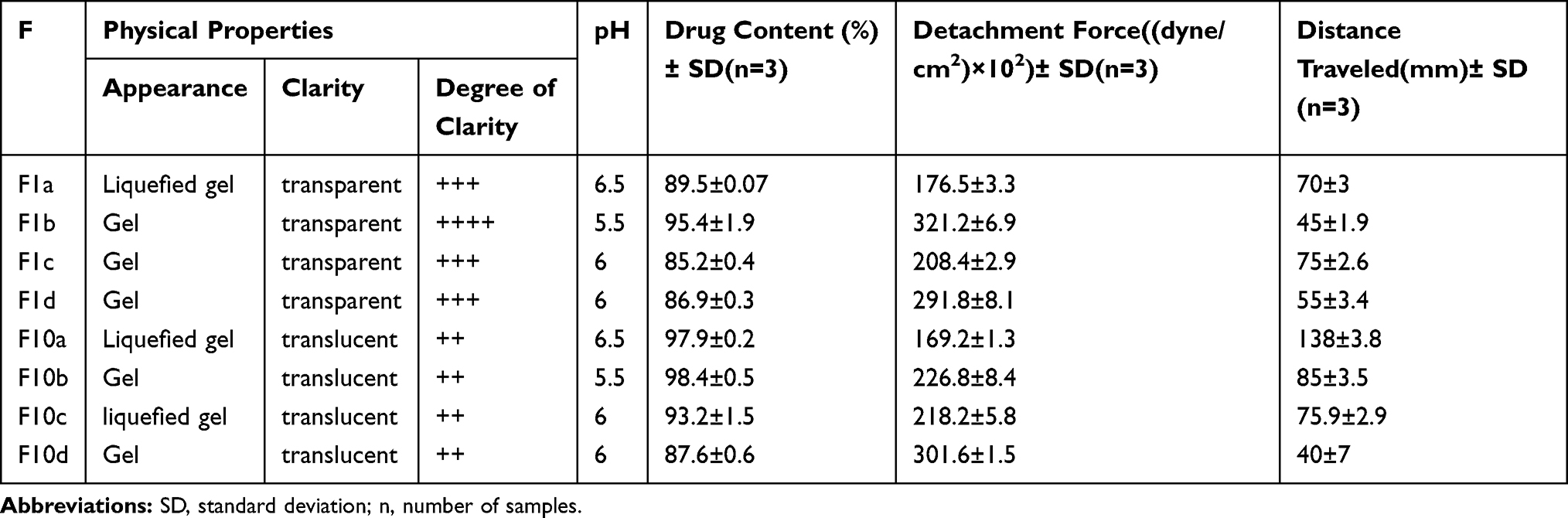 |
Table 4 The Physical Properties, pH, Drug Content, Mucoadhesion, and Spreadability of Cinnarizine Freshly Prepared Aural Transfersomal Gels |
pH Measurement
All formulae exhibited pH values ranging between 5.5 and 6.5, which are suitable and non- irritant to the middle ear mucosal surface;37,38 Table 4.
Determination of Drug Content
The theoretical drug concentration was 0.1 g %, while the drug content values ranged between (85.21%±0.40–98.38% ±0.48); Table 4.
Determination of Mucoadhesive Force
The Mucoadhesive Strength of tested gels after two minutes of contact time is presented in Table 4. It was reported that the higher the concentration of the polymer used, the higher the mucoadhesive strength becomes.39 The detachment force ranged from 169.188±1.26 to 321.212±6.94 in ((dyne/cm2) ×102).
Spreadability
The degree of spreadability of the tested gels was indicated through the distance traveled by them when they were individually compressed between slides. It was noticed that the spreadability was decreasing with the increase in the concentration of the polymer.40 The distance traveled by the gels ranged from 40 ±7.03 mm to 138 ±3.77 mm; Table 4.
