Back to Journals » Drug Design, Development and Therapy » Volume 13
Formulation and bioequivalence studies of choline alfoscerate tablet comparing with soft gelatin capsule in healthy male volunteers
Authors Min MH, Park JH, Hur JH, Shin HC, Cho YJ, Kim DD ![]()
Received 4 November 2018
Accepted for publication 21 January 2019
Published 5 April 2019 Volume 2019:13 Pages 1049—1058
DOI https://doi.org/10.2147/DDDT.S193424
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Qiongyu Guo
Mi-Hong Min,1,2 Jin-Hyong Park,2 Jong-Hyun Hur,2 Ho-Chul Shin,2 YoungJoo Cho,2 Dae-Duk Kim1
1College of Pharmacy and Research Institute of Pharmaceutical Sciences, Seoul National University, Seoul 08826, Republic of Korea; 2Central Research Institute, Whanin Pharmaceutical Company, Suwon 16229, Republic of Korea
Purpose: The aim of this study was to develop a tablet formulation of choline alfoscerate and to assess its bioequivalence by comparing its pharmacokinetic parameters with those of a commercially available soft gelatin capsule (Gliatilin®) in healthy Korean male volunteers.
Materials and methods: Film-coated tablet formulation was optimized to control the hygroscopicity of choline alfoscerate. Bioequivalence study was performed under fasted condition with a randomized, single-dose, two-period crossover design. Subjects were orally treated with 1,200 mg of test or reference choline alfoscerate (400 mg × three doses) formulation. Blood samples were collected up to 12 hours the day before dosing to correct the baseline level of choline and 12 hours after dosing to obtain drug absorption profile. Pharmacokinetic parameters were determined after analyzing plasma concentration of choline by using LC–MS/MS.
Results: Hygroscopicity of choline alfoscerate was successfully controlled by adding suitable amount of Neusilin® (magnesium aluminometasilicate) in the film-coated tablet. Stability of the tablet formulation was also confirmed under the accelerated condition for 3 months. Bioequivalence study showed that the mean area under the plasma concentration–time curve from time 0 to infinity of test tablet and reference soft capsule was 3.428±2.170 and 3.305±1.803 µg·h/mL, respectively; the mean Cmax was 0.365±0.158 and 0.380±0.108 µg/mL, respectively; and the mean Tmax was 3.51±2.57 and 3.85±3.19 hours, respectively. The 90% CIs for geometric mean ratios of test to reference formulation for AUC0–t and Cmax were 84.51%–111.98% and 83.31%–104.10%, respectively, and satisfied the EMA regulatory criteria for bioequivalence.
Conclusion: Pharmacokinetic parameters including the Cmax and AUC0–t determined after oral administration of the two formulations in healthy Korean male volunteers showed that the differences between the formulations (tablet vs soft capsule) were not significant for bioequivalence. Both formulations were well tolerated, with no serious adverse events reported.
Keywords: choline alfoscerate, bioequivalence study, tablet, healthy Korean male volunteers
Introduction
Choline alfoscerate (Figure 1) is hydrolyzed to choline which is the precursor of the neurotransmitter acetylcholine, and is used for the improvement of cognitive dysfunction in patients with dementia of neurodegenerative and vascular origin.1,2 It has an elimination half-life of 0.5–6.2 hours3 and is completely absorbed following oral administration.4 Choline alfoscerate is commercially available as soft capsule and administered at a dose of 400 mg three times a day. However, the physical strength of the gelatin shell could become weaker under high temperature, shell can deform at high temperature. The drug dissolved in soft gelatin can also migrate to the gelatin shell over time.5 Thus, a tablet dosage form of choline alfoscerate was developed to overcome the disadvantages of the soft capsule. Unfortunately, choline alfoscerate is highly hygroscopic, and hence, its powder can turn sticky when exposed to humid air during manufacturing process. This could be the main reason that the first commercial preparation (reference) of choline alfoscerate was launched in the market as a soft capsule, in which choline alfoscerate was dissolved in glycerin. The formulation strategy used in this study for developing choline alfoscerate tablet was selection of suitable excipients that could efficiently surround the surface of the drug to inhibit water absorption. However, the amount of hydrophobic excipients needed to be minimized as they could retard the dissolution of choline alfoscerate,6 thereby negatively influencing the bioequivalence of the tablet.
 | Figure 1 The structure of choline alfoscerate (l-alpha glycerylphosphorylcholine). |
Choline alfoscerate is readily hydrolyzed by phosphodiesterases in the gut mucosa to form free choline.7 The active major metabolite, choline, can be measured in plasma following oral administration of choline alfoscerate, and the increased plasma levels of choline reflect the absorption of choline alfoscerate.4 Choline in plasma can be measured using LC–MS/MS. However, choline is an endogenous material which comes from one of the two sources: the dietary intake and synthesis by de novo pathway from phosphatidylcholine.8 The difference in the concentrations of endogenous choline may cause subject variability in drug absorption and failure in bioequivalence studies. Thus, the absorption of choline after drug administration should be checked under choline-limited diet control in healthy volunteers and the removal of individual interference by endogenous choline.
