")
Back to Journals » International Journal of Nephrology and Renovascular Disease » Volume 9
Formation of tight junctions between neighboring podocytes is an early ultrastructural feature in experimental crescentic glomerulonephritis
Authors Succar L, Boadle RA, Harris DC, Rangan GK
Received 19 May 2016
Accepted for publication 23 July 2016
Published 24 November 2016 Volume 2016:9 Pages 297—312
DOI https://doi.org/10.2147/IJNRD.S113071
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Pravin Singhal
Lena Succar,1 Ross A Boadle,2 David C Harris,1,3 Gopala K Rangan1,3
1Centre for Transplant and Renal Research, The Westmead Institute for Medical Research, The University of Sydney, 2Electron Microscopy Laboratory, Institute of Clinical Pathology and Medical Research, Westmead Hospital, 3Department of Renal Medicine, Westmead Hospital, Western Sydney Local Health District, Westmead, Sydney, NSW, Australia
Purpose: In crescentic glomerulonephritis (CGN), the development of cellular bridges between podocytes and parietal epithelial cells (PECs) triggers glomerular crescent formation. However, the sequential changes in glomerular ultrastructure in CGN are not well defined. This study investigated the time course of glomerular ultrastructure in experimental CGN.
Methods: Transmission electron microscopy (TEM) was performed using kidney samples from rats with nephrotoxic serum nephritis (NSN) from day 1 to day 14. Morphometric analysis was conducted on randomly selected glomeruli captured on TEM digital images.
Results: On day 1 of NSN, there was widespread formation of focal contacts between the cell bodies of neighboring podocytes, and tight junctions were evident at the site of cell-to-cell contact. This was confirmed by the increased expression of the tight junction molecule, zonula occludens-1 (ZO-1), which localized to the points of podocyte cell–cell body contact. On day 2, the interpodocyte distance decreased and the glomerular basement membrane thickness increased. Foot process effacement (FPE) was segmental on day 3 and diffuse by day 5, accompanied by the formation of podocyte cellular bridges with Bowman’s capsule, as confirmed by a decrease in podocyte-to-PEC distance. Fibrinoid necrosis and cellular crescents were evident in all glomeruli by days 7 and 14. In vitro, the exposure of podocytes to macrophage-conditioned media altered cellular morphology and caused an intracellular redistribution of ZO-1.
Conclusion: The formation of tight junctions between podocytes is an early ultrastructural abnormality in CGN, preceding FPE and podocyte bridge formation and occurring in response to inflammatory injury. Podocyte-to-podocyte tight junction formation may be a compensatory mechanism to maintain the integrity of the glomerular filtration barrier following severe endocapillary injury.
Keywords: glomerular, crescent, inflammation, zonula occludens
Introduction
Crescentic glomerulonephritis (CGN) comprises a group of life-threatening immune-initiated diseases that are characterized by glomerular inflammation with crescents and rapid progression to kidney failure.1 Although conventional treatments, using immunosuppressant drugs and monoclonal antibodies, have improved disease outcomes, the development of cell-based approaches in therapy may lead to more targeted and specific treatments.2 In this regard, podocytes have an important role in mediating glomerular crescent formation in human and experimental models of CGN.3–8 Immune-mediated damage to the glomerular basement membrane (GBM) and the glomerular tuft9–11 causes podocyte injury, linking acute disease processes in the endocapillary compartment to the formation of cellular crescents in Bowman’s space.5 Historically, podocytes were excluded as participants of crescent formation, largely because crescentic cells were not positive for podocyte markers.9 However, recent studies show that podocytes form bridges between the glomerular capillary tuft and Bowman’s capsule by detaching from the GBM and migrating between parietal epithelial cells (PECs).5,6,12,13 The latter is thought to initiate the proliferation of PECs and renal progenitor cells,14 triggering glomerular crescent formation.5,6,15
The correct alignment of cytoskeletal proteins is pivotal to the structure and function of podocytes, and the tight junction protein, zonula occludens-1 (ZO-1), is a key regulator of the cytoskeleton.16,17 ZO-1, a member of the membrane-associated guanylate kinases superfamily of proteins, is responsible for sealing and organizing components of tight junctions by linking them to the cortical actin cytoskeleton and associating with F-actin, α-actinin 4, α- and β-catenins, and occludins.18 Interactions between the extracellular domains of podocyte-specific proteins of the slit diaphragm, including Nephrin, Neph1, Fat1, and Fat2,19,20 connect to ZO-1 through actin microfilaments,20,21 giving rise to the structural integrity of the slit diaphragm as well as participating in important signaling events that maintain the physiological glomerular filtration function. Similar changes in ZO-1 expression and delocalization in rats with diabetic nephropathy were shown to associate with proteinuria and loss of glomerular function.22,23 Therefore, glomerular injury disturbing the ZO-1 distribution likely diminishes slit diaphragm and podocyte ultrastructure, mediating glomerular crescent formation.
Understanding the changes in podocyte structure in CGN may yield new approaches for treatment.24 However, the early ultrastructural alterations underlying glomerular crescent formation have not been previously investigated in detail. Therefore, the aim of this study was to determine the timecourse of glomerular ultrastructural changes during the first week of experimental model of CGN. The results of our study demonstrate that in the early phase of an anti-GBM model of CGN, podocytes form tight junctions with each other, and this corresponds with an upregulation in ZO-1. These changes were found to precede foot process effacement (FPE), GBM thickening, and podocyte contact with the parietal epithelium.
Methods
Experimental model
Nephrotoxic serum nephritis (NSN) was induced by preimmunizing adult male Wistar-Kyoto rats (6–8 weeks old, weighing ~225 g) with normal sheep IgG (4 mg by subcutaneous injection) emulsified in complete Freund’s adjuvant (Sigma-Aldrich, Sydney, NSW, Australia) on day 5 and then injected intravenously 5 days later with 1 mL/kg body weight sheep anti-rat GBM serum via the tail vein (termed day 0). Sheep antirat GBM serum was prepared as previously described.25 Groups of animals were sacrificed on days 1, 2, 3, 5, 7, and 14 (N=5 per time point). In addition, control rats were administered a tail-vein injection of saline on day 0 for 5 days (N=3 per time point). All rats were obtained from the Animal Resources Centre (Perth, Australia) and housed in a temperature- and light-controlled environment, with access to food and water ad libitum. The experimental procedures and the study were approved by the Animal Ethics Committee Westmead Hospital (Protocol number 4089) and according to guidelines of the National Health and Medical Research Council of Australia. Animals with NSN developed renal impairment, proteinuria, and histological changes of CGN, and the findings will be described in a future paper.
Transmission electron microscopy (TEM)
Fixation and tissue processing
Coronal kidney sections of 3 mm thickness were fixed overnight in modified Karnovsky fixative (2.5% electron microscopy grade glutaraldehyde, 2.4% formalin in 0.1 M 3-(N-morpholino)propanesulfonic acid) buffer, pH 7.4). The tissue was buffer washed (0.1 M 3-(N-morpholino)propanesulfonic acid)), postfixed in 2% buffered osmium tetroxide, dehydrated, and infiltrated with TAAB epoxy resin (TAAB Laboratories, UK). The tissues were embedded in TAAB epoxy resin and polymerized at 70°C for 10 hours.
Light and transmission electron microscopy
Semithin (0.5 mm) sections were cut on glass knives, stained with alkalized toluidine blue, and examined by light microscopy for the selection of glomeruli. For TEM, ultrathin sections 80–90 nm in thickness were cut and contrasted with uranyl acetate (2% in 50% ethanol) and Reynold’s lead citrate. Sections were then examined using a Philips CM10 transmission electron microscope (FEI, Eindhoven, the Netherlands) at 80 kV. Digital images were acquired using the Megaview G2 digital camera (Olympus, Munster, Germany).
Morphometric analysis of glomerular ultrastructure
For each animal, 10 glomeruli were randomly selected from coronal sections and isolated for morphometric analysis, totaling 50 and 30 glomeruli for the NSN and control groups, respectively. Thus, a total of 480 ultrastructural specimens were used in the morphometric analysis. All image and morphometric analysis of the digital images were performed using the iTEM imaging software (Olympus), and the following parameters were assessed: 1) Interpodocyte distance: the distance between neighboring podocytes was assessed by determining the length between the cell bodies of two nucleated podocytes. At least four pairs of nucleated podocytes per glomerular cross-section (GCS) were assessed. Ten morphometric measurements were taken between each pair of neighboring nucleated podocytes as shown by the red lines in Figure 1A. The mean distance was calculated by averaging the 10 measurements to give a final mean distance in nanometers; 2) Podocyte-to-parietal basement membrane (PBM) distance: similarly, for every nucleated podocyte, 10 measurements of the distance between the cell body and the parietal epithelium were taken (as shown by the blue lines in Figure 1A) and the mean was determined, and on average five nucleated podocytes per glomeruli were assessed; 3) GBM thickness: the GBM thickness was determined by capturing five glomerular capillary loops using the iTEM imaging system at a magnification of 7000× and developed to a final print magnification of ~15000×. For each of the five capillary loops, 20 measurements were obtained by placing the cursor at the beginning of the endothelial surface of the GBM and ending at the outer lining of the lamina rara externa underneath the cytoplasmic membrane of the epithelial foot process, as demonstrated by the distance between the red arrows in Figure 1B; and 4) Filtration slit frequency (FSF): the FSF was used as a measure of podocyte FPE. Images of five glomerular capillary loops were captured at a magnification of 3600× and developed to a final print magnification of ~7000×, and FSF determined as described previously26 by counting the number of epithelial filtration slits divided by the length of the peripheral capillary wall to give FSF/µm GBM length (Figure 1B).