The in-vitro Release Profile of Cinnarizine Aural Transfersomal Gels
The in-vitro released percentages of Cinnarizine from its prepared aural transfersomal gels were ranging between 81.63±1.128% and 97.78±0.102%; Figure 3. The reported results revealed that the top drug release percentages were for gels of CL934 bases followed by that prepared using HPMC. It was noticed that changing the polymer concentration affects Cinnarizine’s extent released from the prepared gel formulae. An inversely proportional relationship was observed between the concentrations of the used polymer and the released amount of drug. The density of the polymer chain structure leads to restricting the active substance movement area, which in turn decreases the release of the active substance.41
 |
Figure 3 In vitro Release profile of Cinnarizine aural transfersomal gels ±SD (n=3). |
Viscosity Measurement
All the prepared formulae possessed thixotropic behavior except F1c, and F10c exhibited a non-thixotropic behavior. Thixotropy is a time-dependent flow that occurs as the gel requires a finite time to rebuild its original structure that was broken down during continuous shear measurement. The hysteresis loop was measured by the trapezoidal rule to determine the degree of thixotropy for each formula. All formulae exhibited non-Newtonian shear-thinning (pseudoplastic) flow since the viscosity decreased with increasing the shear rate. The causes of pseudoplastic flow may be the progressive rupture of the gels’ internal structure by increasing shear and later reconstruction through the Brownian movement. It was clear that the Farrow’s constant (N) is greater than (1) for all of the formulae indicating non-Newtonian shear-thinning pseudoplastic flow behavior. Following the power law, the flow index (n) values were smaller than (1), which indicates a non-Newtonian shear thinning pseudoplastic flow behavior. The lower the flow index (n), the more shear-thinning is.42,43 The gel formulae were arranged in a descending order according to their viscosities at a minimum shear rate as follows: F10d> F1b> F1d> F1a> F10b> F10c> F1c > F10a; Figure 4. F1A showed the highest permeation percentage and acceptable results in other investigations, so it was selected for FTIR analysis and the in-vivo pharmacokinetic study.
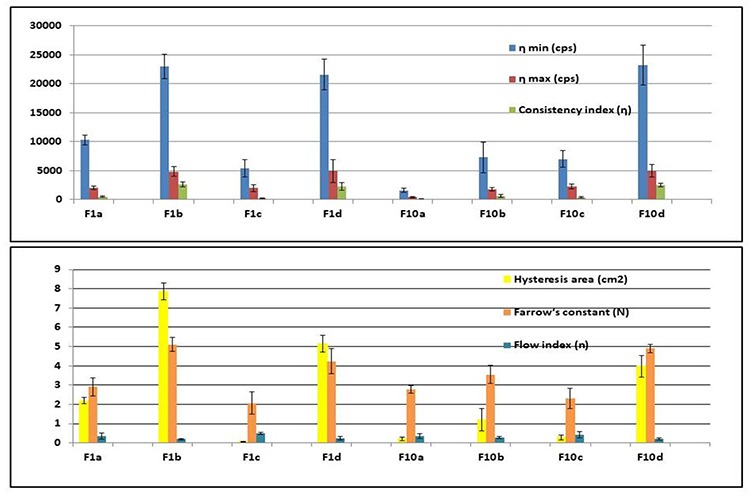 |
Figure 4 Rheological data of Cinnarizine prepared aural transfersomal gels ±SD (n=3). |
Fourier Transform Infrared (FTIR) Analysis
The infra-red spectra of Cinnarizine pure drug (free base) showed significant peaks at the following frequencies in cm−1: 2959, 2936, 1597, 1490, 1448, 1134, 999, and 962. The Frequency of 2959 cm−1 indicates (C-H) stretching (aromatic, alkene, mono-substituted), 2936 cm−1 indicates (C-H) stretch (aliphatic, alkane), 1597 cm−1 indicates (C=C) (aromatic stretch), 1490 and 1448 cm−1 indicates (CH2) (alkane), 1134 cm−1 indicates (C-N) stretching, and 999and 962 cm−1 indicates (=C-H) (aromatic, alkene);44 Figure 5. The infrared spectra of the physical mixture of F1A ingredient (1:1:1:1) (Drug: PC: PF127: CP349) showed similar characteristic peaks at their respective wavelengths, which indicates the compatibility of Cinnarizine with other excipients; Figure 5.