The aim of the present study was to prepare an optimum tablet formulation of choline alfoscerate and to compare the bioequivalence of the newly formulated tablet with the reference soft capsule according to the EMA guidelines9 in healthy Korean male volunteers. The absorbed plasma concentration of choline after drug administration was determined, and the plasma concentration was also calculated after correcting the and then was calculated by subtracting the baseline values determined before dosing at the same plasma sampling time.9,10
Materials and methods
Materials
Choline alfoscerate was purchased from HanseoChem (Pyeongtaek, Republic of Korea). Neusilin® (magnesium aluminometasilicate; Fuji Chemical, Osaka, Japan) was used to control the water absorption of drug and to improve the granule fluidity. Polyvinylpyrrolidone (PVP) K-30 (BASF, Ludwigshafen, Germany) was added as a binder. Microcrystalline cellulose (Mingtai Chemical, Mountainside, Bah-Der, Taiwan), lactose monohydrate and lactose anhydrous (DFE Pharma, Goch, Germany), and dicalcium phosphate anhydrous (A-TAB; Innophos, Cranbury, NJ, USA) were used as excipients for the tablets. Croscarmellose sodium (DFE Pharma), sodium starch glycolate (Yung Zip, Taichung, Taiwan), crospovidone (Polyplasdone XL; Ashland, Covington, KY, USA), magnesium stearate (FACI, Carasco, Italy), and sodium stearyl fumarate (Pruv; JRS Pharma, Rosenberg, Germany) were used as superdisintegrant and lubricant, respectively. Opadry I® and Opadry AMB® were obtained from Colorcon (Shanghai, China). Gliatilin® soft capsules (Daewoong Pharmaceutical, Seoul, Republic of Korea) were purchased from a local pharmacy.
Selection of excipients
Table 1 shows the list of excipients tested for their ability to prevent water uptake of choline alfoscerate. An aliquot amount of choline alfoscerate (1 g) was thoroughly mixed with the same weight of each excipient and was kept in 80% relative humidity (RH) condition for 1 day. Relative moisture uptake (%) was calculated by comparing the weight gain before and after mixing with excipients. The excipients with relatively low moisture uptake were selected for the tablet formulation of choline alfoscerate.
 | Table 1 Selection of excipients for choline alfoscerate tablet formulation (n=3) |
Preparation and evaluation of choline alfoscerate core tablets
Preparation of core tablets
Wet granulation method was applied for preparing the choline alfoscerate tablet. Table 2 provides the composition of core tablets to show the effect of magnesium aluminometasilicate on the protection of moisture and disintegration time. Magnesium aluminometasilicate (5%–30%) was added both in and out of the granules of the tablets in order to surround the drug particles more efficiently.11 Briefly, after preblending choline alfoscerate with magnesium aluminometasilicate and microcrystalline cellulose, the granulation process was performed using a high-speed mixer (PharmaConnect™; GEA, Düsseldorf, Germany) with 70% ethanol binder solution containing PVP K-30. The binder solution was sprayed through a 0.3 mm spray nozzle at 1.5 bar, followed by drying at 50°C in an oven (MOV-212S; Sanyo, Osaka, Japan). Then, the granules were mixed with magnesium aluminometasilicate, dicalcium phosphate, and croscarmellose sodium. After adding lubricant (sodium stearyl fumarate), oblong-shaped core tablets were compressed using a tableting machine (Rimek MINI II SF; Karnavati Engineering, Gujarat, India). Tablet processing problems including sticking, picking, laminating, and punch-filling issue observed for each composition were recorded.