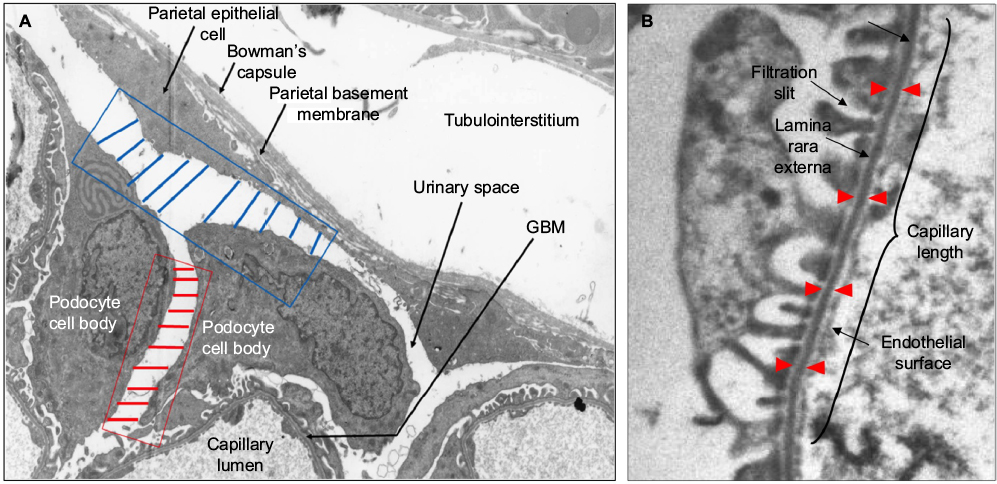 | Figure 1 Method for morphometric measurements of interpodocyte distance, podocyte cell body to PEC distance, GBM thickness, and FSF/µm in TEM images. Notes: (A) The interpodocyte distance was determined by taking 10 measurements between nuclei of neighboring podocyte cell bodies as shown by the red lines. The blue lines represent 10 measurements taken between podocyte cell bodies and the parietal epithelium. Magnification 7000×. (B) GBM thickness was defined by the length between the red arrows. The FSF was determined by dividing the number of filtration slits counted along the GBM of a capillary loop by the length of GBM. Magnification 12000×. Abbreviations: FSF, filtration slit frequency; GBM, glomerular basement membrane; PEC, parietal epithelial cell; TEM, transmission electron microscopy. |
Immunofluorescence for ZO-1
Coronal kidney sections were fixed in 10% neutral-buffered formalin,27 and kidney sections were incubated (overnight at 4°C) with polyclonal rabbit anti-ZO-1 (1:200; Thermo Fisher Scientific, Waltham, MA, USA), followed by incubation for 45 minutes with goat antirabbit Texas Red-conjugated secondary antibody (1:200 dilution; Invitrogen) in a dark chamber at room temperature. After incubation, slides were washed and incubated for 5 minutes in 4′,6-diamidine-2′-phenylindole dihydrochloride (DAPI; Boehringer, Mannheim, Germany) at room temperature in a dark chamber, washed then mounted in fluorescence mounting media (Dako Australia Pty Ltd, Sydney, NSW, Australia). Sections were analyzed using a fluorescence imaging microscope (BX60; Olympus Optical Co., Tokyo, Japan) equipped with a camera, and images were digitally merged using an imaging software (SPOT Advance Version 4.7; Spot Imaging, Sterling Heights, MI, USA) to show colocalization with the nuclear cell marker DAPI and ZO-1. Computer-based image analysis using the ImageJ software (Version 1.43) was performed as described later.
In vitro studies
Murine podocyte cell line
A conditionally immortalized mouse podocyte cell line was kindly provided by Professor Stuart Shankland and Mr Jeffery Pipping (Division of Nephrology, University of Washington, Seattle, WA, USA). Podocytes were maintained in Roswell Park Memorial Institute 1640 medium supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 0.1 mg/mL streptomycin, as previously described.28 To initiate podocyte proliferation, cells were cultured at 33°C (permissive conditions) on bovine collagen type I (354231; BD Biosciences, Sydney, NSW, Australia) coated Primaria® cultureware 75 cm2 vented plastic flasks (353810; BD Biosciences). The medium was supplemented with 10 U/mL mouse recombinant interferon-γ (Roche Life Science, Sydney, NSW, Australia) to enhance expression of the temperature-sensitive large T antigen. To induce differentiation, podocytes were maintained at 37°C without interferon-γ (restrictive conditions) for at least 7 days, supplemented with 10% FBS, 100 U/mL penicillin, and 0.1 mg/mL streptomycin.
Murine macrophage cell line and synthesis of activated macrophage-conditioned media (MCM)
Because macrophages have a key role in mediating podocyte injury in CGN,11 the effect of MCM on ZO-1 expression in podocytes was examined.29 A murine macrophage cell line (J774 cells) was cultured in high glucose Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% fetal calf serum as previously described.30 MCM was produced by growing J774 cells in culture medium containing 10% fetal calf serum and 5 μg/mL of lipopolysaccharide (lps) (Sigma-Aldrich) for 24 hours, rinsed three times in phosphate buffered saline (PBS), and cultured in serum-free DMEM. After 48 hours, the medium was collected and filtered through 0.22 μm filter and designated +lps MCM. DMEM was also collected 48 hours after incubation in macrophages not exposed to lps (designated -lps MCM).
Experimental design of in vitro studies
Podocytes were grown under restrictive conditions for a minimum of 10 days before seeding on coverslips coated with bovine collagen type I at a density of 90,000 cells/well, yielding an initial confluence of 20%. Cells were allowed to adhere and further differentiate for 72 hours, after which culture plates were exposed to equal volumes of normal podocyte growth media, -lps MCM, or +lps (activated) MCM and harvested at 6 hours, 24 hours, 48 hours, and 96 hours for morphological, immunofluorescent, and mRNA analyses.
Semiquantitative assessment of podocyte morphology in vitro
Phase contrast images of control and -lps and +lps MCM-treated podocytes grown in six-well tissue culture plates were taken from a minimum of 10 consecutive fields with total cell count of at least 100 cells/well. Results were obtained in triplicate for each group per experiment and from three independent experiments. Using the ImageJ software, each image was divided into four quadrants, and a semiquantitative score from 0 to 4 was recorded for each quadrant. Determination of semiquantitative scores for morphological changes was based on the classification of podocytes as being a typically homogeneous population of well-differentiated arborized cells with a large cobblestone cell body as described previously.28 Cells presenting normal morphology were given a score of 0, whereas those which appeared to have an abrogated cell body with loss of protruding foot processes were classified as being morphologically abnormal. A score of 1 was given when <25% of the cells appeared morphologically abnormal, a score of 2 was given when 25–50% of the cells appeared abnormal, a score of 3 was given when 50–75% of the cells appeared abnormal, and a score of 4 was given when >75% of the cells appeared abnormal.
Immunofluorescence staining of ZO-1, F-actin, synaptopodin, nephrin, and podocin in cultured podocytes
Cells were fixed with 2% paraformaldehyde plus 4% sucrose for 10 minutes at 37°C and permeabilized with 0.3% Triton X-100 (Sigma-Aldrich) for 4 minutes at room temperature. Nonspecific binding was quenched with blocking solution (2% FBS, 2% bovine serum albumin, and 0.2% bovine gelatin in PBS) for 30 minutes at room temperature. To detect ZO-1, cells were incubated (overnight at 4°C) with polyclonal rabbit anti-ZO-1 (1:200) and rinsed in PBS followed by incubation with goat antirabbit Texas Red-conjugated secondary antibody (1:200 dilution). For F-actin detection, cells were incubated with rhodamine phalloidin (20 U/mL; Molecular Probes; Thermo Fisher Scientific) after permeabilization.
Cells incubated with synaptopodin (1:100; Fitzgerald Laboratories, North Acton, MA, USA) primary antibody were incubated with fluorescein isothiocyanate-conjugated secondary antibody (Invitrogen) for 45 minutes in a dark chamber at room temperature. For the detection of nephrin, three-step immunohistochemistry was conducted. Polyclonal guinea pig antinephrin antibody (1:100; Fitzgerald Laboratories) was incubated overnight at 4°C followed by rabbit antiguinea pig (Dako Laboratories) for 1 hour at room temperature before incubation with Alexa Fluor 688-conjugated secondary antibody.
Quantitation of ZO-1, F-actin, and synaptopodin immunofluorescence in cultured podocytes
Fluorescence was analyzed by an imaging microscope (BX60) equipped with a camera. Images were digitally merged using the imaging software (SPOT Advance Version 4.7) to show colocalization with DAPI followed by computer-based image analysis (ImageJ, Version 1.43). For each stain (ZO-1, F-actin, and synaptopodin), 10 fields (×400) were randomly digitized in a blinded manner. All images were taken at 400× magnification, and a total cell count of at least 100 podocytes per slide per stain was used for the quantitative analysis. Results were obtained from a triplicate from three independent experiments. Quantitation for ZO-1 was determined by counting the number of cells exhibiting fragmentation of ZO-1 at the apical membranes of cell–cell contacts from the total number of ZO-1 expressing cells. For F-actin, each cell displaying reorganization of actin filaments was counted to give a percentage of the total number of F-actin-positive cells.