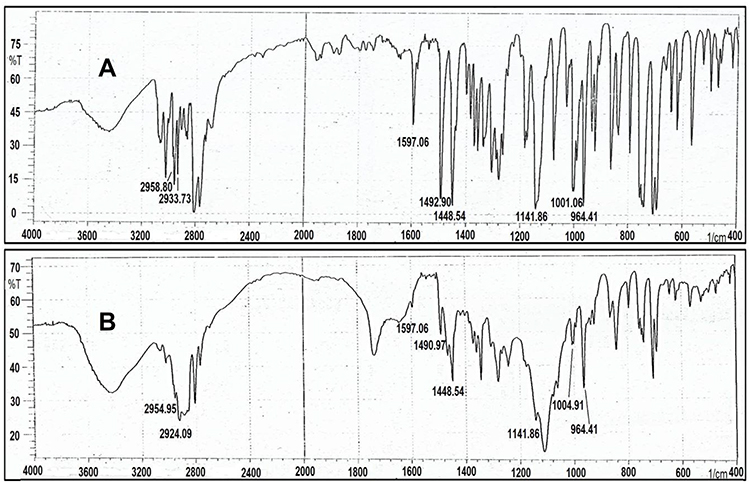 |
Figure 5 (A) FTIR Spectra of pure Cinnarizine powder, (B) FTIR Spectra of the physical mixture of (1:1:1:1) (Cinnarizine: PC: PF127: CL 934). |
In vivo Pharmacokinetic Study
The HPLC standard calibration curve was constructed by plotting Cinnarizine’s peak areas against their corresponding concentrations, where the linear regression R2 equals 0.9995 across the full range of concentrations from 50 to10000 ɲg/mL. The lower limit of quantitation (LLOQ) was 50ɲg/mL. The coefficient of variation percent CV% and the relative error percent RE% averages equal 8.933% and 6.01%, respectively, which indicates an acceptable accuracy and precision of the adopted analytical method. The mean plasma concentration versus time data of the three groups appears graphically in Figure 6, as the solid line data were fitted to one compartment open pharmacokinetic model. The pharmacokinetic parameters of the Cinnarizine® oral tablet were as follow: (Cmax)=129.775±1.144ɲg/mL, (Tmax) =2.995±0.018 hours, (Kabs) =0.478±0.003hr−1, (Kel) = 0.222±0.001hr−1, (T1/2 abs) =1.450±0.009hours, (T1/2 elimination) =3.118±0.017hours. The pharmacokinetic parameters of the aural gel (FIA); were as follows: (Cmax) = 157.162±3.580 ɲg/mL, (Tmax) was 3.868±0.054 hours, (Kabs) =0.329±0.006hr−1, (Kel) = 0.199±0.002 hr−1, (T1/2 abs) =2.109±0.040hours, (T1/2 elimination) =3.479±0.026hours; Table 5. The calculated [AUC]0–24 for FIA was significantly higher than that of Cinnarizine® oral tablet, 703.563±26.470 and 494.256±9.621 ɲg.hr/mL, respectively, at (P< 0.05).45 The relative bioavailability (FRel) of the aural form to the oral standard equals 142.347%, which indicates an increased bioavailability of the prepared aural mucoadhesive transfersomal gel over that of the oral tablets.