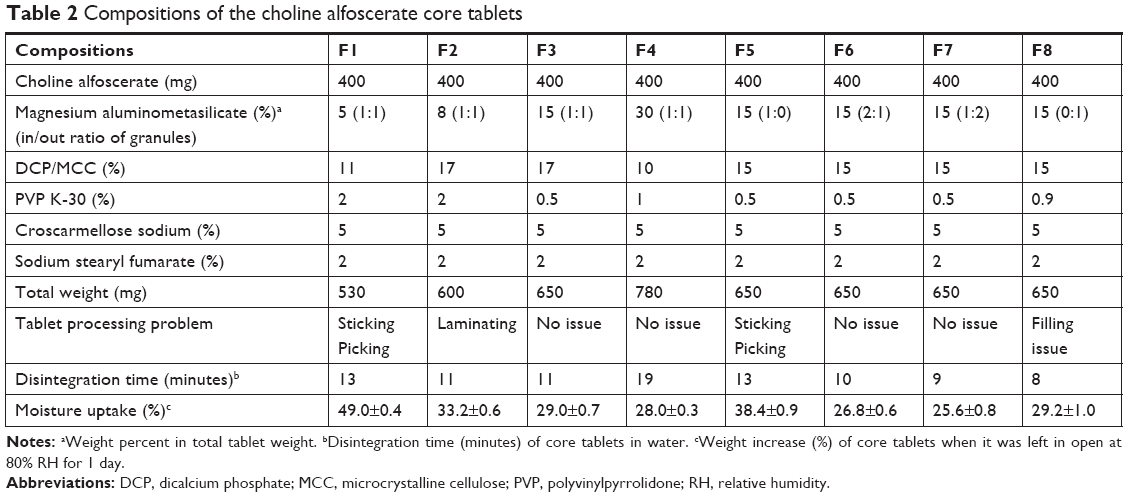 | Table 2 Compositions of the choline alfoscerate core tablets |
Disintegration time of core tablets
Disintegration time for core tablets was determined in water using the USP disintegration method (DIT-200; Labfine, Gunpo, Republic of Korea).
Moisture uptake of core tablets
Weight gain of core tablets by moisture uptake was measured to evaluate the effect of magnesium aluminometasilicate at various contents (F1–F4; Table 2) and in/out ratios of granules (F5–F8; Table 2). Core tablets were put in a Petri dish and left for 1 day in the desiccator with 80% RH, equilibrated with the saturated aqueous solution of ammonium sulfate.12 The weight gain (%) was determined by measuring the weight of tablets before and after storage in the desiccator.
Preparation and evaluation of film-coated tablets
Preparation of film-coated tablets
The core tablets were subcoated with hydroxypropyl methylcellulose-based Opadry I® using organic solvent for 1% weight gain, and followed by coating with polyvinyl alcohol-based Opadry AMB® using aqueous system for 3% weight gain to improve the water stability during storage of tablet.11
Appearance change of film-coated tablets
Appearance of the film-coated tablet was observed at various RH conditions. Desiccators with 22%, 33%, 60%, and 80% RH were prepared using saturated salt solution of potassium acetate, magnesium chloride, sodium bromide, and ammonium sulfate, respectively.12 Film-coated tablet of F7 was placed in desiccators for up to 30 days. The time point at which the appearance began to change due to the uptake of moisture was recorded.
In vitro dissolution test of film-coated tablets
The dissolution of choline alfoscerate was measured using USP apparatus 2 (paddle). The dissolution medium used was 900 mL of distilled water at 37°C±0.5°C and stirred at 50 rpm. Dissolution study was conducted on 12 individual film-coated test tablets or reference soft capsules. At predetermined intervals (0, 5, 10, 15, and 30 minutes), 5 mL of the medium was sampled and filtered through a membrane filter (0.45 μm). Then, the concentration of choline alfoscerate was analyzed using a HPLC system with refractive index detector (Waters 410; Waters, Milford, MA, USA).13 Zorbax SB-CN column (250 × 4.6 mm, 5 μm; Agilent) filled with porous silica particles chemically bonded with nitrile groups was used for analytical assay and maintained at 38°C. The mobile phase used was a mixture of acetonitrile and water (60/40, v/v) at a flow rate of 1.5 mL/min. The injection volume was 20 μL.
Stability test in the accelerated condition
Stability of the film-coated tablets packaged with Zymax blister film (Bilcare, Changi, Singapore) was tested after keeping the tablets in an accelerated chamber (40°C/75% RH) for 3 months (Table 3). Appearance and disintegration time were compared with those of the reference soft capsule. The content of choline alfoscerate was analyzed by HPLC system, as described above. Hardness of the test tablet was also checked using a hardness tester (MT50; Sotax, Aesch, Switzerland), which can be lowered by the moisture uptake. The dissolution test in water was also performed for the test tablets.
 | Table 3 Stability of film-coated test tablet and the reference soft capsule of choline alfoscerate (400 mg) after 3 months of storage under the accelerated condition (40°C/75% RH) |
Bioequivalence study
Subjects
The bioequivalence study was conducted at Yangji Hospital (Seoul, Republic of Korea) with 19–46 aged healthy Korean male volunteers. All subjects were determined to be in good health based on medical history, physical examination, and hematological examination. Subjects were excluded if they had hypersensitivity to any ingredient in the choline alfoscerate tablets, took other drugs that could interfere with the study results within 10 days before the trial, and had taken alcohol or medications that induce or inhibit drug-metabolizing enzymes (eg, barbitals) within 1 month before the study. All subjects signed a written informed consent after being explained the purpose, the methods, and the adverse drug reactions of this study in accordance with the EMA guideline.9 Subjects were monitored by hospital staff during the study period using interview, vital sign measurement, adverse event recording, and physical examination.