Assessment of ZO-1 mRNA in cultured podocytes by real-time PCR
Total RNA was extracted using 350 μL of RLT buffer and homogenized by drawing five times through a 0.5 mL insulin syringe. Extracted RNA was purified using the RNeasy Mini Kit according to the manufacturer’s instructions (Qiagen, Chadstone, VIC, Australia). The yield and purity of RNA was measured with spectrophotometer and considered adequate if the A260:A280 was between 1.8 and 2.0. cDNA was synthesized with 200 ng of RNA in 20 μL reaction buffer by reverse transcription using the Superscript First Strand Synthesis System (Invitrogen) and random hexamer primers. The primer sequence pairs targeting the ZO-1 (primer sequence: forward 5′-AGAACAGAGCTGAACAGTTA-3′; reverse 5′-ACAATGCATCCACGAACAGA-3′, 306 bp) and 18S (primer sequence: forward 5′-AAACGGCTACCACATCCAAG-3′; reverse 5′-CCTCCAATGGATCCTCGTTA-3′, 244 bp) (used as the internal control) were generated using the Invitrogen Primer Design Tool. For reverse transcriptase-polymerase chain reaction, 2 μL of cDNA was used in a 25 μL PCR mixture containing 10 pmol/μL of each primer, 5 U/μL of Red Hot Taq Polymerase (Abgene, Rockford, IL, USA). PCR was performed with 32 cycles at 94°C for 1 minute, 56–62°C for 30 seconds, and 72°C for 1 minute. For negative control, cDNA was replaced with water. PCR products were size fractionated on 2% agarose gel (Promega, Madison, WI, USA) and detected by SYBR Green (AMRESCO) staining. For real-time PCR, the PCR mixture contained 0.5 μL of cDNA, 10 pmol/μL of each primer in a 20 μL final volume of SYBR master mix (Invitrogen). Amplification was performed using the Rotogene-6000 Real-Time Thermo cycler and was cycled for 2 minutes at 50°C, 10 minutes at 95°C followed by 40 cycles at 95°C for 15 seconds, and 1 minute at 60°C.
Statistical analysis
Results are presented as mean ± standard error of the mean. The data were analyzed using the JMP statistical software package (Version 4.04; SAS Institute, Cary, NC, USA) and Prism (Version 5; GraphPad Software, La Jolla, CA, USA). Comparisons between the experimental groups were performed by analysis of variance, followed by a post hoc analysis with the Tukey–Kramer honest significant difference test. A P-value of <0.05 indicated statistical significance.
Results
Glomerular changes in NSN in semithin sections
Examination of toluidine blue-stained semithin sections by light microscopy revealed observable histological changes within glomeruli as early as day 3 in rats with NSN (Figure 2). At day 3, intraluminal mononuclear cells were evident, and at day 5, glomerular capillary loops displayed marked alterations in the endocapillary compartment with advanced fibrin deposition and mesangial expansion, followed by crescent formation in the majority of glomeruli after day 7 (Figure 2).
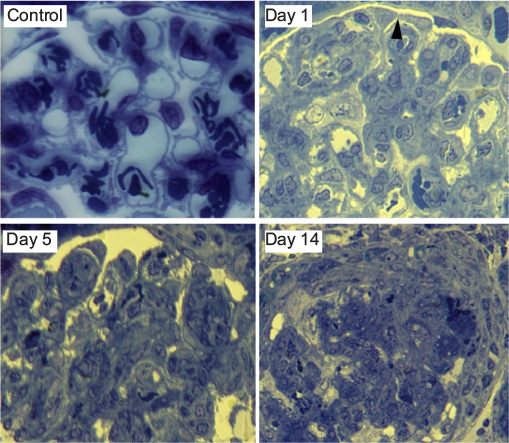 | Figure 2 Toluidine blue-stained glomerular sections from NSN compared to control. Notes: Glomeruli of control rats exhibited normal appearance, whereas glomeruli on day 1 of NSN revealed capillary damage with areas of podocyte cell body–cell body contact (black arrow). On day 5, evidence of podocoytes in contact with the PBM was prominent. Glomeruli were characterized by well-developed crescentic lesions on day 14. Magnification 1000× (oil immersion). Abbreviations: NSN, nephrotoxic serum nephritis; PBM, parietal basement membrane. |
Pathological description of glomerular ultrastructure in NSN
By TEM, podocytes from control animals exhibited healthy and large cell bodies with thick primary processes, which ramified into distinct secondary foot processes and evenly spaced slit diaphragms (Figure 3A). In rats with NSN, abnormalities in podocyte ultrastructure were identified as early as day 1, and this was characterized by the formation of focal contacts between neighboring podocyte cell bodies coupled with microvillus transformation at the site of cell–cell contact (Figure 3B). On day 2 of NSN, GBM thickness increased and podocyte–podocyte cell body contact was frequent with extensive microvillus transformation and an increase in cytoplasmic ribosomes in podocyte cell bodies (Figure 3C). On day 3, most capillary loops were characterized by patchy FPE, particularly where podocytes had formed contact. Podocyte migration to the PBM of Bowman’s capsule was evident on day 5 of NSN (Figure 3D). During this time, podocytes appeared hypertrophic and the interpodocyte distance decreased markedly. On days 7 and 14 of NSN, all glomeruli examined exhibited advanced fibrinoid necrosis and cellular crescents (Figure 3E and F).
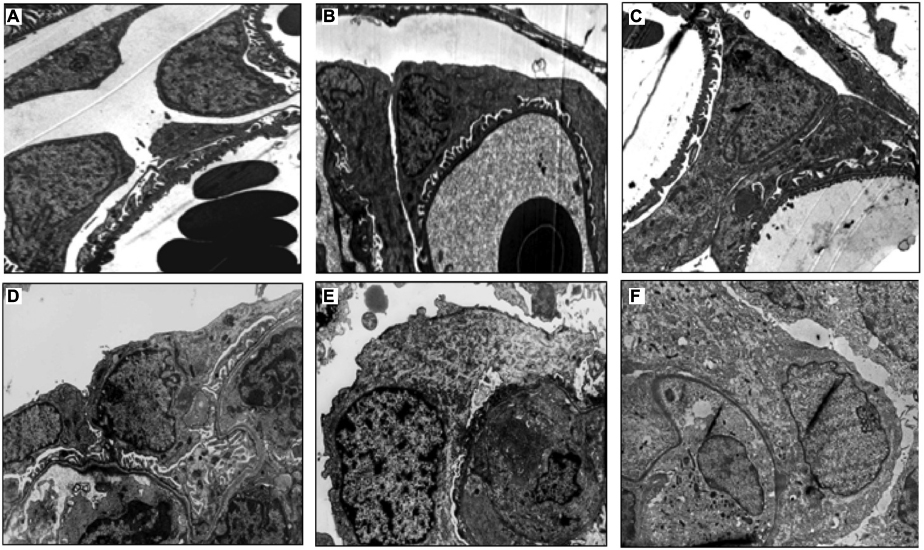 | Figure 3 Electron microscopic images displaying the timecourse of podocytic changes in NSN. Notes: In control (A), the distance between neighboring podocytes was evenly spaced in all glomeruli. On day 1 of NSN (B), the distance between podocytes decreased markedly and neighboring podocytes appeared to be in close apposition making focal contact with each other. On day 2 of NSN (C), the distance between podocytes decreased further and neighboring podocytes were in contact segmentally along their apical membranes. The interpodocyte distance declined further on day 5 (D) and continued to decrease on day 7 (E) and day 14 (F) of NSN. All glomeruli examined on days 7 and 14 exhibited several glomerular injuries and well-established cellular crescents. Magnification 5000×. Abbreviation: NSN, nephrotoxic serum nephritis. |
Morphometric analysis of interpodocyte distance and podocyte tight junction formation
Further studies were undertaken to investigate the characteristics of the podocyte-to-podocyte cell contact. At high magnification, structures resembling tight junctions were visible in podocytes making cell–cell body contact (Figure 4A). Morphometric analysis was undertaken to precisely define the sequential changes in podocyte tight junction formation. The number of neighboring podocytes in contact and exhibiting a tight junction at the site of podocyte cell–cell contact were quantitated per GCS. On day 1 of NSN, 3.29±0.49/GCS podocytes displayed tight junction at cell–cell contacts, reaching peak levels on day 2 with 4.62±0.78/GCS, despite the presence of normal capillary loops, foot processes, and GBM thickness. The incidence of this ultrastructural event decreased on day 5 (2.14±0.30/GCS) and continued to decline until day 14 of NSN (Figure 4B). These changes were associated with a reduction in the interpodocyte distance (Figure 4C). In control animals, no change was observed in distances between neighboring podocytes during the timecourse of the study (Figure 4C). On day 1 of NSN, the interpodocyte distance was 33% lower in rats with NSN compared to the control group (1610.0±484.5 nm and 2418.4±228.9 nm for NSN versus control groups, respectively), and neighboring podocytes appeared to be in close apposition with focal contact (Figures 3 and 4B). On day 2 of NSN, the interpodocyte distance decreased a further 47% compared to control (1181.4±0.2 nm versus 2238.4±372.5 nm for NSN versus control groups, respectively), and neighboring podocytes were in contact segmentally along their apical membranes. The interpodocyte distance declined further (74%) on day 5 compared to control (506.1±100.7 nm versus 2017.2±594.1 nm for NSN versus control groups, respectively). The interpodocyte distance continued to decrease on day 7 (245.8±31.4 nm versus 1734.98±296.7 nm for NSN versus control groups, respectively) and on day 14 (313.4±95.6 versus 2644.5±108.4 for NSN versus control groups, respectively).