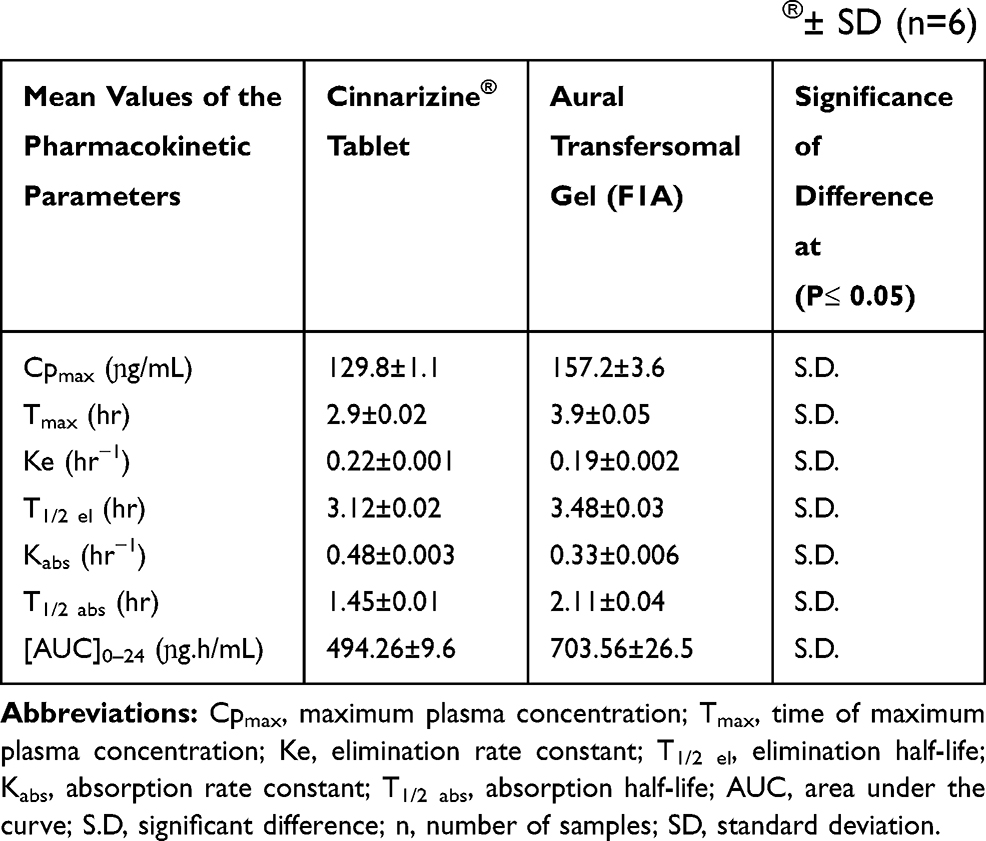 |
Table 5 The Calculated Pharmacokinetic Parameters of Both Aural FIA and the Oral Tablet of Cinnarizine 25mg®± SD (n=6) |
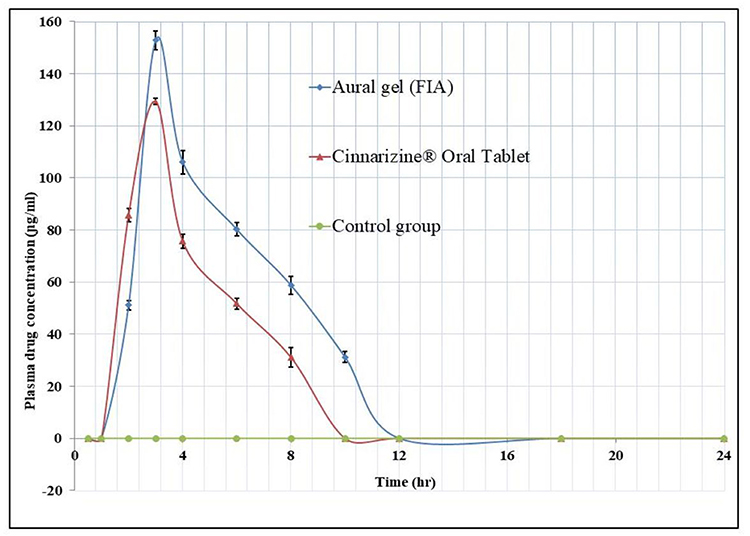 |
Figure 6 The mean plasma concentration profiles of Cinnarizine for eighteen rabbit subjects following administration of no treatment (Control group n=6), oral tablet Cinnarizine® (Second group n=6), and aural transfersomal gel F1A (Third group n=6). The solid line is data fitted to open one-compartment pharmacokinetic model ±SD. |
Conclusion
The optimum Transfersomes of F1 that composed of (1:5) Dipalmitoyl PC: PF127 and showed EE % of 85.51±2.447%, the in-vitro release of 95.92±1.18%, the average size of 295.2±25.61nm, Zeta potential of 2.44±0.516 (mV) and PDI of 0.644±0.137 was incorporated into hydrogel using 1% CL 934. The resultant gel (F1A) was a clear liquefied gel that has a pseudoplastic behavior, pH of 6.5, drug content of 89.52±0.07%, spreadability of 70±3.02mm, detachment stress of 176.544±3.28 (dyne/cm2×102), the in-vitro release of 97.78±0.102%, ingredients compatibility and increased bioavailability of Cinnarizine about 1.4 fold over that of the Cinnarizine 25mg® oral tablets which are available on the market. The in-vivo study revealed that the prepared Transfersomes could carry the Cinnarizine across the tympanic membrane of the albino rabbit subjects to the blood circulation successfully. Moreover, incorporating the Transfersomes into a gel form increased the formula residence over the tympanic membrane and prevented its leakage. Therefore, the recent study provides a promising targeted alternative delivery route of Cinnarizine that could avoid the first-pass metabolism, which potentially occurs with the oral administration.