Study design
This study was performed under fasted conditions with a randomized, single-dose, two-period crossover design.9 All subjects received a single dose of choline alfoscerate in the form of test tablet and reference soft capsule, with 7 days of washout period between treatments. The study protocol was approved by the Yangji Hospital institutional review board, and followed the principles of the Declaration of Helsinki.14 Subjects were hospitalized for 5 days before the study, and exercise, meal, smoking, and consumption of grapefruit juice were restricted from 10 hours before the beginning of the trial to the end of blood collection. During the trial, consumption of food and drink, except water, was controlled and choline-free meals were provided.
Subjects were fasted for 10 hours before and 4 hours after drug administration to exclude the effects of diet. Two groups were treated with 1,200 mg (choline alfoscerate 400 mg × three doses) of the reference capsule or the test tablet orally with 150 mL of water at 8 am. Subjects were not allowed to drink water for 1 hour before and after drug administration. Choline-restricted standard meals were provided for lunch and dinner at 4 and 10 hours after dosing. After the Period I blood collection, subjects returned home and were advised to avoid excessive drinking, taking drugs, and drinking grapefruit juice and to prohibit excessive intake of choline-containing foods (eg, eggs, beans). After 7 days for washout period, all subjects were called up to hospitalize for 5 days before the study and the drug was administered in the same manner as in the Period I.
A total of 24 blood samples were collected at predetermined time points (0, 0.5, 0.75, 1, 1.33, 1.67, 2, 3, 4, 6, 8, and 12 hours) on the day before drug dosing and on the day of drug administration. Before collecting each blood sample, 1 mL of blood was drawn and discarded to completely remove any remaining saline in the catheter. Aliquot (8 mL) of blood was collected into vacutainer with sodium heparin, and then 1 mL of heparinized normal saline was injected into the catheter to prevent blood clotting. Blood samples were centrifuged at 3,000 rpm for 10 minutes. The plasma was transferred to Eppendorf tube and stored at −70°C until analysis.
Determination of plasma choline concentrations
Choline concentration in each plasma sample was determined by a validated LC–MS/MS assay for choline.15 The plasma samples were placed at room temperature to thaw. Aliquot (1 mL) of metformin (20 ng/mL in methanol) was added as internal standard (IS) to 50 μL of plasma. Each sample was vortexed and centrifuged at 12,000 rpm for 5 minutes. The supernatant (2 μL) of the mixture was taken and chromatically analyzed using Shiseido Nanospace SI-2 (Osaka Soda, Osaka, Japan) with a Luna 3 μm HILIC (3 μm, 2.0 mm ID × 150 mm L; Phenomenex, Torrance, CA, USA). The mobile phase consisted of 1 mM ammonium formate and acetonitrile (45:55, v/v). The flow rate was 0.3 mL/min. Column and sample tray temperatures were set at 45°C and 4°C, respectively. Detection and quantification were performed using Triple Quadruple Mass Spectrometer System, API 4000 (AB SCIEX, Framingham, MA, USA) in positive ion electrospray ionization with multiple reaction monitoring mode. The m/z value of the precursor to product for choline and IS were 104.2 → 60.1 and 113.3 → 69.1, respectively. The LC–MS/MS system was controlled by using Analyst software (version 1.4; AB SCIEX), and the results were processed by using Microsoft Office Excel 2007 (Microsoft Corp., Redmond, WA, USA). The validation of this chromatographic analytical method was performed in order to evaluate its specificity, linearity, precision, accuracy, and stability in solution. The calibration curve from the standard choline samples was constructed based on the peak area measurement, which was linear in 0.05–10 μg/mL range.