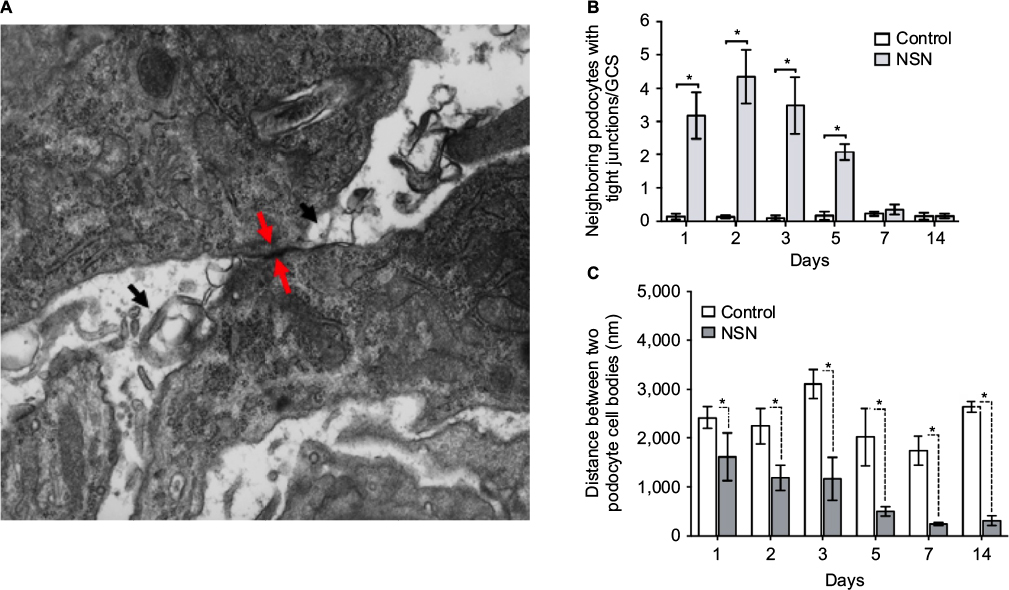 | Figure 4 Tight junction formation between podocytes in rats with NSN. Notes: (A) High power electron microscopic image displaying a tight junction at the site of two podocyte cell bodies in contact at day 1 of NSN. Tight junction formation is shown by the red arrows, and microvillus transformations are indicated by the black arrows. Magnification 20000×. (B) Morphometric analysis of the number of tight junctions between neighboring pdocoytes per GCS. (C) Morphometric analysis of the distance between the cell bodies of neighboring podocytes in rats with NSN compared to control. All data expressed as mean ± SEM; *P<0.05 versus the control group. Abbreviations: GCS, glomerular cross-section; NSN, nephrotoxic serum nephritis; SEM, standard error of the mean. |
Expression and localization of ZO-1 in NSN
To investigate the pattern of tight junction formation, immunofluorescence staining for ZO-1 was performed. In control animals, ZO-1 was limited to the slit diaphragm of glomeruli (Figure 5A). In NSN, ZO-1 expression was elevated on day 1 in 57.3±2.8% of glomeruli compared to control expression levels (Figure 5A and B) particularly at the apical membrane of podocytes in contact (Figure 5B, high power image). These changes were confirmed by quantitative analysis (Figure 5C). On day 5, glomerular ZO-1 expression was diffuse, with 89.5±2.3% (P≤0.05) of glomeruli expressing ZO-1 and then decreasing by 26% (P≤0.05) on day 7, displaying a fragmented pattern of expression (Figure 5). On day 14, ZO-1 expression diminished further in NSN as only 4.7±1.1% (P≤0.01) of glomeruli exhibited an increase in ZO-1 expression (Figure 5C). ZO-1 expression was limited to the glomerular capillary tuft and not detected in crescents.
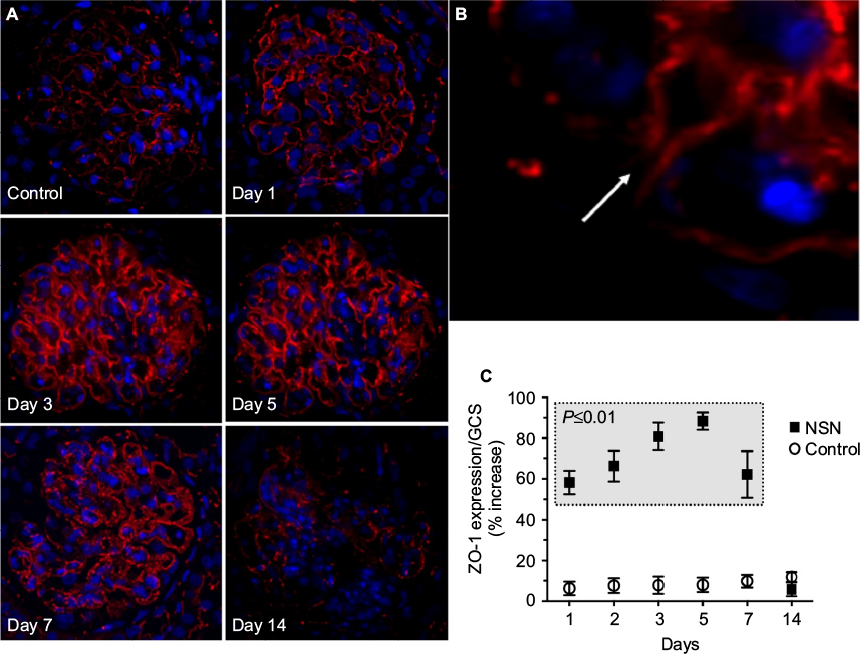 | Figure 5 Expression of the tight junction molecule ZO-1 in glomeruli of rats with NSN compared to control. Notes: (A and B) Immunohistochemical localization of ZO-1 in glomeruli of rats with NSN compared to the control. ZO-1 was localized to the slit diaphragm in the glomeruli of control rats, whereas on day 1 of NSN, the localization pattern was altered and redistributed to the apical membrane of podocytes at the site of cell body contact (higher power view [B]; the arrow designates the point of cell body contact). ZO-1 expression continued to increase on days 3 and 5 and had a fragmented pattern of expression on day 7. There was a reduction in ZO-1 expression in the glomerulus on day 14. Magnification (A) 400×, (B) 1000×. (C) Box–Whisker plot analysis quantifying the timecourse of ZO-1 glomerular expression in NSN compared to the control groups. *P≤0.01 for NSN (in the gray box) versus control at each timepoint. Abbreviations: GCS, glomerular cross-section; NSN, nephrotoxic serum nephritis; ZO-1, zonula occludens-1. |
Timecourse of other glomerular ultrastructural changes in NSN
By morphometric analysis, there was no change in the podocyte–PBM distance on day 1 to day 3 compared to the control group (Figure 6). The distance between nucleated podocyte cell bodies and the PEC of Bowman’s capsule appeared evenly spaced in both the NSN and control groups. On day 5, there was diffuse podocyte contact with the PEC as shown in Figure 6 and as demonstrated by the reduction in the mean podocyte–PEC distance compared to control (1711.38±303.57 nm versus 3462.90±188.36 nm for NSN versus control groups, respectively; P<0.05). The podocyte–PEC distance also decreased on day 7 and further on day 14 (554.73±129.86 nm versus 3594.22±119.52 nm for NSN versus control groups, respectively; P<0.05) (Figure 6).