Acknowledgment
We are so grateful to the staff members of the research lab, Misr University for Science & Technology, for their kind support.
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Disclosure
The authors report no conflicts of interest for this work.
References
1. Mishra N, Pant P, Porwal A, Jaiswal J, Samad MA, Tiwari S. Targeted drug delivery: a review. Am J Pharm Tech Res. 2016;6(1):
2. Patel J, Szczupak M, Rajguru S, Balaban C, Michael EH. Inner ear therapeutics: an overview of middle ear delivery. Front Cell Neurosci. 2019;13(article no.261). doi:10.3389
3. Qureishi A, Lee Y, Belfield K. Update on otitis media – prevention and treatment. Infect Drug Resist. 2014;10(7):15–24.
4. Szymanski A, Toth J, Geiger Z. Anatomy, Head and Neck, Ear Tympanic Membrane. StatPearls Publishing LLC; 2019. Copyright©. (Bookshelf ID: NBK448117PMID: 28846242).
5. Brunaugh AD, Hugh DCS, Williams III RO. Ophthalmic and otic drug delivery. Essential Pharm. pp ©. 2019. 123–130. (AAPSINSTR)
6. Ballachanda B. The Human Ear Canal. San Diego (CA): Plural Publishing. Incorporated©. 2013. ISBN-13: 978-1597564137, ISBN-10: 1597564133.
7. Solanki D, Kushwah L. Vicky chouhan and mohit motiwale. Transferosomes A Review ” World J Pharmacy Pharm Sci. 2016;5(10):435–449.
8. Chaurasiya P, Ganju E, Upmanyu N, Ray SK, Jain P. Transfersomes: a novel technique for transdermal drug delivery. J Drug Delivery Therapeutics. 2019;9(1):279–285. doi:10.22270/jddt.v9i1.2198
9. Kirtane MV, Bhandari A, Narang P, Santani R. Cinnarizine: a contemporary review. Indian J Otolaryngol Head Neck Surgery. 2017;71(Suppl2):S1060–S1068. doi:10.1007/s12070-017-1120-7
10. Scholtz A-W, Ilgner J, Benjamin Loader BW. Pritschow and Gerhard Weisshaar. Cinnarizine and Dimenhydrinate in the Treatment of Vertigo in Medical Practice Wien Klin Wochenschr. 2015;128(910):341–347. doi:10.1007/s00508-015-0905-5
11. Harry GB. Profiles of Drug Substances, Excipients, and Related Methodology. ISBN: 978-0-12-803300-5; ISSN: 1871-5125; Copyright©
12. Shamma RN, Elsayed. Transfersomal lyophilized gel of buspirone HCl: formulation, evaluation, and statistical optimization. J Liposome Res. 2013;23(3):244–254. doi:10.3109/08982104.2013.801489
13. Daneshmand S, Golmohammadzadeh S, Mahmoud RJ, et al. Encapsulation challenges, the substantial issue in solid lipid nanoparticles characterization. Journal of cellular biochemistry. 2018;119(6):4251–4264.