Pharmacokinetic and statistical analysis
The concentration of choline in the plasma before and after drug administration was calculated with the peak area ratio of choline to the IS, metformin. The choline concentration after drug absorption at each time point was calculated by subtracting the endogenous choline level at the same blood collection point of each subject before the drug administration. When a negative value was obtained after correction, it was considered as zero.16
The pharmacokinetic parameters of choline were determined for both test tablet and reference soft capsule using a noncompartmental model with BA Calc 2007 program (version 1.0.0; MFDS, Seoul, Republic of Korea).17 The Cmax and Tmax were determined from the experimental data. The elimination rate constant (ke) was calculated from the least-squares regression slope of the terminal plasma concentration, and then the half-life was calculated as 0.693/ke. The calculated choline plasma concentrations were used to obtain the AUC0–t. The area under the plasma concentration–time curve from time 0 to 12 hours (AUC0–12) was calculated by the linear trapezoidal method.18 AUC0–∞ was calculated as AUC0–12 + C12/ke, where C12 is the choline concentration at the last time point (12 hours). Comparative bioavailability was measured by 90% CIs of the geometric mean ratios of test to reference which were determined using log-transformed data of AUC0–t and Cmax. All statistical calculations were performed using K-BE Test 2007 program (version 1.1.0; MFDS) for bioequivalence analysis recommended by the MFDS.19 The regulatory range of 90% CIs of geometric mean ratios for bioequivalence is 80.00%–125.00%.9
Results and discussion
Selection of excipients for core tablet formulation
Table 1 summarizes the list of excipients tested for the formulation of choline alfoscerate tablets and the weight gain (%) due to moisture uptake when each mixture was left in 80% RH condition for 1 day. Choline alfoscerate powder had good flowability, but was highly hygroscopic and was prone to become sticky under exposure to air. This would cause sticking and picking by adhering to the surface of a tablet-punch face during tablet manufacturing process. When each excipient was mixed with choline alfoscerate, the amount of moisture uptake decreased because the surface area of drug exposed to air decreased. Based on the moisture uptake measurement, the excipients with low moisture uptake were selected which are marked in Table 1. It was notable that magnesium aluminometasilicate showed the lowest moisture uptake among the excipients tested. It is known to have an ultrafine particle size of 15 nm and a high porosity.20 The large surface area of magnesium aluminometasilicate was expected to improve the stability of the drug against moisture by surrounding it in a small amount, thereby minimizing the retardation effect on the dissolution rate.
Preparation and evaluation of core tablet
As shown in Table 2, the average weight of core tablets was in the range of 530–780 mg depending on the compositions and the hardness of the tablets was maintained in the range of 18–20 kp. The effect of magnesium aluminometasilicate at various contents and in/out ratios of granules were evaluated in terms of the tablet processing problems (Table 2). F1 composition containing 5% magnesium aluminometasilicate showed sticking and picking phenomenon during tableting process due to the insufficient amounts of magnesium aluminometasilicate. Although higher content (30%) of magnesium aluminometasilicate (F4) did not cause the tablet processing problem, the disintegration of tablet was retarded up to 19 minutes. Since the disintegration time of the reference soft capsule was 10 minutes, 15% magnesium aluminometasilicate was selected, and was added in and out of granules at various ratios (F5–F8). When magnesium aluminometasilicate was added only in the granules (F5), sticking and picking were observed during the tableting process and weight gain by moisture uptake was relatively high. Moreover, when magnesium aluminometasilicate was added only out of the granules (F8), filling of final mixture into the punch was not smooth enough. Thus, F7 composition was selected for film coating as the disintegration time was <10 minutes without tablet processing problem and moisture uptake was the lowest.
Evaluation of film-coated core tablet
Appearance stability of film-coated tablet
Figure 2 shows the time point when the appearance of tablet began to change due to the uptake of moisture at various RH conditions. When the tablets began to uptake moisture, the film coating layer was broken by the swelling of core or water droplets were formed on the surface of tablets. It is noteworthy that the film-coated tablet of F7 maintained the appearance for up to 30 days in 60% RH condition.
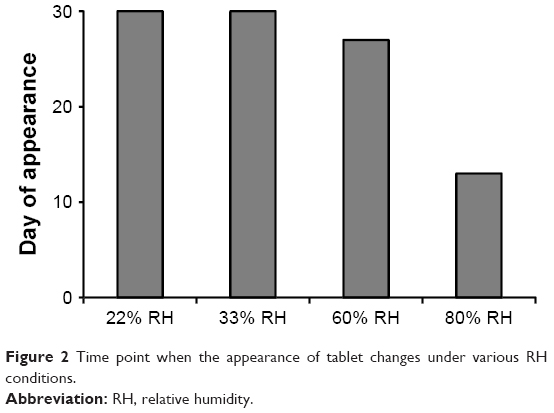 | Figure 2 Time point when the appearance of tablet changes under various RH conditions. |
In vitro dissolution test of film-coated tablets
Figure 3 shows the dissolution profiles of choline alfoscerate (400 mg) from the film-coated test tablet and the reference soft capsules in distilled water. It was notable that the dissolution rate of choline alfoscerate from the test tablet was higher than that from the reference soft capsule for 10 minutes. It was probably due to the slower disintegration of gelatin shell than that of the tablet, and also indicated that magnesium aluminometasilicate in tablet formulation did not affect the dissolution of choline alfoscerate. Moreover, the average dissolution rate of both test and reference reached not less than 85% within 15 minutes, which satisfied the EMA guidelines.9
 | Figure 3 In vitro dissolution profiles of choline alfoscerate (400 mg) from the film-coated test tablet and the reference soft capsules (Gliatilin®) in distilled water (n=6). |
Stability test in the accelerated condition
The stability of the film-coated test tablet and the reference soft capsule stored under the accelerated condition for 3 months is summarized in Table 3. There was no change in appearance, disintegration time, content, and harness of the test tablet during the storage period. Moreover, the dissolution profiles of choline alfoscerate in water were within the acceptance level of similarity with the initial profile for 3 months (Figure 4). Based on the results of the stability studies under the accelerated condition, it was concluded that the hygroscopicity of choline alfoscerate was effectively controlled in the film-coated tablet, and hence, the bioequivalence of the tablet and the soft capsule was tested in human.