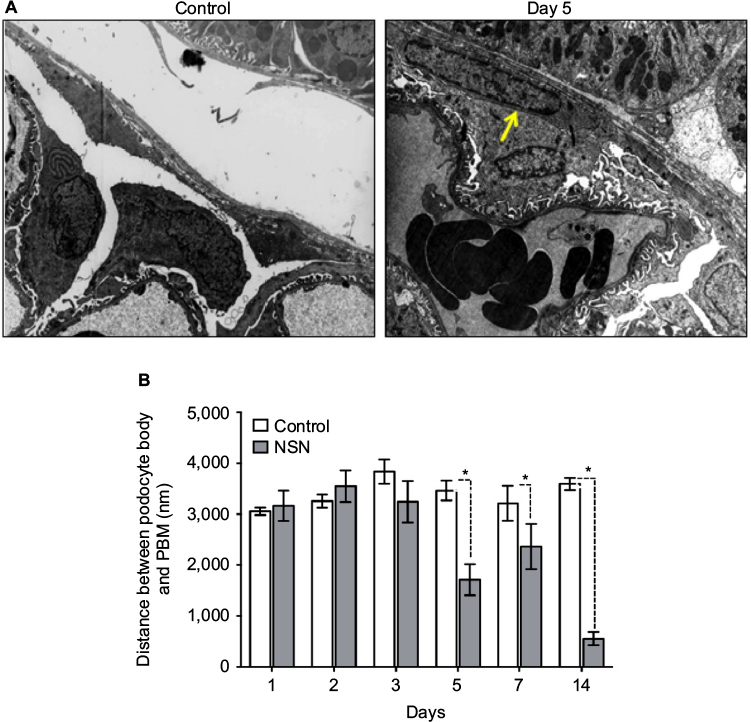 | Figure 6 Timecourse of podocyte to parietal basement membrane distance in NSN. Notes: (A) Representative electron micrographs demonstrating the reduction in distance between podocyte and PBM in control compared to rats with NSN on day 5. The yellow arrow demonstrates the marked reduction in distance between a podocyte and parietal epithelial cell. Magnification 5000×. (B) Morphometric analysis of the distance between podocyte cell bodies and the PBM. All data expressed as mean ± SEM; *P<0.05 versus the control group. Abbreviations: PBM, parietal basement membrane; NSN, nephrotoxic serum nephritis; SEM, standard error of the mean. |
The GBM thickness of control animals remained constant at all time points, with the mean thickness ranging from 500 nm to 165 nm (Figure 7A and B). There was no change in GBM thickness on day 1 in NSN compared to control. GBM thickness increased on day 2 (202.8±6.9 nm versus 165.5±5.1 nm for NSN versus control groups, respectively) and continued to increase progressively until day 14 to almost double that of control (318.6±5.8 nm versus 175.1 ± 5.3 nm) (Figure 7B). In control animals, glomerular capillaries exhibited a consistent FSF/µm GBM length ranging from 2.07±0.07 to 2.30±0.08 as foot processes appeared morphologically normal with structurally continuous filtration slits from day 1 to day 14 (Figure 7A and C). There was no change in FSF/µm GBM on days 1 and 2 of NSN but it was reduced by 10% on day 3 compared to control (1.91±0.07 FSF/µm versus 2.13±0.08 FSF/µm for NSN versus control groups, respectively). Podocyte FPE was focally distributed particularly at the sites of podocyte–podocyte contact. By day 5, FPE was extensive as reflected by the sustained reduction in FSF/µm GBM length (1.5±0.1 FSF/µm versus 2.2±0.07 FSF/µm), which progressed until day 14 (0.49±0.06 FSF/µm versus 2.14±0.05 FSF/µm) (Figure 7C).
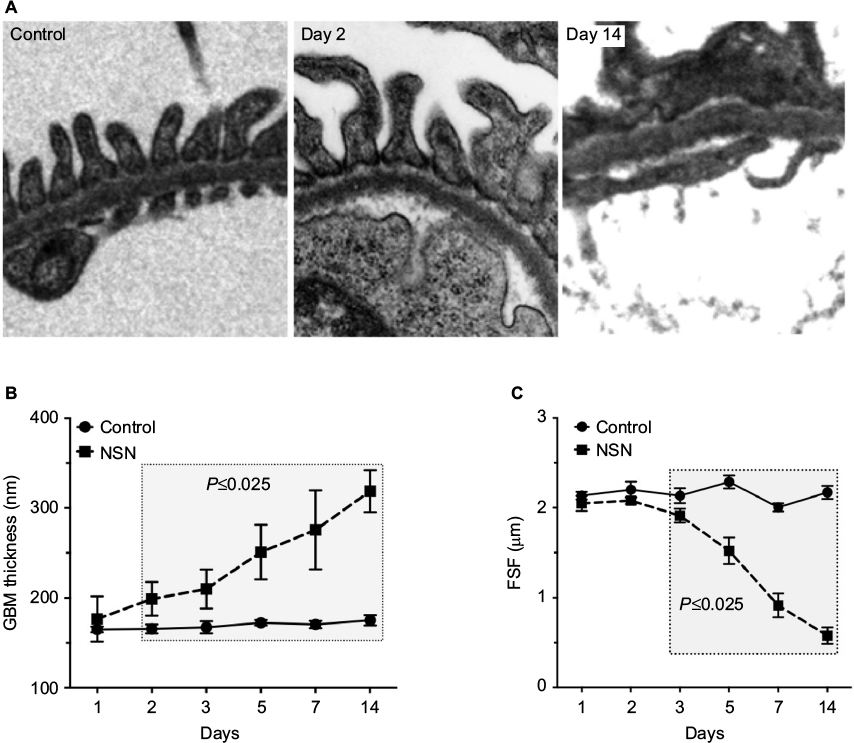 | Figure 7 Timecourse of glomerular basement membrane thickening in NSN. Notes: (A) Representative electron micrographs depicting increases in GBM thickness on days 3 and 5 and FPE on day 5. Magnification 12000×. (B) Morphometric analysis of the GBM thickness in the experimental groups over time. (C) Morphometric analysis of the FSF in the experimental groups over time. All data expressed as mean ± SEM. Abbreviations: FPE, foot process effacement; FSF, filtration slit frequency; GBM, glomerular basement membrane; NSN, nephrotoxic serum nephritis; SEM, standard error of the mean. |
In vitro effect of MCM on podocyte morphology
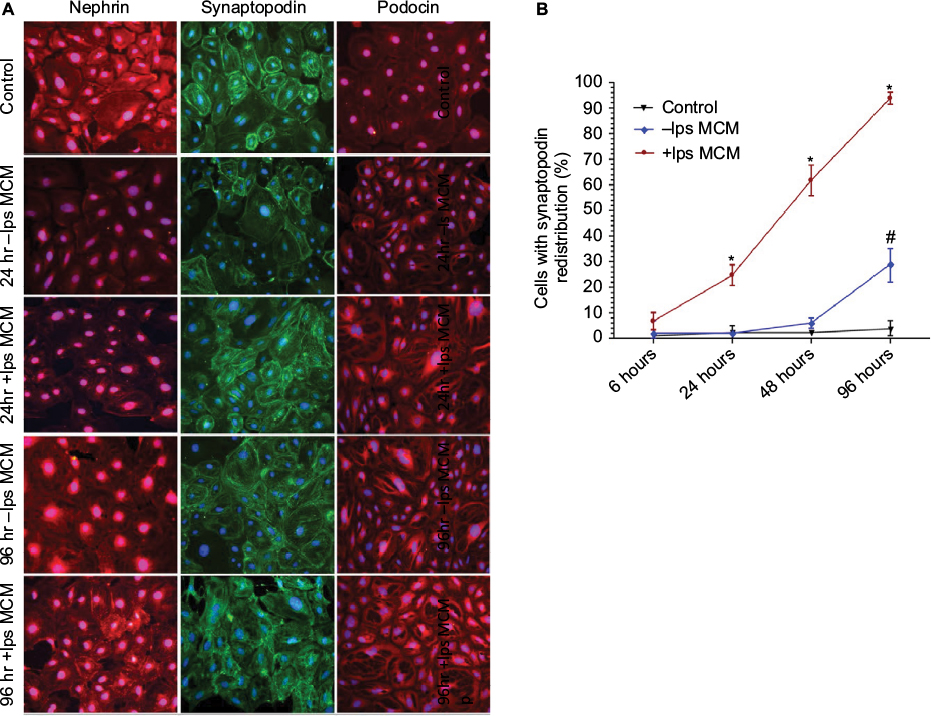
To determine whether the increase in glomerular ZO-1 expression in early NSN is a response to inflammatory injury, an in vitro system was utilized to examine the effect of activated MCM on podocyte morphology. Control cells exhibited normal cell morphology characterized by well-developed interdigitating processes. There were no observable morphological changes detected by light microscopy in podocytes exposed to -lps and +lps MCM at 6 hours and 24 hours. At 48 hours, morphological changes were more obvious in the +lps MCM group (1.8±0.66, P<0.05) and cells appeared flatter, thinner, with arborized cell bodies, and thinner foot processes compared to those in the -lps MCM group (0.52±0.27) and control (0.08±0.01) (Figure 8A). At 96 hours, morphological changes were more widespread and pronounced, as cell bodies appeared fully arborized in the +lps MCM-treated group (3.14±0.36, P<0.05) whereas the -lps MCM-treated podocytes were altered to a lesser extent (1.26±0.29) (Figure 8A).