14. El-Hadidy GN. A pharmaceutical study on topical antifungal drug [M.Sc. Thesis]. Cairo Univ: Fac. Pharm; 2010
15. Al-Mahallawi AM, Khowessah OM, Shoukri RA. Nano-transfersomal ciprofloxacin loaded vesicles for non-invasive trans-tympanic ototopical delivery: in-vitro optimization, ex-vivo permeation studies, and in-vivo assessment. Int J-Pharm. 2014;472(12):304–314. doi:10.1016/j.ijpharm.2014.06.041
16. Shen J, Burgess DJ. In vitro dissolution testing strategies for nanoparticulate drug delivery systems: recent developments and challenges. Drug Deliv Transl Res. 2013;3(5):409–415. doi:10.1007/s13346-013-0129-z
17. Susan D. A review of in vitro drug release test methods for nano-sized dosage forms- review article. Adv Pharm. 2014;2014(Article ID 304757):12. doi:10.1155/2014/304757
18. Scognamiglio I, De Stefano D, Campani V, et al. Nanocarriers for topical administration of resveratrol: a comparative study. Int J Pharm. 2013;440(2):179–187. doi:10.1016/j.ijpharm.2012.08.009
19. Danaei M, Dehghankhold M, Ataei S, et al. Impact of particle size and polydispersity index on the clinical applications of lipidic nanocarrier systems. Pharmaceutics. 2018;10(2):57. doi:10.3390/pharmaceutics10020057
20. Shak. Tzeyung A, Md S, Bhattamisra SK, et al. Fabrication, optimization, and evaluation of rotigotine-loaded chitosan nanoparticles for nose-to-brain delivery. Pharmaceutics 11010026. 2019;11(1):26. doi:10.3390/pharmaceutics11010026
21. Patil Sonali K, Dhage Azad N, Sachinkumar P, Patil Shitalkumar S. Formulation and evaluation of nasal in situ gel for Alzheimer’s disease. Int Res J Pharm Biosci. 2015;2(2):41–58.
22. Qushawy M, Nasr A, Abd-Alhaseeb M, Swidan S. Design, optimization, and characterization of a transfersomal gel using miconazole nitrate for the treatment of candida skin infections. Pharmaceutics. 2018;10(1):2. doi:10.3390/pharmaceutics10010026
23. Sailaja K, Supraja R. Formulation of mefenamic acid loaded transfersomal gel by thin-film hydration technique and handshaking method. Nano Medicin J. 2017;4(2):126–134.
24. Thakur N, Jain P, Jain V. Formulation development and evaluation of transferosomal gel. J Drug Delivery Therapeutics. 2018;8(5):168–177. doi:10.22270/jddt.v8i5.1826
25. Gandhi K, Maganti RS, Kaur H, Vinod KS, Verma P. Formulation and evaluation of sol-gel drug delivery system for intracanal ph-sensitive controlled delivery of chlorhexidine. J Clin Diagn Res. 2017;11(4):ZC68–ZC72.
26. Dantas MGB, Reis SAGB, Damasceno CMD, et al.. Development and evaluation of stability of a gel formulation containing the monoterpene borneol. Scientific World J.2016;2016:4. doi:10.1155/2016/7394685
27. Fatma -SA-S, Seham AE, Azza AM, Hussein OA. Diflucortolone valerate loaded solid lipid nanoparticles as a semisolid topical delivery system. Bulletin Faculty Pharm. 2016;54(1):1–7.
28. Shirbhate MP, Chavan MJ. Effect of polymer concentration on drug release kinetics of Etodolac. J Innovations Pharm Biol Sci. 2017;4(4):190–197.