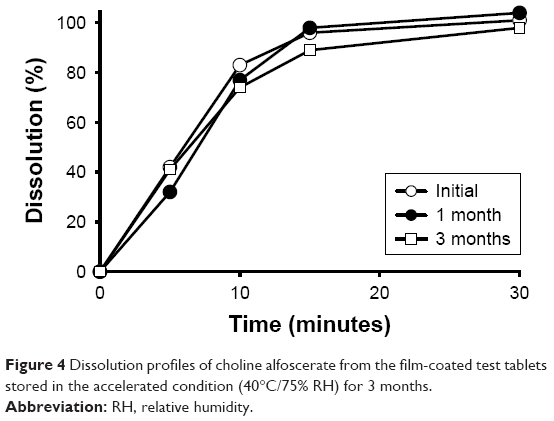 | Figure 4 Dissolution profiles of choline alfoscerate from the film-coated test tablets stored in the accelerated condition (40°C/75% RH) for 3 months. |
Bioequivalence study
Subjects
Fifty healthy Korean male volunteers aged 19–46 years (mean ± SD: 27.7±6.5), with a height of 158–188 cm (mean ± SD: 173.3±6.6) and a weight of 55–96 kg (mean ± SD: 69.4±8.3), were enrolled in this study. All the subjects completed the study in Period 1, but two subjects withdrew from the study during Period 2. One subject withdrew because of acute upper respiratory tract infections, and the other subject withdrew because of a bruised leg. Thus, 48 subjects completed the study and were included in the pharmacokinetic analysis.
Pharmacokinetic analysis
Figure 5 shows the mean plasma concentration profiles of choline before and after the administration of test tablet and reference soft capsule. The corrected plasma concentration–time curve after subtracting the endogenous choline level is shown in Figure 6, which suggests that test tablet had a similar bioavailability to the reference soft capsule. The major mean pharmacokinetic parameters such as AUC0–t, Cmax, and Tmax for the two preparations are summarized in Table 4. The mean AUC0–∞ of the test tablet was 3.428±2.170 μg·h/mL and was not significantly different from that of the reference soft capsule (3.305±1.803 μg·h/mL). The mean Cmax of the test tablet (0.365±0.158 μg/mL) was also not significantly different from that of the reference soft capsule (0.380±0.108 μg/mL). The mean Tmax of the test tablet and reference soft capsule was 3.51±2.57 and 3.85±3.19 hours, respectively, and was not significantly different, although the initial dissolution rate of choline alfoscerate from the reference soft capsule was slightly slower than that from the test tablet (Figure 3).