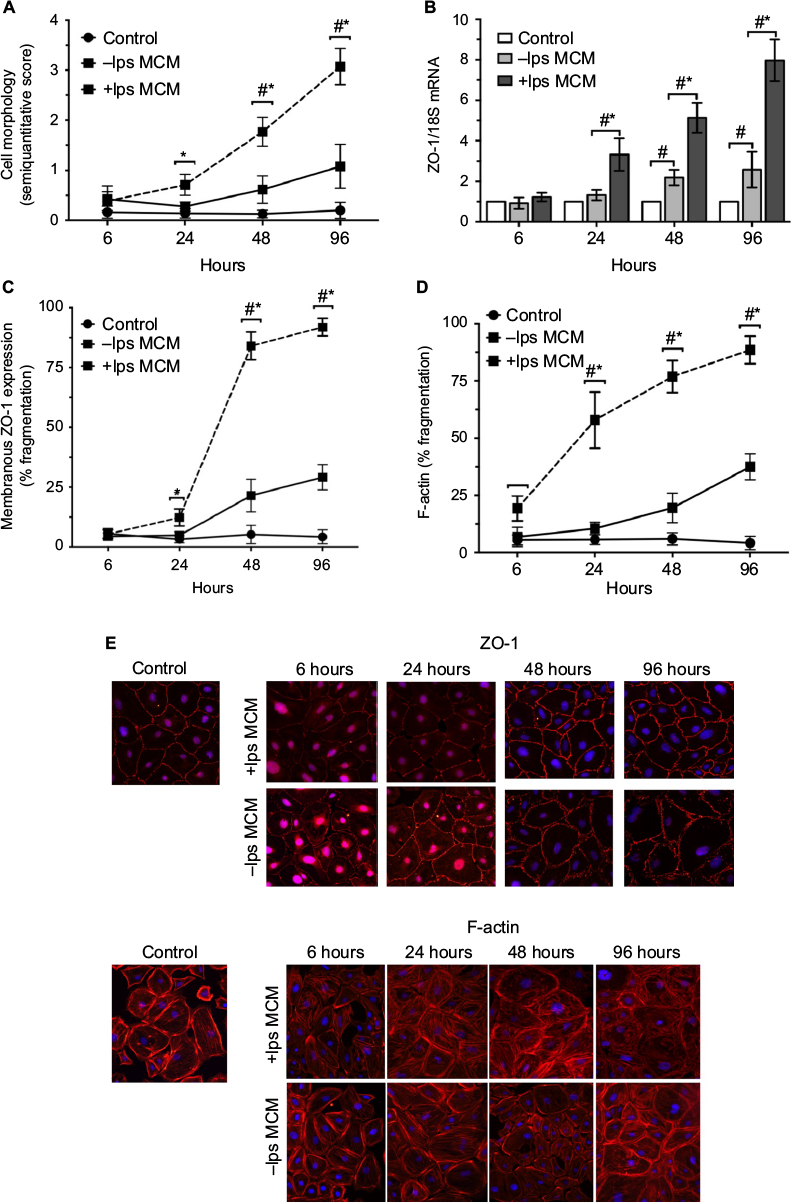 | Figure 8 Effects of -lps and +lps MCM on cell morphology, ZO-1 in podocytes in vitro. Notes: (A) Quantitative analysis of cell morphology. Data expressed as mean ± SEM; *P<0.05 for +lps MCM versus -lps MCM; #P<0.05 for +lps and -lps MCM versus control. (B) Timecourse of ZO-1 mRNA postexposure to control (medium), -lps MCM and +lps MCM from 24 hours to 96 hours. (C) Timecourse of fragmentation in ZO-1 expression. (D) Timecourse of F-actin fragmentation. Data expressed as mean ± SEM; *P<0.01 compared to control and -lps MCM; #P<0.05 compared to control. (E) Immunofluorescent microscopic images of podocytes stained (red) for ZO-1 (upper panels) or F-actin (lower panels) and blue staining indicating DAPI. Magnification 400×. Abbreviations: DAPI, 4′,6-diamidine-2′-phenylindole dihydrochloride; lps, lipopolysaccharide; MCM, macrophage-conditioned media; SEM, standard error of the mean; ZO-1, zonula occludens-1. |
Effect of MCM on podocyte expression of ZO-1 and F-actin
ZO-1 mRNA was significantly upregulated postexposure to +lps MCM at 24 hours (3.39±0.52, P<0.01), 48 hours (5.67±0.46, P<0.01), and 96 hours (7.92±0.8, P<0.01) compared to control (Figure 8B). ZO-1 mRNA was also increased in the -lps MCM group at 48 hours (2.28±0.31, P<0.01) and 96 hours (2.61 ± 0.68, P<0.01), but the increase was to a lesser degree than podocytes exposed to +lps MCM (Figure 8B). By immunofluorescence, ZO-1 was expressed in three cellular compartments, apical membrane, nucleus, and cytoplasm. In control, ZO-1 expression in podocytes was peripherally distributed (at contacts and process interdigitations of adjacent cells) as continuous and circumferential fine segments in all cell nuclei of examined podocytes (Figure 8E). There was an increased nuclear expression of ZO-1 at 6 hours and 24 hours in -lps and +lps MCM-treated groups, which declined at 48 hours and 96 hours (Figure 8E). The cytoplasmic expression of ZO-1 was increased at 24 hours in the +lps MCM group and decreased at 48 hours and 96 hours, whereas these changes were not observed in the -lps MCM group (Figure 8E). At 24 hours, fragmentation in ZO-1 expression was evident as the density increased between cell-to-cell contacts postexposure to +lps MCM (11.6±1.5%; P<0.01) compared to -lps MCM (4.3±1.1%) and control (0.07±0.00%) (Figure 8C). At 48 hours, the percentage of cells with fragmented ZO-1 expression increased markedly postexposure to +lps MCM (85.2±3.1%, P<0.01) compared to -lps MCM (24.66±3.48%; P<0.05) and control (4.36±1.84%; P<0.01). These changes increased further at 96 hours (94.72±2.08%; P<0.01), with cell-to-cell contacts exhibiting a distinct pattern of ZO-1 expression as structures resembling finger-like protrusions were diffuse, features not seen in the -lps MCM and control groups (Figure 8C). Fragmentation in F-actin microfilaments was detected in the +lps MCM group at 6 hours (18.3±2.6%) and 24 hours (61.6±3.7%), whereas cells of the -lps MCM and control groups displayed normal actin cytoskeletal arrangement at these time points (Figure 8D). At 96 hours, perturbation of F-actin increased with >90% of cells displaying complete derangement of actin microfilaments. The latter was characterized by coarse and short clumps of F-actin fibers lacking any specific organization (Figure 8D and E).
Effect of MCM on podocyte expression of synaptopodin, nephrin, and podocin
Synaptopodin
In control cells, synaptopodin, which binds both to F-actin and ZO-1, was normally distributed, localizing to cell nuclei, extending to the cytoskeleton, and finishing at cell–cell contacts (Figure S1A and B) as previously described.31 Cells exposed to +lps MCM displayed disrupted organization of synaptopodin at 24 hours (24.66±3.95%) compared to those exposed to –lps MCM, and this continued to increase at 48 hours (Figure S1A and B). At 96 hours, perturbation of synaptopodin organization occurred in >90% of the cells treated with +lps MCM characterized by punctuated pattern of expression, loss of peripheral staining, and formation of clumped protein structures in respect to control podocytes and -lps MCM groups (Figure S1A and B). On the other hand, the majority of cells pertaining to the -lps MCM group maintained a normal synaptopodin distribution, with only 28.07±3.25% of cells displaying signs of distorted synaptopodin organization at 96 hours (Figure S1A and B).
Nephrin
Similarly, +lps MCM altered podocyte expression of the slit diaphragm protein nephrin. In control, nephrin localized densely to the nucleus, perinuclear region, and apical membranes of cell–cell contacts (Figure S1A). The distribution of nephrin appeared to be normally distributed in podocytes exposed to -lps and +lps MCM at the 6 hours and 24 hours time points. At the 48 hours and 96 hours time points, podocytes of both -lps and +lps MCM groups displayed increased cytoplasmic nephrin expression and reduced expression in the perinuclear area (Figure S1A).
Podocin
Podocin expression was also altered in podocytes exposed to +lps MCM, yet this time, alterations were displayed also in the -lps MCM group and as early as 6 hours time point. In control, podocin localized to the nucleus and to cell–cell contacts (Figure S1A). There were a gradual decline in the nuclear expression and an increase in the cytoplasmic expression of podocin from 24 hours to 96 hours postexposure to -lps and +lps MCM. The cytoplasmic podocin was markedly punctuated and distorted at 48 hours and 96 hours in both -lps and +lps groups, characterized by large pockets of cleaved podocin expression (Figure S1A).
Discussion
The most important finding of this study is that tight junctions form between the cell bodies of neighboring podocytes during the early stages of a rodent model of CGN. To our knowledge, this ultrastructural abnormality has not been noted in previous studies, and it preceded the development of other well-known morphological changes, including podocyte–PBM formation, FPE, and GBM thickening. The formation of tight junctions in podocytes was associated with the upregulation in ZO-1 at the apical membrane in podocytes. It is postulated that podocytes respond by redistributing ZO-1 to the cell membrane to preserve cell–cell contacts, and this can be in part explained by a mechanism dependent on actin rearrangement as demonstrated in cultured podocytes postexposure to +lps MCM.
The formation of cell–cell contacts and tight junctions between the cell bodies of neighboring podocytes on day 1 of NSN was the first ultrastructural alteration observed in NSN and occurred despite healthy secondary foot processes. Podocyte cell bodies subsequently exhibited extensive formation of microvilli at the sites of cell–cell contact. Of note, focal FPE became apparent from day 2, as seen by the reduction in FSF/µm, and upon closer examination, these focally effaced areas presented adjacent to podocyte cell bodies that had formed contact. On day 3, podocytes appeared to extend toward the parietal epithelium and all capillary loops possessed patchy FPE. On day 5, podocytes made full contact with PECs coupled with advanced GBM thickening and diffuse FPE. Glomeruli analyzed from day 7 to day 14 were predominantly characterized by cellular crescents and represented the end point of this analysis. The timecourse of these ultrastructural alterations suggests that the formation of tight junctions at the site of podocytes in contact could be an early response to macrophage infiltration following NSN challenge. These ultrastructural changes may be important for intracellular signaling for foot processes to undergo effacement, potentially an adaptive response to maintain the filtration barrier.
The increased expression of ZO-1 in experimental anti-GBM CGN has not been previously described. It is speculated that the glomerular upregulation of ZO-1 early in NSN, particularly at the site of podocyte–podocyte contact, can be likened to a wound-healing process as the visceral epithelium prepares to seal and prevent the inflammatory injury occurring at the endocapillary compartment from spreading to Bowman’s space. In control animals, ZO-1 was distributed linearly along the slit diaphragm of the GBM wall, consistent with previous studies describing the slit diaphragm as a tenuous-modified tight junction barrier.16,32–34 Consistent with Kim et al22 who report an upregulation of ZO-1 in puromycin nephrosis, in NSN, ZO-1 expression was increased in >50% of glomeruli examined on day 1, with peak expression levels occurring in 80% of glomeruli on day 5 when glomeruli were undergoing advanced inflammation, fibrinoid deposition, and subsequent obliteration to the GBM. It is possible that podocytes increase their production of ZO-1 to prepare for FPE and subsequent shedding from the GBM capillary wall. Experimentally induced epithelial wounds have been shown to cause F-actin and myosin-II to accumulate at tight junctions and form a “purse-string” that closes the wound to restore epithelial barrier function.35,36 This supports a potentially important role for ZO-1 during barrier maintenance by mediating visceral epithelial wound healing in NSN.