29. Basha M, Abd El-Alim SH, Shamma RN, Awad GE. Design, and optimization of surfactant-based nanovesicles for ocular delivery of clotrimazole. J Liposome Res. 2013;23(3):203–210. doi:10.3109/08982104.2013.788025
30. Gabriela E, Antonio J, Cecilia K, Manuel J. Design of Experiments for Chemical, Pharmaceutical, Food, and Industrial Applications; published in the United States of America by IGI Global, Book Series Advances in Chemical and Materials Engineering (ACME). copyright ©. 2020. (ISSN:2327-5448;eISSN: 2327-5456)
31. Zaru M, Mourtas S, Klepetsanis P, Fadda AM, Antimisiaris SG. Liposomes for drug delivery to the lungs by nebulization. Eur J Pharm Biopharm. 2007;67(3):655–666. doi:10.1016/j.ejpb.2007.04.005
32. Bnyan R, Khan I, Ehtezazi T, et al. Surfactant effects on lipid-based vesicles properties. J Pharm Sci. 2018;107:5. doi:10.1016/j.xphs.2018.01.005
33. Ola MEL-H, Zawilla NH, Mohammad A. Development and Validation of Rp-Lc method for the determination of Cinnarizine/piracetam and Cinnarizine/Heptaminol Acefyllinate in presence of Cinnarizine Reported Degradation products. Anal Chem Insights. 2013;8:99–106. doi:10.4137/ACI.S12478
34. Ogiso T, Yamaguchi T, Iwaki M, Miyake Y. Effect of positively and negatively charged liposomes on skin permeation of drugs. J Drug Target. 2001;9(1):49–59. doi:10.3109/10611860108995632
35. Moawad FA, Ali AA, Salem HF. NanoTransfersomes-loaded thermosensitive in situ gel as a rectal delivery system of tizanidine HCl: preparation, in vitro, and in vivo performance. Drug Deliv. 2017;24(1):252–260. doi:10.1080/10717544.2016.1245369
36. El Sayyad MK, Zaky AA, Samy AM. Fabrication and characterization of sildenafil citrate loaded Transfersomes as a carrier for transdermal drug delivery. Pharmacy Pharmacol Int J. 2017;5(2). doi:10.15406/ppij.2017.05.00113
37. Crouch J. Functional Human Anatomy. Lea and Febiger; 1984. Publisher: Lea & Febiger ©. (ISBN 10: 0812109309 ISBN 13: 9780812109306).
38. Wiegand S, Berner R, Schneider A, Lundershausen E, Dietz A. Otitis externa: investigation and evidence-based treatment. Dtsch Arztebl Int. 2019;116(13):224–234. doi:10.3238/arztebl.2019.0224
39. José Das N, Bruno S. Mucosal Delivery of Biopharmaceuticals: Biology, Challenges, and Strategies. SPRINGER©; 2014:601, 73.
40. Garala K, Joshi P, Shah M, Ramkishan A, Patel J. Formulation and evaluation of periodontal in situ gel. Int J Pharm Investig. 2013;3(1):29–41. doi:10.4103/2230-973X.108961
41. Sutapa B. M. Emerging concepts in analysis and applications of hydrogels, BoD InTech Open©2016. Science. 2016;266, 183.
42. Hao Z-Q, Chen Z-J, Chang M-C, Meng J-L, Liu J-Y, Feng C-P. Rheological properties and gel characteristics of polysaccharides from fruit-bodies of Sparassis crispa. Int J Food Properties. 2018;21(1):2283–2295. doi:10.1080/10942912.2018.1510838
43. Rahman MNA, Qader OAJA, Sukmasari S, Ismail AF, Doolaanea AA. Rheological characterization of different gelling polymers for dental gel formulation. J Pharm Sci Res. 2017;9(12):2633–2640.
44. Yeo LK, Temidayo OB, Olusanya C, Chaw S, Elkordy AA. Brief effect of a small hydrophobic drug (cinnarizine) on the physicochemical characterisation of niosomes produced by thin-film hydration and microfluidic methods. Pharmaceutics. 2018;10(4):
45. Nasir B, Ranjha NM, Hanif M. Pharmacokinetic studies of 5-fluorouracil in rabbit plasma. Acta Poloniae Pharmaceutica Ñ Drug Res. 2017;74(5):
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.