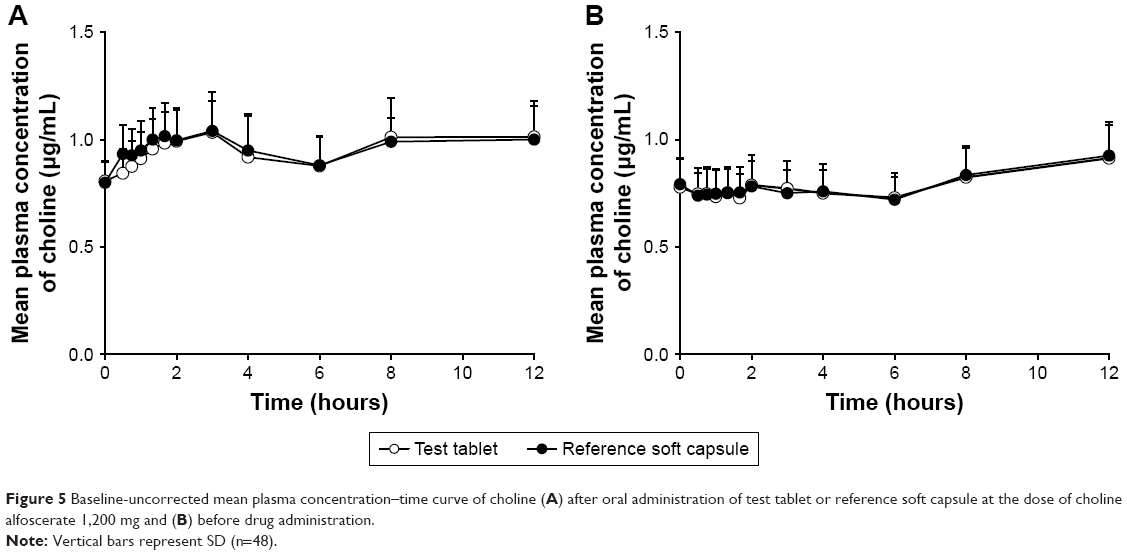 | Figure 5 Baseline-uncorrected mean plasma concentration–time curve of choline (A) after oral administration of test tablet or reference soft capsule at the dose of choline alfoscerate 1,200 mg and (B) before drug administration. |
 | Figure 6 Baseline-corrected mean plasma concentration–time curve of choline after oral administration of test tablet or reference soft capsule (Gliatilin®) at the dose of choline alfoscerate 1,200 mg. |
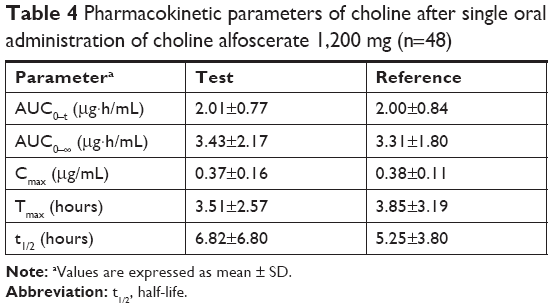 | Table 4 Pharmacokinetic parameters of choline after single oral administration of choline alfoscerate 1,200 mg (n=48) |
Statistical analysis and bioequivalence evaluation
Statistical results of the bioequivalence evaluation of the two formulations of choline alfoscerate 400 mg in healthy Korean male volunteers are summarized in Table 5. The calculated 90% CIs of geometric mean ratios of test to reference for AUC0–t and Cmax were 84.51%–111.98% and 83.31%–104.10%, respectively, and satisfied the accepted bioequivalence criteria of 80.00%–125.00%. Moreover, AUC0–t and Cmax of the test tablet and the reference soft capsule were not significantly different.
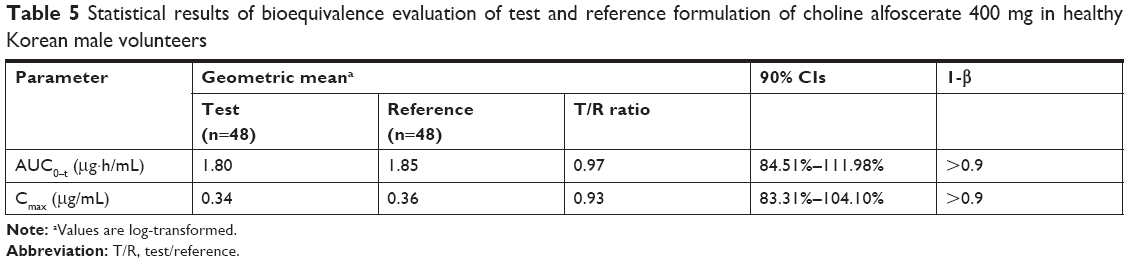 | Table 5 Statistical results of bioequivalence evaluation of test and reference formulation of choline alfoscerate 400 mg in healthy Korean male volunteers |
Tolerability
Tolerability was assessed by vital sign, adverse event, and physical examination; no adverse events were reported during this study. Two subjects were excluded from the trial because of an acute upper respiratory tract infection (runny nose, cough) and a bruised leg with external impact during washout period but later fully recovered. These findings were considered to have a little causal relationship with the drug.
Conclusion
The hygroscopicity of choline alfoscerate was successfully controlled by using magnesium aluminometasilicate in the preparation of a film-coated tablet. In the bioequivalence study in healthy Korean male volunteers, no significant differences in pharmacokinetic parameters were found between the test tablet and the reference soft capsule as 90% CIs for AUC0–t and Cmax were within the regulatory acceptance criteria. These results indicated that the new tablet formulation can be prescribed as an alternative to choline alfoscerate soft capsule.
Abbreviations
AUC0–t, area under the plasma concentration–time curve from time 0 to the last measurement; AUC0–∞, area under the plasma concentration–time curve from time 0 to infinity; Cmax, maximum plasma concentration; EMA, European Medicines Agency; LC–MS/MS, liquid chromatography–tandem mass spectrometry; MFDS, Ministry of Food and Drug Safety; Tmax, time to reach Cmax.