In contrast, glomerular expression of ZO-1 decreased from day 7 of NSN, as glomeruli exhibited a punctuated pattern of expression limited to the glomerular tuft without localizing to crescentic lesions. It might be that upregulation of ZO-1 early in NSN serves an important protective role to initial insult, while late-phase downregulation is a maladaptive response that leads to advanced FPE, scarring, and fibrosis. The loss of ZO-1 expression at the slit diaphragm in a profibrotic ischemic model of renal injury, where ZO-1 was reduced to sub-basal levels in rats’ post-ATP depletion leading to podocyte damage and proteinuria, is consistent with the reduction in ZO-1 expression after day 7 in NSN.37 Rincon-Choles et al23 also demonstrated a decrease in ZO-1 expression in glomeruli of rats with type 1 and type 2 diabetes, suggesting that alterations in the localization of ZO-1 may be relevant to the pathogenesis of proteinuria and fibrosis in diabetes as well. Hence, reduction in ZO-1 expression appears to associate with worsening of renal injury and proteinuria. Therefore, strategies aimed at stabilizing ZO-1 production at podocyte–podocyte contacts may potentially be a relevant therapeutic tool for treating anti-GBM CGN.
A severe macrophage infiltrate is likely to be mediating the early podocyte changes seen, and therefore an in vitro system was designed to investigate the effects of activated MCM on the podocyte expression of ZO-1. Our findings showed that fragmentation at the apical membrane and increased nuclear expression of ZO-1 was observed alongside the derangement of cytoskeletal (F-actin and synaptopodin) and slit diaphragm (nephrin and podocin) proteins in podocytes 24 hours postexposure to +lps MCM and not -lps MCM. Profound changes in podocyte shape and morphology were also observed, which could be likened to FPE in vivo. Consistent with Ikezumi et al, these changes are not simply a passive process following chemokine injury but are due to a complex interplay of proteins that comprise the molecular anatomy of the different cytoskeletal protein domains in podocytes.28,29 Decreased F-actin as well as nephrin and podocin expression levels in podocytes have been attributed to the secretion of TNF-α by activated macrophages,29 changes shown to be mediated via p38 MAPK and JNK signaling29 in proliferative glomerulonephritis.38–40 Although the effects of activated macrophages on podocytes in vitro damage are documented, the effect on ZO-1 expression in podocytes, or any other type of epithelial cell for that matter, has not been investigated to date. These findings have novel implications for the role of ZO-1 in podocyte response to renal injury, consistent with the ultrastructural changes described. We infer that in response to cytoskeletal injury, podocytes increase the synthesis of ZO-1 and accumulate it at cell–cell contacts to form tight junctions in an attempt to create a barrier to combat the spread of chemokine injury. Further functional in vitro and in vivo studies using siRNA and CRISPR technology as well as investigation of the role of TNF-α as a paracrine effector of ZO-1 induction are required to further substantiate the new hypotheses raised by the current study.
Finally, the observations of this study expand on the previous studies by Le Hir et al5 who were one of the first to investigate the ultrastructural changes in podocytes in crescent formation in CGN. Based on the observations of this study, a hypothetical sequence of events summarizing the alterations in podocyte ultrastructure starting from initial response to GBM injury to the development of a necrotizing crescent is proposed in Figure 9. First, a normal situation is depicted (Figure 9A), which post-initiation of NSN on day 0 results in stressed podocyte cell bodies. Podocytes make contact and exhibit extensive microvillous transformation, which may represent a stressed cell state with extensive membrane dynamics.41,42 Here, the expression of ZO-1 is upregulated and potentially leads to the formation of tight junctions between podocytes (day 1, Figure 9B), suggestive of intercellular cross talk, which may mediate signaling down to the foot processes via actin filaments. The ongoing endocapillary inflammation leads to GBM thickening (and lysis), FPE becomes apparent (day 2, Figure 9C), and ZO-1 upregulation continues, perhaps in an attempt to seal off injury from spreading to Bowman’s space. As the inflammatory process advances, the glomerular tuft expands causing podocytes to form contact with the parietal epithelium, thereby instigating the proliferation of PECs (day 5, Figure 9D), culminating in an advanced cellular crescentic lesion as shown in Figure 9E.
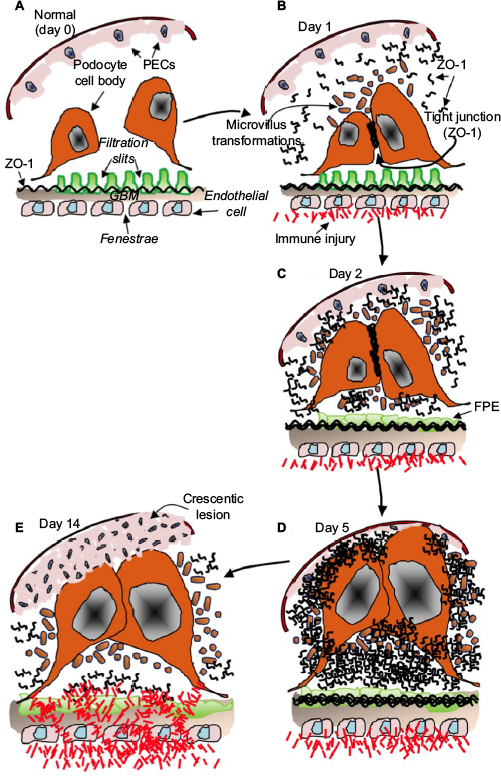 | Figure 9 Schematic diagram depicting the early response of podocytes in the pathogenesis of glomerular crescent formation. Notes: (A) Podocyte cell bodies respond to immune injury to the GBM by making contact and forming tight junctions between cell bodies at the apical membranes. Microvillus transformations (brown circles) appear at the site of cell–cell contact, and this is followed by increased expression of ZO-1 at the site of podocyte cell–cell contact (B). ZO-1 expression increases, FPE progresses (C), and podocytes affix to the PBM (D), leading to the proliferation of PECs and resulting in an irreversible crescentic lesion (E). Abbreviations: GBM, glomerular basement membrane; FPE, foot process effacement; PBM, parietal basement membrane; PECs, parietal epithelial cells; ZO-1, zonula occludens-1. |
Conclusion
This study documents new data on the early ultrastructural alterations of podocytes in an in vivo model of anti-GBM CGN. Our findings imply that the formation of tight junctions between the cell bodies of neighboring podocytes may be an important early event that underlies the development of glomerular crescents. It is postulated that podocytes respond by redistributing ZO-1 early to form cell–cell contacts, which potentially serve an important role to combat injury occurring within the glomerular endocapillary compartment from spreading to Bowman’s space. This study prompts further investigation using experimental studies, which aim to stabilize ZO-1 in macrophage-mediated models of podocyte injury to help elucidate whether stabilizing ZO-1 is a therapeutic target in immune-mediated CGN.
Acknowledgments
The authors are grateful to Dr David Nikolic-Paterson (Monash Medical Centre, Monash University, Melbourne) for providing anti-GBM serum and also comments on the manuscript. The authors thank Professor Stuart Shankland and Mr Jeffrey Pippin (University of Washington, Seattle, WA, USA) for providing the podocyte cell line. Dr Lena Succar was supported by a Kidney Health Australia Biomedical Research Scholarship. This study was funded by Australian Government research grants from the National Health and Medical Research Council of Australia (230500, 457575, and 632647) (85%) and, in part, by an investigator-initiated grant from Wyeth Australia (now Pfizer) (15%) to Dr Rangan.