Disclosure
Mi-Hong Min, Jin-Hyong Park, Jong-Hyun Hur, Ho-Chul Shin, and YoungJoo Cho are employed by Whanin Pharmaceutical Company. The authors report no other conflicts of interest in this work.
References
Parnetti L, Mignini F, Tomassoni D, Traini E, Amenta F. Cholinergic precursors in the treatment of cognitive impairment of vascular origin: ineffective approaches or need for re-evaluation? J Neurol Sci. 2007;257(1–2):264–269. | ||
De Jesus Moreno Moreno M. Cognitive improvement in mild to moderate Alzheimer’s dementia after treatment with the acetylcholine precursor choline alfoscerate: a multicenter, double-blind, randomized, placebo-controlled trial. Clin Ther. 2003;25(1):178–193. | ||
Gatti G, Barzaghi N, Acuto G, Abbiati G, Fossati T, Perucca E. A comparative study of free plasma choline levels following intramuscular administration of L-alpha-glycerylphosphorylcholine and citicoline in normal volunteers. Int J Clin Pharmacol Ther Toxicol. 1992;30(9):331–335. | ||
Kang H-A, Kim S-M, Kang S-R, et al. Bioequivalence of Cholicerin soft capsule to Gliatilin soft capsule (choline Alphoscerate 400 Mg). J Pharm Investig. 2010;40(2):109–115. | ||
Choi S-U, Cho S-W. Formulation of liquid choline Alphoscerate as a solid dosage form. J Korea Acad Industr Coop Soc. 2013;14(12):6324–6329. | ||
Helmis M, Mohamad B, Kumpugdee-Vollrath M. Influence of several excipients on drug release of tablets containing resveratrol. M J Phar. 2016;1(2):1–7. | ||
Traini E, Bramanti V, Amenta F. Choline alphoscerate (alpha-glyceryl-phosphoryl-choline) an old choline-containing phospholipid with a still interesting profile as cognition enhancing agent. Curr Alzheimer Res. 2013;10(10):1070–1079. | ||
Zeisel SH. Phosphatidylcholine: endogenous precursor of choline. Lecithin. Boston, MA: Springer; 1987:107–120. | ||
EMA. Guideline on the investigation of bioequivalence. 2010. Doc. Ref.: CPMP/EWP/QWP/1401/98 Rev.1. Available from: https://www.ema.europa.eu/investigation-bioequivalence. Accessed November 4, 2018. | ||
Korea Food & Drug Administration (KFDA). Guideline for management of bioequivalence test (KFDA notification No. 2013-115). 2013. Available from: www.bio-core.com/UpFiles/NewsLetter/201308/5.pdf. Accessed November 4, 2018. | ||
An BN, Min MH, Park JH. Tablets containing choline alfoscerate and methods for making the same. Korean patent 10-1628937. 2014 November 4, 2018. | ||
Greenspan L. Humadity fixed points of binary saturated aqueous solutions. J Res Natl Bur Stand. 1977;81(1):89–96. | ||
The Korean Pharmacopoeia eleventh edition. Choline alfoscerate capsules. P1269. 2014, P1269. Available from: https://nedrug.mfds.go.kr/bbs/5/1/. Accessed February 11, 2019. | ||
World Medical Association. WMA Declaration of Helsinki Ethical Principles for Medical Research Involving Human Subjects; 2008. Available from: https://www.wma.net/wp-content/uploads/2018/07/DoH-Oct2008.pdf. Accessed January 4, 2019. | ||
Holm PI, Ueland PM, Kvalheim G, Lien EA. Determination of choline, betaine, and dimethylglycine in plasma by a high-throughput method based on normal-phase chromatography-tandem mass spectrometry. Clin Chem. 2003;49(2):286–294. | ||
FDA Guidance. Updated draft guidance on progesterone. 2011. Available from: https://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm209294.pdf. Accessed December 22, 2018. | ||
Lee YJ, Kim YG, Lee MG, et al. Analysis of bioequivalence study using log-transformed model. Yakhakhoeji. 2000;44(4):308–314. | ||
Takka S, Sakr A, Goldberg A. Development and validation of an in vitro-in vivo correlation for buspirone hydrochloride extended release tablets. J Control Release. 2003;88(1):147–157. | ||
Lee YJ, Choi JH, Song SH, et al. Development of K-BE test, a computer program for the analysis of bioequivalence. J Kor Pharm Sci. 1998;28:223–229. | ||
Rowe RC, Sheskey PJ, Quinn ME. Handbook of Pharmaceutical Excipients. 6th ed. London: Pharmaceutical Press. 2009. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.