Author contributions
All authors contributed toward data analysis, drafting and critically revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Jennette JC. Rapidly progressive crescentic glomerulonephritis. Kidney Int. 2003;63(3):1164–1677. | ||
Li X, Chen N. Management of crescentic glomerulonephritis: what are the recent advances? Contrib Nephrol. 2013;181:229–239. | ||
Churg J, Morita T, Suzuki Y. Glomerulonephritis with fibrin and crescent formation. Perspect Nephrol Hypertens. 1973;1(pt 2):677–694. | ||
Ohnuki T. Crescentic glomerulonephritis induced in the goat by immunization with homologous or heterologous glomerular basement membrane antigen. Acta Pathol Jpn. 1975;25(3):319–331. | ||
Le Hir M, Keller C, Eschmann V, Hahnel B, Hosser H, Kriz W. Podocyte bridges between the tuft and Bowman’s capsule: an early event in experimental crescentic glomerulonephritis. J Am Soc Nephrol. 2001;12(10):2060–2071. | ||
Moeller MJ, Soofi A, Hartmann I, et al. Podocytes populate cellular crescents in a murine model of inflammatory glomerulonephritis. J Am Soc Nephrol. 2004;15(1):61–67. | ||
Bariety J, Bruneval P, Meyrier A, Mandet C, Hill G, Jacquot C. Podocyte involvement in human immune crescentic glomerulonephritis. Kidney Int. 2005;68(3):1109–1119. | ||
Thorner PS, Ho M, Eremina V, Sado Y, Quaggin S. Podocytes contribute to the formation of glomerular crescents. J Am Soc Nephrol. 2008;19(3):495–502. | ||
Boucher A, Droz D, Adafer E, Noel LH. Relationship between the integrity of Bowman’s capsule and the composition of cellular crescents in human crescentic glomerulonephritis. Lab Invest. 1987;56(5):526–533. | ||
Atkins RC, Nikolic-Paterson DJ, Song Q, Lan HY. Modulators of crescentic glomerulonephritis. J Am Soc Nephrol. 1996;7(11):2271–2278. | ||
Han Y, Ma FY, Tesch GH, Manthey CL, Nikolic-Paterson DJ. c-fms blockade reverses glomerular macrophage infiltration and halts development of crescentic anti-GBM glomerulonephritis in the rat. Lab Invest. 2011;91(7):978–991. | ||
Besse-Eschmann V, Le Hir M, Endlich N, Endlich K. Alteration of podocytes in a murine model of crescentic glomerulonephritis. Histochem Cell Biol. 2004;122(2):139–149. | ||
Smeets B, Uhlig S, Fuss A, et al. Tracing the origin of glomerular extracapillary lesions from parietal epithelial cells. J Am Soc Nephrol. 2009;20(12):2604–2615. | ||
Singh SK, Jeansson M, Quaggin SE. New insights into the pathogenesis of cellular crescents. Curr Opin Nephrol Hypertens. 2011;20(3):258–262. | ||
El Machhour F, Keuylian Z, Kavvadas P, Dussaule JC, Chatziantoniou C. Activation of Notch3 in glomeruli promotes the development of rapidly progressive renal disease. J Am Soc Nephrol. 2015;26(7):1561–1575. | ||
Reiser J, Kriz W, Kretzler M, Mundel P. The glomerular slit diaphragm is a modified adherens junction. J Am Soc Nephrol. 2000;11(1):1–8. | ||
Koda R, Yoshino A, Imanishi Y, et al. Expression of tight junction protein claudin-1 in human crescentic glomerulonephritis. Int J Nephrol. 2014;2014:598670. | ||
Fanning AS, Ma TY, Anderson JM. Isolation and functional characterization of the actin binding region in the tight junction protein ZO-1. FASEB J. 2002;16(13):1835–1837. | ||
Yaoita E, Kurihara H, Yoshida Y, et al. Role of Fat1 in cell-cell contact formation of podocytes in puromycin aminonucleoside nephrosis and neonatal kidney. Kidney Int. 2005;68(2):542–551. | ||
Liu G, Kaw B, Kurfis J, Rahmanuddin S, Kanwar YS, Chugh SS. Neph1 and nephrin interaction in the slit diaphragm is an important determinant of glomerular permeability. J Clin Invest. 2003;112(2):209–221. | ||
Verma R, Kovari I, Soofi A, Nihalani D, Patrie K, Holzman LB. Nephrin ectodomain engagement results in Src kinase activation, nephrin phosphorylation, Nck recruitment, and actin polymerization. J Clin Invest. 2006;116(5):1346–1359. | ||
Kim BS, Park HC, Kang SW, et al. Impact of cyclosporin on podocyte ZO-1 expression in puromycin aminonucleoside nephrosis rats. Yonsei Med J. 2005;46(1):141–148. | ||
Rincon-Choles H, Vasylyeva TL, Pergola PE, et al. ZO-1 expression and phosphorylation in diabetic nephropathy. Diabetes. 2006;55(4):894–900. | ||
Henique C, Bollee G, Lenoir O, et al. Nuclear factor erythroid 2-related factor 2 drives podocyte-specific expression of peroxisome proliferator-activated receptor gamma essential for resistance to crescentic GN. J Am Soc Nephrol. 2016;27(1):172–188. | ||
Ma FY, Flanc RS, Tesch GH, Bennett BL, Friedman GC, Nikolic-Paterson DJ. Blockade of the c-Jun amino terminal kinase prevents crescent formation and halts established anti-GBM glomerulonephritis in the rat. Lab Invest. 2009;89(4):470–484. | ||
El-Aouni C, Herbach N, Blattner SM, et al. Podocyte-specific deletion of integrin-linked kinase results in severe glomerular basement membrane alterations and progressive glomerulosclerosis. J Am Soc Nephrol. 2006;17(5):1334–1344. | ||
Ikezumi Y, Hurst LA, Masaki T, Atkins RC, Nikolic-Paterson DJ. Adoptive transfer studies demonstrate that macrophages can induce proteinuria and mesangial cell proliferation. Kidney Int. 2003;63(1):83–95. | ||
Shankland SJ, Pippin JW, Reiser J, Mundel P. Podocytes in culture: past, present, and future. Kidney Int. 2007;72(1):26–36. | ||
Ikezumi Y, Suzuki T, Karasawa T, Kawachi H, Nikolic-Paterson DJ, Uchiyama M. Activated macrophages down-regulate podocyte nephrin and podocin expression via stress-activated protein kinases. Biochem Biophys Res Commun. 2008;376(4):706–711. | ||
Tan TK, Zheng G, Hsu TT, et al. Macrophage matrix metalloproteinase-9 mediates epithelial-mesenchymal transition in vitro in murine renal tubular cells. Am J Pathol. 2010;176(3):1256–1270. | ||
Schiwek D, Endlich N, Holzman L, Holthofer H, Kriz W, Endlich K. Stable expression of nephrin and localization to cell-cell contacts in novel murine podocyte cell lines. Kidney Int. 2004;66(1):91–101. | ||
Fukasawa H, Bornheimer S, Kudlicka K, Farquhar MG. Slit diaphragms contain tight junction proteins. J Am Soc Nephrol. 2009;20(7):1491–1503. | ||
Kurihara H, Anderson JM, Kerjaschki D, Farquhar MG. The altered glomerular filtration slits seen in puromycin aminonucleoside nephrosis and protamine sulfate-treated rats contain the tight junction protein ZO-1. Am J Pathol. 1992;141(4):805–816. | ||
Schnabel E, Anderson JM, Farquhar MG. The tight junction protein ZO-1 is concentrated along slit diaphragms of the glomerular epithelium. J Cell Biol. 1990;111(3):1255–1263. | ||
Clark AG, Miller AL, Vaughan E, Yu HY, Penkert R, Bement WM. Integration of single and multicellular wound responses. Curr Biol. 2009;19(16):1389–1395. | ||
Bement WM, Mandato C, Kirsch MN. Wound-induced assembly and closure of an actomyosin purse string in Xenopus oocytes. Curr Biol. 1999;11(9):579–587. | ||
Wagner MC, Rhodes G, Wang E, et al. Ischemic injury to kidney induces glomerular podocyte effacement and dissociation of slit diaphragm proteins Neph1 and ZO-1. J Biol Chem. 2008;283(51):35579–35589. | ||
Stambe C, Atkins RC, Hill PA, Nikolic-Paterson DJ. Activation and cellular localization of the p38 and JNK MAPK pathways in rat crescentic glomerulonephritis. Kidney Int. 2003;64(6):2121–2132. | ||
Iwata Y, Wada T, Furuichi K, et al. p38 Mitogen-activated protein kinase contributes to autoimmune renal injury in MRL-Fas lpr mice. J Am Soc Nephrol. 2003;14(1):57–67. | ||
Sheryanna A, Bhangal G, McDaid J, et al. Inhibition of p38 mitogen-activated protein kinase is effective in the treatment of experimental crescentic glomerulonephritis and suppresses monocyte chemoattractant protein-1 but not IL-1beta or IL-6. J Am Soc Nephrol. 2007;18(4):1167–1179. | ||
Matsui K, Breiteneder-Geleff S, Kerjaschki D. Epitope-specific antibodies to the 43-kD glomerular membrane protein podoplanin cause proteinuria and rapid flattening of podocytes. J Am Soc Nephrol. 1998;9(11):2013–2026. | ||
Hara M, Yanagihara T, Kihara I, Higashi K, Fujimoto K, Kajita T. Apical cell membranes are shed into urine from injured podocytes: a novel phenomenon of podocyte injury. J Am Soc Nephrol. 2005;16(2):408–416. |
Supplementary material
 | Figure S1 Timecourse of lps on slit diaphragm and cytoskeletal protein expression in podocytes in vitro. Notes: (A) Effect of -lps and +lps MCM (24 hours and 96 hours) on nephrin, synaptopodin, and podocin expressions in podocytes. Representative fluorescence images are depicted for nephrin (red), synaptopodin (green), and podocin (red) colabeled with the nuclear marker DAPI (blue). Magnification 400×. (B) Effect of -/+lps MCM on synaptopodin distribution in podocytes. At 24 hours, there was an increase in podocytes displaying disrupted organization of synaptopodin postexposure to +lps MCM compared to <5% in the -lps MCM group. The percentage of podocytes with alterations in synaptopodin distribution increased at 48 hours and 96 hours in the +lps MCM group and to a lesser degree in the -lps MCM group. Data expressed as mean ± SEM; *P<0.01 compared to control and -lps MCM; #P<0.05 compared to control. Abbreviations: DAPI, 4′,6-diamidine-2′-phenylindole dihydrochloride; lps, lipopolysaccharide; MCM, macrophage-conditioned media; SEM, standard error of the mean. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.